 Mina Takahashi
Mina Takahashi Richard C. Shelton
Richard C. Shelton- 1Department of Psychiatry, Heersink School of Medicine, University of Alabama at Birmingham, Huntsville, AL, United States
- 2Department of Psychiatry and Behavioral Neurobiology, Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
Major depressive disorder (MDD) is a complex and debilitating condition with high global prevalence. While pharmacological treatments are available, the long-term biological underpinnings – especially those linked to adverse childhood experiences (ACEs), remain incompletely understood. ACEs, including physical, sexual, and emotional abuse, neglect, and other traumas, significantly increase lifelong vulnerability to depression and reduce responsive to treatment. Epigenetic mechanisms such as DNA methylation and altered expression of mRNA and short and long non-coding RNAs such as microRNAs (miRNAs) are emerging as key mediators of the relationship between early environmental adversity and brain development and function. Specific miRNAs (e.g., miR-124, miR-135) influence neuroinflammation, and affect synaptic plasticity and monoaminergic signaling. Concurrently, DNA methylation in promoter regions can silence genes critical for stress regulation. For example, hypermethylation of the NR3C1 gene (encoding the glucocorticoid receptor) has been linked with altered HPA axis feedback and cortisol imbalance following ACEs. These epigenetic changes, together with trauma-induced microglial activation and neuroinflammation, may create lasting neural vulnerability. This paper explores how the interplay between childhood trauma, hormonal dysregulation, microglial activation, and epigenetic modification contributes to the pathophysiology of depression. Synthesizing evidence across epigenetic networks and neurobiological systems can deepen an understanding of trauma-related mood disorders. This may inform targeted interventions, identify biomarkers for diagnosis and treatment, and support personalized approaches to care and suicide prevention.
Introduction
Adverse childhood experiences (ACEs) such as abuse, neglect, loss, and other traumas, represent significant risk factors for subsequent mental illness and suicide. There is now strong evidence that childhood adversity, especially those that are experienced very early in life, induce longstanding physiological changes that underlie this risk. The purpose of this article is to review known physiological adaptations to adversity that are associated with these negative outcomes.
Methods
This article is intended as an narrative review, providing and overview and summary of existing relevant literature. We searched the MEDLINE database (PubMed) for articles published before the original submission date of July 7, 2025. Searches included the following terms: adverse childhood experiences, childhood trauma, abuse (including physical, sexual and emotional), neglect AND the following: adrenocorticotrophic hormone (ACTH); α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor; amygdala; apoptosis; corticotropin releasing hormone (CRH), CRH receptor 1 (CRHR1); cortisol (and corticosterone); dendritic spine; depression/major depression (MDD); DNA methylation; epidemiology; epigenetics; excitotoxicity; FK506 binding protein 5 (FKBP5); functional magnetic resonance imaging (fMRI); glucorticoid; glutamate; glycogen synthase kinse 3 (GSK3); hippocampus; hypothalamic-pituitary-adrenal (HPAA) axis; hypothalamus; hypothalamus; inflammation (neuroinflammation); interleukin; interneuron; ketamine; locus coeruleus; micro RNA (miRNA); microglia; n-methyl-D-aspartic acid (NMDA) receptor; neural circuit; neuroplasticity; neurotrophin (including brain derived neurotrophic factor [BDNF]); post-traumatic stress disorder (PTSD); postmortem brain study; pruning; pyramidal cell; synapse; triggering receptor expressed on myeloid cells 2 (TREM2); tropomyosin-related kinase B (TrkB) receptors; and ventromedial prefrontal cortex (vmPFC). We also examined reference lists of relevant review papers, noted in the text. Appropriate papers were then selected by the authors for inclusion.
Adverse childhood experiences: long-term risk factors for depression
ACEs refer to a range of serious challenges a child may face during critical developmental periods, including neglect; physical, sexual, or emotional abuse; poverty; discrimination; exposure to violence; and various forms of household dysfunction, such as living with family members who have substance use or mental health disorders, incarceration, or exposure to intimate partner violence (1–3). These adverse experiences can significantly shape brain and behavioral development, with consequences that extend well into adulthood (4, 5). Approximately one in five adolescents in the United States have experienced ACEs at some point during childhood (6, 7). The Centers for Disease Control and Prevention (CDC) defines ACEs as potentially traumatic events occurring before the age of 18 (8). Approximately two-thirds of U.S. adults report having experienced at least one ACE, and one in six reports experiencing four or more. Adults who experienced four or more ACEs were 4.6 times more likely to report depression in the past year, compared to those with no ACEs and even experiencing just one ACE increased the risk of depression by 50% (8). Felitti et al. (9) identified a dose-response relationship: the more adverse experiences a person had in childhood, the more likely they were to suffer from serious health issues in adulthood, including cardiovascular disease (CVD) (10–13), diabetes (14), high cholesterol, a known contributor to CVD (15), cognitive and other neurological disorders (16), substance use disorders, and internalizing problems such as anxiety and depression (17, 18). Evidence also shows that preventing or addressing ACEs early could reduce the overall prevalence of depression in the population by up to 44.1% (18).
The impact of ACEs varies depending on factors such as the timing of exposure (e.g., early vs. later childhood), the nature of the adversity (e.g., abuse vs. neglect), and individual characteristics like sex (5, 19). Yet, trauma that occurs during sensitive periods of brain development tends to have more severe and enduring effects. Cumulative exposure to multiple adversities further compounds psychological and physiological burdens, leading to elevated risks for depression, suicidality, and other mental health conditions (20–22). ACEs can keep the brain’s stress-response system in a prolonged state of activation, which can lead to changes in gene expression (23). Rather than returning to baseline after a threat passes, children repeatedly exposed to trauma continue to secrete stress hormones, such as corticotropin-releasing hormone (CRH), leading to chronic hyperarousal (2, 24, 25) (Figure 1). This persistent stress disrupts the development of key brain regions involved in memory, attention, emotional regulation, and learning (2).
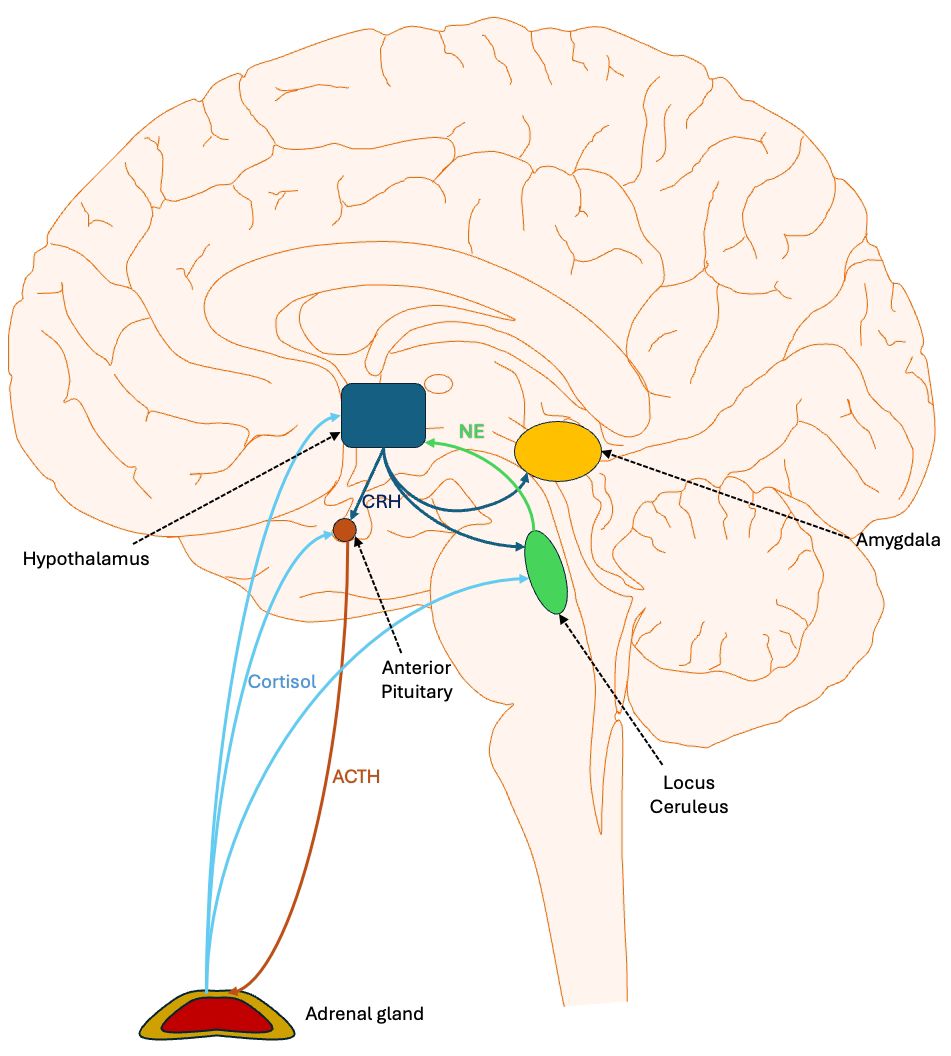
Figure 1. Central regulation of stress response: the hypothalamic pituitary adrenal axis and related structures. Stress activates the ventromedial nucleus of the hypothalamus to release corticotropin-releasing hormone (CRH). CRH travels through the hypophyseal portal system, a network of blood vessels connecting the hypothalamus and the anterior pituitary gland, to stimulate the release of adrenocorticotropic hormone (ACTH). This hormone then travels in the peripheral circulation to the adrenal cortex, where it triggers the release of cortisol. The latter binds to glucocorticoid receptors in various brain areas, thereby dampening the stress response. In particular, it inhibits the production of CRH and ACTH by neurons. CRH also projects to the locus coeruleus, which returns norepinephrine innervation to the hypothalamus, forming a positive feedback loop that is dampened by cortisol. CRH also innervates the amygdala, which mediates some of the emotional response to stress.
Chronic stress alters neurochemical pathways; for example, it increases dynorphin levels, which activate kappa opioid receptors (KORs), which inhibit dopamine release and contribute to depressive symptoms, particularly anhedonia (26–28). In addition to impairing brain development, prolonged early stress can trigger neuroinflammation, microglial activation, and epigenetic changes such as DNA methylation and altered microRNA (miRNA) expression (29). These biological mechanisms contribute not only to mental illness but also to substance abuse, high-risk behavior, chronic disease (9, 11, 18), and suicide.
In summary, chronic stress during early developmental stages leads to long-lasting disruptions in neurobiological systems that regulate emotion, cognition, and stress. These changes underlie an increased vulnerability to depression and other health outcomes through mechanisms involving hormonal imbalance, immune dysregulation, neuroinflammation, and structural brain alterations.
Childhood trauma and the escalating mental health crisis in the United States
According to the CDC (30), between August 2021 and August 2023, 13.1% of individuals aged 12 and older in the U.S. experienced depressive symptoms during any given two-week period. This means that approximately 1 in 8 people met criteria for depression within that timeframe. Rates were especially high among females, younger individuals, and those living below the poverty line; 22.1% of those in poverty reported depressive symptoms, suggesting a strong link between economic hardship and mental health.
Over the past decade, depression has risen sharply, increasing from 8.2% in 2013–2014 to 13.1% in 2021–2023, a nearly 60% increase. Among those affected, 87.9% reported that their symptoms interfered with work, home, or social functioning. Despite this growing burden, more than 60% of individuals with depression did not receive treatment in the past year. These trends highlight an urgent need to expand access to mental health services and preventive care (30).
A growing body of research has identified ACEs as significant risk factors for both depression and suicidal behavior (31–34). ACEs are alarmingly common, with roughly one-third of the North American population reporting some form of childhood maltreatment (35, 36). ACEs have been associated with the earlier onset, severity, and chronicity of multiple psychiatric disorders, as well as higher rates of comorbid conditions and lower treatment response (37–39). Additionally, ACEs increase the risk of suicide attempts in adulthood by two to three times (40). Importantly, these effects may be mediated not only through the development of mental illness but also through intermediate disruptions in emotional development, such as attachment insecurity (32, 41, 42). ACEs are therefore considered an early risk factor, meaning its influence extends long after the trauma itself, unlike more immediate triggers (43, 44). The suicide model proposed by Turecki and Brent (43) underscores this, identifying childhood trauma as a persistent individual-level risk factor for suicide through mechanisms such as heightened emotional dysregulation and stress sensitivity.
Merrick et al. (45) conducted a multivariate analysis demonstrating that specific ACEs, including sexual abuse, physical abuse, household substance abuse, household mental illness, and emotional neglect, were all associated with greater risk of adult depression, suicide attempts, substance use, and alcohol misuse. Notably, sexual abuse emerged as the most impactful predictor, being significantly linked to all four adverse adult mental health outcomes (45). ACEs are strongly associated with long-term disruptions in emotional regulation, cognitive functioning, and neurodevelopment, significantly increasing the risk of psychiatric and physical health disorders across the lifespan (46–49). These findings emphasize the cumulative and long-lasting emotional toll of childhood trauma and highlight the need for early identification and trauma-informed care.
Early-life adversity and dysregulation of the stress response system
Stress, particularly when experienced during early development, can induce lasting alterations in the amygdala, the brain region central to processing fear and emotional responses (50, 51). Stress activates the hypothalamic-pituitary-adrenal axis (HPA). The hypothalamus releases corticotropin releasing hormone (CRH), which travels to the pituitary that releases adrenocorticotrophic hormone (ACTH), which stimulates the adrenals to release cortisol (52, 53). The activation of the hypothalamus occurs primarily via norepinephrine (NE) originating in the locus ceruleus (LC) (54). While we commonly think of the actions of CRH being limited to the HPA, in fact, CRH receptors (e.g., CRHR1) are widely distributed across the brain, particularly in stress-responsive regions including the hypothalamus, hippocampus, amygdala, and locus ceruleus (55) (Figure 1). CRH projections originating in the hypothalamus innervate the LC and enhance NE release. This, then, forms a positive feedback loop with the hypothalamus (56). Early maternal separation, a model of early life stress in mice, permanently increases the density of CRHR1 in the amygdala, prefrontal cortex, and hippocampus (57). CRH makes the amygdala, particularly the central nucleus (CeA), more sensitive to emotional and threatening situations and it is involved in fear-related behavior, memory of aversive events, and fear conditioning (58).
Stressful experiences initially activate the limbic system, which signals the hypothalamus to release CRH and vasopressin (53, 59). Cortisol helps the body manage prolonged stress by regulating metabolism, immune function, and energy use (53). Simultaneously, the sympathetic nervous system is activated, which releases epinephrine and norepinephrine (NE) from the adrenal medulla (60). These hormones initiate the fight-or-flight response, increasing heart rate, alertness, and blood flow to muscles (60). This response also temporarily boosts brain function, particularly in the prefrontal cortex (PFC), which is involved in decision-making and emotion regulation (61, 62).
Even after the stressor resolves, the amygdala may continue to overreact to minor triggers, increasing the risk for anxiety, emotional dysregulation, and depression (23, 63, 2011). Animal studies confirm that early or chronic exposure to glucocorticoids leads to structural and functional changes in the amygdala that persist into adulthood (64–66). For example, research on children adopted after spending early childhood in orphanages found enlarged amygdala volumes, consistent with findings from animal models (67, 68). Interestingly, these studies did not report corresponding changes in the hippocampus (67, 68). Even after adoption into nurturing families, these children retained enlarged amygdala volumes (68, 69), suggesting that early adversity can produce enduring changes in amygdala structure that may not be fully reversed by later positive environments. Supporting this, animal studies have shown that stress-induced growth in the amygdala tends to persist, while other regions, such as the hippocampus, often exhibit recovery after the stress ends (68). One hypothesis is that this enduring amygdala enlargement reflects a biological adaptation to early danger, essentially keeping the brain on high alert in anticipation of future threats (68, 69). Over time, this heightened sensitivity can lower the threshold for fear responses, particularly when stress is frequent or occurs during critical developmental periods. Even after the stressor is removed, the amygdala may remain hyperreactive to minor triggers, increasing vulnerability to anxiety, emotional dysregulation, and other mental health conditions (68, 70). This persistent hyperreactivity may be partly explained by stress-induced impairments in GABAergic inhibition within the amygdala, especially a reduction in tonic inhibition, which usually helps suppress excessive neuronal firing and maintain emotional balance (71).
The amygdala is also part of a broader neural network that includes the ventromedial prefrontal cortex (vmPFC) and the hippocampus, regions that collaborate to regulate emotional responses and learning (72–75). The vmPFC exerts inhibitory control over the amygdala, while the hippocampus and amygdala co-regulate each other during emotionally salient experiences, allowing contextual memory to shape emotional responses (70, 76–78). However, stress – especially chronic or experienced early in life – can disrupt this finely tuned system (79). In adults, stress weakens the vmPFC’s capacity to regulate fear, thereby impairing processes such as fear extinction learning. This may occur through mechanisms such as decreased levels of brain-derived neurotrophic factor (BDNF), resulting in reduced synaptic plasticity and structural remodeling of neurons (80, 81). As a result, the amygdala may remain hyper-responsive to perceived threats, even in the absence of actual danger (82). Moreover, early life adversity appears to alter the structural and functional development of the vmPFC, suggesting that stress during sensitive developmental windows can lead to long-term disruptions in the connectivity and balance within the emotion-regulation network (83).
Dopamine, originating predominantly in the ventral tegmental area (VTA), is an important component of reward-related behavior, particularly incentive motivation, which is the drive to pursue reward, by altering the perceived valuation of a reward cue (84). Both acute and persistent stressors affect dopamine dynamics, but in opposing manners. An acute stress increases dopamine release, which helps to motivate the fight-or-flight response; that is, exerting energy to reduce or eliminate the stressor. By contrast, chronic stress dampens dopamine release by at least two mechanisms. Persisting stressors increase the release of striatal dynorphin, which activates presynaptic kappa receptors on striatal dopamine neurons (85). Stress also stimulates the amygdala to activate CRH projections to both the VTA and striatum, dampening dopamine release (86). ACEs are associated with altered striatal dopamine regulation, including an increase in striatal dopamine release (87), a blunted reward response to dopamine signals, and a blunted connectivity between VTA and striatum (88). This may underlie trait anhedonia, a key risk factor for depression (89).
ACEs similarly impact central serotonin (5-HT) function. For example, mice pups exposed to early life stressors exhibited an increase in anxiety-related behaviors and reduced functional connectivity of raphe 5-HT neurons (90). Prior trauma exposure, including early life trauma, is associated with altered 5-HT receptor expression, particularly 5-HT1A receptors, which may alter subsequent risk of mood and trauma-related disorders (91). Alterations in 5-HT dynamics related to ACEs may contribute to heightened risk for mood, anxiety, and trauma disorders.
Structural and functional brain changes following early-life and chronic stress: insights from human and animal studies
In parallel with amygdala alterations, early life adversity also affects other key brain regions involved in mood regulation, particularly the prefrontal cortex (PFC) and hippocampus (92, 93). Both individuals with major depressive disorder (MDD) and animal models of early or chronic stress exhibit reduced volume and neuronal atrophy in these areas (94–96). Functional imaging often shows decreased connectivity between the PFC, hippocampus, and associated regions (97), although increased connectivity in some areas suggests a more complex pattern of dysregulation. For example, adults with PTSD related to childhood maltreatment show heightened amygdala and hippocampal activation during emotional tasks, reflecting increased engagement of affective networks (98). Resting-state studies indicate that childhood abuse correlates with increased connectivity within the salience network (SN) and enhanced coupling between SN and the default mode network (DMN) (99). Additionally adverse childhood experiences are associated with stronger functional connectivity within the medial prefrontal cortex (mPFC) and between mPFC and other frontal regions, which may relate to repetitive self-referential processing, such as rumination (100). On the other hand, in animal models, in the Unpredictable Postnatal Stress (UPS) model, mice show reduced PFC volume and disrupted connectivity in the corpus callosum – findings that mirror structural abnormalities seen in trauma-exposed humans, including reduced corpus callosum volume and fractional anisotropy (FA) (19, 96).
Chronic stress induces dendritic atrophy and reduces spine density primarily in the apical dendrites of pyramidal neurons in layers II/III and V of the medial prefrontal cortex (mPFC), a brain region essential for decision-making, emotional regulation, and executive function (101–106). This structural remodeling impairs synaptic signaling by weakening excitatory postsynaptic potentials, partly due to the disruption of neurotransmitter systems, including serotonin (5-HT) and hypocretin (orexin), which are critical for maintaining synaptic strength (97, 107). The resulting reduction in synaptic connectivity and neurotransmitter sensitivity may contribute to the decreased size of the PFC and hippocampus observed in chronic stress. It may underlie the limited efficacy of selective serotonin reuptake inhibitors (SSRIs) in some patients, particularly those with a history of ACEs (97). Even short-term stress has measurable effects; just one week of restraint stress (20–30 minutes per day) can shrink pyramidal neurons in the PFC (61). Such rapid changes demonstrate how even brief stress exposures can compromise PFC integrity.
Postmortem studies provide further evidence of structural damage. Neurons often show signs of atrophy rather than a reduction in number in the dorsolateral PFC (dlPFC) and hippocampus of individuals with MDD (97, 108). Pyramidal neurons exhibit shrinkage, and GABAergic interneurons, which regulate excitatory signaling, are often reduced in number (97, 109). These findings point to a pattern of synaptic loss, neurotransmitter imbalance, and glial cell reduction underlying MDD. In humans, prolonged exposure to chronic stressors, whether social, environmental, or occupational, may lead to comparable atrophy, contributing to the impaired emotion regulation and decision-making commonly observed in depression (97).
Nonetheless, discrepancies between species and methodological variability highlight the need for cautious interpretation and more refined cross-species approaches. Longitudinal human studies, combined with advances in neuroimaging and molecular techniques, are essential for identifying sensitive developmental windows and potential mechanisms of resilience (110). By deepening our understanding of how stress shapes the brain, we can more effectively develop targeted interventions to prevent or mitigate psychiatric disorders stemming from early adversity.
Chronic stress, synapse loss, and depression
Brain-derived neurotrophic factor (BDNF) is a crucial extracellular signaling protein belonging to the neurotrophin family, which supports neuronal development, survival, and plasticity (111). It is the most abundant neurotrophin in the central nervous system, highly expressed in key brain regions involved in mood regulation such as the hippocampus, amygdala, and prefrontal cortex (112, 113). BDNF primarily acts by binding to and activating the tropomyosin-related kinase B (TrkB) receptors, triggering pathways that promote neuronal growth, differentiation, and synaptic plasticity, promoting stress resilience (114, 115). These mechanisms are essential for neuronal survival, synaptic plasticity, maintenance, and growth. Under conditions of chronic stress, BDNF expression declines markedly in the PFC and hippocampus, leading to synaptic atrophy and behavioral symptoms of depression (116, 117). The effects of BDNF are region-specific: while reduced levels in the cortex and hippocampus are detrimental, increased BDNF in the mesolimbic dopamine system may worsen mood-related symptoms (116). The resulting synaptic impairment involves multiple interrelated mechanisms. One is the suppression of BDNF–TrkB signaling; under normal conditions, BDNF binds to TrkB receptors to activate pathways such as ERK and Akt, activating the mammalian target of rapamycin complex 1 (mTORC1), which drives synaptic protein synthesis, promoting dendritic spine and synapse formation, and maintaining synaptic structures (118). Chronic stress disrupts this signaling, compromising synaptic integrity (119, 120). Stress also dysregulates glutamate transmission by accelerating the degradation of glutamate receptors in the PFC, impairing excitatory neurotransmission and cognitive function (121). In addition, stress increases transcriptional repressors that suppress the expression of genes involved in synaptic maintenance and plasticity, further contributing to the loss of synapses (122). The molecular disruptions do not act in isolation. Instead, they are part of a broader cascade involving neuroendocrine imbalances and abnormalities in excitatory signaling. Chronic stress exerts profound effects on glutamatergic neurotransmission and adrenal steroid release, two closely linked systems that jointly shape synaptic plasticity and neuronal health.
Glutamate, glucocorticoids, and the neurochemical cascade of chronic stress
In the early stage of acute stress, glutamate release in the prefrontal cortex (PFC) enhances focus, vigilance, and executive functioning (123). However, with prolonged or repeated exposure to stress, this same excitatory signaling becomes maladaptive (123, 124). Persistently elevated glutamate levels disrupt neural homeostasis, weaken synaptic integrity, and impair cognitive function (123, 124). Over time, sustained glutamate elevation becomes neurotoxic, resulting in dendritic atrophy, synaptic loss, and cognitive decline, hallmarks of depression-related neuropathology (97, 124).
Concurrently, the HPA axis becomes dysregulated, failing to shut off in over half of individuals with depression. This results in persistent high levels of stress hormones that contribute to further brain injury and depressive symptoms (97). Exposure to chronic or repeated stress leads to the release of adrenal steroids that affect the shape and structure of neurons, particularly in the hippocampus, a region essential for memory and emotion (97). Notably, studies show that blocking the n-methyl-D-aspartic acid (NMDA) receptors or stopping adrenal steroid production can prevent stress-related brain changes (125).
Normal neurotransmission also becomes disrupted as extracellular glutamate levels rise under chronic stress (126). To counteract potential excitotoxicity, Astrocytes play a major role in removing excess glutamate from the extracellular space through glutamate transporters that re-uptake the neurotransmitter, while glutamate receptors on cell surfaces detect and respond to extracellular glutamate (127). However, when glutamate accumulates outside the synapse, it activates extrasynaptic NMDA receptors, which suppress CREB signaling and initiate neurotoxic cascades, in contrast to synaptic NMDA receptors that promote survival and plasticity (128, 129), This receptor-location-dependent signaling shift contributes to dendritic atrophy and functional decline observed in stress-related disorders such as depression.
The CA3 region of the hippocampus offers particularly compelling evidence of how chronic stress alters glutamate transmission and neuronal structure (130). In the CA3 region of the hippocampus, glutamate release by mossy fibers – a key excitatory pathway – plays a pivotal role in stress-induced neuronal remodeling (131). This process is tightly regulated by adrenal steroids, such as cortisol, which modulate glutamate release (130). After 21 days of chronic restraint stress (CRS), animal studies show several markers of enhanced glutamatergic activity: (1) depletion of clear synaptic vesicles in mossy fiber terminals, suggesting excessive neurotransmitter release; (2) increased expression of presynaptic proteins involved in vesicle trafficking and release; and (3) greater mitochondrial density within terminals, indicating elevated energy demands to sustain neurotransmission (132). High levels of glucocorticoids synergize with excitatory amino acids, such as glutamate, to exacerbate excitotoxic damage —a process linked to impaired glucose uptake and reduced energy metabolism in neurons (133). This metabolic disruption limits the brain’s capacity to handle prolonged excitation, making neurons more vulnerable. Interestingly, this relationship follows an inverted U-shaped dose-response curve: at physiological levels, glucocorticoids translocate to mitochondria, where they reduce oxidative stress and support cellular resilience. However, during prolonged or excessive stress, this protective mechanism fails, leading to increased oxidative and excitotoxic damage (134). Mitochondria play a central role in both energy production and cellular stress responses, and their sensitivity to glucocorticoid signaling is crucial in determining whether the brain adapts successfully or deteriorates under chronic stress (135).
The glutamate transporter-1 (Glt-1), which clears excess glutamate from synapses, is upregulated in the hippocampus under stress (136). However, this regulation is biphasic: low to moderate glucocorticoid levels may enhance Glt-1 expression, but very high or prolonged exposure can suppress it (137). This highlights how timing and intensity of glucocorticoid exposure produce different neurological outcomes (137).
Interestingly, glucocorticoids can exert beneficial effects under certain conditions. For instance, they increase levels of cocaine amphetamine regulated transcript (CART) in the hippocampus during chronic stress – a change linked to reduced anxiety, possibly through modulation of hypothalamic stress circuits (138). This highlights the complex and context-dependent nature of glucocorticoid activity in the brain. In addition to synaptic regulation, glucocorticoids activate microglia, the brain’s resident immune cells, contributing to neuroinflammation in stress-sensitive regions such as the hippocampus, PFC, and amygdala (53).
Other molecules such as corticotropin-releasing factor (CRF), tissue plasminogen activator (tPA), and BDNF also play key roles in stress induced brain remodeling (139). CRF increases the expression of tPA in the amygdala, a change linked to anxiety-like behaviors and dendritic spine loss in both the amygdala and hippocampus. Notably, mice lacking tPA are protected from these stress-induced changes, suggesting that elevated tPA contributes to cognitive and synaptic impairments under chronic stress (140–143). Despite its harmful effects under excessive stress, tPA also plays a physiological role in neuronal plasticity. Specifically, it catalyzes the conversion of pro-BDNF to mature BDNF, which activates TrkB receptors and downstream pathways such as MAPK/Erk1/2 – promoting synaptic remodeling and modulating emotional behavior (144, 145).
GSK3: A central player in synaptic pruning and depression
While tPA promotes the maturation of BDNF, chronic stress often leads to impaired BDNF signaling despite this compensatory mechanism. One key downstream target affected by reduced BDNF-Akt signaling is glycogen synthase kinase 3 (GSK3), an intracellular enzyme critical for synaptic plasticity and neuronal function (146–148). GSK3 plays a central role in synaptic deconsolidation or pruning by regulating the cycling of glutamate receptors, particularly the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and NMDA receptors (149). Under normal physiological conditions, GSK3 is inhibited by Akt and activated by protein phosphatase 1 (PP1), maintaining synaptic stability (97). However, in chronic stress and depressive states, this inhibition is lost, leading to GSK3 overactivation, which contributes to dendritic spine loss in regions such as the prefrontal cortex and nucleus accumbens (150). Postmortem studies show elevated GSK3 activity in individuals with MDD and bipolar disorder, and animal models demonstrate that GSK3 inhibition in the nucleus accumbens produces antidepressant effects (150, 151). Lithium, a mood stabilizer widely used in bipolar disorder, is a potent inhibitor of GSK3 and has been shown to enhance the efficacy of some other antidepressants in MDD (152). These findings highlight GSK3’s role in depression-related neuroplastic changes and its promise as a therapeutic target. Interestingly, lithium has little if any antidepressant effects on its own, suggesting that just inhibiting GSK3 may not be sufficient for an antidepressant action. However, since antidepressants act via neurotrophic mechanisms, inhibition of the antineurotrophic effects of GSK3 may enhance antidepressant effects.
Beyond these synaptic and intracellular alterations, chronic stress also activates neuroimmune pathways. Microglia, the brain’s resident immune cells (153), play a pivotal role in mediating inflammation and structural remodeling, especially in stress-sensitive brain regions.
Microglia in development and disorder: sensing stress and pruning synapses
As the brain’s resident immune cells, microglia are essential for healthy development and function (154). They contribute to neurogenesis, apoptosis, axonal growth, myelination, and immune responses to injury and infection (19, 155–158). A key function is synaptic pruning – the elimination of unnecessary or damaged synapses – which supports synaptic plasticity, refines neural circuits during development, and helps maintain cognitive function throughout life (159, 160). In addition to these structural roles, microglia secrete signaling molecules such as BDNF, transforming growth factor-beta (TGF-β), and complement proteins that regulate synaptic plasticity and behavior by shaping neural circuits (53, 158). However, this same sensitivity to external stimuli – such as sensory deprivation or early life stress – can render them vulnerable to dysfunction (19, 155).
Disruptions in microglial function may contribute to long-term cognitive and mental health challenges in individuals exposed to childhood adversity (19, 155). Early life stress has been shown to impair microglial pruning, resulting in excess synapses and abnormal brain connectivity – changes that likely underlie persistent cognitive and behavioral difficulties (19, 96, 161–163).
Chronic stress can drive excessive microglial activation, shifting these cells from a supportive to a pro-inflammatory, neurotoxic state. This dysregulated activation is linked to synaptic loss, reduced dendritic spine density, and impairments in memory and emotional regulation (164). Notably, interventions that restore IL-10 levels (an anti-inflammatory interleukin) or deplete microglia using PLX5622 reverse these memory and synaptic deficits, highlighting the critical role of microglia in depression-related cognitive impairment (164).
Microglia also contribute to the cognitive and emotional symptoms observed in PTSD. Both human patients and animal models show increased brain inflammation, including elevated TNF-α and IL-1β levels in the hippocampus. In a PTSD mouse model, microglia proliferate and adopt an activated morphology characterized by enlarged cell bodies, simplified branching, and dendritic spine loss in the hippocampal CA1 region – changes that likely underlie memory deficits and emotional dysregulation (165, 166). Importantly, these same studies suggest that chronic or dysregulated activation disrupts this balance – underscoring microglia’s dual role as both protectors and potential drivers of neuropathology.
Mechanisms and functions of microglial synaptic pruning
Microglia contribute to synaptic plasticity by regulating key processes such as long-term potentiation (LTP) and long-term depression (LTD), which respectively strengthen or weaken synapses to encode learning and experience (167, 168). Through continuous monitoring of synaptic activity, microglia selectively preserve or eliminate synapses based on their functional relevance (158), thereby enabling dynamic synaptic remodeling that supports essential cognitive processes such as learning, memory, and behavioral adaptation (169, 170).
One major mechanism underlying microglia-mediated synaptic pruning is the classical complement cascade. In this pathway, proteins such as C1q, C3, and CR3 tag weak or redundant synapses for elimination, facilitating activity-dependent refinement of neural circuits (171–173). The C1q-dependent mechanism is especially important in memory-encoding engram neurons; its inhibition preserves synapses and enhances memory retrieval (158, 174). Blocking C1q signaling in the hippocampus also prevents synaptic degradation and reduces depressive-like behaviors in chronically stressed mice (175).
Complement component C3 is similarly pivotal in both developmental and stress-related pruning. Chronic stress increases C3 expression in the medial prefrontal cortex (mPFC), leading to excessive synaptic elimination by microglia. This disrupts connectivity between the mPFC and key mood regulating areas such as the anterior cingulate cortex, septum, and dorsal raphe nucleus, contributing to depression like behaviors (97). Notably, C3 knockout mice retain synaptic integrity and display resilience to stress-induced behavioral deficits – through complete C3 loss may also impair normal immune and synaptic functions (175–178). These findings highlight the pathological role of dysregulated complement signaling and suggest that targeted inhibition of C3 could offer therapeutic potential for stress-related mood disorders.
Beyond the classical complement cascade, other signaling pathways regulate microglial pruning. One such mechanism involves CD47 and SIRPα, which act as inhibitory signals that protect healthy synapses from inappropriate elimination by microglia (171–173). CD55, an inhibitor of this protective pathway, promotes pruning between engram cells in the dentate gyrus by allowing microglia to eliminate synapses that would otherwise be spared (158, 174). This leads to increased synaptic turnover and may influence memory retrieval by modifying engram reactivation (158, 174).
Microglial synaptic pruning and stress-induced dysregulation
TREM 2 – phosphatidylserine signaling and stress-induced impairment
Microglia eliminate dysfunctional synapses by recognizing phosphatidylserine (PS), a lipid exposed on compromised neuronal membranes, through the triggering receptor expressed on myeloid cells 2 (TREM2) (19, 158, 162, 179). The PS-TREM2 signaling pathway is crucial for synaptic refinement in brain regions such as the hippocampus and dorsal lateral geniculate nucleus, where pruning is essential for proper circuit maturation (158, 179). Notably, early-life stress disrupts this mechanism. Dayananda et al. (19) demonstrated that limited bedding (LB) and unpredictable postnatal stress (UPS) decrease surface TREM2 protein levels in microglia without affecting TREM2 mRNA expression, indicating post-transcriptional regulation. Consequently, impaired TREM2 signaling results in deficient synaptic pruning and an excess of dendritic spines on CA1 pyramidal neurons in the hippocampus, potentially leading to reduced neural efficiency and cognitive deficits later in life (19).
Stress, microglial immune activation and neuroinflammation
In addition to synaptic pruning, microglia serve as the brain’s primary immune sentinels (154). They phagocytose cellular debris, respond to injury, and express immune markers such as MHC-II, CD80, and CD86, facilitating communication with peripheral immune cells (180). However, when excessively activated, microglia can drive a proinflammatory state that impairs neural function, highlighting their dual role as both protectors and potential contributors to neurodegeneration (53).
Stress activates the sympathetic nervous system, releasing norepinephrine, which binds to β2- adrenergic receptors on microglia. This activation triggers the production of inflammatory cytokines such as interleukin-1β (IL-1β) (181) and CCL2, which further recruit peripheral immune cells into the brain, amplifying neuroinflammation (53, 182). This inflammatory cascade disrupts synaptic function and plasticity. Notably, pharmacological blocking β-adrenergic receptors has been shown to reduce IL-1β expression and microglial activation, suggesting a promising strategy for mitigating stress-induced neuroinflammation (53, 182).
Molecular regulation of microglial synaptic pruning
Microglial synaptic pruning and phagocytosis are partly regulated by the CX3CR1/CX3CL1 signaling pathway, in which the microglial receptor CX3CR1 binds its neuronal ligand CX3CL1 (fractalkine) to enable direct neuron–microglia communication (158, 183). This signaling axis plays a critical role in guiding microglia to eliminate synapses selectively during brain development and in response to environmental changes. Disruptions in this pathway impair synaptic refinement and may contribute to abnormal neural connectivity in both neurodevelopmental and stress-related disorders (158).
In addition to synaptic pruning, CX3CR1/CX3CL1 signaling shapes microglial interactions with oligodendrocyte progenitor cells (OPCs) (184). In CX3CR1 knockout mice, microglia retain contact with OPCs but exhibit reduced phagocytic activity, suggesting that CX3CR1 is crucial for engulfment rather than target detection alone (158, 185–187). Deleting either CX3CR1 or its ligand CX3CL1 also impairs synapse elimination following sensory deprivation, such as whisker cauterization in rodents (188), further underscoring the importance of this pathway in activity dependent neural remodeling (186, 189).
Early-life stress models, such as unpredictable postnatal stress (UPS) and limited bedding (LB), differentially influence microglial function through distinct molecular mechanisms. In the UPS model, microglia exhibit increased expression of pruning-related genes – including C1q, MERTK, and SIRPα – which correlates with enhanced synaptic pruning, increased microglial volume, and greater synaptic engulfment (19). In contrast, the LB model is associated with elevated TNF and reduced NGF, which may disrupt synaptic development and impair cognitive outcomes.
Although both models increase CX3CR1 expression, possibly as a compensatory mechanism, this response appears insufficient: both UPS and LB ultimately lead to increased spine density. Notably, the UPS model also reduces IL-1α and IL-1β levels, further impairing neurogenesis and synaptic signaling. These distinct molecular profiles may help explain the divergent effects of early-life stress on hippocampal development and function (19).
To prevent excessive or inappropriate pruning, microglial phagocytic activity is tightly regulated by inhibitory signals, including CD47/SIRPα and CD200/CD200R, which protect functional synapses during development (173, 190–192). CD47, a protein expressed on neurons, binds to SIRPα receptor on microglia, suppressing phagocytosis and protecting synapses from inappropriate elimination.
Similarly, the neuronal glycoprotein CD200 binds to the CD200R receptor on microglia to suppress their phagocytic activity (192, 193). In CD200 knockout mice, microglia exhibit increased synaptic engulfment, particularly in response to amyloid beta, highlighting the loss of inhibitory control in the absence of this signaling pathway (192, 193).
Another important regulator is high mobility group box 1 (HMGB1), a nuclear protein released during stress or injury. Acting as a damage-associated molecular pattern (DAMP), HMGB1 triggers inflammation and microglial activation, making it a potential therapeutic target in neuroinflammatory and stress-related conditions (194, 195).
MicroRNA as potential regulators of long-lasting neuroinflammatory and structural brain changes
MicroRNAs (miRNAs) are small, single-stranded RNA molecules that regulate gene expression post-transcriptionally by binding to messenger RNAs (mRNAs), thereby blocking their translation or promoting their degradation (196). Importantly, miRNA activity is also modulated by other non-coding RNAs, particularly long non-coding RNAs (lncRNAs), which can sequester miRNAs and prevent them from interacting with target mRNAs – thus indirectly preserving gene expression (197–199). Another class of small RNAs, small interfering RNAs (siRNAs), similarly mediate gene silencing through RNA interference (RNAi), although they differ from miRNAs in terms of biogenesis and target specificity (196, 200, 201).
miRNAs play essential roles in the central nervous system, contributing to neurogenesis, axonal guidance, synaptic development, and neuronal plasticity. Through these functions, they shape neuronal growth, connectivity, and long-term adaptability (198, 202–205).
To carry out these regulatory roles, miRNAs undergo a multistep biogenesis process. They are transcribed from miRNA genes in the nucleus by RNA polymerase II into primary transcripts (pri-miRNAs), which form characteristic hairpin loop structures (196). These pri-miRNAs are cleaved by the DROSHA-DGCR8 complex into precursor miRNAs (pre-miRNAs), which are then exported to the cytoplasm via Exportin-5 (206, 207).
Once in the cytoplasm, the pre-miRNA is further processed by the enzyme Dicer and its cofactor TRBP into a short RNA duplex. In some instances, Argonaute 2 (AGO2) may perform additional cleavage (206, 208, 209). The mature strand of this duplex is then incorporated into the RNA-induced silencing complex (RISC), while the passenger strand is degraded. Guided by the mature miRNA, the RISC complex binds to complementary target messenger RNAs (mRNAs), leading to translational repression and mRNA degradation (206, 208, 209).
MicroRNA regulation of synaptic plasticity in major depressive disorder
MicroRNAs (miRNAs) are critical post-transcriptional regulators of synaptic plasticity. Disruption of key processing enzymes such as Dicer and DGCR8 impairs synaptic remodeling in mouse models, leading to structural abnormalities – such as reduced dendritic branching and elongated dendritic spines – accompanied by cognitive and behavioral deficits (198, 210).
Specific miRNAs, including miR-124, miR-132, miR-134, and miR-138, fine-tune the expression of plasticity-related genes such as ARC, CaMKIIα, LimK1, FMRP, CREB, and BDNF (198, 211). For instance, miR-134 negatively regulates LimK1, disrupting dendritic spine structure and weakening excitatory synaptic transmission (198, 212). Elevated miR-134 expression – observed in the hippocampal CA1 region of Sirt1-deficient mice – reduces CREB and BDNF levels, highlighting the role of miRNAs in neurotrophin-mediated synaptic regulation (198, 213).
Other miRNAs such as miR-9, miR-125a/b, and miR-188 epigenetically influence genes including REST, FXR1P, CAMKK2-AMPK, PSD-95, BCL-W, SYN-2, NRP-2, 2-AG, and BACE1, demonstrating the broad regulatory role of miRNAs in maintaining neuronal plasticity and cognitive function (198, 212).
More than 25 studies have reported altered miRNA expression in key brain areas affected by MDD, including the anterior cingulate cortex and Brodmann Areas (BA) 9, 10, 44, and 46, as well as the locus coeruleus (198). For example, Smalheiser et al. (214) identified 21 miRNAs significantly downregulated in the prefrontal cortex of individuals with MDD who died by suicide, many of which target genes critical for synaptic plasticity.
Extending these postmortem findings, later studies reported significant reductions in synaptosomal miRNAs – especially miR-508-3p and miR-152-3p – with miR-508-3p levels particularly diminished in suicide cases (198). These results support the hypothesis that suicidal depression is associated with widespread miRNA downregulation in brain circuits governing mood and cognition (214, 215).
Similarly, Lopez et al. (216) reported decreased expression of miR-1202 in Brodmann are (BA) 44 of individuals with MDD. Additional evidence implicates miR-124-3p, which was significantly reduced in BA 46 of MDD patients (217). Interestingly, fluoxetine treatment further reduced miR-124-3p expression, suggesting a potential role in antidepressant mechanisms (198).
In a study of the locus coeruleus in MDD patients who died by suicide, Roy et al. (217) identified 10 upregulated and 3 downregulated miRNAs. Predicted targets of these upregulated miRNAs included RELN, GSK-3β, MAOA, CHRM1, PLCB1, and GRIK1 – genes previously implicated in psychiatric disorders (198). Notably, several of these targets, particularly RELN, GSK-3β, and MAOA, showed reduced expression, supporting their potential involvement in the molecular mechanisms underlying depression and suicide (198).
Using microarray analysis, researchers identified several genes whose expression was inversely correlated with miR-1202, including metabotropic glutamate receptor 4 (GRM4), a gene implicated in antidepressant response (216). These results suggest that miR- 1202 may serve as a predictive biomarker for treatment outcomes in MDD.
Azevedo et al. (218) further investigated miRNA expression and found that miR-34a and miR-184 were significantly downregulated in individuals with MDD. They focused on miR-34a, which regulates genes such as NCOA1, NCOR2, and PDE4B – key players in glucocorticoid receptor signaling and synaptic cAMP regulation. In vitro studies confirmed that miR-34a overexpression reduced NCOA1 and PDE4B levels, supporting its role in disrupting stress hormone signaling and synaptic function, thereby contributing to the pathophysiology of MDD (198).
miRNAs as biomarkers in depression, treatment response, and suicide risk
Many miRNAs are closely involved in regulating synaptic plasticity, neuroinflammation, and glucocorticoid signaling, all of which are disrupted in depression (219). Because of their specificity and regulatory power, these small non-coding RNAs are gaining attention not only as mechanistic contributors but also as potential biomarkers of psychiatric illness, particularly, MDD, bipolar, and schizophrenia (SZ) (220). Among the most studied are miR-124, miR-125, miR-29, miR-16, and miR-200, which are consistently associated with stress-related psychiatric conditions (206).
A notable study by Cattane et al. (221) found elevated levels of miR-125b-1-3p in both healthy individuals with a history of early-life stress and in patients with SZ. These results, validated in animal models and in vitro, suggest that early changes in miR-125b may lead to lasting disruptions in neuronal signal transduction via NMDA receptor subunit targeting – contributing to altered brain plasticity and psychiatric vulnerability (198, 222).
More recently, miRNA profiling has been used to differentiate between psychiatric conditions and predict treatment response. For example, Maffioletti et al. (223) analyzed blood samples from patients with MDD, bipolar disorder (BD), and healthy controls, identifying distinct expression patterns across groups. miR-24-3p, miR-425-3p, and members of the let-7 family were significantly altered in MDD, while miR-30e, miR-21– 3p, and miR-140–3p were associated with BD. KEGG pathway analysis identified Wnt and mTOR signaling as key biological targets of these dysregulated miRNAs.
Building on this, Lopez et al. (216) examined miRNA expression following duloxetine treatment using next-generation sequencing. They found that miRNAs involved in Wnt and MAPK signaling – including miR-146b-5p, miR-24-3p, and miR-425-3p – were downregulated post treatment but elevated in postmortem brains of suicide victims, suggesting these miRNAs may be linked to treatment resistance or elevated suicide risk.
miRNAs in treatment response and peripheral biomarker potential
Further evidence from Roy et al. (224) and Lopez et al. (216) supports the role of several miRNAs–miR-146b-5p, miR-24-3p, and miR-425-3p-as potential treatment-response markers. These miRNAs decrease with escitalopram treatment but are elevated in postmortem brain samples of individuals who died by suicide. Functionally, they are linked to Wnt and MAPK pathways, which regulate synaptic plasticity, stress response, and mood. Consistent with this, rats with depression-like behavior showed reduced expression of Wnt pathway genes targeted by miR-128-3p, further emphasizing the significance of miRNA-pathway interactions in depression biology (224).
Importantly, some miRNA changes in the central nervous system are mirrored in peripheral blood, especially through exosomes (174). For example, miR-330-3p is upregulated in both the locus coeruleus and blood samples of MDD patients (198). Similarly, miR-124-3p and miR-19a-3p display comparable expression in brain regions and blood (217, 225) suggesting their potential as accessible biomarkers that reflect CNS pathology.
miR-124, one of the most abundant brain microRNAs, arises from three distinct genes encoding the same mature sequence and is essential for neurogenesis, neuronal differentiation, and synaptic plasticity (226–228).
Evidence suggests a context-dependent function in stress-related behaviors. In the hippocampus, miR-124 enhances resilience to chronic stress through posttranscriptional regulation of HDAC4/5 and GSK3β expression (229). In contrast, other studies show that miR-124a overexpression promotes depression-like behaviors, whereas its inhibition yields antidepressant-like effects, potentially via CREB1 and BDNF upregulation (230, 231).
Likewise, miR-19a-3p has been implicated in neuroinflammation through its interaction with tumor necrosis factor-alpha (TNF- α) (225, 232). In the dorsolateral prefrontal cortex (dlPFC) of individuals who died by suicide, both TNF-α and miR-19a-3p were elevated. Notably, similar increases were observed in the blood of depressed individuals with suicidal ideation, suggesting that miR-19a-3p may reflect central immune activation in peripheral samples (232).
Although miRNAs usually suppress gene expression, Wang et al. (225) showed a nonlinear interaction: the RNA-binding protein HuR binds to the 3′ untranslated region (3′UTR) of TNF-α mRNA, shielding it from miR-19a-3p-mediated repression. This may explain persistently high TNF-α levels despite elevated miR-19a-3p, emphasizing the context-dependent complexity of miRNA regulation.
Despite these regulatory intricacies, circulating miRNAs also hold promise as accessible biomarkers. A serum-based case-control study demonstrated that a panel of miR-16, miR-135a, and miR-1202 could distinguish individuals with MDD from healthy controls (233). The combined panel outperformed individual miRNAs in diagnostic accuracy.
This potential is reinforced by the role of extracellular vesicles (EVs) – membranous carriers of nucleic acids and proteins involved in intercellular communication (234). Because EVs can cross the blood brain barrier and reflect CNS pathology, they represent a promising platform for non-invasive diagnosis of neuropsychiatric conditions (235, 236).
However, a significant challenge lies in ensuring EV purity. EVs are inherently heterogeneous, and contamination or inconsistent isolation can compromise the specificity and reproducibility of miRNA profiling (237). To realize the full diagnostic utility of EV-based miRNA biomarkers, rigorous standardization in EV isolation and characterization is imperative.
Despite over a decade of research, the clinical application of miRNAs as diagnostic or prognostic tools remains in its early stages (198, 238). Genetic markers such as the 5-HT2A receptor and SKA2 gene have also been explored in relation to suicidal behavior (239, 240). While numerous biomarkers have been investigated, only a few – such as prolactin and thyroid hormone levels – have demonstrated consistent reliability in predicting behavioral outcomes like suicide attempts (241).
DNA methylation and suicide risk in MDD
Building on the critical role of miRNAs as biomarkers for diagnosis and treatment, DNA methylation has also emerged as a key epigenetic mechanism implicated in the pathophysiology of trauma-related MDD and suicidal behavior (242, 243). Like miRNAs, DNA methylation modulates gene expression without altering the genetic code and may offer novel biomarkers for early detection and personalized treatment (244, 245).
DNA methylation refers to the addition of methyl groups to cytosine bases, particularly at CpG sites, leading to transcriptional repression when occurring near promoter regions (246). Abnormal methylation patterns have been increasingly associated with psychiatric conditions, especially MDD and suicidality (244, 247).
Early-life stressors, such as childhood abuse or neglect, can induce long-lasting epigenetic modifications, leaving molecular “scars” that increase lifelong vulnerability to depression and suicidal tendencies (64, 248). These epigenetic changes are particularly evident in brain regions involved in emotion regulation and stress processing, including the prefrontal cortex, hippocampus, and amygdala (249).
Specific methylation changes have been found in genes critical to neurotransmission and neuroplasticity, including SLC6A4 (serotonin transporter), BDNF (brain-derived neurotrophic factor), and GABA A receptor subunits (245, 249, 250). For example, hypermethylation of the SLC6A4 promoter is linked to reduced serotonin reuptake, while decreased BDNF expression through promoter hypermethylation is associated with impaired synaptic function (249). Hypermethylation of TrkB1 and BDNF in suicide victims’ prefrontal cortices further supports this association (249). In addition, genome-wide studies have identified broader methylation alterations across hundreds of promoters involved in neurodevelopment, immune function, and synaptic signaling (251, 252). These epigenetic modifications predominantly localize near gene promoters and regulatory elements, including transcription start sites (TSS), 5′ untranslated regions (5′UTR), and 3′UTRs.
DNA methylation was first recognized in CpG-rich regions known as CpG islands – segments of DNA approximately 1,000 base pairs in length with a high frequency of cytosine-guanine dinucleotides (253–255). Roughly 70% of gene promoters are located within these islands, making them critical regions for transcriptional regulation. Because promoter methylation typically leads to transcriptional repression, CpG islands in promoters of actively transcribed genes are usually unmethylated to allow gene expression (253). Elevated methylation in these regions has been consistently observed in individuals with MDD and heightened suicide risk, implicating the silencing of genes critical for stress adaptation and emotional regulation (242, 245).
To further investigate these mechanisms, researchers performed protein–protein interaction (PPI) analyses on the top 100 differentially methylated CpG sites, identifying CDH5, ACTN1, and GNA12 as hub genes in individuals with MDD, regardless of suicide history (245, 256, 257). These hub genes regulate key downstream targets including RAPTOR, ADAMTS17, and HSP90AA1. Specifically, RAPTOR is involved in excitatory neuronal activity and synaptic remodeling; ADAMTS17 contributes to cognitive function through synaptic plasticity; and HSP90AA1 modulates the HPA axis (178, 258–261). Notably, both RAPTOR and ADAMTS17 are integral components of the mTOR signaling pathway, which interacts with AMPA receptors to support stress resilience (262, 263). Epigenetic silencing of these genes may impair neuroplasticity and reduce the ability to adapt to chronic stress, thereby increasing suicide risk among trauma-exposed individuals (264, 265).
Chromosome-specific methylation patterns have also emerged as significant in MDD and suicide. Alterations in methylation are particularly prominent on chromosomes 2, 3, and 4 (245). Chromosome 2 is primarily associated with increased gene silencing, whereas chromosome 3 shows more gene activation, offering insights into chromosome-level vulnerability to environmental and epigenetic influences (245, 266, 267). Some studies have reported reduced or unchanged methylation levels, likely reflecting differences in CpG site selection, analytic methods, and sample characteristics such as ancestry and tissue type (268).
Trauma-associated methylation of glucocorticoid signaling genes
In addition to genes involved in synaptic plasticity, trauma-related methylation patterns frequently affect key components of the stress response system, particularly NR3C1 and FKBP5, which modulate glucocorticoid signaling (269, 270). Increased methylation of NR3C1, which encodes the glucocorticoid receptor, has been consistently observed in children exposed to early-life adversity, suggesting disrupted stress feedback mechanisms and heightened emotional vulnerability (269, 271). In adults, findings have been more variable: some studies have reported increased NR3C1 methylation following early-life adversity, while others have found hypomethylation associated with suicide or post-traumatic stress disorder (PTSD) (251, 272, 273).
IL-6, a cytokine induced by stress, is another molecule closely linked to the epigenetic mechanisms underlying depression. IL-6 promoter methylation in DNA extracted from buccal swabs was found to be lower in older adults (≥65 years) with major depressive disorder (MDD) compared to healthy controls (274). Notably, antidepressant use was independently associated with higher IL-6 methylation, supporting the potential role of IL-6 as a biomarker for both MDD and antidepressant response.
FK506 binding protein 5 (FKBP5) is a chaperone protein that protects the glucocorticoid receptor in the cytosol and modulates its sensitivity (275). Klengel et al. (276) were the first to report that childhood maltreatment is linked to decreased FKBP5 methylation in individuals carrying the T risk allele. This gene–environment interaction was replicated in a follow-up study (277). However, other research has found no significant effect of maltreatment or genotype, and one study in young adults observed that childhood stressful events were associated with increased FKBP5 methylation, which in turn mediated alterations in prefrontal brain activity (278). These mixed findings highlight the complexity of trauma-related epigenetic responses and reinforce the potential of NR3C1 and FKBP5 methylation as biomarkers of sustained stress exposure and elevated suicide risk.
Beyond specifying which genes or gene regions are hyper- or hypomethylated, several important considerations must be addressed. First, prenatal exposures can exert epigenetic effects on stress-related biological systems, potentially confounding associations observed in childhood (279). For example, intimate partner violence during pregnancy has been linked to increased NR3C1 methylation in offspring during late childhood and adolescence (280). Similarly, maternal smoking and depression during pregnancy have been associated with altered methylation of placental stress-regulatory genes, including NR3C1 and HSD11B2, the latter encoding an enzyme that inactivates cortisol (281–284). These findings suggest that associations between childhood maltreatment and DNA methylation may, in part, reflect prenatal environmental influences.
Second, sex differences may moderate the effects of trauma on DNA methylation. Future research should carefully consider the role of the sex of a child to avoid overlooking critical gene-by-sex interaction effects (268).
Finally, variation in tissue types used for methylation analysis also warrants attention. While blood, saliva, and buccal cells have all been utilized, most pediatric studies rely on saliva or buccal samples, with fewer examining methylation patterns in blood (285). This methodological heterogeneity may affect the consistency and comparability of results across studies.
Paoli et al. (286) reviewed and analyzed published studies investigating DNA methylation in depression, emphasizing its potential for future biomarker development and pharmacological intervention. Their meta-analysis identified four genes – BDNF, SLC6A4, FKBP5, and NR3C1 – with the highest consistency of methylation changes. Specifically, most studies reported increased methylation at BDNF, SLC6A4, and NR3C1, while FKBP5 typically showed decreased methylation associated with depression. DNA methylation thus represents a promising avenue for future research and clinical application. However, like miRNAs, its diagnostic and predictive utility remains under investigation. Continued research is essential, but current findings provide considerable hope for advancing precision medicine in psychiatry and stress-related disorders.
Synaptic dysregulation in depression and suicide: the role of stress, epigenetics, and rapid-acting antidepressants
According to recent CDC data (287), depression affects 19.2% of adolescents aged 12 to 19, compared to just 8.7% of adults over 60. Certain groups, like adults with disabilities, are disproportionately affected – 28.2% take medication for depression, nearly three times higher than those without disabilities. In 2023, over 49,000 Americans died by suicide – one every 11 minutes (288). These alarming statistics make clear the urgent need for improved early detection and intervention strategies. Suicide risk is strongly associated with depression, substance use disorders – particularly alcohol use – and other comorbid psychiatric conditions (289).
Mounting evidence highlights ACEs as foundational contributors to long-term psychiatric vulnerability (290, 291). The effects of ACEs appear cumulative, with higher exposure linked to an increased risk of depression, anxiety, self-harm, and suicidality in adulthood (292–294).
In psychiatric clinical practice, assessing a patient’s suicidal ideation is ideal, but not always possible. According to Obegi (295), nearly 50% of individuals with suicidal ideation withheld this information from their healthcare providers. Given this barrier, systematically screening for psychiatric risk based on ACE history may help identify individuals at elevated biological and psychological risk – without relying on verbal disclosure. This emphasizes the need for objective, biologically based tools to support suicide prevention efforts (296).
MDD is increasingly understood as a condition rooted in disrupted synaptic function (108, 122, 297). Neuroimaging studies consistently demonstrate reduced volume in the prefrontal cortex and hippocampus – regions essential for emotion regulation, memory, and cognition – suggesting stress-related neuronal atrophy, particularly in cases of prolonged illness or delayed treatment (92, 93). Functional imaging further reveals altered connectivity between these regions and broader disruptions across neural circuits, potentially reflecting dysregulated reciprocal signaling (94, 95). These anatomical and functional abnormalities are now believed to arise from a cascade of molecular and cellular alterations induced by chronic stress. This cascade begins with glutamatergic overactivation and is compounded by glucocorticoid dysregulation, resulting in widespread disturbances to neuronal architecture and synaptic signaling (298, 299). In addition to impairing mitochondrial integrity and immune function, these changes promote region-specific remodeling of brain structures (300, 301). A key driver of this pathology is neuroinflammation: stress activates microglia and increases pro-inflammatory cytokine levels, leading to synaptic dysfunction and reduced neurogenesis (53). Together, these molecular disruptions converge to produce the brain atrophy and weakened synaptic connectivity consistently observed in MDD.
Treatment
Early life trauma tends to reduce the responsiveness to standard antidepressant therapies (302). ACEs appear to induce anti-neurotrophic effects that are sufficient to counteract the pro-neurotrophic effects of regular oral antidepressants like the serotonin selective reuptake inhibitors. Over the last two decades, efforts have been made to develop a new generation of antidepressants with enhanced neurotrophic effects. The first of these, has been shown to improve the treatment of many patients with treatment-resistant major depression, including those with histories of trauma (303). Some more recent research suggests that ketamine may also be effective for PTSD (304).
In recent years, ketamine, an FDA-approved anesthetic and pain management agent, has emerged as a rapid-acting antidepressant by targeting the mTORC1 signaling pathway and downstream plasticity pathways (97). Ketamine rapidly induces robust antidepressant effects by blocking NMDA receptors, which triggers a surge in glutamate release and subsequent activation of AMPA receptors (305) (Figure 2). One of ketamine’s metabolites, N-desmethylketamine (norketamine), also directly activates AMPA receptors (306). AMPA receptor activation promotes the rapid release of BDNF, which then binds to its receptor, tropomyosin receptor kinase B (TrkB), initiating downstream signaling through the mTORC1 pathway that leads to spine and synapse formation (307). As a noncompetitive NMDA receptor antagonist, ketamine can produce antidepressant effects within hours, particularly in patients resistant to traditional treatments (303, 308).
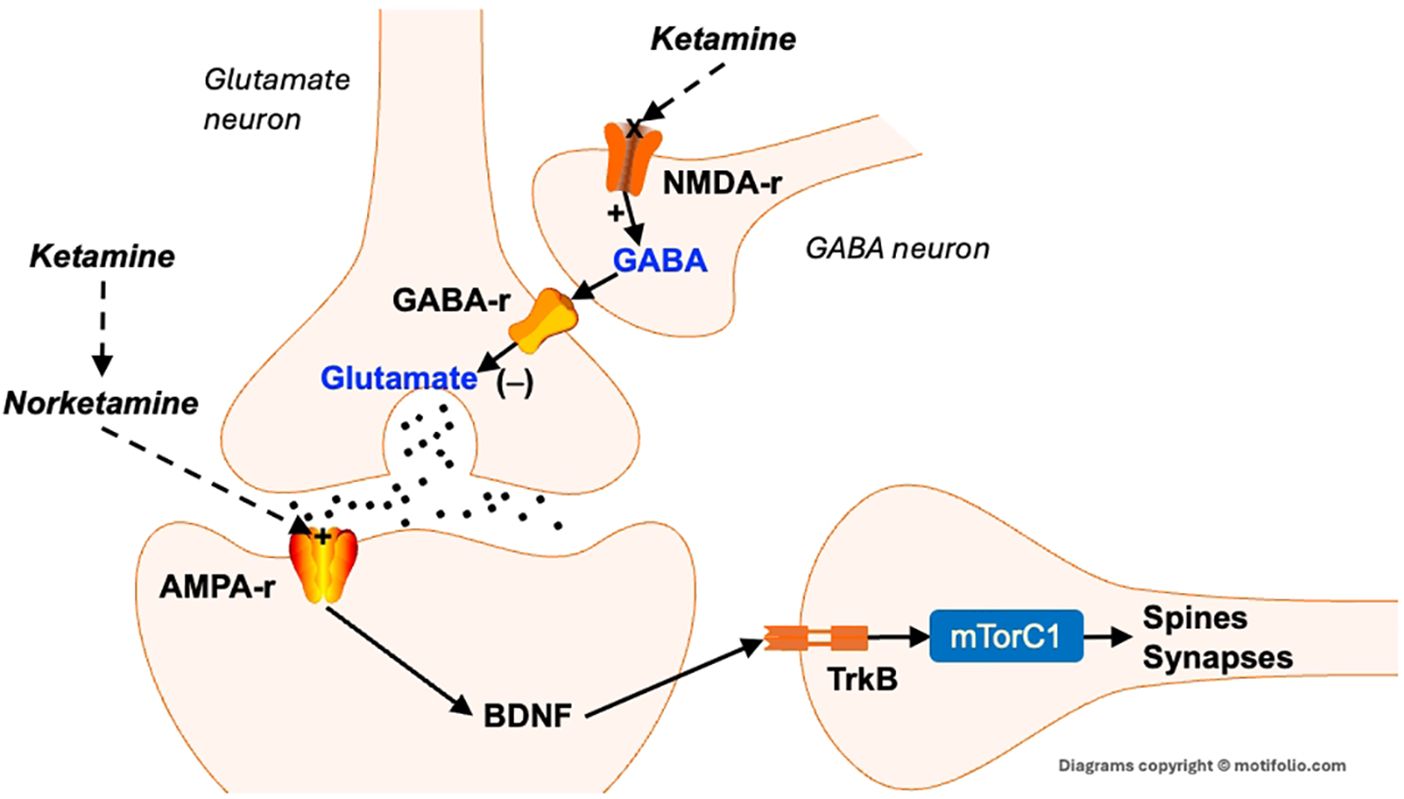
Figure 2. Neurotrophic effects of ketamine. Excess glutamate activates NMDA receptors on GABA-containing interneurons. This stimulates the release of GABA, which binds to GABAA receptors that inhibit the release of glutamate. The blockade of NMDA receptors reduces GABA’s inhibition of glutamate neurons, leading to a rapid release of glutamate. This, in turn, activates post-synaptic AMPA receptors that, among other actions, cause the rapid release of BDNF. BDNF then binds to its receptor trk-B, which activates mTORC1, resulting in the rapid synthesis of spine and synaptic proteins. This then results in the formation (or maintenance) of dendritic spines and synapses. AMPA-r, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor. BDNF, Brain-derived neurotrophic factor; GABA-r, Gamma amino butyric acid receptor; NMDA-r, N-methyl-D-aspartic acid receptor; TrkB, tropomyosin receptor kinase B.
In addition to promoting BDNF release, ketamine induces the expression of activity-regulated cytoskeleton-associated protein (Arc), inhibits glycogen synthase kinase-3 (GSK3), and reverses stress-induced synaptic loss in the medial prefrontal cortex (PFC) and hippocampus (149, 150, 309). These effects are dependent on intact BDNF and GSK3 signaling pathways – blocking either pathway eliminates ketamine’s behavioral and synaptic benefits in animal models (310–313). The resulting synaptogenesis enhances emotional regulation and is considered a core mechanism behind ketamine’s rapid antidepressant effects. However, despite its efficacy, ketamine’s clinical use is constrained by short-lived benefits, the need for repeated dosing, and side effects such as dissociation and psychosis (314). To address these challenges, several novel therapeutic agents are being developed to replicate ketamine’s rapid antidepressant properties while minimizing its adverse effects. These include AMPA receptor positive allosteric modulators (AMPA-PAMs), which enhance glutamatergic transmission and synaptic plasticity (315, 316) and NMDA subunit modulators.
The NMDA receptor is composed of four protein subunits, two NR1 and two NR2 (which can be further classified as NR2A, B, C, and D). There are also NR3 subunits that can be assembled into a complete NMDA receptor. NMDA receptor modulators such as apimostinel and zelquistinel also produce rapid antidepressant effects in animal models and early-stage human studies, but with improved safety profiles. Unlike ketamine – which directly blocks the NMDA receptor ion channel – these compounds selectively inhibit the NR2B subunit, a mechanism that may account for their reduced risk of dissociative side effects (317). Additional agents targeting the NMDA receptor or downstream effectors such as mTORC1 are also under development (315, 316). These next-generation, rapid-acting antidepressants represent a paradigm shift – moving beyond traditional monoaminergic strategies to directly enhance synaptic resilience.
Conclusions
Collectively, the evidence presented in this paper demonstrates how adverse early experiences interact with neurobiological processes to increase susceptibility to depression and suicidality. A thorough understanding of these mechanisms is critical for designing more effective, biologically informed interventions. Looking ahead, integrating pharmacological advances with emerging epigenetic tools – such as microRNA profiling and DNA methylation analysis – holds promise for identifying at-risk individuals even before clinical symptoms manifest. These personalized strategies could transform psychiatric care from reactive crisis management into proactive prevention.
There are several limitations to this review. Although this review is broad in scope, it does not cover every possible mechanism. This was not a systematic review. We acknowledge that, even within the specific topics covered in this paper, there may be relevant information that was not addressed. We also did not cover the important area of biomarkers, given the length and complexity of the issues covered.
This review highlights the fact that emotional traumas experienced early in life have far-reaching effects that may last a lifetime. An integrative view is that many possible mechanisms, including DNA methylation, altered expression of both protein-coding genes and short regulatory RNAs like miRNAs, enhancement of pro-apoptotic mechanisms, synaptic remodeling, and others, have anti-neurotrophic effects that predispose to mood, anxiety, and trauma-related disorders and suicide across the lifespan. Novel treatments like ketamine must have profound pro-neurotrophic effects that supersede the blocks introduced by traumas. While ketamine is a very effective treatment, there are distinct disadvantages to ketamine, including the intravenous mode of delivery and significant side effects. Several NMDA-selective antagonists, including NR2B-selective NAMs, have failed in clinical trials for various reasons. A dextromethorphan and bupropion combination drug, which ostensibly targets NMDA receptors, has been approved for treatment-resistant major depression. Still, it is unclear if it is as effective as ketamine, particularly in people with a history of trauma. Time will tell if newer compounds like the “stinels” (apomostinel and zelquistinel) or onfasprodil that target the NMDA receptor, or mefluleucine (NV-5138) that completely bypasses upstream signal transduction mechanisms by directly activating mTORC1, will eventually make it through clinical trials for depression or other conditions.
Author contributions
MT: Writing – original draft. RS: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ondeck L, Combe L, Feeser CJ, and King R. Care of victims of child maltreatment: the school nurse's role. Position statement. Natl Assoc School Nurses (NASN). (2014).
2. Tsehay M, Necho M, and Mekonnen W. The role of adverse childhood experience on depression symptom, prevalence, and severity among school going adolescents. Depression Res Treat. (2020) 2020:5951792. doi: 10.1155/2020/5951792
3. Donovan A, Assari S, Grella C, Shaheen M, Richter L, and Friedman. TC. Neuroendocrine mechanisms in the links between early life stress, affect, and youth substance use: A conceptual model for the study of sex and gender differences. Front Neuroendocrinol. (2024) 73:101121. doi: 10.1016/j.yfrne.2024.101121
4. Teicher MH and Samson JA. Annual research review: enduring neurobiological effects of childhood abuse and neglect. J Child Psychol Psychiatry. (2016) 57:241–66. doi: 10.1111/jcpp.12507
5. White JD and Kaffman A. The moderating effects of sex on consequences of childhood maltreatment: from clinical studies to animal models. Front Neurosci. (2019) 13:1082. doi: 10.3389/fnins.2019.01082
6. Hoffman EA, Clark DB, Orendain N, Hudziak J, Squeglia LM, and Dowling GJ. Stress exposures, neurodevelopment and health measures in the abcd study. Neurobiol Stress. (2019) 10:100157. doi: 10.1016/j.ynstr.2019.100157
7. Bomysoad RN and Francis LA. Adverse childhood experiences and mental health conditions among adolescents. J Adolesc Health. (2020) 67:868–70. doi: 10.1016/j.jadohealth.2020.04.013
8. Desch J, Mansuri F, Tran D, Schwartz SW, and Bakour C. The association between adverse childhood experiences and depression trajectories in the add health study. Child Abuse Negl. (2023) 137:106034. doi: 10.1016/j.chiabu.2023.106034
9. Felitti VJ, Anda RF, Nordenberg D, Williamson DF, Spitz AM, Edwards V, et al. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults: the adverse childhood experiences (Ace) study. Am J Prev Med. (1998) 14:245–58. doi: 10.1016/S0749-3797(98)00017-8
10. Wade JR, Cronholm PF, Fein JA, Forke CM, Davis MB, Harkins-Schwarz M, et al. Household and community-level adverse childhood experiences and adult health outcomes in a diverse urban population. Child Abuse Negl. (2016) 52:135–45. doi: 10.1016/j.chiabu.2015.11.021
11. Hughes K, Bellis MA, Hardcastle KA, Sethi D, Butchart A, Mikton C, et al. The effect of multiple adverse childhood experiences on health: A systematic review and meta-analysis. Lancet Public Health. (2017) 2:e356–e66. doi: 10.1016/S2468-2667(17)30118-4
12. Chanlongbutra A, Singh GK, and Mueller CD. Adverse childhood experiences, health-related quality of life, and chronic disease risks in rural areas of the United States. J Environ Public Health. (2018) 2018:7151297. doi: 10.1155/2018/7151297
13. Dong M, Giles WH, Felitti VJ, Dube SR, Williams JE, Chapman DP, et al. Insights into causal pathways for ischemic heart disease: adverse childhood experiences study. Circulation. (2004) 110:1761–66. doi: 10.1161/01.CIR.0000143074.54995.7F
14. Gilbert LK, Breiding MJ, Merrick MT, et al. Childhood adversity and adult chronic disease: an update from ten states and the district of Columbia 2010. Am J Prev Med. (2015) 48:345–49. doi: 10.1016/j.amepre.2014.09.006
15. Kopin L and Lowenstein C. In the clinic® Dyslipidemia. Ann Internal Med. (2017) 167:ITC81–95. doi: 10.7326/AITC201712050
16. Short AK and Baram TZ. Early-life adversity and neurological disease: age-old questions and novel answers. Nat Rev Neurol. (2019) 15(11):657–69. doi: 10.1038/s41582-019-0246-5
17. Whitaker RC, Dearth-Wesley T, Gooze RA, Becker BD, Gallagher KC, and McEwen BS. Adverse childhood experiences, dispositional mindfulness, and adult health. Prev Med. (2014) 67:147–53. doi: 10.1016/j.ypmed.2014.07.029
18. Merrick MT, Ford DC, Ports KA, Guinn AS, Chen J, Klevens J, et al. Estimated proportion of adult health problems attributable to adverse childhood experiences and implications for prevention-25 states 2015-2017. MMWR: Morbid Mortal Wkly Rep. (2019) 68:999–1005. doi: 10.15585/mmwr.mm6844e1
19. Dayananda KK, Ahmed S, Wang D, Polis B, Islam R, and Kaffman A. Early life stress impairs synaptic pruning in the developing hippocampus. Brain Behav Immun. (2023) 107:16–31. doi: 10.1016/j.bbi.2022.09.014
20. Chen LP, Murad MH, Paras ML, Colbenson KM, Sattler AL, Goranson EN, et al. Sexual Abuse and Lifetime Diagnosis of Psychiatric Disorders: Systematic Review and Meta-Analysis. Mayo Clinic Proceedings Vol. 85. Elsevier (2010).
21. Evans GW, Li D, and Whipple SS. Cumulative risk and child development. psychol Bull. (2013) 139:1342. doi: 10.1037/a0031808
22. McLaughlin KA, Colich NL, Rodman AM, and Weissman DG. Mechanisms linking childhood trauma exposure and psychopathology: A transdiagnostic model of risk and resilience. BMC Med. (2020) 18:1–11. doi: 10.1186/s12916-020-01561-6
23. Tottenham N, Hare TA, Quinn BT, McCarry TW, Nurse M, Gilhooly T, et al. Prolonged institutional rearing is associated with atypically large amygdala volume and difficulties in emotion regulation. Dev Sci. (2010) 13:46–61. doi: 10.1111/j.1467-7687.2009.00852.x
24. Shonkoff JP. Capitalizing on advances in science to reduce the health consequences of early childhood adversity. JAMA Pediatr. (2016) 170:1003–07. doi: 10.1001/jamapediatrics.2016.1559
25. Everly GS, Lating JM, Everly GS, and Lating JM. The anatomy and physiology of the human stress response. In: A clinical guide to the treatment of the human stress response Cham, Switzerland (Springer International Publishing): Springer (2019). p. 19–56.
26. Knoll AT and Carlezon WA Jr. Dynorphin, stress, and depression. Brain Res. (2010) 1314:56–73. doi: 10.1016/j.brainres.2009.09.074
27. Jalewa J, Wong-Lin K, Martin McGinnity T, Prasad G, and Hölscher C. Increased number of orexin/hypocretin neurons with high and prolonged external stress-induced depression. Behav Brain Res. (2014) 272:196–204. doi: 10.1016/j.bbr.2014.05.030
28. Serova LI, Lauková M, Alaluf LG, Pucillo L, and Sabban EL. Intranasal neuropeptide Y reverses anxiety and depressive-like behavior impaired by single prolonged stress ptsd model. Eur Neuropsychopharmacol. (2014) 24:142–47. doi: 10.1016/j.euroneuro.2013.11.007
29. Anda RF, Butchart A, Felitti VJ, and Brown DW. Building a framework for global surveillance of the public health implications of adverse childhood experiences. Am J Prev Med. (2010) 39:93–8. doi: 10.1016/j.amepre.2010.03.015
30. Brody DJ and Hughes JP. Depression Prevalence in Adolescents and Adults: United States, August 2021–August 2023. NCHS Data Brief no. 527. National Center for Health Statistics (2025). Available online at: https://www.cdc.gov/nchs/products/databriefs/db527.htm.
31. Angst J, Hengartner MP, Rogers J, Schnyder U, Steinhausen H-C, Ajdacic-Gross V, et al. Suicidality in the prospective Zurich study: prevalence, risk factors and gender. Eur Arch Psychiatry Clin Neurosci. (2014) 264:557–65. doi: 10.1007/s00406-014-0500-1
32. Turecki G. The molecular bases of the suicidal brain. Nat Rev Neurosci. (2014) 15:802–16. doi: 10.1038/nrn3839
33. Liu RT, Trout ZM, Hernandez EM, Cheek SM, and Gerlus N. A behavioral and cognitive neuroscience perspective on impulsivity, suicide, and non-suicidal self-injury: meta-analysis and recommendations for future research. Neurosci Biobehav Rev. (2017) 83:440–50. doi: 10.1016/j.neubiorev.2017.09.019
34. O'Connor RC and Kirtley OJ. The integrated motivational–volitional model of suicidal behaviour. Philos Trans R Soc B: Biol Sci. (2018) 373:20170268. doi: 10.1098/rstb.2017.0268
35. Hussey JM, Chang JJ, and Kotch JB. Child maltreatment in the United States: prevalence, risk factors, and adolescent health consequences. Pediatrics. (2006) 118:933–42. doi: 10.1542/peds.2005-2452
36. Finkelhor D, Turner HA, Shattuck A, and Hamby SL. Violence, crime, and abuse exposure in a national sample of children and youth: an update. JAMA Paediatr. (2013) 167:614–21. doi: 10.1001/jamapediatrics.2013.42
37. Collishaw S, Pickles A, Messer J, Rutter M, Shearer C, and Maughan B. Resilience to adult psychopathology following childhood maltreatment: evidence from a community sample. Child Abuse Negl. (2007) 31:211–29. doi: 10.1016/j.chiabu.2007.02.004
38. Mandelli L, Petrelli C, and Serretti A. The role of specific early trauma in adult depression: A meta-analysis of published literature. Childhood Trauma Adult Depress Eur Psychiatry. (2015) 30:665–80. doi: 10.1016/j.eurpsy.2015.04.007
39. de Codt A, Monhonval P, Bongaerts X, Belkacemi I, and Tecco JM. Bipolar disorder and early affective trauma. Psychiatria Danubina. (2016) 28:4–8.
40. Angelakis I, Gillespie EL, and Panagioti M. Childhood maltreatment and adult suicidality: A comprehensive systematic review with meta-analysis. psychol Med. (2019) 49:1057–78. doi: 10.1017/S0033291718003823
41. Brezo J, Paris J, Vitaro F, Hebert M, Tremblay RE, and Turecki G. Predicting suicide attempts in young adults with histories of childhood abuse. Br J Psychiatry. (2008) 193:134–39. doi: 10.1192/bjp.bp.107.037994
42. Nock MK, Hwang I, Sampson NA, and Kessler RC. Mental disorders, comorbidity and suicidal behavior: results from the national comorbidity survey replication. Mol Psychiatry. (2010) 15:868–76. doi: 10.1038/mp.2009.29
43. Turecki G and Brent DA. Suicide and suicidal behaviour. Lancet. (2016) 387:1227–39. doi: 10.1016/S0140-6736(15)00234-2
44. Yao K, Chen P, Zhou H, Ruan J, Chen D, Yang X, et al. The effect of childhood trauma on suicide risk: the chain mediating effects of resilience and mental distress. BMC Psychiatry. (2023) 23:865. doi: 10.1186/s12888-023-05348-w
45. Merrick MT, Ports KA, Ford DC, Afifi TO, Gershoff ET, and Grogan-Kaylor A. Unpacking the impact of adverse childhood experiences on adult mental health. Child Abuse Negl. (2017) 69:10–9. doi: 10.1016/j.chiabu.2017.03.016
46. Miller GE, Chen E, and Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. psychol Bull. (2011) 137:959. doi: 10.1037/a0024768
47. Evans GW and Kim P. Childhood poverty, chronic stress, self-regulation, and coping. Child Dev Perspect. (2013) 7:43–8. doi: 10.1111/cdep.12013
48. Tzouvara V, Kupdere P, Wilson K, Matthews L, Simpson A, and Foye U. Adverse childhood experiences, mental health, and social functioning: A scoping review of the literature. Child Abuse Negl. (2023) 139:106092. doi: 10.1016/j.chiabu.2023.106092
49. Irshad S and Lone A. Adverse childhood experiences and their influence on psychological well-being and emotional intelligence among university students. BMC Psychol. (2025) 13:255. doi: 10.1186/s40359-025-02565-8
50. McEwen BS and Gianaros PJ. Stress-and allostasis-induced brain plasticity. Annu Rev Med. (2011) 62:431–45. doi: 10.1146/annurev-med-052209-100430
51. McEwen BS and Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. (2013) 79:16–29. doi: 10.1016/j.neuron.2013.06.028
52. Spencer RL and Deak T. A users guide to hpa axis research. Physiol Behav. (2017) 178:43–65. doi: 10.1016/j.physbeh.2016.11.014
53. Schramm E and Waisman A. Microglia as central protagonists in the chronic stress response. Neurol: Neuroimmunol Neuroinflamm. (2022) 9:e200023. doi: 10.1212/NXI.0000000000200023
54. Godoy LD, Rossignoli MT, Delfino-Pereira P, Garcia-Cairasco N, and de Lima Umeoka EH. A comprehensive overview on stress neurobiology: basic concepts and clinical implications. Front Behav Neurosci. (2018) 12:127. doi: 10.3389/fnbeh.2018.00127
55. Domin H and Śmiałowska M. The Diverse Role of Corticotropin-Releasing Factor (Crf) and Its Crf1 and Crf2 Receptors under Pathophysiological Conditions: Insights into Stress/Anxiety, Depression, and Brain Injury Processes. Neurosci Biobehav Rev. (2024) 105748. doi: 10.1016/j.neubiorev.2024.105748
56. Gold PW. The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry. (2015) 20:32–47. doi: 10.1038/mp.2014.163
57. Anisman H, Zaharia MD, Meaney MJ, and Merali Z. Do early-life events permanently alter behavioral and hormonal responses to stressors? Int J Dev Neurosci. (1998) 16:149–64. doi: 10.1016/s0736-5748(98)00025-2
58. Li J, Li Z-a, Tian H-m, Tao S, Zhang Q, Li F, et al. Corticotropin-releasing hormone neurons in the central amygdala: an integrative hub for pain, emotion, and addiction neurobiology. Brain Res. (2025), 149753. doi: 10.1016/j.brainres.2025.149753
59. Miller DB and O'Callaghan JP. Neuroendocrine aspects of the response to stress. Metabo Clin Exp. (2002) 51:5–10. doi: 10.1053/meta.2002.33184
60. Thau L, Gandhi J, and Sharma S. Physiology, cortisol. In: Statpearls. Treasure Island, Florida, USA: StatPearls Publishing (2023).
61. Liu R-J and Aghajanian GK. Stress blunts serotonin-and hypocretin-evoked epscs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci. (2008) 105:359–64. doi: 10.1073/pnas.0706679105
62. McEwen BS, Eiland L, Hunter RG, and Miller MM. Stress and anxiety: structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology. (2012) 62:3–12. doi: 10.1016/j.neuropharm.2011.07.014
63. Heim C, Newport DJ, Mletzko T, Miller AH, and Nemeroff CB. The link between childhood trauma and depression: insights from hpa axis studies in humans. Psychoneuroendocrinology. (2008) 33:693–710. doi: 10.1016/j.psyneuen.2008.03.008
64. Mehta MA, Golembo NI, Nosarti C, Colvert E, Mota A, Williams SCR, et al. Amygdala, hippocampal and corpus callosum size following severe early institutional deprivation: the English and Romanian adoptees study pilot. J Child Psychol Psychiatry. (2009) 50:943–51. doi: 10.1111/j.1469-7610.2009.02084.x
65. McCrory E, Brito SAD, and Viding E. The link between child abuse and psychopathology: A review of neurobiological and genetic research. J R Soc Med. (2012) 105:151–56. doi: 10.1258/jrsm.2011.110222
66. Malter Cohen M, Jing D, Yang RR, Tottenham N, Lee FS, and Casey BJ. Early-life stress has persistent effects on amygdala function and development in mice and humans. Proc Natl Acad Sci. (2013) 110:18274–78. doi: 10.1073/pnas.1310163110
67. Vyas A, Mitra R, Shankaranarayana Rao BS, and Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. (2002) 22:6810–18. doi: 10.1523/JNEUROSCI.22-15-06810.2002
68. Vyas A, Pillai AG, and Chattarji S. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience. (2004) 128:667–73. doi: 10.1016/j.neuroscience.2004.07.013
69. Yang J, Hou C, Ma N, Liu J, Zhang Y, Zhou J, et al. Enriched environment treatment restores impaired hippocampal synaptic plasticity and cognitive deficits induced by prenatal chronic stress. Neurobiol Learn Memory. (2007) 87:257–63. doi: 10.1016/j.nlm.2006.09.001
70. LeDoux JE. Emotion circuits in the brain. Fear Anxiety. (2013), 259–88. doi: 10.4324/9780203825266
71. Zhang X, Ge TT, Yin G, Cui R, Zhao G, and Yang W. Stress-Induced functional alterations in amygdala: implications for neuropsychiatric diseases. Front Neurosci. (2018) 12:367. doi: 10.3389/fnins.2018.00367
72. Amaral DG. The amygdale: neurobiological aspects emotion memory and mental dysfunction. Anatomical Organ Primate Amygdaloid Complex. (1992).
73. Milad MR and Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. (2002) 420:70–4. doi: 10.1038/nature01138
74. Ghashghaei HT, Hilgetag CC, and Barbas H. Sequence of information processing for emotions based on the anatomic dialogue between prefrontal cortex and amygdala. Neuroimage. (2007) 34:905–23. doi: 10.1016/j.neuroimage.2006.09.046
75. Yang Y and Wang J-Z. From structure to behavior in basolateral amygdala-Hippocampus circuits. Front Neural Circuits. (2017) 11:86. doi: 10.3389/fncir.2017.00086
76. Maren S. Neurobiology of pavlovian fear conditioning. Annu Rev Neurosci. (2001) 24:897–931. doi: 10.1146/annurev.neuro.24.1.897
77. Medina JF, Repa JC, Mauk MD, and LeDoux JE. Parallels between cerebellum-and amygdala-dependent conditioning. Nat Rev Neurosci. (2002) 3:122–31. doi: 10.1038/nrn728
78. Paré D, Quirk GJ, and Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. (2004) 92:1–9. doi: 10.1152/jn.00153.2004
79. Phelps EA. Human emotion and memory: interactions of the amygdala and hippocampal complex. Curr Opin Neurobiol. (2004) 14:198–202. doi: 10.1016/j.conb.2004.03.015
80. Maroun M and Richter-Levin G. Exposure to acute stress blocks the induction of long-term potentiation of the amygdala–prefrontal cortex pathway in vivo. J Neurosci. (2003) 23:4406–09. doi: 10.1523/JNEUROSCI.23-11-04406.2003
81. Izquierdo A, Wellman CL, and Holmes A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci. (2006) 26:5733–38. doi: 10.1523/JNEUROSCI.0474-06.2006
82. Correll CM, Rosenkranz JA, and Grace AA. Chronic cold stress alters prefrontal cortical modulation of amygdala neuronal activity in rats. Biol Psychiatry. (2005) 58:382–91. doi: 10.1016/j.biopsych.2005.04.009
83. Ovtscharoff W Jr. and Braun K. Maternal separation and social isolation modulate the postnatal development of synaptic composition in the infralimbic cortex of octodon degus. Neuroscience. (2001) 104:33–40. doi: 10.1016/S0306-4522(01)00059-8
84. Westbrook A and Braver TS. Dopamine does double duty in motivating cognitive effort. Neuron. (2016) 89(4):695–710. doi: 10.1016/j.neuron.2015.12.029
85. Tejeda HA and Bonci A. Dynorphin/kappa-opioid receptor control of dopamine dynamics: Implications for negative affective states and psychiatric disorders. Brain Res. (2019) 1713:91–101. doi: 10.1016/j.brainres.2018.09.023
86. Dedic N, Kühne C, Jakovcevski M, Hartmann J, Genewsky AJ, Gomes KS, et al. Chronic CRH depletion from GABAergic, long-range projection neurons in the extended amygdala reduces dopamine release and increases anxiety. Nat Neurosci. (2018) 21(6):803–7. doi: 10.1038/s41593-018-0151-z
87. Dahoun T, Nour MM, McCutcheon RA, Adams RA, Bloomfield MAP, and Howes OD. The relationship between childhood trauma, dopamine release and dexamphetamine-induced positive psychotic symptoms: a [11C]-(+)-PHNO PET study. Transl Psych. (2019) 9(1):287. doi: 10.1038/s41398-019-0627-y
88. Park AT, Tooley UA, Leonard JA, Boroshok AL, McDermott CL, Tisdall MD, et al. Early childhood stress is associated with blunted development of ventral tegmental area functional connectivity. Dev Cogn. Neurosci. (2021) 47:100909. doi: 10.1016/j.dcn.2020.100909
89. Fan J, Liu W, Xia J, Li S, Gao F, Zhu J, et al. Childhood trauma is associated with elevated anhedonia and altered core reward circuitry in major depression patients and controls. Hum Brain Mapping. (2021) 42(2):286–97. doi: 10.1002/hbm.25222
90. Ramkumar R, Edge-Partington M, Terstege DJ, Adigun K, Ren Y, Khan NS, et al. Long-term impact of early-life stress on serotonin connectivity. Biol Psychiatry. (2024) 96:287–99. doi: 10.1016/j.biopsych.2024.01.024
91. Lewis MW, Jones RT, and Davis MT. Exploring the impact of trauma type and extent of exposure on posttraumatic alterations in 5-ht1a expression. Trans Psychiatry. (2020) 10:237. doi: 10.1038/s41398-020-00915-1
92. Price JL and Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. (2010) 35:192–216. doi: 10.1038/npp.2009.104
93. MacQueen G and Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry. (2011) 16:252–64. doi: 10.1038/mp.2010.80
94. Perlman G, Simmons AN, Wu J, Hahn KS, Tapert SF, Max JE, et al. Amygdala response and functional connectivity during emotion regulation: A study of 14 depressed adolescents. J Affect Disord. (2012) 139:75–84. doi: 10.1016/j.jad.2012.01.044
95. Zeng L-L, Shen H, Liu L, Wang L, Li B, Fang P, et al. Identifying major depression using whole-brain functional connectivity: A multivariate pattern analysis. Brain. (2012) 135:1498–507. doi: 10.1093/brain/aws059
96. White JD, Arefin TM, Pugliese A, Lee CH, Gassen J, Zhang J, et al. Early life stress causes sex-specific changes in adult fronto-limbic connectivity that differentially drive learning. Elife. (2020) 9:e58301. doi: 10.7554/eLife.58301.sa2
97. Duman RS and Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. (2012) 338:68–72. doi: 10.1126/science.1222939
98. Bremner JD, Narayan M, Staib LH, Southwick SM, McGlashan T, and Charney DS. Neural correlates of memories of childhood sexual abuse in women with and without posttraumatic stress disorder. Am J Psychiatry. (1999) 156:1787–95. doi: 10.1176/ajp.156.11.1787
99. Fadel E, Boeker H, Gaertner M, Richter A, Kleim B, Seifritz E, et al. Differential alterations in resting state functional connectivity associated with depressive symptoms and early life adversity. Brain Sci. (2021) 11:591. doi: 10.3390/brainsci11050591
100. Makovac E, Fagioli S, Rae CL, Critchley HD, and Ottaviani C. Can't get it off my brain: meta-analysis of neuroimaging studies on perseverative cognition. Psychiatry Res: Neuroimaging. (2020) 295:111020. doi: 10.1016/j.pscychresns.2019.111020
101. Cook SC and Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. (2004) 60:236–48. doi: 10.1002/neu.20025
102. Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. (2006) 26:7870–74. doi: 10.1523/JNEUROSCI.1184-06.2006
103. Radley JJ, Rocher AB, Miller M, Janssen WGM, Liston C, Hof PR, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. (2006) 16:313–20. doi: 10.1093/cercor/bhi104
104. Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, et al. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. (2009) 164:798–808. doi: 10.1016/j.neuroscience.2009.08.053
105. Hercher C, Canetti L, Turecki G, and Mechawar N. Anterior cingulate pyramidal neurons display altered dendritic branching in depressed suicides. J Psychiatr Res. (2010) 44:286–93. doi: 10.1016/j.jpsychires.2009.08.011
106. Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science. (2019) 364:eaat8078. doi: 10.1126/science.aat8078
107. Li N, Liu R-J, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. (2011) 69:754–61. doi: 10.1016/j.biopsych.2010.12.015
108. Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. (1999) 45:1085–98. doi: 10.1016/S0006-3223(99)00041-4
109. Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, and Miguel-Hidalgo JJ. Gabaergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. (2007) 32:471–82. doi: 10.1038/sj.npp.1301234
110. Herzog JI and Schmahl C. Adverse childhood experiences and the consequences on neurobiological, psychosocial, and somatic conditions across the lifespan. Front Psychiatry. (2018) 9:420. doi: 10.3389/fpsyt.2018.00420
111. Zuccato C and Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. (2009) 5:311–22. doi: 10.1038/nrneurol.2009.54
112. Binder DK and Scharfman HE. Brain-derived neurotrophic factor. Growth Factors (Chur Switzerland). (2004) 22:123. doi: 10.1080/08977190410001723308
113. Tsai S-J. Critical issues in bdnf val66met genetic studies of neuropsychiatric disorders. Front Mol Neurosci. (2018) 11:156. doi: 10.3389/fnmol.2018.00156
114. Leal G, Comprido D, and Duarte CB. Bdnf-Induced local protein synthesis and synaptic plasticity. Neuropharmacology. (2014) 76:639–56. doi: 10.1016/j.neuropharm.2013.04.005
115. Gelle T, Samey RA, Plansont B, Bessette B, Jauberteau-Marchan M-O, Lalloué F, et al. Bdnf and pro-bdnf in serum and exosomes in major depression: evolution after antidepressant treatment. Prog Neuropsychopharmacol Biol Psychiatry. (2021) 109:110229. doi: 10.1016/j.pnpbp.2020.110229
116. Krishnan V and Nestler EJ. The molecular neurobiology of depression. Nature. (2008) 455:894–902. doi: 10.1038/nature07455
117. Duman RS and Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. (2012) 35(1):47–56. doi: 10.1016/j.tins.2011.11.004
118. Sasi M, Vignoli B, Canossa M, and Blum R. Neurobiology of local and intercellular bdnf signaling. Pflügers Archiv European J Physiol. (2017) 469:593–610. doi: 10.1007/s00424-017-1964-4
119. Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, et al. A negative regulator of map kinase causes depressive behavior. Nat Med. (2010) 16:1328–32. doi: 10.1038/nm.2219
120. Hoeffer CA and Klann E. Mtor signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. (2010) 33:67–75. doi: 10.1016/j.tins.2009.11.003
121. Yuen EY, Wei J, Liu W, Zhong P, Li X, and Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. (2012) 73:962–77. doi: 10.1016/j.neuron.2011.12.033
122. Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. (2012) 18:1413–17. doi: 10.1038/nm.2886
123. Popoli M, Yan Z, McEwen BS, and Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. (2012) 13:22–37. doi: 10.1038/nrn3138
124. Licznerski P and Duman RS. Remodeling of axo-spinous synapses in the pathophysiology and treatment of depression. Neuroscience. (2013) 251:33–50. doi: 10.1016/j.neuroscience.2012.09.057
125. Magarin AM and McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal ca3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. (1995) 69:89–98. doi: 10.1016/0306-4522(95)00259-L
126. Pal MM. Glutamate: the master neurotransmitter and its implications in chronic stress and mood disorders. Front Hum Neurosci. (2021) 15:722323. doi: 10.3389/fnhum.2021.722323
127. Mahmoud S, Gharagozloo M, Simard C, and Gris D. Astrocytes maintain glutamate homeostasis in the cns by controlling the balance between glutamate uptake and release. Cells. (2019) 8:184. doi: 10.3390/cells8020184
128. Hardingham GE, Fukunaga Y, and Bading H. Extrasynaptic nmdars oppose synaptic nmdars by triggering creb shut-off and cell death pathways. Nat Neurosci. (2002) 5:405–14. doi: 10.1038/nn835
129. Lewerenz J and Maher P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front Neurosci. (2015) 9:469. doi: 10.3389/fnins.2015.00469
130. McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. (1999) 22:105–22. doi: 10.1146/annurev.neuro.22.1.105
131. Wiera G and Mozrzymas JW. Extracellular proteolysis in structural and functional plasticity of mossy fiber synapses in hippocampus. Front Cell Neurosci. (2015) 9:427. doi: 10.3389/fncel.2015.00427
132. Magariños AM, Verdugo JMG, and Mcewen BS. Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci. (1997) 94:14002–08. doi: 10.1073/pnas.94.25.14002
133. Sapolsky RM. Glucocorticoids, Stress and Exacerbation of Excitotoxic Neuron Death Vol. 6. Elsevier (1994).
134. Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci. (2009) 106:3543–48. doi: 10.1073/pnas.0812671106
135. Picard M, McEwen BS, Epel ES, and Sandi C. An energetic view of stress: focus on mitochondria. Front Neuroendocrinol. (2018) 49:72–85. doi: 10.1016/j.yfrne.2018.01.001
136. Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, et al. Localization of neuronal and glial glutamate transporters. Neuron. (1994) 13:713–25. doi: 10.1016/0896-6273(94)90038-8
137. Autry AE, Grillo CA, Piroli GG, Rothstein JD, McEwen BS, and Reagan LP. Glucocorticoid regulation of glt-1 glutamate transporter isoform expression in the rat hippocampus. Neuroendocrinology. (2006) 83:371–79. doi: 10.1159/000096092
138. Hunter RG, Bellani R, Bloss E, Costa A, Romeo RD, and McEwen. BS. Regulation of cart mrna by stress and corticosteroids in the hippocampus and amygdala. Brain Res. (2007) 1152:234–40. doi: 10.1016/j.brainres.2007.03.042
139. Mennesson M and Revest J-M. Glucocorticoid-responsive tissue plasminogen activator (Tpa) and its inhibitor plasminogen activator inhibitor-1 (Pai-1): relevance in stress-related psychiatric disorders. Int J Mol Sci. (2023) 24:4496. doi: 10.3390/ijms24054496
140. Matys T, Pawlak R, Matys E, Pavlides C, McEwen BS, and Strickland S. Tissue plasminogen activator promotes the effects of corticotropin-releasing factor on the amygdala and anxiety-like behavior. Proc Natl Acad Sci. (2004) 101:16345–50. doi: 10.1073/pnas.0407355101
141. Pawlak R, Magarinos AM, Melchor J, McEwen B, and Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. (2003) 6:168–74. doi: 10.1038/nn998
142. Pawlak R, Rao BSS, Melchor JP, Chattarji S, McEwen B, and Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci. (2005) 102:18201–06. doi: 10.1073/pnas.0509232102
143. Bennur S, Shankaranarayana Rao BS, Pawlak R, Strickland S, McEwen BS, Chattarji S, et al. Stress-induced spine loss in the medial amygdala is mediated by tissue-plasminogen activator. Neuroscience. (2007) 144:8–16. doi: 10.1016/j.neuroscience.2006.08.075
144. Revest J-M, Blasi FD, Kitchener P, Rougé-Pont F, Desmedt A, Turiault M, et al. The mapk pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci. (2005) 8:664–72. doi: 10.1038/nn1441
145. Sarrazin N, Blasi FD, Roullot-Lacarrière V, Rougé-Pont F, Le Roux A, Costet P, et al. Transcriptional effects of glucocorticoid receptors in the dentate gyrus increase anxiety-related behaviors. PloS One. (2009) 4:e7704. doi: 10.1371/journal.pone.0007704
146. Frame S and Cohen P. Gsk3 takes centre stage more than 20 years after its discovery. Biochem J. (2001) 359:1–16. doi: 10.1042/bj3590001
147. Doble BW and Woodgett JR. Gsk-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. (2003) 116:1175–86. doi: 10.1242/jcs.00384
148. Jope RS and Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. (2004) 29:95–102. doi: 10.1016/j.tibs.2003.12.004
149. Collingridge GL, Peineau S, Howland JG, and Wang YT. Long-term depression in the Cns. Nat Rev Neurosci. (2010) 11:459–73. doi: 10.1038/nrn2867
150. Li X and Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology. (2010) 35:2143–54. doi: 10.1038/npp.2010.105
151. Wilkinson MB, Dias C, Magida J, Mazei-Robison M, Lobo MK, Kennedy P, et al. A novel role of the wnt-dishevelled-gsk3β Signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J Neurosci. (2011) 31:9084–92. doi: 10.1523/JNEUROSCI.0039-11.2011
152. Timothy O'Brien W and Klein PS. Validating gsk3 as an in vivo target of lithium action. Biochem Soc Trans. (2009) 37:1133–38. doi: 10.1042/BST0371133
153. Qin J, Ma Z, Chen X, and Shu S. Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front Neurol. (2023) 14:1103416. doi: 10.3389/fneur.2023.1103416
154. Wang J, Chen H-S, Li H-H, Wang H-J, Zou R-S, Lu X-J, et al. Microglia-dependent excessive synaptic pruning leads to cortical underconnectivity and behavioral abnormality following chronic social defeat stress in mice. Brain Behav Immun. (2023) 109:23–36. doi: 10.1016/j.bbi.2022.12.019
155. Johnson FK and Kaffman A. Early life stress perturbs the function of microglia in the developing rodent brain: new insights and future challenges. Brain Behav Immun. (2018) 69:18–27. doi: 10.1016/j.bbi.2017.06.008
156. Borst K, Dumas AA, and Prinz M. Microglia, immune and non-immune functions. Immunity. (2021) 54:2194–208. doi: 10.1016/j.immuni.2021.09.014
157. Hammond BP, Manek R, Kerr BJ, Macauley MS, and Plemel JR. Regulation of microglia population dynamics throughout development, health, and disease. Glia. (2021) 69:2771–97. doi: 10.1002/glia.24047
158. Cornell J, Salinas S, Huang H-Y, and Zhou M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen Res. (2022) 17:705–16. doi: 10.4103/1673-5374.322423
159. Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, et al. Negative feedback control of neuronal activity by microglia. Nature. (2020) 586:417–23. doi: 10.1038/s41586-020-2777-8
160. Geloso MC and D’Ambrosi N. Microglial pruning: relevance for synaptic dysfunction in multiple sclerosis and related experimental models. Cells. (2021) 10. doi: 10.3390/cells10030686
161. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. (2014) 17:400–06. doi: 10.1038/nn.3641
162. Filipello F, Morini R, Corradini I, et al. The microglial innate immune receptor Trem2 is required for synapse elimination and normal brain connectivity. Immunity. (2018) 48:979–91.e8. doi: 10.1016/j.immuni.2018.04.016
163. Rocha M, Wang D, Avila-Quintero V, Bloch MH, and Kaffman A. Deficits in hippocampal-dependent memory across different rodent models of early life stress: systematic review and meta-analysis. Trans Psychiatry. (2021) 11:231. doi: 10.1038/s41398-021-01352-4
164. Worthen RJ, Zighelboim SSG, Jaramillo CST, and Beurel. E. Anti-inflammatory il-10 administration rescues depression-associated learning and memory deficits in mice. J Neuroinflamm. (2020) 17:1–16. doi: 10.1186/s12974-020-01922-1
165. Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: A systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. (2015) 2:1002–12. doi: 10.1016/S2215-0366(15)00309-0
166. Li S, Liao Y, Dong Y, et al. Microglial deletion and inhibition alleviate behavior of post-traumatic stress disorder in mice. J Neuroinflamm. (2021) 18:1–14. doi: 10.1186/s12974-020-02040-8
167. Raghuraman R, Karthikeyan A, Wei WL, Thameem Dheen S, and Sajikumar S. Activation of microglia in acute hippocampal slices affects activity-Dependent long-Term potentiation and synaptic tagging and capture in area Ca1. Neurobiol Learn Memory. (2019) 163:107039. doi: 10.1016/j.nlm.2019.107039
168. Sa de Almeida J, Vargas M, Fonseca-Gomes J, Tanqueiro SR, Belo RF, Miranda-Lourenço C, et al. Microglial sirtuin 2 shapes long-term potentiation in hippocampal slices. Front Neurosci. (2020) 14:614. doi: 10.3389/fnins.2020.00614
169. Tremblay M-È, Lowery RL, and Majewska AK. Microglial interactions with synapses are modulated by visual experience. PloS Biol. (2010) 8:e1000527. doi: 10.1371/journal.pbio.1000527
170. Pan Y and Monje M. Activity shapes neural circuit form and function: A historical perspective. J Neurosci. (2020) 40:944–54. doi: 10.1523/JNEUROSCI.0740-19.2019
171. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun. (2016) 7:12540. doi: 10.1038/ncomms12540
172. Sellgren CM, Sheridan SD, Gracias J, Xuan D, Fu T, and Perlis RH. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol Psychiatry. (2017) 22:170–77. doi: 10.1038/mp.2016.220
173. Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, et al. Cd47 protects synapses from excess microglia-mediated pruning during development. Neuron. (2018) 100:120–34.e6. doi: 10.1016/j.neuron.2018.09.017
174. Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science. (2020) 367:688–94. doi: 10.1126/science.aaz2288
175. Tillmon H, Soteros BM, Shen L, Cong Q, Wollet M, General J, et al. Complement and microglia activation mediate stress-induced synapse loss in layer 2/3 of the medial prefrontal cortex in male mice. Nat Commun. (2024) 15:9803. doi: 10.1038/s41467-024-54007-5
176. Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, et al. Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci. (2015) 35:13029–42. doi: 10.1523/JNEUROSCI.1698-15.2015
177. Crider A, Feng T, Pandya CD, Davis T, Nair A, Ahmed AO, et al. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain Behav Immun. (2018) 70:246–56. doi: 10.1016/j.bbi.2018.03.004
178. Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, et al. Complement C3 is activated in human ad brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. (2019) 28:2111–23.e6. doi: 10.1016/j.celrep.2019.07.060
179. Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. (2020) 39:e105380. doi: 10.15252/embj.2020105380
180. Jurga AM, Paleczna M, and Kuter KZ. Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci. (2020) 14:198. doi: 10.3389/fncel.2020.00198
181. Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, et al. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. (2005) 135:1295–307. doi: 10.1016/j.neuroscience.2005.06.090
182. Sugama S, Takenouchi T, Hashimoto M, Ohata H, Takenaka Y, and Kakinuma Y. Stress-induced microglial activation occurs through β-adrenergic receptor: noradrenaline as a key neurotransmitter in microglial activation. J Neuroinflamm. (2019) 16:1–16. doi: 10.1186/s12974-019-1632-z
183. Bolós M, Perea JR, Terreros-Roncal J, Pallas-Bazarra N, Jurado-Arjona J, Ávila J, et al. Absence of microglial Cx3cr1 impairs the synaptic integration of adult-Born hippocampal granule neurons. Brain Behav Immun. (2018) 68:76–89. doi: 10.1016/j.bbi.2017.10.002
184. Hagemeyer N, Hanft K-M, Akriditou M-A, Unger N, Park ES, Stanley ER, et al. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. (2017) 134:441–58. doi: 10.1007/s00401-017-1747-1
185. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
186. Basilico B, Pagani F, Grimaldi A, Cortese B, Di Angelantonio S, Weinhard L, et al. Microglia shape presynaptic properties at developing glutamatergic synapses. Glia. (2019) 67:53–67. doi: 10.1002/glia.23508
187. Nemes-Baran AD, White DR, and DeSilva TM. Fractalkine-dependent microglial pruning of viable oligodendrocyte progenitor cells regulates myelination. Cell Rep. (2020) 32. doi: 10.1016/j.celrep.2020.108047
188. Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, et al. Sensory lesioning induces microglial synapse elimination via adam10 and fractalkine signaling. Nat Neurosci. (2019) 22:1075–88. doi: 10.1038/s41593-019-0419-y
189. D'Haese JG, Friess H, and Ceyhan GO. Therapeutic potential of the chemokine–receptor duo fractalkine/Cx3cr1: an update. Expert Opin Ther Targets. (2012) 16:613–18. doi: 10.1517/14728222.2012.682574
190. Elberg G, Liraz-Zaltsman S, Reichert F, Matozaki T, Tal M, and Rotshenker S. Deletion of Sirpα (Signal regulatory protein-α) promotes phagocytic clearance of myelin debris in Wallerian degeneration, axon regeneration, and recovery from nerve injury. J Neuroinflamm. (2019) 16:1–14. doi: 10.1186/s12974-019-1679-x
191. Sato-Hashimoto M, Nozu T, Toriba R, Horikoshi A, Akaike M, Kawamoto K, et al. Microglial Sirpα Regulates the emergence of Cd11c+ Microglia and demyelination damage in white matter. Elife. (2019) 8:e42025. doi: 10.7554/eLife.42025
192. Sun H, He X, Tao X, Hou T, Chen M, He M, et al. The Cd200/Cd200r signaling pathway contributes to spontaneous functional recovery by enhancing synaptic plasticity after stroke. J Neuroinflamm. (2020) 17:1–15. doi: 10.1186/s12974-020-01845-x
193. Lyons A, Minogue AM, Jones RS, Fitzpatrick O, Noonan J, Campbell VA, et al. Analysis of the impact of Cd200 on phagocytosis. Mol Neurobiol. (2017) 54:5730–39. doi: 10.1007/s12035-016-0223-6
194. Pellegrini L, Foglio E, Pontemezzo E, Germani A, Russo MA, and Limana F. Hmgb1 and repair: focus on the heart. Pharmacol Ther. (2019) 196:160–82. doi: 10.1016/j.pharmthera.2018.12.005
195. Chen R, Huang Y, Quan J, Liu J, Wang H, Billiar TR, et al. Hmgb1 as a potential biomarker and therapeutic target for severe Covid-19. Heliyon. (2020) 6. doi: 10.1016/j.heliyon.2020.e05672
196. Ranganathan K and Sivasankar V. Micrornas-biology and clinical applications. J Oral Maxillofac Pathol. (2014) 18:229–34. doi: 10.4103/0973-029X.140762
197. Paraskevopoulou MD and Hatzigeorgiou AG. Analyzing mirna–lncrna interactions. Long Non-Coding RNAs: Methods Protoc. (2016) 1402:271–86. doi: 10.1007/978-1-4939-3378-5_21
198. Roy B, Yoshino Y, Allen L, Prall K, Schell G, and Dwivedi Y. Exploiting circulating micrornas as biomarkers in psychiatric disorders. Mol Diagn Ther. (2020) 24:279–98. doi: 10.1007/s40291-020-00464-9
199. Chao H-M, Wang T-W, Chern E, and Hsu S-h. Regulatory rnas, microrna, long-non coding rna and circular rna roles in colorectal cancer stem cells. World J Gastrointest Oncol. (2022) 14:748. doi: 10.4251/wjgo.v14.i4.748
200. Cheng JC, Moore TB, and Sakamoto KM. Rna interference and human disease. Mol Genet Metab. (2003) 80:121–28. doi: 10.1016/j.ymgme.2003.08.011
201. MacFarlane L-A and Murphy PR. Microrna: biogenesis, function and role in cancer. Curr Genomics. (2010) 11:537–61. doi: 10.2174/138920210793175895
202. Pittenger C and Duman RS. Stress, depression, and neuroplasticity: A convergence of mechanisms. Neuropsychopharmacology. (2008) 33:88–109. doi: 10.1038/sj.npp.1301574
203. Follert P, Cremer H, and Béclin C. Micrornas in brain development and function: A matter of flexibility and stability. Front Mol Neurosci. (2014) 7:5. doi: 10.3389/fnmol.2014.00005
204. Deraredj Nadim W, Simion V, Benedetti H, Pichon C, Baril P, and Morisset-Lopez S. Micrornas in neurocognitive dysfunctions: new molecular targets for pharmacological treatments? Curr Neuropharmacol. (2017) 15:260–75. doi: 10.2174/1570159x14666160709001441
205. Nowakowski TJ, Rani N, Golkaram M, Zhou HR, Alvarado B, Huch K, et al. Regulation of cell-type-specific transcriptomes by microrna networks during human brain development. Nat Neurosci. (2018) 21:1784–92. doi: 10.1038/s41593-018-0265-3
206. Allen L and Dwivedi Y. Microrna mediators of early life stress vulnerability to depression and suicidal behavior. Mol Psychiatry. (2020) 25:308–20. doi: 10.1038/s41380-019-0597-8
207. Davis-Dusenbery BN and Hata A. Smad-mediated mirna processing: A critical role for a conserved rna sequence. RNA Biol. (2011) 8:71–6. doi: 10.4161/rna.8.1.14299
208. Diederichs S and Haber DA. Dual role for argonautes in microrna processing and posttranscriptional regulation of microrna expression. Cell. (2007) 131:1097–108. doi: 10.1016/j.cell.2007.10.032
209. Wilson RC, Tambe A, Kidwell MA, Noland CL, Schneider CP, and Doudna JA. Dicer-trbp complex formation ensures accurate mammalian microrna biogenesis. Mol Cell. (2015) 57:397–407. doi: 10.1016/j.molcel.2014.11.030
210. Devaraju P, Yu J, Eddins D, Mellado-Lagarde MM, Earls LR, Westmoreland JJ, et al. Haploinsufficiency of the 22q11. 2 microdeletion gene Mrpl40 disrupts short-term synaptic plasticity and working memory through dysregulation of mitochondrial calcium. Mol Psychiatry. (2017) 22:1313–26. doi: 10.1038/mp.2016.75
211. Aksoy-Aksel A, Zampa F, and Schratt G. Micrornas and synaptic plasticity—a mutual relationship. Philos Trans R Soc B: Biol Sci. (2014) 369:20130515. doi: 10.1098/rstb.2013.0515
212. Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, et al. A brain-specific microrna regulates dendritic spine development. Nature. (2006) 439:283–89. doi: 10.1038/nature04367
213. Gao J, Wang W-Y, Mao Y-W, Gräff J, Guan J-S, Pan L, et al. A novel pathway regulates memory and plasticity via Sirt1 and Mir-134. Nature. (2010) 466:1105–09. doi: 10.1038/nature09271
214. Smalheiser NR, Lugli G, Rizavi HS, Torvik VI, Turecki G, and Dwivedi Y. Microrna expression is down-regulated and reorganized in prefrontal cortex of depressed suicide subjects. PloS One. (2012) 7:e33201. doi: 10.1371/journal.pone.0033201
215. Serafini G, Pompili M, Hansen KF, Obrietan K, Dwivedi Y, Shomron N, et al. The involvement of micrornas in major depression, suicidal behavior, and related disorders: A focus on mir-185 and mir-491-3p. Cell Mol Neurobiol. (2014) 34:17–30. doi: 10.1007/s10571-013-9997-5
216. Lopez JP, Pereira F, Richard-Devantoy S, Berlim M, Chachamovich E, Fiori LM, et al. Co-variation of peripheral levels of mir-1202 and brain activity and connectivity during antidepressant treatment. Neuropsychopharmacology. (2017) 42:2043–51. doi: 10.1038/npp.2017.9
217. Roy B, Wang Q, Palkovits M, Faludi G, and Dwivedi Y. Altered mirna expression network in locus coeruleus of depressed suicide subjects. Sci Rep. (2017) 7:4387. doi: 10.1038/s41598-017-04300-9
218. Azevedo JA, Carter BS, Meng F, Turner DL, Dai M, Schatzberg AF, et al. The microrna network is altered in anterior cingulate cortex of patients with unipolar and bipolar depression. J Psychiatr Res. (2016) 82:58–67. doi: 10.1016/j.jpsychires.2016.07.012
219. Ye Y, Xu H, Su X, and He X. Role of microrna in governing synaptic plasticity. Neural Plasticity. (2016) 2016:4959523. doi: 10.1155/2016/4959523
220. Maffioletti E, Tardito D, Gennarelli M, and Bocchio-Chiavetto L. Micro spies from the brain to the periphery: new clues from studies on micrornas in neuropsychiatric disorders. Front Cell Neurosci. (2014) 8:75. doi: 10.3389/fncel.2014.00075
221. Cattane N, Mora C, Lopizzo N, Borsini A, Maj C, Pedrini L, et al. Identification of a miRNAs signature associated with exposure to stress early in life and enhanced vulnerability for schizophrenia: New insights for the key role of miR-125b-1-3p in neurodevelopmental processes. Schizophr Res. (2019) 205:63–75. doi: 10.1016/j.schres.2018.07.030
222. Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, et al. Regulation of synaptic structure and function by fmrp-associated micrornas mir-125b and mir-132. Neuron. (2010) 65:373–84. doi: 10.1016/j.neuron.2010.01.005
223. Maffioletti E, Cattaneo A, Rosso G, Maina G, Maj C, Gennarelli M, et al. Peripheral whole blood microrna alterations in major depression and bipolar disorder. J Affect Disord. (2016) 200:250–58. doi: 10.1016/j.jad.2016.04.021
224. Roy B, Dunbar M, Agrawal J, Allen L, and Dwivedi Y. Amygdala-based altered mirnome and epigenetic contribution of mir-128-3p in conferring susceptibility to depression-like behavior via wnt signaling. Int J Neuropsychopharmacol. (2020) 23:165–77. doi: 10.1093/ijnp/pyz071
225. Wang Q, Roy B, Turecki G, Shelton RC, and Dwivedi Y. Role of complex epigenetic switching in tumor necrosis factor-α Upregulation in the prefrontal cortex of suicide subjects. Am J Psychiatry. (2018) 175:262–74. doi: 10.1176/appi.ajp.2017.16070759
226. Makeyev EV, Zhang J, Carrasco MA, and Maniatis T. The microrna mir-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mrna splicing. Mol Cell. (2007) 27:435–48. doi: 10.1016/j.molcel.2007.07.015
227. Cheng L-C, Pastrana E, Tavazoie M, and Doetsch F. Mir-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. (2009) 12:399–408. doi: 10.1038/nn.2294
228. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, and Tuschl. T. Identification of tissue-specific micrornas from mouse. Curr Biol. (2002) 12:735–39. doi: 10.1016/S0960-9822(02)00809-6
229. Higuchi F, Uchida S, Yamagata H, Abe-Higuchi N, Hobara T, Hara K, et al. Hippocampal microrna-124 enhances chronic stress resilience in mice. J Neurosci. (2016) 36:7253–67. doi: 10.1523/JNEUROSCI.0319-16.2016
230. Bahi A, Chandrasekar V, and Dreyer J-L. Selective lentiviral-mediated suppression of microrna124a in the hippocampus evokes antidepressants-like effects in rats. Psychoneuroendocrinology. (2014) 46:78–87. doi: 10.1016/j.psyneuen.2014.04.009
231. Yang W, Liu M, Zhang Q, Zhang J, Chen J, Chen Q, et al. Knockdown of mir-124 reduces depression-like behavior by targeting Creb1 and Bdnf. Curr Neurovasc Res. (2020) 17:196–203. doi: 10.2174/1567202617666200319141755
232. Dwivedi Y. Micrornas in depression and suicide: recent insights and future perspectives. J Affect Disord. (2018) 240:146–54. doi: 10.1016/j.jad.2018.07.075
233. Gheysarzadeh A, Sadeghifard N, Afraidooni L, Pooyan F, Mofid MR, Valadbeigi H, et al. Serum-based microrna biomarkers for major depression: mir-16, mir-135a, and mir-1202. J Res Med Sci. (2018) 23:69. doi: 10.4103/jrms.JRMS_879_17
234. Sun D-S and Chang H-H. Extracellular vesicles: function, resilience, biomarker, bioengineering, and clinical implications. Tzu Chi Med J. (2024) 36:251–59. doi: 10.4103/tcmj.tcmj_28_24
235. Tsilioni I, Panagiotidou S, and Theoharides TC. Exosomes in neurologic and psychiatric disorders. Clin Ther. (2014) 36:882–88. doi: 10.1016/j.clinthera.2014.05.005
236. Fries GR and Quevedo J. Exosomal micrornas as potential biomarkers in neuropsychiatric disorders. MicroRNA Protoc. (2018) 1733:79–85. doi: 10.1007/978-1-4939-7601-0_6
237. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (Misev2018): A position statement of the international society for extracellular vesicles and update of the misev2014 guidelines. J Extracell Vesicles. (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
238. Perkins DO, Jeffries CD, Fredrik Jarskog L, Thomson JM, Woods K, Newman MA, et al. Microrna expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol. (2007) 8:1–11. doi: 10.1186/gb-2007-8-2-r27
239. Pandey GN, Dwivedi Y, Rizavi HS, Ren X, Pandey SC, Pesold C, et al. Higher expression of serotonin 5-ht2a receptors in the postmortem brains of teenage suicide victims. Am J Psychiatry. (2002) 159:419–29. doi: 10.1176/appi.ajp.159.3.419
240. Kaminsky Z, Wilcox HC, Eaton WW, Van Eck K, Kilaru V, Jovanovic T, et al. Epigenetic and genetic variation at Ska2 predict suicidal behavior and post-traumatic stress disorder. Trans Psychiatry. (2015) 5:e627–e27. doi: 10.1038/tp.2015.105
241. Pompili M, Gibiino S, Innamorati M, Serafini G, Del Casale A, De Risio L, et al. Prolactin and thyroid hormone levels are associated with suicide attempts in psychiatric patients. Psychiatry Res. (2012) 200:389–94. doi: 10.1016/j.psychres.2012.05.010
242. Haghighi F, Xin Y, Chanrion B, O'Donnell AH, Ge Y, Dwork AJ, et al. Increased DNA methylation in the suicide brain. Dialogues Clin Neurosci. (2014) 16:430–38. doi: 10.31887/DCNS.2014.16.3/jmann
243. Peng H, Zhu Y, Strachan E, Fowler E, Bacus T, Roy-Byrne P, et al. Childhood trauma, dna methylation of stress-related genes, and depression: findings from two monozygotic twin studies. Psychosom Med. (2018) 80:599–608. doi: 10.1097/PSY.0000000000000604
244. Malki K, Koritskaya E, Harris F, Bryson K, Herbster M, and Tosto MG. Epigenetic differences in monozygotic twins discordant for major depressive disorder. Trans Psychiatry. (2016) 6:e839–e39. doi: 10.1038/tp.2016.101
245. Dwivedi Y, Roy B, and Korla PK. Genome-wide methylome-based molecular pathologies associated with depression and suicide. Neuropsychopharmacology. (2025) 50:705–16. doi: 10.1038/s41386-024-02040-9
246. Turecki G and Meaney MJ. Effects of the social environment and stress on glucocorticoid receptor gene methylation: A systematic review. Biol Psychiatry. (2016) 79:87–96. doi: 10.1016/j.biopsych.2014.11.022
247. Numata S, Ishii K, Tajima A, Iga J, Kinoshita M, Watanabe S, et al. Blood diagnostic biomarkers for major depressive disorder using multiplex dna methylation profiles: discovery and validation. Epigenetics. (2015) 10:135–41. doi: 10.1080/15592294.2014.1003743
248. Smart C, Strathdee G, Watson S, Murgatroyd C, and McAllister-Williams RH. Early life trauma, depression and the glucocorticoid receptor gene–an epigenetic perspective. psychol Med. (2015) 45:3393–410. doi: 10.1017/S0033291715001555
249. Ernst C, Deleva V, Deng X, Sequeira A, Pomarenski A, Klempan T, et al. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch Gen Psychiatry. (2009) 66:22–32. doi: 10.1001/archpsyc.66.1.22
250. Poulter MO, Du L, Weaver ICG, Palkovits M, Faludi G, Merali Z, et al. Gabaa receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol Psychiatry. (2008) 64:645–52. doi: 10.1016/j.biopsych.2008.05.028
251. Labonté B, Suderman M, Maussion G, Lopez JP, Navarro-Sánchez L, Yerko V, et al. Genome-wide methylation changes in the brains of suicide completers. Am J Psychiatry. (2013) 170:511–20. doi: 10.1176/appi.ajp.2012.12050627
252. Policicchio S, Washer S, Viana J, Iatrou A, Burrage J, Hannon E, et al. Genome-wide dna methylation meta-analysis in the brains of suicide completers. Trans Psychiatry. (2020) 10:69. doi: 10.1038/s41398-020-0752-7
253. Moore LD, Le T, and Fan G. DNA methylation and its basic function. Neuropsychopharmacology. (2013) 38:23–38. doi: 10.1038/npp.2012.112
254. Menke A and Binder EB. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin Neurosci. (2014) 16:395–404. doi: 10.31887/DCNS.2014.16.3/amenke
255. Skvortsova K, Iovino N, and Bogdanović O. Functions and mechanisms of epigenetic inheritance in animals. Nat Rev Mol Cell Biol. (2018) 19:774–90. doi: 10.1038/s41580-018-0074-2
256. Zeng D, He S, Ma C, Wen Y, Song W, Xu Q, et al. Network-based approach to identify molecular signatures in the brains of depressed suicides. Psychiatry Res. (2020) 294:113513. doi: 10.1016/j.psychres.2020.113513
257. Peng S, Zhou Y, Xiong L, and Wang Q. Identification of novel targets and pathways to distinguish suicide dependent or independent on depression diagnosis. Sci Rep. (2023) 13:2488. doi: 10.1038/s41598-023-29101-1
258. Coles CH and Bradke F. Coordinating neuronal actin–microtubule dynamics. Curr Biol. (2015) 25:R677–91. doi: 10.1016/j.cub.2015.06.020
259. Mahajan GJ, Vallender EJ, Garrett MR, Challagundla L, Overholser JC, Jurjus G, et al. Altered neuro-inflammatory gene expression in hippocampus in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 82:177–86. doi: 10.1016/j.pnpbp.2017.11.017
260. Powell V, Martin J, Thapar A, Rice F, and Anney RJL. Investigating regions of shared genetic variation in attention deficit/hyperactivity disorder and major depressive disorder: A gwas meta-analysis. Sci Rep. (2021) 11:7353. doi: 10.1038/s41598-021-86802-1
261. Tao Y, Zhang H, Jin M, Xu H, Zou S, Deng F, et al. Co-expression network of mrna and dna methylation in first-episode and drug-naive adolescents with major depressive disorder. Front Psychiatry. (2023) 14:1065417. doi: 10.3389/fpsyt.2023.1065417
262. Réus GZ, Quevedo J, and Rodrigues ALS. Mtor signaling in the neuropathophysiology of depression: current evidence. J Receptor Ligand Channel Res. (2015) 8:65–74. doi: 10.2147/JRLCR.S70351
263. De Berardis D, Fornaro M, Valchera A, Cavuto M, Perna G, Di Nicola M, et al. Eradicating suicide at its roots: preclinical bases and clinical evidence of the efficacy of ketamine in the treatment of suicidal behaviors. Int J Mol Sci. (2018) 19:2888. doi: 10.3390/ijms19102888
264. He J-G, Zhou H-Y, Wang F, and Chen J-G. Dysfunction of glutamatergic synaptic transmission in depression: focus on ampa receptor trafficking. Biol Psychiatry Global Open Sci. (2023) 3:187–96. doi: 10.1016/j.bpsgos.2022.02.007
265. Maitra M, Mitsuhashi H, Rahimian R, Chawla A, Yang J, Fiori LM, et al. Cell type specific transcriptomic differences in depression show similar patterns between males and females but implicate distinct cell types and genes. Nat Commun. (2023) 14:2912. doi: 10.1038/s41467-023-38530-5
266. Kimbrel NA, Ashley-Koch AE, Qin XJ, Lindquist JH, Garrett ME, Dennis MF, et al. Identification of novel, replicable genetic risk loci for suicidal thoughts and behaviors among US military veterans. JAMA Psychiatry. (2023) 80:135–45. doi: 10.1001/jamapsychiatry.2022.3896
267. Li QS, Shabalin AA, DiBlasi E, Gopal S, Canuso CM, FinnGen, et al. Genome-wide association study meta-analysis of suicide death and suicidal behavior. Mol Psychiatry. (2023) 28:891–900. doi: 10.1038/s41380-022-01828-9
268. Parade SH, Huffhines L, Daniels TE, Stroud LR, Nugent NR, and Tyrka AR. A systematic review of childhood maltreatment and dna methylation: candidate gene and epigenome-wide approaches. Trans Psychiatry. (2021) 11:134. doi: 10.1038/s41398-021-01207-y
269. Perroud N, Paoloni-Giacobino A, Prada P, Olié E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (Nr3c1) in adults with a history of childhood maltreatment: A link with the severity and type of trauma. Trans Psychiatry. (2011) 1:e59. doi: 10.1038/tp.2011.60
270. Miller O, Shakespeare-Finch J, Bruenig D, and Mehta D. DNA methylation of Nr3c1 and Fkbp5 is associated with posttraumatic stress disorder, posttraumatic growth, and resilience. psychol Trauma: Theory Res Pract Policy. (2020) 12:750. doi: 10.1037/tra0000574
271. Tyrka AR, Ridout KK, Parade SH, Paquette A, Marsit CJ, and Seifer R. Childhood maltreatment and methylation of fk506 binding protein 5 gene (Fkbp5). Dev Psychopathol. (2015) 27:1637–45. doi: 10.1017/S0954579415000991
272. Labonte B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M, et al. Differential glucocorticoid receptor exon 1b, 1c, and 1h expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry. (2012) 72:41–8. doi: 10.1016/j.biopsych.2012.01.034
273. Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F, et al. Lower methylation of glucocorticoid receptor gene promoter 1f in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry. (2015) 77:356–64. doi: 10.1016/j.biopsych.2014.02.006
274. Ryan J, Pilkington L, Neuhaus K, Ritchie K, Ancelin M-L, and Saffery R. Investigating the epigenetic profile of the inflammatory gene Il-6 in late-Life depression. BMC Psychiatry. (2017) 17:1–7. doi: 10.1186/s12888-017-1515-8
275. Binder EB. The role of Fkbp5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. (2009) 34:S186–S95. doi: 10.1016/j.psyneuen.2009.05.021
276. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific Fkbp5 dna demethylation mediates gene–childhood trauma interactions. Nat Neurosci. (2013) 16:33–41. doi: 10.1038/nn.3275
277. Tozzi L, Farrell C, Booij L, Doolin K, Nemoda Z, Szyf M, et al. Epigenetic changes of fkbp5 as a link connecting genetic and environmental risk factors with structural and functional brain changes in major depression. Neuropsychopharmacology. (2018) 43:1138–45. doi: 10.1038/npp.2017.290
278. Harms MB, Birn R, Provencal N, Wiechmann T, Binder EB, Giakas SW, et al. Early life stress, Fk506 binding protein 5 gene (Fkbp5) methylation, and inhibition-related prefrontal function: A prospective longitudinal study. Dev Psychopathol. (2017) 29:1895–903. doi: 10.1017/S095457941700147X
279. Dieckmann L and Czamara D. Epigenetics of prenatal stress in humans: the current research landscape. Clin Epigenet. (2024) 16:20. doi: 10.1186/s13148-024-01635-9
280. Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, et al. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Trans Psychiatry. (2011) 1:e21. doi: 10.1038/tp.2011.21
281. Conradt E, Lester BM, Appleton AA, Armstrong DA, and Marsit CJ. The roles of dna methylation of Nr3c1 and 11β-Hsd2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics. (2013) 8:1321–29. doi: 10.4161/epi.26634
282. Stroud LR, Papandonatos GD, Rodriguez D, McCallum M, Salisbury AL, Phipps MG, et al. Maternal smoking during pregnancy and infant stress response: test of a prenatal programming hypothesis. Psychoneuroendocrinology. (2014) 48:29–40. doi: 10.1016/j.psyneuen.2014.05.017
283. Stroud LR, Papandonatos GD, Parade SH, Salisbury AL, Phipps MG, Lester BM, et al. Prenatal major depressive disorder, placenta glucocorticoid and serotonergic signaling, and infant cortisol response. Psychosom Med. (2016) 78:979–90. doi: 10.1097/PSY.0000000000000410
284. Stroud LR, Papandonatos GD, Salisbury AL, Phipps MG, Huestis MA, Niaura R, et al. Epigenetic regulation of placental nr3c1: mechanism underlying prenatal programming of infant neurobehavior by maternal smoking? Child Dev. (2016) 87:49–60. doi: 10.1111/cdev.12482
285. Rylander-Rudqvist T, Hakansson N, Tybring G, and Wolk A. Quality and quantity of saliva dna obtained from the self-administrated oragene method—a pilot study on the cohort of Swedish men. Cancer Epidemiol Biomarkers Prev. (2006) 15:1742–45. doi: 10.1158/1055-9965.EPI-05-0706
286. Paoli C, Misztak P, Mazzini G, and Musazzi L. DNA methylation in depression and depressive-like phenotype: biomarker or target of pharmacological intervention? Curr Neuropharmacol. (2022) 20:2267–91. doi: 10.2174/1570159X20666220201084536
287. Centers for Disease Control and Prevention. New Reports Highlight Depression Prevalence Medication Use in the U.S (2025). Available online at: https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2025/20250416.htm (Accessed April 16, 2025).
288. Centers for Disease Control and Prevention. Suicide Data and Statistics (2025). Available online at: https://www.cdc.gov/suicide/facts/data.html (Accessed March 26, 2025).
289. Bertolote JM and Fleischmann A. Suicide and psychiatric diagnosis: A worldwide perspective. World Psychiatry. (2002) 1:181.
290. Croft J, Heron J, Teufel C, Cannon M, Wolke D, Thompson A, et al. Association of trauma type, age of exposure, and frequency in childhood and adolescence with psychotic experiences in early adulthood. JAMA Psychiatry. (2019) 76:79–86. doi: 10.1001/jamapsychiatry.2018.3155
291. Petruccelli K, Davis J, and Berman T. Adverse childhood experiences and associated health outcomes: A systematic review and meta-analysis. Child Abuse Negl. (2019) 97:104127. doi: 10.1016/j.chiabu.2019.104127
292. Cavanagh JTO, Carson AJ, Sharpe M, and Lawrie SM. Psychological autopsy studies of suicide: a systematic review. psychol Med. (2003) 33:395–4055. doi: 10.1017/S0033291702006943
293. Holmstrand C, Bogren M, Mattisson C, and Brådvik L. Long-term suicide risk in no, one or more mental disorders: the Lundby study 1947–1997. Acta Psychiatr Scand. (2015) 132:459–69. doi: 10.1111/acps.12506
294. Bachmann S. Epidemiology of suicide and the psychiatric perspective. Int J Environ Res Public Health. (2018) 15:1425. doi: 10.3390/ijerph15071425
295. Obegi JH. How common is recent denial of suicidal ideation among ideators, attempters, and suicide decedents? A literature review. Gen Hosp Psychiatry. (2021) 72:92–5. doi: 10.1016/j.genhosppsych.2021.07.009
296. Brådvik L. Suicide Risk and Mental Disorders. International Journal of Environmental Research and Public Health. Vol. 15. MDPI (2018).
297. Drevets WC. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res. (2000) 126:413–31. doi: 10.1016/S0079-6123(00)26027-5
298. Wang S, Liu X, Shi W, Qi Q, Zhang G, Li Y, et al. Mechanism of chronic stress-induced glutamatergic neuronal damage in the basolateral amygdaloid nucleus. Anal Cell Pathol. (2021) 2021:8388527. doi: 10.1155/2021/8388527
299. Pluma-Pluma A, Tovias-Sanchez L, Romero-Sandoval EA, and Murbartián J. Glucocorticoid receptor dynamics and neuroinflammation in chronic restraint stress-induced mechanical allodynia in female rats. J Pain. (2025) 309:105473. doi: 10.1016/j.jpain.2025.105473
300. Bansal Y and Kuhad A. Mitochondrial dysfunction in depression. Curr Neuropharmacol. (2016) 14:610–18. doi: 10.2174/1570159X14666160229114755
301. Khan M, Baussan Y, and Hebert-Chatelain E. Connecting dots between mitochondrial dysfunction and depression. Biomolecules. (2023) 13:695. doi: 10.3390/biom13040695
302. Williams LM, Debattista C, Duchemin AM, Schatzberg AF, and Nemeroff CB. Childhood trauma predicts antidepressant response in adults with major depression: data from the randomized international study to predict optimized treatment for depression. Trans Psychiatry. (2016) 6:e799–e99. doi: 10.1038/tp.2016.61
303. Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. (2006) 63:856–64. doi: 10.1001/archpsyc.63.8.856
304. Fremont R, Brown O, Feder A, and Murrough J. Ketamine for treatment of posttraumatic stress disorder: state of the field. Focus. (2023) 21:257–65. doi: 10.1176/appi.focus.20230006
305. Deyama S and Duman RS. Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol Biochem Behav. (2020) 188:172837. doi: 10.1016/j.pbb.2019.172837
306. Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, et al. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev. (2018) 70:621–60. doi: 10.1124/pr.117.015198
307. Kato T. Role of Mtor1 signaling in the antidepressant effects of ketamine and the potential of Mtorc1 activators as novel antidepressants. Neuropharmacology. (2023) 223:109325. doi: 10.1016/j.neuropharm.2022.109325
308. MaChado-Vieira R, Ibrahim L, Henter ID, and Zarate CA Jr. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. (2012) 100:678–87. doi: 10.1016/j.pbb.2011.09.010
309. Beurel E, Song L, and Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. (2011) 16:1068–70. doi: 10.1038/mp.2011.47
310. Bramham CR. Local protein synthesis, actin dynamics, and Ltp consolidation. Curr Opin Neurobiol. (2008) 18:524–31. doi: 10.1016/j.conb.2008.09.013
311. Jourdi H, Hsu Y-T, Zhou M, Qin Q, Bi X, and Baudry M. Positive ampa receptor modulation rapidly stimulates Bdnf release and increases dendritic mrna translation. J Neurosci. (2009) 29:8688–97. doi: 10.1523/JNEUROSCI.6078-08.2009
312. Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P-f, et al. Nmda receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. (2011) 475:91–5. doi: 10.1038/nature10130
313. Liu R-J, Lee FS, Li X-Y, Bambico F, Duman RS, and Aghajanian GK. Brain-derived neurotrophic factor Val66met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. (2012) 71:996–1005. doi: 10.1016/j.biopsych.2011.09.030
314. Sanacora G and Katz R. Ketamine: A review for clinicians. Focus. (2018) 16:243–50. doi: 10.1176/appi.focus.20180012
315. MaChado-Vieira R, Henter ID, and Zarate CA Jr. New targets for rapid antidepressant action. Prog Neurobiol. (2017) 152:21–37. doi: 10.1016/j.pneurobio.2015.12.001
316. Wang Q and Dwivedi Y. Advances in novel molecular targets for antidepressants. Prog Neuropsychopharmacol Biol Psychiatry. (2021) 104:110041. doi: 10.1016/j.pnpbp.2020.110041
Keywords: abuse, adverse childhood experiences, a-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, genomics, glutamate, hypothalamic-pituitary adrenal axis, micro RNAs
Citation: Takahashi M and Shelton RC (2025) From trauma to depression: structural, synaptic, epigenetic, and molecular pathways linking early stress to lifelong vulnerability. Front. Psychiatry 16:1666599. doi: 10.3389/fpsyt.2025.1666599
Received: 15 July 2025; Accepted: 16 September 2025;
Published: 16 October 2025.
Edited by:
Gustavo E. Tafet, Texas A and M University, United StatesReviewed by:
Sebastian Luca D’Addario, Santa Lucia Foundation (IRCCS), ItalySonia Tabares, Institución Universitaria de Envigado, Colombia
Copyright © 2025 Takahashi and Shelton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mina Takahashi, TWluYTYzMzdAdWFibWMuZWR1