 Simone Hauck1,2*
Simone Hauck1,2*- 1Graduate Program in Psychiatry and Behavioral Sciences, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 2Psychodynamic Psychiatry Lab, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
Burnout and trauma are often framed as psychosocial conditions or as dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis. Yet across more than two decades of clinical observation, I have repeatedly encountered a recurring metabolic signature that does not fit existing frameworks: persistent hyperferritinemia without hemochromatosis or overt inflammation, coexisting with low dehydroepiandrosterone-sulfate (DHEA-S) and preserved but gradually declining cortisol dynamics. This constellation is frequently observed in neurodivergent individuals and their families, with early signs already visible in childhood as mild anemia, elevated ferritin, low vitamin D, and behavioral hypervigilance. I propose that this pattern reflects a functional iron blockade (FIB), in which low-grade interleukin-6 signaling upregulates hepcidin, degrades ferroportin, and traps iron intracellularly. While protective against oxidative stress by reducing labile Fe²+, the adaptive cost is functional iron deficiency, impaired mitochondrial efficiency, refractory fatigue, and cognitive rigidity. Recognizing this mechanism may refine the understanding of stress-related fatigue and autistic burnout, prevent misdiagnosis as hemochromatosis or incidental hyperferritinemia, and guide research into integrative pathways linking iron metabolism, vitamin D status, and HPA dynamics. This perspective highlights FIB as a potential adaptive but costly response of stress physiology, disproportionately affecting neurodivergent phenotypes.
Introduction
Burnout and trauma are often discussed as psychosocial conditions or as dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis (1). However, persistent fatigue in these patients often remains unexplained by conventional markers. In long-term clinical practice, I have repeatedly observed a metabolic signature that diverges from existing frameworks: ferritin levels elevated well above expected ranges, in the absence of systemic inflammation, hepatic disease, or genetic hemochromatosis, coexisting with low DHEA-S and preserved morning cortisol.
Clinical observations
● Adult men: ferritin often 500–700 µg/L+, hemoglobin low-normal, transferrin saturation normal.
● Women of reproductive age: ferritin typically ranges 150–300 µg/L, elevated relative to expected menstrual losses, yet often accompanied by low-normal hemoglobin and hematocrit with normal transferrin saturation—a pattern suggestive of functional iron sequestration rather than true sufficiency.
● Children: early signs include mild anemia, elevated ferritin for age, low 25(OH)D, and behavioral hypervigilance.
● Adults with neurodivergent phenotypes: in some, vitamin D deficiency is persistent despite supplementation, suggesting higher metabolic consumption (2).
These findings are consistent across multiple families, suggesting a trait-like susceptibility. Importantly, they persist despite repeated negative evaluations for C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR), homeostatic iron regulator (HFE) gene mutations, or hepatic disease. They also appear disproportionately frequent in neurodivergent populations—autism spectrum, ADHD, giftedness, and dyslexia—although not exclusive to them (3). Typical laboratory profiles are listed in Table 1.
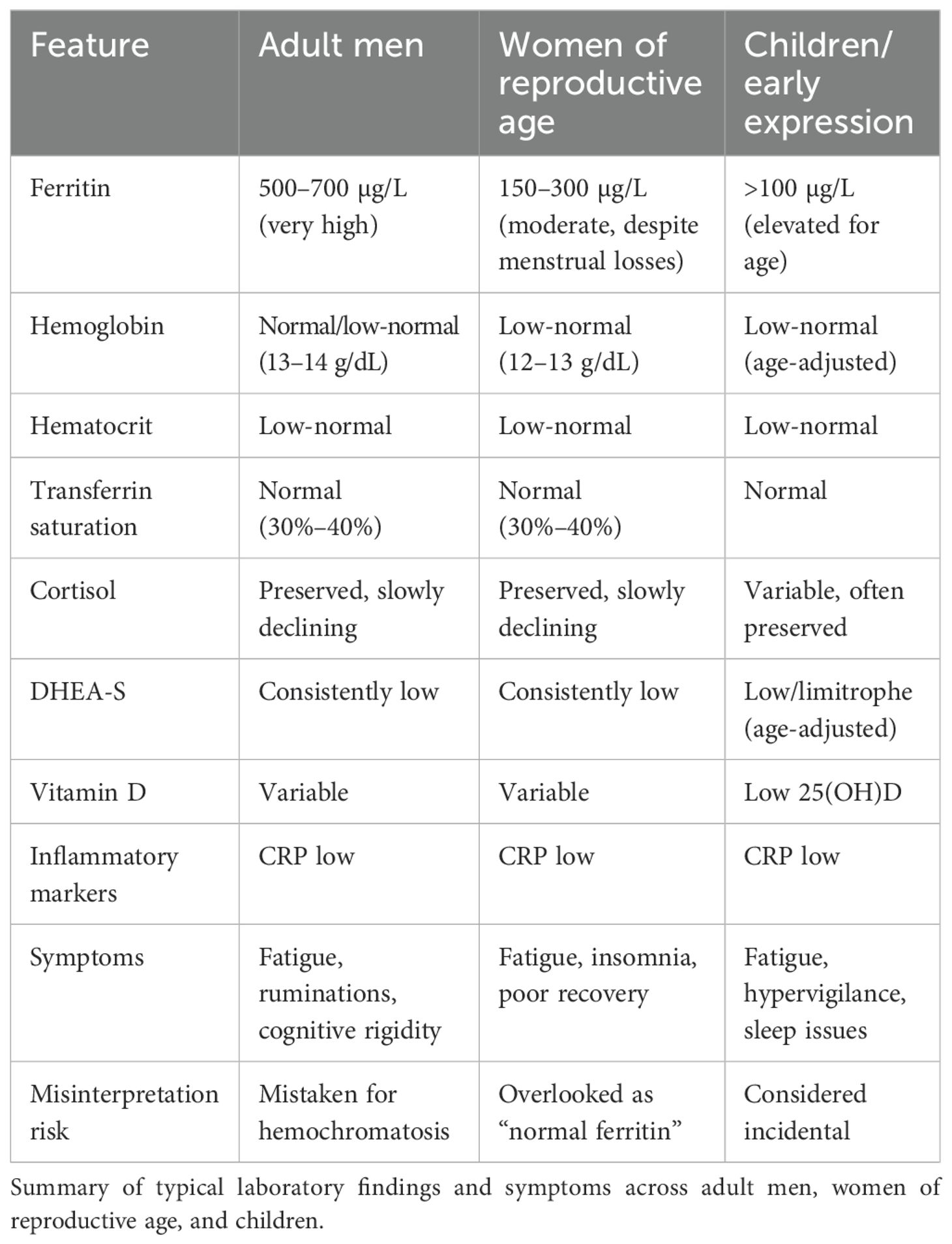
Table 1. Clinical profiles suggestive of functional iron blockade in chronic stress.
Proposed mechanism
Chronic stress maintains low-grade cytokine signaling (IL-6), upregulating hepcidin. Hepcidin degrades ferroportin, preventing iron export from hepatocytes, enterocytes, and macrophages (4). Iron accumulates as ferritin, whereas serum iron and transferrin saturation remain normal (5, 6). Experimental studies have shown that IL-6 directly induces hepcidin expression through activation of the STAT3 pathway in hepatocytes (7), and this same IL-6/STAT3 axis can be pharmacologically modulated—ferulic acid, for instance, downregulates IL-6 and HAMP expression, reducing hepcidin secretion (8). These findings substantiate the biological plausibility of the proposed mechanism.
● Adaptive function: oxidative protection—reducing circulating Fe²+ limits hydroxyl radical formation (Fenton reaction) under chronic stress.
● Clinical cost: functional iron deficiency—impaired mitochondrial efficiency, anemia-like fatigue despite “normal” indices, cognitive rigidity, poor recovery.
Chronic stress maintains low-grade IL-6 signaling, which upregulates hepatic hepcidin. Hepcidin degrades ferroportin, blocking iron export and leading to intracellular iron sequestration and increased ferritin (functional iron blockade, FIB). This adaptive mechanism protects against oxidative stress by reducing labile Fe2+ and limiting hydroxyl radical production (Fenton reaction). However, the clinical cost includes functional iron deficiency, mitochondrial inefficiency, refractory fatigue, and cognitive rigidity. This model illustrates FIB as an adaptive yet costly pathway of stress physiology. The adaptive mechanism is summarized in Figure 1.
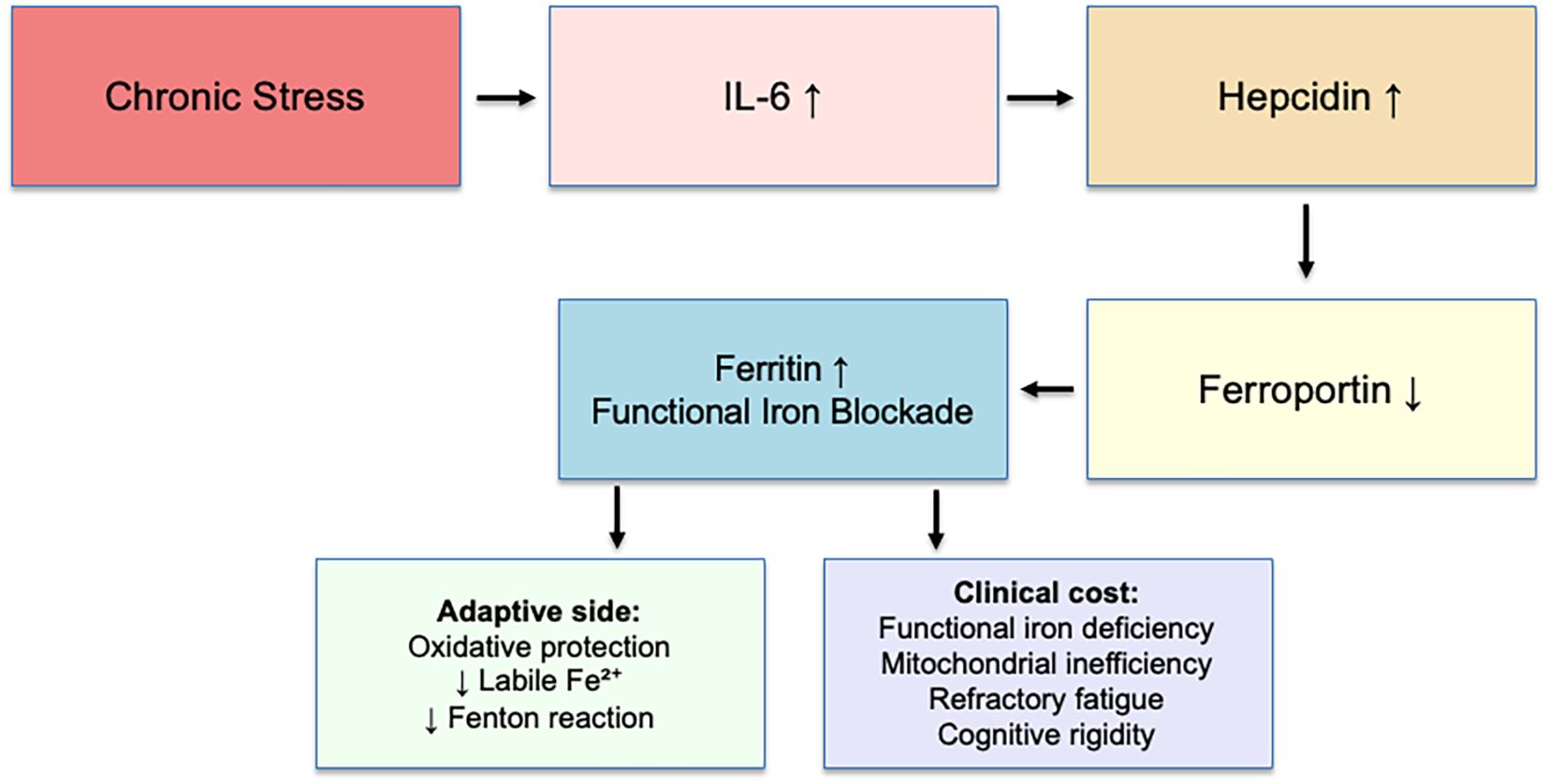
Figure 1. Functional iron blockade mechanism.
Preliminary operationalization of FIB
Based on recurring clinical and laboratory constellations, Functional Iron Blockade may be provisionally characterized by:
1. Ferritin ≥150 μg/L in women or ≥300 μg/L in men, persisting for ≥3 months in the absence of systemic inflammation (C-reactive protein <3 mg/L, ESR <15 mm/h) or HFE variants causing hemochromatosis;
2. Transferrin saturation within or below the normal range (20%–40%);
3. Low or low-normal hemoglobin/hematocrit relative to ferritin level, suggesting intracellular sequestration rather than true sufficiency;
4. Low DHEA-S (below the age-adjusted 25th percentile) and gradually declining cortisol dynamics despite preserved morning values; and
5. Clinical phenotype of refractory fatigue, cognitive rigidity, and poor stress recovery.
These parameters are intended for exploratory use in research and may evolve with longitudinal validation studies integrating ferritin, hepcidin, HPA hormones, and neurobehavioral outcomes.
Relationship between FIB and ferroptosis
The IL-6–hepcidin–ferroportin axis central to FIB closely interfaces with ferroptosis, an iron-dependent, lipid-peroxidation-driven form of regulated cell death. Chronic IL-6 signaling increases hepcidin transcription through the STAT3 pathway, promoting ferroportin degradation and intracellular Fe²+ retention (7, 8). This adaptive sequestration limits redox-active iron but simultaneously expands the labile iron pool susceptible to Fenton chemistry under oxidative stress (9, 10). Within neurons and other metabolically active cells, this milieu mirrors the pre-ferroptotic state described by Alves et al. (11): Mitochondrial efficiency is downregulated to contain oxidative flux, whereas GPX4-dependent defenses remain functional but strained. In this context, FIB can be interpreted as a sub-threshold, reversible restraint on ferroptotic potential—preserving oxidative safety at the expense of energetic capacity. Persistent IL-6–driven hepcidin expression, if unrelieved, may tilt this adaptive blockade toward maladaptive ferroptosis, as described in chronic stress and neuropsychiatric phenotypes (12).
Vitamin D–hepcidin axis
Vitamin D not only influences bone and immune homeostasis but also directly regulates iron trafficking by suppressing hepcidin transcription through vitamin D receptor (VDR) binding to a response element in the HAMP promoter (13). This mechanism rapidly decreases circulating hepcidin, thereby facilitating ferroportin-mediated iron export from monocytes and hepatocytes. Human trials confirm that vitamin D3 supplementation can reduce hepcidin concentrations within 24–48 h (14, 15), and translational studies in monocytes corroborate that VDR activation downregulates HAMP expression and restores iron efflux capacity (16). These effects reinforce the role of vitamin D deficiency as a facilitator of chronic intracellular iron sequestration under IL-6–mediated signaling—particularly relevant to neurodivergent profiles, where low 25(OH)D levels are consistently reported (2).
Implications and discussion
The pattern described here—ferritin elevation without inflammation or genetic iron overload—suggests a distinct adaptive mechanism rather than pathology. The model posits that under chronic stress, mild IL-6 activity increases hepcidin, reducing iron export and promoting intracellular storage. The trade-off protects against oxidative damage but leads to functional iron deficiency and energy inefficiency. The intersection between psychological stress, inflammation, and iron metabolism has been recently emphasized in human studies: Psychological stress activates the HPA axis, elevates IL-6, and induces hepcidin, resulting in functional iron deficiency even with adequate intake (17). This provides further support for interpreting FIB as a psychoneuroendocrine adaptation rather than a static hematologic condition.
Clinical recognition of this pattern may explain refractory fatigue in burnout and NDV profiles where conventional iron indices appear normal. Misdiagnosis remains common, as elevated ferritin can be mistaken for hemochromatosis or incidental finding. Integrating biochemical markers (ferritin, hepcidin, transferrin saturation, DHEA-S, 25(OH)D, and cortisol dynamics) into stress-related research could clarify adaptive versus maladaptive stages of this mechanism.
Elucidating the fine boundary between adaptive regulation and the subsequent failure of these compensatory systems is crucial—not only for advancing research and mechanistic understanding but also for designing individualized clinical interventions. Poorly timed or miscalibrated therapeutic strategies may inadvertently increase oxidative load on an already strained system, collapsing rather than supporting the remaining adaptive capacity. Recognizing this risk is particularly relevant for neurodivergent individuals, whose biological regulation may operate closer to the threshold of energetic collapse. Such consideration aligns with the emerging concept of autistic burnout, in which sustained physiological stress responses eventually erode compensatory mechanisms, leading to exhaustion and functional shutdown.
Future directions
Prospective studies should evaluate FIB-related biomarkers—ferritin, hepcidin, cortisol (including diurnal profile and CAR), DHEA-S, and 25(OH)D—in parallel with neurocognitive and autonomic measures in burnout, trauma, and NDV cohorts. The relationship between IL-6/STAT3 activity, vitamin D status, and ferroptotic vulnerability warrants exploration using integrated endocrine and redox biomarkers. Translational research may clarify how adaptive mechanisms (iron sequestration and mitochondrial downregulation) evolve into clinical exhaustion or neuroinflammatory states. Understanding this adaptive-to-maladaptive continuum may lead to preventive strategies integrating light exposure, nutrition, hormonal modulation, and antioxidant support.
Conclusion
Functional iron blockade represents an adaptive stress response to chronic overload of the HPA and immune systems, disproportionately affecting neurodivergent phenotypes. Recognizing this mechanism may refine our understanding of refractory fatigue, prevent misdiagnosis, and guide new preventive strategies— linking iron metabolism, vitamin D status, and HPA dynamics into psychoneuroendocrinology research.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
SH: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. During the preparation of this work the author used ChatGPT (OpenAI) to assist in expanding literature searches, articulating the clinical hypothesis more clearly, and improving the English language and readability. After using this tool, the author carefully reviewed, edited, and approved the content, and takes full responsibility for the integrity and accuracy of the manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Abell JG, Shipley MJ, Ferrie JE, Kivimäki M, Kumari M, and Malik M. Recurrent short sleep, chronic insomnia symptoms and salivary cortisol: A 10-year follow-up in the Whitehall II study. Psychoneuroendocrinology. (2016) 68:91–9. doi: 10.1016/j.psyneuen.2016.02.021
2. Wang T, Shan L, Du L, Feng J, Xu Z, Staal WG, et al. Serum concentration of 25-hydroxyvitamin D in autism spectrum disorder: A systematic review and meta-analysis. Eur Child Adolesc Psychiatry. (2016) 25:341–50. doi: 10.1007/s00787-015-0786-1
3. Quadt L, Csecs J, Bond R, Harrison NA, Critchley HD, Davies KA, et al. Childhood neurodivergent traits, inflammation and chronic disabling fatigue in adolescence: a longitudinal case–control study. BMJ Open. (2024) 14:e084203. doi: 10.1136/bmjopen-2024-084203
4. Weiss G and Goodnough LT. Anemia of chronic disease. New Engl J Med. (2005) 352:1011–23. doi: 10.1056/NEJMra041809
5. Ganz T. Systemic iron homeostasis. Physiol Rev. (2013) 93:1721–41. doi: 10.1152/physrev.00008.2013
6. Nemeth E and Ganz T. Hepcidin–ferroportin interaction controls systemic iron homeostasis. Int J Mol Sci. (2021) 22:6493. doi: 10.3390/ijms22126493
7. Wrighting DM and Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. (2006) 108:3204–9. doi: 10.1182/blood-2006-06-027631
8. Al-Sanabra OM, Abu-Qatouseh LF, Ahmad MIA, Al-Khreisat MJ, and Alsaleh MM. Diminishing hepcidin via reducing IL-6/STAT3 pathway by utilizing ferulic acid: An in vitro study. Biomedicines. (2025) 13:923. doi: 10.3390/biomedicines13040923
9. Galaris D, Barbouti A, and Pantopoulos K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim Biophys Acta (BBA) - Mol Cell Res. (2019) 1866:118535. doi: 10.1016/j.bbamcr.2019.118535
10. Mollica MP, Catapano A, Cimmino F, Petrella L, Pizzella A, D’Angelo M, et al. Iron metabolism and ferroptosis in health and diseases: the crucial role of mitochondria in metabolically active tissues. J Nutr Biochem. (2025) 140:109888. doi: 10.1016/j.jnutbio.2025.109888
11. Alves F, Lane D, Nguyen TPM, Bush AI, and Ayton S. In defence of ferroptosis. Signal Transduction Targeted Ther. (2025) 10:2. doi: 10.1038/s41392-024-02088-5
12. Pawłowska M, Nuszkiewicz J, and Jarek DJ. Ferroptosis and metabolic dysregulation: Emerging chemical targets in cancer and infection. Molecules. (2025) 30:3020. doi: 10.3390/molecules30143020
13. Bacchetta J, Zaritsky JJ, Sea JL, Chun RF, Lisse TS, Zavala K, et al. Suppression of iron-regulatory hepcidin by vitamin D. J Am Soc Nephrol. (2014) 25:564–72. doi: 10.1681/ASN.2013040355
14. Smith EM, Alvarez JA, Kearns MD, Hao L, Sloan JH, Konrad RJ, et al. High-dose vitamin D3 reduces circulating hepcidin concentrations: A pilot, randomized, double-blind, placebo-controlled trial in healthy adults. Clin Nutr. (2017) 36:980–5. doi: 10.1016/j.clnu.2016.06.015
15. Greenwood A, von Hurst PR, Beck KL, Mazahery H, Lim K, and Badenhorst CE. Relationship between vitamin D, iron, and hepcidin in premenopausal females, potentially confounded by ethnicity. Eur J Nutr. (2023) 62:3361–8. doi: 10.1007/s00394-023-03240-7
16. Zughaier SM, Alvarez JA, Sloan JH, Konrad RJ, and Tangpricha V. The role of vitamin D in regulating the iron–hepcidin–ferroportin axis in monocytes. J Clin Trans Endocrinol. (2014) 1:19–25. doi: 10.1016/j.jcte.2014.01.003
Keywords: ferritin, hepcidin, neurodivergence, burnout, stress physiology, HPA axis, ferroptosis, vitamin D
Citation: Hauck S (2025) Functional iron blockade in chronic stress and neurodivergence: a perspective on adaptive stress physiology. Front. Psychiatry 16:1701625. doi: 10.3389/fpsyt.2025.1701625
Received: 08 September 2025; Accepted: 20 October 2025;
Published: 03 November 2025.
Edited by:
Parsa Ravanfar, Boston Children’s Hospital and Harvard Medical School, United StatesReviewed by:
Liu Yueying, Affiliated Hospital of Jiangnan University, ChinaCopyright © 2025 Hauck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simone Hauck, c2hhdWNrQGhjcGEuZWR1LmJy