 Rachel Montel Hayes
Rachel Montel Hayes Christopher E. Mason
Christopher E. Mason John J. Miller
John J. Miller- 1Department of Physiology and Biophysics, Weill Cornell Medicine, New York, NY, United States
- 2The His Royal Highness (HRH) Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine, Weill Cornell Medicine, New York, NY, United States
- 3The Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, New York, NY, United States
- 4WorldQuant Initiative for Quantitative Prediction, Weill Cornell Medicine, New York, NY, United States
- 5Brain Health, Exeter, NH, United States
The etiology of psychiatric disorders is complex, involving both genetic and environmental factors with emerging evidence suggesting that epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNA regulation, significantly contribute to mental health. The epigenome influences the development of psychiatric disorders and human behavior and may be considered in clinical observations. Epigenetic changes have been well-established in BDNF, COMT, FKBP5, NR3C1, SLC6A4, and DRD2, genes associated with psychiatric disorders, including schizophrenia, major depressive disorder (MDD), bipolar disorder (BP), post-traumatic stress disorder (PTSD), and autism spectrum disorder (ASD). Therefore, these epigenetic marks have the potential to be suitable biomarkers for diagnostics, as predictors of prognosis, and for the development of personalized treatments. By exploring the role of clinically relevant epigenetic genes, we review the role of the epigenome in the context of psychiatric disorders and human behavior; and we consider that these changes may be observed in the context of precision psychiatry. This review synthesizes findings from over 100 original research articles and reviews spanning a range of clinical studies. Despite promising associations, challenges in the onset of precision psychiatry, such as tissue heterogeneity, small sample sizes, and lack of replication, are likely to limit translation into clinical practice. Future research in precision psychiatry will help identify clinically actionable epigenetic biomarkers, ushering in an era of genomic medicine in psychiatry.
1 Introduction
Mental health is rooted in biological predispositions and experiences with genetic factors contributing to psychiatric disorders but not being able to fully explain differences in symptom onset, severity, and treatment response (1). The concept that external influences such as stress, trauma, in-utero exposures (medications, substances of abuse, starvation, viral exposure, toxoplasmosis), early-life adversity, social relationships, and lifestyle can alter gene expression without changing the DNA sequence and affects brain function and behavior is the basis for epigenetics (2). These changes to the DNA sequence include epigenetic mechanisms that are readily reversible spanning from DNA methylation, histone modifications to non-coding RNAs and chromatin remodeling (3, 4).
In psychiatry, epigenetics is not only useful for understanding how life experiences can leave molecular marks on the genome that shape behavior but has expanded in its use as biomarkers to being able to predict risk, diagnose or characterize disease subtypes, aid in treatment response, and predict prognosis and relapse risk (5). With the advent of precision psychiatry and personalized medicine, understanding an individual’s epigenetic profile is essential, and when used in combination with genetic factors and their environmental history can open the door to targeted interventions and aid in better treatment response (6).
This review explores the major epigenetic mechanisms including DNA methylation, histone modifications, non-coding RNAs, and chromatin remodeling, and how each mechanism can contribute to gene-environment interactions in the brain (7). By examining well studied genes (including BDNF, COMT, FKBP5, NR3C1, SLC6A4, and DRD2) implicated in psychiatry disorders, including major depressive disorder (MDD), bipolar disorder (BP), schizophrenia, post-traumatic stress disorder (PTSD), attention deficit/hyperactivity disorder (ADHD), autism spectrum disorder (ASD), obsessive compulsive disorder (OCD), alcohol use disorder (AUD), and substance use disorder (SUD), and how their expression is regulated epigenetically, we gain insight into the molecular pathways linking lifetime experiences to lifelong mental health outcomes. This review also considers how epigenetic biomarkers and precision psychiatry are reshaping the future of diagnosis, prognosis, and personalized treatment, as well as the limitations hindering the onset of precision psychiatry.
2 Epigenetic mechanisms in psychiatry
Epigenetic modifications are readily reversible changes that influence gene expression without altering the DNA sequence (3). These mechanisms act as molecular switches or dimmers, turning genes on or off in response to internal and external stimuli (7). In psychiatry, epigenetic mechanisms play a crucial role in regulating neural pathways that underlie emotion, cognition, stress response, and behavior; and provide a compelling framework for understanding how environmental factors such as early-life adversity, trauma, social experiences, nutrition, disease, lifestyle (physical activity and quality of sleep), and therapeutic interventions, can shape brain function and contribute to the development of mental illness (5).
2.1 DNA methylation
DNA methylation is the process by which DNA methyltransferases (DNMTs) catalyze the addition of a methyl group (-CH3) to cytosine residues on the 5-carbon of the cytosine ring in CpG dinucleotides, cytosine-rich regions often near gene promoters, between genes (referred to as intergenic regions), or in gene bodies (8). When methylation occurs within promoter regions, transcription factors are blocked from binding and gene expression is reduced or silenced (9). In psychiatry, this dynamic process where methylation is readily added or removed occurs in response to developmental cues and environmental exposures such as stress, trauma, and aging (10). Because methylation patterns are inherited through cell divisions, DNA methylation is said to affect cellular memory with gene expression states preserved across time and development (11). In the context of the epigenome, DNA methylation plays a central role in important biological processes including embryonic development, X-chromosome inactivation, imprinting, tissue-specific gene expression, and brain plasticity (12).
In MDD, hypermethylation of BDNF promoters, especially exon IV, reduces BDNF expression and impairs neuroplasticity whereas hypermethylation of the NR3C1 promoter impairs hypothalamic-pituitary-adrenal (HPA)-axis regulation following childhood trauma (13, 14). Studies in schizophrenia show that RELN hypermethylation reduces reelin expression and disrupts synaptic plasticity and neuronal migration; COMT methylation alters dopamine metabolism; and BDNF methylation is associated with cognitive impairment and negative symptoms (13, 15). In BD, epigenetic signatures are often state-dependent, with methylation differing in manic and depressive states compared to euthymic states (16). Likewise, increased BDNF methylation is reported during mood episodes, where it correlates with symptom severity (17). In ASD, global methylation differences are observed in neuronal development genes. In MECP2, both mutations and methylation abnormalities interact to disrupt synaptic development (18). In addiction and substance use disorders, hypermethylation of the mu-opioid receptor (OPRM1) is linked to heroin and alcohol dependence, and these epigenetic changes can persist to influence relapse risk (19, 20). However, OPRM1 is not in the discussion of this review.
2.2 Histone modifications
Histone modifications are chemical changes to histone proteins that influence how DNA is packaged. DNA is wrapped around histone (H) octamers (with the core units made of H2A, H2B, H3, and H4) to form nucleosomes (21). Most histone modifications occur on histone tails (mainly on H3 and H4) and by chemical modifications including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation. Histone acetyltransferases (HATs) add acetyl groups to lysine residues on histone tails, a modification referred to as acetylation (22). Acetylation activates gene expression by loosening chromatin and is reversed by histone deacetylases (HDACs) in a process known as deacetylation. Histones methylation is catalyzed by histone methyltransferases, which add methyl groups to lysine or arginine residues on histone tails, and reversed by histone demethylases (23). In this context, gene expression is activated or silenced depending on the specific sites and number of methyl groups (24). Other forms of histone modifications include phosphorylation, which is often associated with DNA repair and chromatin remodeling, as well as ubiquitination and sumoylation, which influence chromatin compaction and can contribute to gene repression (25).
These epigenetic changes modulate the chromatin structure and influence transcription factor accessibility and RNA polymerase binding, often interacting with other epigenetic mechanisms like DNA methylation to synergistically promote or repress gene expression (for example, methylated DNA recruiting histone-modifying enzymes) (26). In the context of psychiatry, abnormal histone modifications have been linked to multiple psychiatric disorders. Studies in depression show decreased H2K9ac at BDNF, in schizophrenia where H3K27me3 is modified at GAD1, in PTSD with repressive marks on FKBP5, and in addiction where changes in H3 acetylation impact reward circuits (27). HDAC inhibitors such as valproic acid and sodium butyrate can restore normal gene expression in psychiatric conditions further validating that histone marks may serve as biomarkers or targets for personalized therapies in mood and anxiety disorders (28).
2.3 MicroRNA regulation
MicroRNAs (miRNAs) are small (~22 nucleotides), non-coding RNA molecules that regulate gene expression post-transcriptionally by binding to target messenger RNAs (mRNAs) and suppress translation or promote mRNA degradation (29). miRNAs are both targets and regulators of epigenetic processes since they exist within a feedback loop with DNA methylation and histone modifications (30). As epigenetic targets, miRNA genes can be silenced or activated by DNA methylation or histone modifications, and as epigenetic regulators, miRNAs can target enzymes like DNMTs, HDACs, or histone methyltransferases for silencing, thereby influencing epigenetic landscapes. miRNAs are context-dependent, so their effect may vary by cell type, developmental stage, and environment, and they are also dynamic, changing in response to stress, drugs, experiences, and hormones (31). miRNAs are abundant in the brain and have crucial roles in neuronal development and differentiation, synaptic plasticity and learning, stress responses, and emotional regulation.
Changes in miRNA expression are clinically relevant across multiple psychiatric disorders. Genome-wide association studies (GWAS) in schizophrenia have implicated miR-137 as one of the most significant risk loci for schizophrenia (32). This miRNA regulates neurodevelopment and synaptic signaling genes, such as CACNA1C and TCF4, and has been proposed as a biomarker, with therapeutic modulation offering a potential means to stabilize synaptic function (32, 33). In depression, miR-124 is abundant in the brain and regulates neurogenesis and stress response; dysregulated levels have been associated with hippocampal neuroplasticity deficits (34). Other studies have reported that plasma miR-124 levels increase after citalopram treatment relative to untreated controls (35). In similar studies, miR-16-5p is reduced in peripheral blood of individuals with MDD and BD (36). Despite this findings, a major limitation is that miRNA expression is often assessed in peripheral blood rather than the brain directly. In schizophrenia, miR-132, miR-134, miR-1271, miR-664, miR-200c, and miR-432 are significantly decreased; however, antipsychotic treatment increases miR-132, miR-664, and miR-1271 compared with pre-treatment levels (37). In other psychiatric disorders, notable miRNAs include miR-34a and miR-181, which are significantly altered in schizophrenia; miR-146a and miR-134, which are altered in ASD, and miR-121 and miR-132, which are associated with addiction (38).
2.4 Chromatin remodeling
Chromatin remodeling is the dynamic structural change in chromatin which regulates access to DNA for transcription, replication, and repair. Chromatin remodeling controls the transition between euchromatin and heterochromatin thereby allowing or blocking gene expression (39). This process is mediated by ATP-dependent chromatin remodeling complexes, which move, eject, or restructure nucleosomes to increase DNA accessibility. Key ATP-dependent chromatin remodeling complexes include: (1) SWI/SNF, which slides or ejects nucleosomes to open chromatin (e.g., BRG1, BAF complex); (2) ISWI, which spaces nucleosomes for transcriptional regulation (e.g., SNF2H); (3) CHD, which contributes to nucleosome assembly and remodeling (e.g., CHD1, CHD8); and (4) INO80, which couples remodeling to DNA repair and replication (e.g., INO80, SRCAP) (40). Chromatin remodeling works with other epigenetic mechanisms by directly interacting with histone modifiers, DNA methylation machinery, and non-coding RNAs (41). In the context of psychiatric and neurological disorders, mutations in chromatin complexes have been observed in schizophrenia where synaptic gene regulation is disrupted, and these mutations are also high-confidence genetic risk factors for ASD (42). In depression, altered chromatin accessibility have been observed at BDNF, GAD1, NR3C1 in response to stress (43).
Currently, chromatin accessibility profiling (e.g., ATAC-seq) is being explored as a biomarker tool in psychiatry (44). Because most psychiatric GWAS findings lie in noncoding regions (i.e., enhancers or promoters), ATAC-seq helps identify which of these regions are open and accessible in brain cells. For example, ATAC-seq can reveal which accessible regions are specific to particular cell types, such as excitatory neurons, interneurons, or microglia. Therefore, single-cell chromatin accessibility profiling (scATAC-seq), together with gene expression assays, is used to identify disease-relevant fetal and adult brain cell types implicated in major depressive disorder, body mass index (BMI), ADHD, and schizophrenia (45). This approach has been applied to the identification of single nucleotide polymorphisms (SNPs) with schizophrenia risk in accessible enhancers of glutamatergic neurons, notably within genes such as CACNA1C, which plays a key role in synaptic function (46). Studies employing ATAC-seq have revealed increased accessibility of glucocorticoid-responsive enhancers in the brains of stressed individuals. These insights have driven the development of FKBP5 inhibitors, which are now being explored in clinical trials for PTSD and alcohol use disorder (47, 48).
2.5 Environmental influence on epigenetic changes
Environmental epigenetics studies how external factors like stress, diet, toxins, infections, relationships, and experiences, can lead to long-lasting changes in gene expression by altering the epigenome (49). For example, nutrition provides methyl donors and cofactors for epigenetic enzymes such as folate, B12, and choline which influence DNA methylation and for which undernutrition affects IGF2 expression (50). Exercise promotes histone acetylation and BDNF expression with aerobic activity increasing BDNF (51). Toxins and pollutants can alter the epigenome by inducing oxidative stress and disrupting methylation machinery, and in the brain, bisphenol A (BPA), lead, and air pollution alter methylation in brain development genes (52–54). However, the evidence surrounding these external factors is less robust than that for early-life stress and warrants further investigation to become clinically actionable. In the case of early-life stress, commonly experienced through childhood abuse, DNA methylation of stress-regulating genes (i.e. NR3C1, FKBP5) increases, hypermethylating the glucocorticoid receptor and altering the HPA axis (10). This effect stems into social environments that affect methylation of social, stress, and emotion-related genes; and, in this context, studies demonstrate that having a high-quality rearing lowers OXTR and NR3C1 methylation (55). Despite these modifications, maladaptive epigenetic changes can be reversed or normalized by psychological interventions such as cognitive behavioral therapy (CBT) and mindfulness, and studies have examined the potential of these interventions to reduce methylation of FKBP5 and SLC6A4 and for psychiatric medications like SSRIs, antipsychotics and the HDAC inhibitor valproate to alter epigenetic landscapes (56, 57).
3 Clinically relevant epigenetically regulated genes in psychiatry
3.1 Brain-derived neurotrophic factor
The brain-derived neurotrophic factor (BDNF) is an essential protein in the brain and nervous system. BDNF is a growth factor that supports neuroplasticity, neuronal survival, neurogenesis, and contributes to neural repair and recovery after brain injuries like stroke or trauma (58). BDNF is highly expressed throughout the brain including the hippocampus, prefrontal cortex, amygdala, cortex, and basal forebrain (59). In the context of cognitive function, higher BDNF levels are associated with improved cognitive performance including memory, attention, and problem-solving (60). In mood regulation, low BDNF levels are linked to depression and anxiety. The BDNF gene has multiple promoters which are prone to epigenetic regulation. For example, exon IV of BDNF is highly sensitive to environmental signals and hypermethylation of BDNF promoter regions, especially exon IV, reduces BDNF expression (61).
Studies have reported reduced BDNF expression in people with MDD, PTSD, schizophrenia, and after early-life stress or childhood trauma (62). In depression, studies report low peripheral BDNF levels and higher promoter methylation, while antidepressants and electroconvulsive therapy (ECT) increase BDNF expression and reverse methylation (63, 64). In BD, BDNF epigenetic changes are state-dependent, with lower levels during manic and depressive episodes compared with euthymia (65). Studies in PTSD and suicidality demonstrate that early-life trauma is associated with long-lasting BDNF promoter methylation, affecting stress reactivity and fear extinction, and for suicide victims, BDNF promoter hypermethylation is observed in the prefrontal cortex and hippocampus (66–68).
BDNF is subject to other forms of epigenetic regulation, including histone modifications, whereby acetylation enhances gene expression and histone deacetylation represses expression (69). Selective serotonin reuptake inhibitors (SSRIs) and HDAC inhibitors can increase histone acetylation at the BDNF loci therefore boosting expressions (70). In addition to DNA and histone modifications, BDNF expression is epigenetically regulated by miRNAs (including miR-132 and miR-206), which can bind BDNF mRNA and suppress translation. Dysregulation of these miRNAs is linked to mood disorder and schizophrenia (71)Although important progress has been made, notable limitations remain. The epigenetic regulation of BDNF varies by isoform and promoter, but this distinction is often overlooked, and most studies are further limited by the absence of rigorously controlled longitudinal cohorts. A summary of these epigenetic alterations has been captured in Table 1.
3.2 Catechol-O-methyltransferase
The catechol-O-methyltransferase (COMT) enzyme metabolizes catecholamines including the neurotransmitters dopamine, norepinephrine, and epinephrine by transferring a methyl group from S-adenosylmethionine (SAM) to these catecholamines thus facilitating catecholamine degradation and regulating their levels in various tissues including the brain (72). COMT exists in two primary forms: soluble COMT (S-COMT) and membrane-bound COMT (MB-COMT). S-COMT is predominantly found in peripheral tissues like the liver, kidneys, and blood and plays a critical role in metabolizing catecholamines circulating in the body. MB-COMT is mainly produced by nerve cells in the brain and is integral to the degradation of neurotransmitters within the central nervous system (73). In the brain, COMT regulates dopamine levels in regions crucial for cognitive functions like the prefrontal cortex. Based on the concept that COMT regulates dopamine metabolism, epigenetic regulation of COMT influences not only gene expression and enzyme activity but neurotransmitter metabolism and alters dopamine signaling, key features in schizophrenia and cognitive dysfunction (74). In fact, methylation of the COMT promoter has been associated with reduced gene expression in both brain and peripheral tissues, leading to increased dopamine availability in the prefrontal cortex (75).
Studies in schizophrenia support that hypomethylation of the COMT promoter increases COMT activity and enhances dopamine degradation in the prefrontal cortex. This dopamine deficit could contribute to the cognitive impairment and negative symptoms which are 2 of the three main sub-syndromes in individuals with schizophrenia (76). In BD studies, dysregulation of dopamine metabolism may contribute to the signature mood instability of this disorder (77). Studies in depression show that hypermethylation of COMT is associated with MDD, and often correlated with stress exposure and treatment response (78). In PTSD, increased COMT methylation is associated with impaired fear inhibition, which affects stress responsivity via dopamine regulation (79). In the context of environmental interactions, cigarette smoking is associated with hypermethylation of the MB-COMT promoter, while cannabis use interacts with the COMT Val158Met genotype/epigenotype to influence psychosis risk (80, 81). Although less well characterized, histone modifications also affect COMT expression, and the general mechanisms such as histone acetylation and methylation are known to influence gene expression in neuropsychiatric contexts (82). Several limitations remain in these studies, notably the requirement for larger cohorts, independent study replication, and efforts to disentangle genotype-epigenotype interactions, such as demonstrating how Val/Met allele effects differ according to methylation status. A summary of these epigenetic alterations has been captured in Table 1.
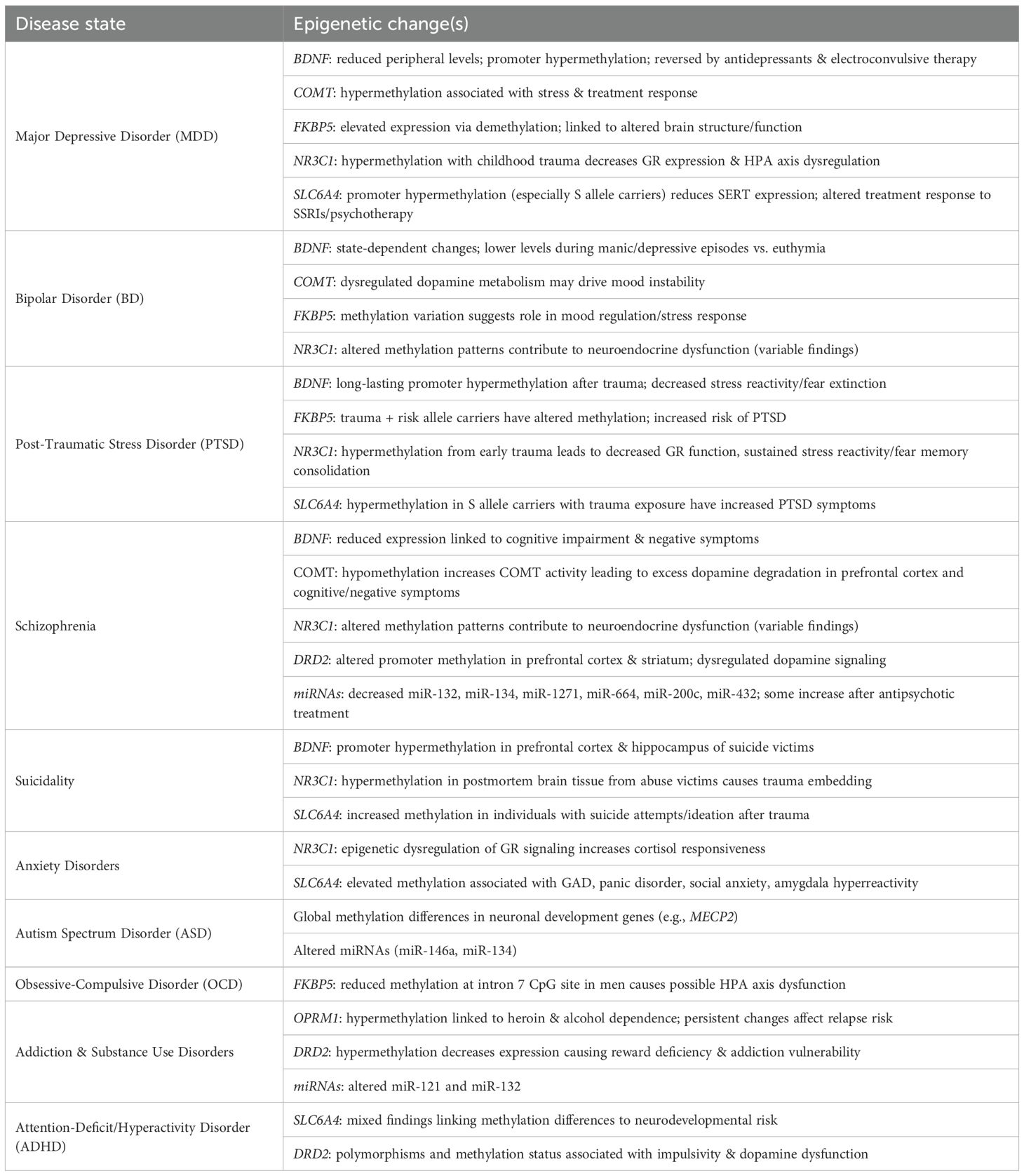
Table 1. Epigenetic alterations across psychiatric disorders.
3.3 FK506 binding protein 5
The FK506 Binding Protein 5 (FKBP5) gene encodes FKBP51, a co-chaperone protein that modulates the glucocorticoid receptor (GR) complex and affects cortisol signaling (83). By altering GR sensitivity, FKBP5 plays a critical role in stress hormone regulation, influencing the HPA axis, and impacting various psychiatric conditions (84). Stress exposure such as early-life stress decreases methylation of FKBP5 and upregulates FKBP5 gene expression which reduces GR sensitivity and may cause an exaggerated stress response (85). In an interplay between the genome and epigenome, polymorphisms in the FKBP5 gene (such as rs1360780) are linked to differential methylation patterns in response to stress that influence gene expression and stress reactivity (56).
In individuals with PTSD, carriers of FKBP5 polymorphisms who experience childhood trauma exhibit altered FKBP5 methylation, which increases their risk of developing the disorder (86). Stress has also been shown to influence chromatin accessibility at the FKBP5 locus, although this area remains less well characterized than DNA methylation (87). Additionally, microRNAs such as miR-15 and miR-511 have been proposed to regulate FKBP5 expression, linking it to pathways of inflammation and stress response (88). MDD studies show that FKBP5 expression is epigenetically elevated and associated with structural and functional alterations in brain regions involved in emotional processing further contributing to MDD pathophysiology (89). With BD, methylation variations in FKBP5 expression are observed which suggests a role in mood regulation and stress response mechanisms (90). In OCD, reduced DNA methylation at the FKBP5 intron 7 CpG site has been reported in male patients relative to controls. While these findings suggest a potential role for FKBP5 methylation in the pathogenesis of OCD, additional studies are required to confirm whether altered DNA methylation at intron 7 contributes to disease mechanisms in concert with HPA axis dysregulation (91). Therapeutic strategies shown to normalize FKBP5 methylation include antidepressants, cognitive behavioral therapy (CBT), and mindfulness practices (92–94). Importantly, FKBP5 alterations are not observed in all trauma survivors, underscoring limitations in biomarker specificity and sensitivity that warrant further investigation. A summary of these epigenetic alterations has been captured in Table 1.
3.4 Glucocorticoid receptor
The NR3C1 gene encodes the glucocorticoid receptor (GR), the main receptor for cortisol and a key regulator of the HPA axis (95). As the body’s primary stress hormone, cortisol binds to GR allowing GR to translocate to the nucleus and regulate the expression of stress-responsive genes. GR signaling is critical for regulating the HPA axis feedback loop and controls inflammation, immune responses, and metabolism while modulating neurodevelopment, mood, and cognitive function (96). Based on the concept that NR3C1 is critical for glucocorticoid signaling and stress regulation, hypermethylation of NR3C1 in individuals exposed to childhood adversity correlates with increased vulnerability to PTSD, depression, and suicide risk (2). Targeting NR3C1 methylation patterns may provide therapeutic potential for stress-related psychiatric disorders (97).
The NR3C1 gene is said to be environmentally programmed by stress reactivity because early-life adversity such as childhood abuse, neglect, and maternal stress are strongly associated with hypermethylation of exon 1F. The NR3C1 exon 1F is a critical promoter region that undergoes epigenetic regulation via DNA methylation where hypermethylation of this region reduces NR3C1 expression, decreases GR availability, impairs cortisol feedback sensitivity, and heightens or prolongs stress responses (97). In MDD, hypermethylation of NR3C1 promoter regions in individuals with childhood trauma is associated with reduced GR expression and dysregulation of the HPA axis, a hallmark of depression (14). PTSD studies show NR3C1 hypermethylation from early trauma leads to decreased GR function, sustained stress reactivity, and fear memory consolidation (56). In studies of post-mortem brain tissue from suicide completers with a history of abuse, NR3C1 methylation is increased, suggesting a biological embedding of trauma at the molecular level (43). Several studies on anxiety disorders link epigenetic dysregulation of GR signaling with generalized anxiety and panic disorder, likely through heightened cortisol responsiveness (98). NR3C1 methylation patterns are also altered in schizophrenia and BD, which may contribute to neuroendocrine dysfunction in these disorders, although these findings are more variable (99).
3.5 Serotonin transporter (SLC6A4/5-HTTLPR)
The SLC6A4 gene encodes the serotonin transporter (SERT), a membrane protein responsible for the reuptake of serotonin (5-HT) from the synaptic cleft back into presynaptic neurons (100). SLC6A4 regulates serotonin availability in the brain, impacts mood, emotion, anxiety, and stress regulation, and is the main target of SSRIs used in treating MDD and anxiety disorders (101). The serotonin-transporter-linked polymorphic region (5-HTTLPR) is a region in the SLC6A4 promoter with two main alleles: the short (s) allele and the long (l) allele. The short allele has lower transcriptional efficiency and reduced SERT expression and is often associated with increased emotional reactivity and greater susceptibility to stress whereas the long allele has higher transcription and increased SERT expression (102). SLC6A4 is epigenetically regulated by DNA methylation influencing SLC6A4 expression independently or interactively with 5-HTTLPR genotype (103). Methylation of CpG sites in the SLC6A4 promoter, especially near exon 1, and the 5-HTTLPR region reduces SLC6A4 expression lowering SERT expression, SERT availability, and increasing extracellular serotonin. This effect is often independent of genotype and can result from environmental exposures such as early-life trauma or stress. S allele carriers that experience early-life stress or trauma tend to show higher SLC6A4 promoter methylation suggesting that stress programs serotonin signaling via epigenetic changes (104). Childhood adversity, maternal depression, or prenatal stress increase SLC6A4 methylation and cause impaired stress resilience and altered emotional regulation later in life (105).
SLC6A4 hypermethylation, together with the presence of the S allele, is linked to an increased risk of depression because SERT expression and serotonin uptake are reduced, especially in those with childhood adversity (105). SSRIs may normalize some of these methylation changes and are often examined within gene x environment (GxE) models in psychiatric research, which also suggests that SLC6A4 methylation can be used as an indicator of treatment response to SSRIs or psychotherapy (106). Elevated SLC6A4 methylation is also associated with generalized anxiety disorder (GAD) and panic disorder, correlates with increased anxiety traits, social anxiety, and amygdala hyperreactivity, and may explain how altered serotonin signaling affects fear processing and threat detection (107). With PTSD, SLC6A4 methylation is increased in S allele carriers with trauma exposure. Several studies in veterans and trauma survivors show higher methylation at SLC6A4 CpGs in patients with PTSD symptoms, demonstrating that these epigenetic changes reflect persistent alterations in stress reactivity pathways and can serve as biomarkers of stress vulnerability (105). In suicide studies, individuals with a history of suicide attempts or ideation that have been exposed to trauma have increased SLC6A4 methylation (43). Some studies link neurodevelopmental risk for attention deficit hyperactivity disorder (ADHD) to differences in SLC6A4 methylation patterns although these findings are mixed (108). A summary of these epigenetic alterations has been captured in Table 1.
3.6 Dopamine Receptor D2
The DRD2 gene encodes the D2 subtype of the five dopamine receptors. As a G-protein-coupled receptor (GPCR), DRD2 is involved in modulating dopaminergic neurotransmission and plays a role as an inhibitory receptor by reducing neuronal excitability and dopamine synthesis via Gi/o protein coupling (109, 110). DRD2 expression is observed at high levels in the striatum, prefrontal cortex, and limbic regions to regulate reward processing, motivation and reinforcement, cognitive control, movement and motor coordination (111). There are two main D2 receptor isoforms that are synthesized by differential splicing of introns and exons form the D2 gene. The short isoform (D2S) is a presynaptic auto-receptor on dopamine neurons that regulates dopamine release, and the long isoform (D2L) is a postsynaptic receptor involved in postsynaptic signaling (112).
DRD2 is subject to epigenetic regulation and changes in DRD2 expression may impact dopamine signaling and contribute to neuropsychiatric disorders (113). For example, DNA hypermethylation of DRD2 promoter regions, such as in exon 1 or upstream CpG islands, downregulates receptor expression; on the contrary, hypomethylation increases DRD2 levels thereby altering dopamine sensitivity (114). Histone deacetylase inhibitors (HDACi) can increase DRD2 expression in certain contexts, suggesting chromatin remodeling as a DRD2 epigenetic regulator (28). MicroRNAs, such as miR-9 and miR-326, are another example of DRD2 epigenetic regulation since they target DRD2 mRNA and reduce translation. Dysregulation of these miRNAs is linked to changes in DRD2 signaling in psychiatric illness (115).
Postsynaptic D2 receptor overactivation, resulting from increased presynaptic dopamine release in the nucleus accumbens (located in the ventral striatum), is central to the dopamine hypothesis of schizophrenia (116). Postmortem studies show altered DRD2 methylation in the prefrontal cortex and striatum of individuals with schizophrenia (117). Studies in substance use disorders including cocaine, alcohol, and nicotine show that substance use can alter DRD2 methylation in animal and human models. In this context, reduced DRD2 expression from DNA hypermethylation is associated with reward deficiency and addiction vulnerability (118). In ADHD, DRD2 polymorphisms and methylation status are linked to impulsivity and dopaminergic dysfunction (119). A summary of these epigenetic alterations has been captured in Table 1.
4 Clinical implications of epigenetics in psychiatry
4.1 Epigenetic-based therapies
Epigenetic-based therapies are designed to regulate gene expression by targeting epigenomic processes while leaving DNA sequence itself unchanged. An example of an epigenetic-based therapy is HDAC inhibition, which increase histone acetylation thereby relaxing the chromatic and increasing gene expression (27). Valproic acid, sodium butyrate, and Vorinostat (SAHA) are examples of HDAC inhibitors that are currently used to target depression, schizophrenia, BD, PTSD and are being preclinically explored for ASD (120). Several therapies are in experimental stages including bromodomain and extra-terminal domain (BET) inhibitors, which bind to BET proteins and prevent them from interacting with acetylated histones modulating gene transcription. JQ1 is an investigational BET inhibitor for addiction, mood disorders, and preclinical neuroinflammation (121).
In addition to HDAC inhibitors, several experimental miRNA-based therapies for psychiatric disorders like schizophrenia, MDD, anxiety, and neurodevelopmental disorders are currently under investigation (122). These miRNA-based therapies include miR-124, miR-132, miR-135 and miR-146a (123). Other epigenetic-based therapies include epigenetic editing approaches using CRISPR/dCas9 for targeted DNA methylation or demethylation, histone editing by CRISPR-dCas9-TET1 for demethylation, and CRISPR-dCas9-HDAC for repression. These therapies focus on reversing stress-induced gene silencing, and studies demonstrate that such interventions can alleviate symptoms of MDD, anxiety, PTSD, while enhancing cognitive resilience (124). They are currently being evaluated in preclinical models of PTSD, addiction, and MDD (125, 126).
4.2 Precision psychiatry and future directions
In the era of precision psychiatry, understanding epigenetic patterns can refine diagnosis and improve clinical subtyping. For example, methylation of NR3C1, BDNF, and FKBP5 can differentiate subtypes of MDD, PTSD, or schizophrenia, help to identify stress-responsiveness compared with neurodevelopmental forms of illness, and be used as predictors of risk and resilience (5). Epigenetic markers like SLC6A4 or OXTR methylation can predict risk for MDD or anxiety after trauma and individual resilience profiles (127). This epigenetic information may be useful for detecting at-risk individuals such as trauma-exposed youth and providing early intervention (128). In the context of pharmaco-epigenetics, methylation of genes like BDNF, SLC6A4, and FKBP5 may predict response to SSRIs, psychotherapy, or electroconvulsive therapy (129). For example, clinical studies suggest that low BDNF methylation correlates with better SSRI response and high NR3C1 methylation predicts poorer CBT response in PTSD (56). Additionally, epigenetic biomarkers could be used to guide novel epigenetic therapies in patients with hypermethylated GAD1 or RELN who may benefit from HDAC inhibitors or DNMT inhibitors to restore expression of key neuroplasticity genes (130).
5 Limitations
Several limitations hinder the application of epigenetic findings as biomarkers in psychiatry. First, access to brain tissue is limited, and sampling relies on that from blood, saliva, or postmortem samples, which may not reflect central nervous system changes. Cell type specificity varies since epigenetic marks vary dramatically between neurons, glia, and other brain cells (45). Therefore, bulk tissue analyses can obscure important differences at the cellular level. As discussed, epigenetic modifications are dynamic and can change rapidly. Therefore, stress, diet, and/or medications allow for temporal variability, which may not be reflected in the long-term pattern. Different assays can yield inconsistent results. For example, bisulfite sequencing vs. ChIP-seq (chromatin immunoprecipitation followed by sequencing) can complicate reproducibility across studies. Lastly, most large-scale genome and epigenome reference datasets, including those used for methylation and chromatin analyses, are derived predominantly from individuals of European ancestry. Therefore, the current body of research lacks ancestral diversity among study populations and may obscure ancestry-specific epigenetic signatures or gene-environment interactions that contribute to psychiatric risk. Future research efforts should focus on expanding cohort diversity to improve the equity, accuracy, and translational relevance of epigenetic discoveries.
In addition to these limitations, there are several confounding effects, including lifestyle, socioeconomic factors, substance use, and comorbid medical conditions, that need to be considered, as they can shape the epigenome, making it difficult to isolate psychiatry-specific signatures. It is important to emphasize that epigenetic findings have not yet been validated for routine use in psychiatric practice. Lastly, because psychiatric disorders are influenced by thousands of genetic and environmental factors, an exclusive focus on epigenetics risks oversimplifying the complexity of disease etiology.
6 Conclusion
In this review, we have outlined the epigenetic mechanisms implicated in psychiatric disorders and their relation to mental health conditions. Extensive studies demonstrate that epigenetic modifications influence the development and progression of psychiatric disorders as well as human behavior. With ongoing research into the clinical applications of epigenetically informed interventions, it is plausible that such treatments will become part of routine psychiatric care. As highlighted, the transition toward precision psychiatry requires overcoming several limitations. Key challenges include the need for standardization across cell type specificity and assay protocols, rigorous control of confounding factors that may bias epigenetic findings, and integration of epigenetic data within the broader context of gene-environment interactions and systems biology. Future research should include large, longitudinal cohorts, tissue-specific analyses, and multi-omics integration, considering not only genomics but transcriptomics and proteomics as well. As investigation into epigenetic biomarkers continues to progress, and the understanding of the role of the epigenome alongside epigenome-targeting treatments develops, we look forward to the advances made in genomic medicine and the field of psychiatry.
Author contributions
RM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. CM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. JM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
CM is the Co-Founder of Biotia and is compensated by Thorne HealthTech. JM serves on the speakers’ bureau for Otsuka/Lundbeck, Teva Pharmaceuticals, Neurocrine Biosciences, Janssen, Intra-Cellular Therapies, Axsome Therapeutics, and Bristol Myer Squibb formerly Karuna Therapeutics and as a consultant/advisory board member for Axsome and Karuna Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nestler EJ and Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. (2010) 13:1161–9. doi: 10.1038/nn.2647
2. Turecki G and Meaney MJ. Effects of the social environment and stress on glucocorticoid receptor gene methylation: A systematic review. Biol Psychiatry. (2016) 79:87–96. doi: 10.1016/j.biopsych.2014.11.022
3. Schuebel K, Gitik M, Domschke K, and Goldman D. Making sense of epigenetics. IJNPPY. (2016) 19:pyw058. doi: 10.1093/ijnp/pyw058
4. Qureshi IA and Mehler MF. Epigenetic mechanisms underlying human epileptic disorders and the process of epileptogenesis. Neurobiol Disease. (2010) 39:53–60. doi: 10.1016/j.nbd.2010.02.005
5. Klengel T and Binder EB. Epigenetics of stress-related psychiatric disorders and gene × Environment interactions. Neuron. (2015) 86:1343–57. doi: 10.1016/j.neuron.2015.05.036
6. Peña CJ, Nestler EJ, and Bagot RC. Environmental programming of susceptibility and resilience to stress in adulthood in male mice. Front Behav Neurosci. (2019) 13:40. doi: 10.3389/fnbeh.2019.00040
7. Jaenisch R and Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. (2003) 33:245–54. doi: 10.1038/ng1089
8. Moore LD, Le T, and Fan G. DNA methylation and its basic function. Neuropsychopharmacol. (2013) 38:23–38. doi: 10.1038/npp.2012.112
9. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. (2002) 16:6–21. doi: 10.1101/gad.947102
10. McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. (2009) 12:342–8. doi: 10.1038/nn.2270
11. Cedar H and Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. (2009) 10:295–304. doi: 10.1038/nrg2540
12. Feng J and Nestler EJ. Epigenetic mechanisms of drug addiction. Curr Opin Neurobiology. (2013) 23:521–8. doi: 10.1016/j.conb.2013.01.001
13. Ferrer A, Labad J, Salvat-Pujol N, Barrachina M, Costas J, Urretavizcaya M, et al. BDNF genetic variants and methylation: effects on cognition in major depressive disorder. Transl Psychiatry. (2019) 9:265. doi: 10.1038/s41398-019-0601-8
14. Perroud N, Paoloni-Giacobino A, Prada P, Olié E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry. (2011) 1:e59–9. doi: 10.1038/tp.2011.60
15. Zhou J, Zhou D, Yan T, Chen W, Xie H, and Xiong Y. Association between CpG island DNA methylation in the promoter region of RELN and positive and negative types of schizophrenia. J Int Med Res. (2022) 50:3000605221100345. doi: 10.1177/03000605221100345
16. Dell׳Osso B, D׳Addario C, Palazzo MC, Benatti B, Camuri G, Galimberti D, et al. Epigenetic modulation of BDNF gene: Differences in DNA methylation between unipolar and bipolar patients. J Affect Disord. (2014) 166:330–3. doi: 10.1016/j.jad.2014.05.020
17. Schröter K, Brum M, Brunkhorst-Kanaan N, Tole F, Ziegler C, Domschke K, et al. Longitudinal multi-level biomarker analysis of BDNF in major depression and bipolar disorder. Eur Arch Psychiatry Clin Neurosci. (2020) 270:169–81. doi: 10.1007/s00406-019-01007-y
18. Nagarajan R, Hogart A, Gwye Y, Martin MR, and LaSalle JM. Reduced meCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. (2006) 1:172–82. doi: 10.4161/epi.1.4.3514
19. Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, Ott J, et al. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacol. (2009) 34:867–73. doi: 10.1038/npp.2008.108
20. Zhang H, Herman AI, Kranzler HR, Anton RF, Simen AA, and Gelernter J. Hypermethylation of OPRM1 promoter region in European Americans with alcohol dependence. J Hum Genet. (2012) 57:670–5. doi: 10.1038/jhg.2012.98
21. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
22. Renthal W and Nestler EJ. Histone acetylation in drug addiction. Semin Cell Dev Biol. (2009) 20:387–94. doi: 10.1016/j.semcdb.2009.01.005
23. Xia C, Tao Y, Li M, Che T, and Qu J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp Ther Med. (2020) 20(4):2923–40. doi: 10.3892/etm.2020.9073
24. Greer EL and Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. (2012) 13:343–57. doi: 10.1038/nrg3173
25. Rossetto D, Avvakumov N, and Côté J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics. (2012) 7:1098–108. doi: 10.4161/epi.21975
26. Du Q, Luu PL, Stirzaker C, and Clark SJ. Methyl-cpG-binding domain proteins: readers of the epigenome. Epigenomics. (2015) 7:1051–73. doi: 10.2217/epi.15.39
27. Grayson DR, Kundakovic M, and Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. (2010) 77:126–35. doi: 10.1124/mol.109.061333
28. Covington HEIII, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. (2009) 29:11451–60. doi: 10.1523/JNEUROSCI.1758-09.2009
30. Krol J, Loedige I, and Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. (2010) 11:597–610. doi: 10.1038/nrg2843
31. Im HI and Kenny PJ. MicroRNAs in neuronal function and dysfunction. Trends Neurosciences. (2012) 35:325–34. doi: 10.1016/j.tins.2012.01.004
32. The Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. (2011) 43:969–76. doi: 10.1038/ng.940
33. Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, et al. MiR-137: an important player in neural development and neoplastic transformation. Mol Psychiatry. (2017) 22:44–55. doi: 10.1038/mp.2016.150
34. He C, Wang Q, Fan D, Liu X, Bai Y, Zhang H, et al. MicroRNA-124 influenced depressive symptoms via large-scale brain connectivity in major depressive disorder patients. Asian J Psychiatry. (2024) 95:104025. doi: 10.1016/j.ajp.2024.104025
35. Fang Y, Qiu Q, Zhang S, Sun L, Li G, Xiao S, et al. Changes in miRNA-132 and miR-124 levels in non-treated and citalopram-treated patients with depression. J Affect Disord. (2018) 227:745–51. doi: 10.1016/j.jad.2017.11.090
36. Ni L, Zhu Y, Lv L, Zhang R, Xie S, and Zhang X. Peripheral blood miR-16-5p as a potential biomarker for distinguishing unmedicated bipolar disorder type II from major depressive disorder. J Affect Disord. (2025) 382:453–61. doi: 10.1016/j.jad.2025.04.126
37. Yu H-C, Wu J, Zhang H-X, Zhang G-L, Sui J, Tong W-W, et al. Alterations of miR-132 are novel diagnostic biomarkers in peripheral blood of schizophrenia patients. Prog Neuropsychopharmacol Biol Psychiatry. (2015) 63:23–9. doi: 10.1016/j.pnpbp.2015.05.007
38. Issler O, Haramati S, Paul ED, Maeno H, Navon I, Zwang R, et al. MicroRNA 135 is essential for chronic stress resiliency, antidepressant efficacy, and intact serotonergic activity. Neuron. (2014) 83:344–60. doi: 10.1016/j.neuron.2014.05.042
39. Clapier CR, Iwasa J, Cairns BR, and Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. (2017) 18:407–22. doi: 10.1038/nrm.2017.26
40. Hargreaves DC and Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. (2011) 21:396–420. doi: 10.1038/cr.2011.32
41. Ho L and Crabtree GR. Chromatin remodelling during development. Nature. (2010) 463:474–84. doi: 10.1038/nature08911
42. Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. (2012) 485:242–5. doi: 10.1038/nature11011
43. Labonté B, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I, et al. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry. (2012) 69(7):722–31. doi: 10.1001/archgenpsychiatry.2011.2287
44. Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. (2016) 48:1193–203. doi: 10.1038/ng.3646
45. Kim SS, Truong B, Jagadeesh K, Dey KK, Shen AZ, Raychaudhuri S, et al. Leveraging single-cell ATAC-seq and RNA-seq to identify disease-critical fetal and adult brain cell types. Nat Commun. (2024) 15:563. doi: 10.1038/s41467-024-44742-0
46. Bryois J, Garrett ME, Song L, Safi A, Giusti-Rodriguez P, Johnson GD, et al. Evaluation of chromatin accessibility in prefrontal cortex of schizophrenia cases and controls. Genomics. (2017) 9(1):3121. doi: 10.1101/141986
47. Duttke SH, Montilla-Perez P, Chang MW, Li H, Chen H, Carrette LLG, et al. Glucocorticoid receptor-regulated enhancers play a central role in the gene regulatory networks underlying drug addiction. Front Neurosci. (2022) 16:858427. doi: 10.3389/fnins.2022.858427
48. Cruz B, Vozella V, Carper BA, Xu JC, Kirson D, Hirsch S, et al. FKBP5 inhibitors modulate alcohol drinking and trauma-related behaviors in a model of comorbid post-traumatic stress and alcohol use disorder. Neuropsychopharmacol. (2023) 48:1144–54. doi: 10.1038/s41386-022-01497-w
49. Feil R and Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. (2012) 13:97–109. doi: 10.1038/nrg3142
50. Waterland RA and Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. (2003) 23:5293–300. doi: 10.1128/MCB.23.15.5293-5300.2003
51. Gomez-Pinilla F, Zhuang Y, Feng J, Ying Z, and Fan G. Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur J Neurosci. (2011) 33:383–90. doi: 10.1111/j.1460-9568.2010.07508.x
52. Byun HM and Baccarelli AA. Environmental exposure and mitochondrial epigenetics: study design and analytical challenges. Hum Genet. (2014) 133:247–57. doi: 10.1007/s00439-013-1417-x
53. Kundakovic M, Gudsnuk K, Herbstman JB, Tang D, Perera FP, and Champagne FA. DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci USA. (2015) 112:6807–13. doi: 10.1073/pnas.1408355111
54. Senut M-C, Cingolani P, Sen A, Kruger A, Shaik A, Hirsch H, et al. Epigenetics of early-life lead exposure and effects on brain development. Epigenomics. (2012) 4:665–74. doi: 10.2217/epi.12.58
55. Krol KM, Puglia MH, Morris JP, Connelly JJ, and Grossmann T. Epigenetic modification of the oxytocin receptor gene is associated with emotion processing in the infant brain. Dev Cogn Neurosci. (2019) 37:100648. doi: 10.1016/j.dcn.2019.100648
56. Yehuda R, Daskalakis NP, Desarnaud F, Makotkine I, Lehrner AL, Koch E, et al. Epigenetic biomarkers as predictors and correlates of symptom improvement following psychotherapy in combat veterans with PTSD. Front Psychiatry. (2013) 4:118. doi: 10.3389/fpsyt.2013.00118
57. Nasca C, Bigio B, Lee FS, Young SP, Kautz MM, Albright A, et al. Acetyl-L-carnitine deficiency in patients with major depressive disorder. Proc Natl Acad Sci USA. (2018) 115:8627–32. doi: 10.1073/pnas.1801609115
58. Binder DK and Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. (2004) 22:123–31. doi: 10.1080/08977190410001723308
59. Lu B and Martinowich K. Cell biology of BDNF and its relevance to schizophrenia. Novartis Found Symp. (2008) 289:119–29. doi: 10.1002/9780470751251.ch10
60. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. (2003) 112:257–69. doi: 10.1016/S0092-8674(03)00035-7
61. Lubin FD, Roth TL, and Sweatt JD. Epigenetic regulation of bdnf gene transcription in the consolidation of fear memory. J Neurosci. (2008) 28:10576–86. doi: 10.1523/JNEUROSCI.1786-08.2008
62. Keller S, Sarchiapone M, Zarrilli F, Videtic A, Ferraro A, Carli V, et al. Increased BDNF promoter methylation in the wernicke area of suicide subjects. Arch Gen Psychiatry. (2010) 67:258. doi: 10.1001/archgenpsychiatry.2010.9
63. Fuchikami M, Morinobu S, Segawa M, Okamoto Y, Yamawaki S, Ozaki N, et al. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PloS One. (2011) 6:e23881. doi: 10.1371/journal.pone.0023881
64. Kleimann A, Kotsiari A, Sperling W, Gröschl M, Heberlein A, Kahl KG, et al. BDNF serum levels and promoter methylation of BDNF exon I, IV and VI in depressed patients receiving electroconvulsive therapy. J Neural Transm. (2015) 122:925–8. doi: 10.1007/s00702-014-1336-6
65. Simões Fernandes B, Gama CS, Ceresér KM, Yatham LN, Fries GR, Colpo G, et al. Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: A systematic review and meta-regression analysis. J Psychiatr Res. (2011) 45:995–1004. doi: 10.1016/j.jpsychires.2011.03.002
66. Botella-López A, Burgaya F, Gavín R, García-Ayllón MS, Gómez-Tortosa E, Peña-Casanova J, et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc Natl Acad Sci USA. (2006) 103:5573–8. doi: 10.1073/pnas.0601279103
67. Kouter K, Zupanc T, and Videtič Paska A. Genome-wide DNA methylation in suicide victims revealing impact on gene expression. J Affect Disord. (2019) 253:419–25. doi: 10.1016/j.jad.2019.04.077
68. Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, and Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. (2003) 60:804. doi: 10.1001/archpsyc.60.8.804
69. Tsankova NM, Kumar A, and Nestler EJ. Histone Modifications at Gene Promoter Regions in Rat Hippocampus after Acute and Chronic Electroconvulsive Seizures. J Neurosci. (2004) 24:5603–10. doi: 10.1523/JNEUROSCI.0589-04.2004
70. Schroeder FA, Lin CL, Crusio WE, and Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. (2007) 62:55–64. doi: 10.1016/j.biopsych.2006.06.036
71. Gao J, Wang W-Y, Mao Y-W, Gräff J, Guan J-S, Pan L, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. (2010) 466:1105–9. doi: 10.1038/nature09271
72. Männistö PT and Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. (1999) 51:593–628. doi: 10.1016/S0031-6997(24)01423-6
73. Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. (2004) 75:807–21. doi: 10.1086/425589
74. Abdolmaleky HM, Cheng K-H, Faraone SV, Wilcox M, Glatt SJ, Gao F, et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. (2006) 15:3132–45. doi: 10.1093/hmg/ddl253
75. Drabe M, Rullmann M, Luthardt J, Boettcher Y, Regenthal R, Ploetz T, et al. Serotonin transporter gene promoter methylation status correlates with in vivo prefrontal 5-HTT availability and reward function in human obesity. Transl Psychiatry. (2017) 7:e1167–7. doi: 10.1038/tp.2017.133
76. Nohesara S, Ghadirivasfi M, Mostafavi S, Eskandari M-R, Ahmadkhaniha H, Thiagalingam S, et al. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J Psychiatr Res. (2011) 45:1432–8. doi: 10.1016/j.jpsychires.2011.06.013
77. Lachman HM, Papolos DF, Saito T, Yu YM, Szumlanski CL, and Weinshilboum RM. Human catechol-O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics. (1996) 6:243–50. doi: 10.1097/00008571-199606000-00007
78. Wiegand A, Blickle A, Brückmann C, Weller S, Nieratschker V, and Plewnia C. Dynamic DNA methylation changes in the COMT gene promoter region in response to mental stress and its modulation by transcranial direct current stimulation. Biomolecules. (2021) 11:1726. doi: 10.3390/biom11111726
79. Norrholm SD, Jovanovic T, Smith AK, Binder E, Klengel T, Conneely K, et al. Differential genetic and epigenetic regulation of catechol-O-methyltransferase is associated with impaired fear inhibition in posttraumatic stress disorder. Front Behav Neurosci. (2013) 7:30. doi: 10.3389/fnbeh.2013.00030
80. Xu Q, Ma JZ, Payne TJ, and Li MD. Determination of methylated cpG sites in the promoter region of catechol-O-methyltransferase (COMT) and their involvement in the etiology of tobacco smoking. Front Psychiatry. (2010) 1:16. doi: 10.3389/fpsyt.2010.00016
81. Van Der Knaap LJ, Schaefer JM, Franken IHA, Verhulst FC, Van Oort FVA, and Riese H. Catechol-O-methyltransferase gene methylation and substance use in adolescents: the TRAILS study. Genes Brain Behavior. (2014) 13:618–25. doi: 10.1111/gbb.12147
82. Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. (2016) 19:40–7. doi: 10.1038/nn.4181
83. Zannas AS, Wiechmann T, Gassen NC, and Binder EB. Gene–stress–epigenetic regulation of FKBP5: clinical and translational implications. Neuropsychopharmacol. (2016) 41:261–74. doi: 10.1038/npp.2015.235
84. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. (2009) 34:S186–95. doi: 10.1016/j.psyneuen.2009.05.021
85. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nat Neurosci. (2013) 16:33–41. doi: 10.1038/nn.3275
86. Klengel T and Binder EB. FKBP5 allele-specific epigenetic modification in gene by environment interaction. Neuropsychopharmacol. (2015) 40:244–6. doi: 10.1038/npp.2014.208
87. Caradonna SG, Paul MR, and Marrocco J. An allostatic epigenetic memory on chromatin footprints after double-hit acute stress. Neurobiol Stress. (2022) 20:100475. doi: 10.1016/j.ynstr.2022.100475
88. Maurel OM, Torrisi SA, Barbagallo C, Purrello M, Salomone S, Drago F, et al. Dysregulation of miR-15a-5p, miR-497a-5p and miR-511-5p Is Associated with Modulation of BDNF and FKBP5 in Brain Areas of PTSD-Related Susceptible and Resilient Mice. Int J Mol Sci. (2021) 22:5157. doi: 10.3390/ijms22105157
89. Höhne N, Poidinger M, Merz F, Pfister H, Brückl T, Zimmermann P, et al. FKBP5 genotype-dependent DNA methylation and mRNA regulation after psychosocial stress in remitted depression and healthy controls. Int J Neuropsychopharmacol. (2015) 18:pyu087–7. doi: 10.1093/ijnp/pyu087
90. Menke A, Arloth J, Pütz B, Weber P, Klengel T, Mehta D, et al. Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacol. (2012) 37:1455–64. doi: 10.1038/npp.2011.331
91. Seo JH, Kim ST, Jeon S, Kang JI, and Kim SJ. Sex-dependent association of DNA methylation of HPA axis-related gene FKBP5 with obsessive-compulsive disorder. Psychoneuroendocrinology. (2023) 158:106404. doi: 10.1016/j.psyneuen.2023.106404
92. Zou Z, Huang Y, Maes M, Wang J, He Y, Min W, et al. Effects of antidepressant on FKBP51 mRNA expression and neuroendocrine hormones in patients with panic disorder. BMC Psychiatry. (2024) 24:269. doi: 10.1186/s12888-024-05704-4
93. Roberts S, Keers R, Breen G, Coleman JRI, Jöhren P, Kepa A, et al. DNA methylation of FKBP5 and response to exposure-based psychological therapy. Am J Med Genet Pt B. (2019) 180:150–8. doi: 10.1002/ajmg.b.32650
94. Bishop JR, Lee AM, Mills LJ, Thuras PD, Eum S, Clancy D, et al. Methylation of FKBP5 and SLC6A4 in relation to treatment response to mindfulness based stress reduction for posttraumatic stress disorder. Front Psychiatry. (2018) 9:418. doi: 10.3389/fpsyt.2018.00418
95. Oakley RH and Cidlowski JA. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J Allergy Clin Immunol. (2013) 132:1033–44. doi: 10.1016/j.jaci.2013.09.007
96. McEwen BS and Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. (2013) 79:16–29. doi: 10.1016/j.neuron.2013.06.028
97. Palma-Gudiel H, Córdova-Palomera A, Leza JC, and Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci Biobehav Rev. (2015) 55:520–35. doi: 10.1016/j.neubiorev.2015.05.016
98. Tyrka AR, Price LH, Marsit C, Walters OC, and Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PloS One. (2012) 7:e30148. doi: 10.1371/journal.pone.0030148
99. Tyrka AR, Price LH, Marsit C, Walters OC, and Carpenter LL. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J Am Acad Child Adolesc Psychiatry. (2014) 53:417–424.e5. doi: 10.1016/j.jaac.2013.12.025
100. Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. (1996) 274:1527–31. doi: 10.1126/science.274.5292.1527
101. Canli T and Lesch KP. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci. (2007) 10:1103–9. doi: 10.1038/nn1964
102. Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, et al. Serotonin transporter genetic variation and the response of the human amygdala. Science. (2002) 297:400–3. doi: 10.1126/science.1071829
103. Booij L, Szyf M, Carballedo A, Frey E-M, Morris D, Dymov S, et al. DNA methylation of the serotonin transporter gene in peripheral cells and stress-related changes in hippocampal volume: A study in depressed patients and healthy controls. PloS One. (2015) 10:e0119061. doi: 10.1371/journal.pone.0119061
104. Van IJzendoorn MH, Caspers K, Bakermans-Kranenburg MJ, Beach SRH, and Philibert R. Methylation matters: interaction between methylation density and serotonin transporter genotype predicts unresolved loss or trauma. Biol Psychiatry. (2010) 68:405–7. doi: 10.1016/j.biopsych.2010.05.008
105. Kang H-J, Kim J-M, Stewart R, Kim S-Y, Bae K-Y, Kim S-W, et al. Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog Neuropsychopharmacol Biol Psychiatry. (2013) 44:23–8. doi: 10.1016/j.pnpbp.2013.01.006
106. Okada S, Morinobu S, Fuchikami M, Segawa M, Yokomaku K, Kataoka T, et al. The potential of SLC6A4 gene methylation analysis for the diagnosis and treatment of major depression. J Psychiatr Res. (2014) 53:47–53. doi: 10.1016/j.jpsychires.2014.02.002
107. Furmark T, Appel L, Henningsson S, Ahs F, Faria V, Linnman C, et al. A link between serotonin-related gene polymorphisms, amygdala activity, and placebo-induced relief from social anxiety. J Neurosci. (2008) 28:13066–74. doi: 10.1523/JNEUROSCI.2534-08.2008
108. Mooney MA, Ryabinin P, Wilmot B, Bhatt P, Mill J, and Nigg JT. Large epigenome-wide association study of childhood ADHD identifies peripheral DNA methylation associated with disease and polygenic risk burden. Transl Psychiatry. (2020) 10:8. doi: 10.1038/s41398-020-0710-4
109. Beaulieu JM and Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. (2011) 63:182–217. doi: 10.1124/pr.110.002642
110. Missale C, Nash SR, Robinson SW, Jaber M, and Caron MG. Dopamine receptors: from structure to function. Physiol Rev. (1998) 78:189–225. doi: 10.1152/physrev.1998.78.1.189
111. Volkow ND, Wang GJ, Fowler JS, and Telang F. Overlapping neuronal circuits in addiction and obesity: evidence of systems pathology. Phil Trans R Soc B. (2008) 363:3191–200. doi: 10.1098/rstb.2008.0107
112. Usiello A, Paladini AA, Gargiulo A, Di Benedetto M, Florio E, Di Cunto F, et al. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. (2000) 408:199–203. doi: 10.1038/35041572
113. Abdolmaleky HM, Smith CL, Faraone SV, Shafa R, Stone W, Glatt SJ, et al. Methylomics in psychiatry: Modulation of gene–environment interactions may be through DNA methylation. Am J Med Genet Pt B. (2004) 127B:51–9. doi: 10.1002/ajmg.b.20142
114. Viana J, Hannon E, Dempster E, Pidsley R, Macdonald R, Knox O, et al. Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum Mol Genet. (2016) 26(1):210–25. doi: 10.1093/hmg/ddw373
115. Shi S, Leites C, He D, Schwartz D, Moy W, Shi J, et al. MicroRNA-9 and microRNA-326 regulate human dopamine D2 receptor expression, and the microRNA-mediated expression regulation is altered by a genetic variant. J Biol Chem. (2014) 289:13434–44. doi: 10.1074/jbc.M113.535203
116. Howes OD and Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bulletin. (2009) 35:549–62. doi: 10.1093/schbul/sbp006
117. Zhang TY and Meaney MJ. Epigenetics and the environmental regulation of the genome and its function. Annu Rev Psychol. (2010) 61:439–66. doi: 10.1146/annurev.psych.60.110707.163625
118. Nielsen DA, Utrankar A, Reyes JA, Simons DD, and Kosten TR. Epigenetics of drug abuse: predisposition or response. Pharmacogenomics. (2012) 13:1149–60. doi: 10.2217/pgs.12.94
119. Suderman M, Borghol N, Pappas JJ, Pinto Pereira SM, Pembrey M, Hertzman C, et al. Molecular genetics of attention-deficit/hyperactivity disorder: an overview. Eur Child Adolesc Psychiatry. (2010) 19:237–57. doi: 10.1007/s00787-010-0090-z
120. Tsankova N, Renthal W, Kumar A, and Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. (2007) 8:355–67. doi: 10.1038/nrn2132
121. Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. (2004) 47:24–32. doi: 10.1016/j.neuropharm.2004.06.031
122. Mellios N and Sur M. The emerging role of microRNAs in schizophrenia and autism spectrum disorders. Front Psychiatry. (2012) 3:39. doi: 10.3389/fpsyt.2012.00039
123. Bocchio-Chiavetto L, Maffioletti E, Bettinsoli P, Giovannini C, Bignotti S, Tardito D, et al. Blood microRNA changes in depressed patients during antidepressant treatment. Eur Neuropsychopharmacol. (2013) 23:602–11. doi: 10.1016/j.euroneuro.2012.06.013
124. Rahman MF and McGowan PO. Cell-type-specific epigenetic effects of early life stress on the brain. Transl Psychiatry. (2022) 12:326. doi: 10.1038/s41398-022-02076-9
125. Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA methylation in the mammalian genome. Cell. (2016) 167:233–247.e17. doi: 10.1016/j.cell.2016.08.056
126. Thakore PI, D'Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. (2015) 12:1143–9. doi: 10.1038/nmeth.3630
127. Provençal N and Binder EB. The effects of early life stress on the epigenome: From the womb to adulthood and even before. Exp Neurology. (2015) 268:10–20. doi: 10.1016/j.expneurol.2014.09.001
128. Cicchetti D and Handley ED. Child maltreatment and the development of substance use and disorder. Neurobiol Stress. (2019) 10:100144. doi: 10.1016/j.ynstr.2018.100144
129. Mohammadi S, Beh-Pajooh A, Ahmadimanesh M, Amini M, Ghazi-Khansari M, Moallem SA, et al. Evaluation of DNA methylation in BDNF, SLC6A4, NR3C1 and FKBP5 before and after treatment with selective serotonin-reuptake inhibitor in major depressive disorder. Epigenomics. (2022) 14:1269–80. doi: 10.2217/epi-2022-0246
Keywords: precision psychiatry, epigenetics, genomics, personalized medicine, psychiatric disorders
Citation: Montel Hayes R, Mason CE and Miller JJ (2025) The clinical use of epigenetics in psychiatry: a narrative review of epigenetic mechanisms, key candidate genes, and precision psychiatry. Front. Psychiatry 16:1671122. doi: 10.3389/fpsyt.2025.1671122
Received: 22 July 2025; Accepted: 16 October 2025;
Published: 29 October 2025.
Edited by:
Roseann E. Peterson, Suny Downstate Health Sciences University, United StatesReviewed by:
Le Wang, University of California, San Diego, United StatesTamer A. Addissouky, University of Menoufia, Egypt
Copyright © 2025 Montel Hayes, Mason and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel Montel Hayes, Y29udGFjdEByYWNoZWxtb250ZWxwaGQuY29t