 Andrew A. Davinack
Andrew A. Davinack- Department of Biological, Chemical and Environmental Sciences, Wheaton College Massachusetts, Norton, MA, United States
Population geneticists increasingly confront a paradox: even with genome-scale datasets and advanced machine learning models, subtle population structure often remains undetected, particularly in systems with low diversity, high dispersal, or recent divergence. This Opinion article argues that quantum computing and quantum machine learning (QML) offer a fundamentally different computational paradigm that may overcome these limitations. By leveraging principles such as superposition, entanglement, and high-dimensional Hilbert space embeddings, quantum systems can represent and analyze complex genetic relationships in ways that classical tools cannot. I outline how QML approaches such as quantum support vector machines, clustering algorithms, and optimization frameworks can be applied to detect cryptic population structure, optimize model selection, and reveal hidden patterns in genomic data. I also propose a conceptual pipeline for integrating quantum tools into molecular ecology and offer a roadmap for interdisciplinary collaboration. As quantum computing advances rapidly across the sciences, now is the time for evolutionary biologists and ecologists to engage with this emerging frontier. Quantum approaches may not only increase computational power, but also shift how we interrogate biological data, and reframe our understanding of population structure and diversity.
1 Introduction
Despite the explosion of genomic data in recent decades, population geneticists still struggle to detect structure in certain systems. Early analyses using mitochondrial DNA and Sanger-sequenced loci offered coarse insights into divergence and connectivity (Avise et al., 1987; Avise, 1991; Zhang and Hewitt, 2003; Avise et al., 2009), but many studies, particularly on high-dispersal taxa, returned “flat” results interpreted as panmixia. The rise of genome-wide approaches like RAD-seq and the integration of machine learning (ML) promised greater resolution by increasing marker density and computational power (Davey and Blaxter, 2010; Lowry et al., 2016; Hodel et al., 2017; Li and Ralph, 2019; Andrews et al., 2016). Yet in many cases, even thousands of SNPs and advanced clustering algorithms fail to reveal meaningful patterns (Meirmans, 2015; David, 2018; Hohenlohe et al., 2020). This persistent opacity raises a key question: is the problem in the data, or in the tools we use to analyze it? Many conventional methods reduce high-dimensional genetic data into linear projections or simplified models, potentially discarding subtle, nonlinear signals of structure. While ML approaches such as random forests or neural networks have improved detection in some cases, they too rely on classical assumptions, feature engineering, and dimensionality reduction that may miss complex relationships.
Quantum computing offers a fundamentally different paradigm—one that may open new doors for detecting structure in noisy, weakly differentiated, or cryptically admixed populations. In this article, I argue for integrating quantum machine learning (QML) into population genetics, outlining how quantum tools might expose hidden diversity where classical methods plateau.
2 Current bottlenecks in population genetics
Even with genome-scale data, molecular ecologists continue to face challenges in inferring population structure, especially in taxa with high dispersal, low diversity, or recent divergence. Classical methods such as FST, PCA, STRUCTURE, and AMOVA often fail to resolve differentiation in these systems, producing ambiguous results even when ecological or biogeographic barriers suggest otherwise (Lowe and Allendorf, 2010; Kelly and Palumbi, 2010; David and Loveday, 2018). Genomic approaches like RAD-seq, and the use of thousands of SNPs, were expected to overcome these issues. Machine learning methods—support vector machines, random forests, deep learning—have pushed the field further, especially in identifying subtle structure and loci under selection (Schrider and Kern, 2018; Sheehan and Song, 2016). However, these approaches still rely on classical computation and statistical frameworks. They require extensive training data, are sensitive to noise and missing data, and often project information into lower-dimensional spaces for tractability. Critically, they may overlook nonlinear relationships among loci, or signals obscured by admixture, gene flow, or founder effects. In invasive species, for instance, repeated introductions and high admixture often create complex patterns that defy clean clustering (see Huang et al., 2024). Even in well-sampled, high-resolution datasets, structure may remain “hidden.” not because it doesn’t exist, but because our tools aren’t built to find it.
The core issue isn’t always data quantity. It is representational capacity. When we compress complex genetic variation into classical spaces, we risk losing the very patterns we seek. A new computational paradigm may be required, one capable of handling high-dimensional, nonlinear, and noisy biological data in fundamentally different ways.
3 Quantum computing basics
Quantum computing departs fundamentally from classical computation. Instead of binary bits (0 or 1), quantum computers use qubits, units that exist in superposition, capable of being 0 and 1 simultaneously. This allows quantum systems to explore multiple states at once, vastly expanding the space of possible solutions. Qubits can also become entangled, meaning the state of one qubit is intrinsically linked to another, regardless of distance. Together, superposition and entanglement enable quantum systems to operate in exponentially larger, high-dimensional spaces known as Hilbert spaces. This makes quantum algorithms particularly powerful for pattern recognition, optimization, and classification in complex datasets.
Two concepts are especially relevant to population genetics. First, quantum feature mapping allows classical data, such as SNP genotypes or haplotype frequencies, to be embedded into quantum state space, potentially making weak or nonlinear patterns more separable. Second, quantum machine learning (QML) models, such as quantum support vector machines (QSVMs) and quantum k-means, can operate in these spaces to detect structure that classical models might miss. While still in early development, QML is increasingly being applied to real-world problems in fields like drug discovery, molecular design, and bioinformatics. These use cases suggest that quantum systems are not just faster—they may offer qualitatively different ways of understanding data.
4 Why this matters for ecology?
Many biological systems pose exactly the kinds of challenges quantum computing is suited to address: low-variation datasets, weak signals, and nonlinear interactions. In molecular ecology, these issues are common. Consider mitochondrial DNA from a species with global dispersal, or genome-wide SNPs from recently diverged populations. Classical analyses may detect no structure, but that doesn’t mean none exists. Quantum computing could shift this paradigm. For example, quantum feature mapping may embed genetic data into a space where structure becomes linearly separable, even if it appears undifferentiated in PCA. Quantum classifiers, like QSVMs, could be used to assign individuals to weakly distinct populations, even in cases of high admixture or recent introduction events. In systems with reticulate evolution, cryptic speciation, or asymmetric gene flow, quantum models may better capture the underlying complexity.
Beyond classification, quantum optimization algorithms could enhance model selection and parameter tuning. In seascape or landscape genetics, where researchers often model genetic differentiation as a function of multiple, correlated environmental variables, quantum optimization may more efficiently explore the solution space—avoiding overfitting and identifying the most explanatory variables. These tools are not just theoretical. Quantum algorithms have already been used in genome assembly, drug target prediction, and peptide design (Boev et al., 2021; Dhoriyani et al., 2025; Vakili et al., 2025). As population genetics grapples with ever-larger datasets and increasingly subtle patterns, the adoption of quantum tools is a logical extension. Quantum approaches will likely not replace classical methods outright, but they may provide complementary insight, especially in systems where traditional approaches return ambiguity. For population genetics, quantum computing offers not just more power, but a fundamentally new lens through which to view biological complexity.
It should be noted that many quantum machine learning models are quantum analogs of classical AI methods, such as QSVMs derived from support vector machines, or quantum neural networks adapted from deep learning. As such, progress in quantum approaches is often informed by advances in classical AI, underscoring the need for cross-training in both fields. Developing robust hybrid models will require expertise in both quantum algorithms and traditional machine learning frameworks.
5 A conceptual framework and roadmap
To realize the potential of quantum computing in population genetics, a coherent pipeline is essential—one that bridges classical genetic data and quantum machine learning models. Figure 1 outlines a six-step conceptual framework designed to help researchers integrate quantum methods into population structure analysis.
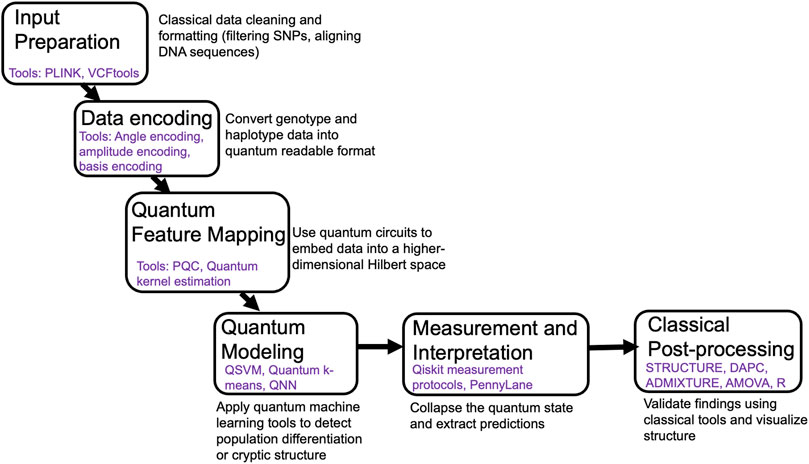
Figure 1. Conceptual pipeline for integrating quantum computing into population genetic analysis. Abbreviations: PQC: Parameterized quantum circuits, QSVM: Quantum Support Vector Machines, QNN: Quantum Neural Networks, DAPC: Discriminant Analysis of Principal Components, AMOVA: Analysis of Molecular Variance.
5.1 Input preparation
This first step begins with standard classical processing: filtering SNPs, aligning sequences, removing low-quality data, and formatting inputs using tools such as PLINK, VCFtools, or BCFtools. Clean, structured data is essential for downstream encoding and modeling.
5.2 Data encoding
Genotype or haplotype information must be converted into a quantum-readable format. Techniques like angle encoding (mapping genotypes to rotational angles on qubits) or amplitude encoding (embedding entire genotype vectors into quantum state amplitudes) translate biological data into a format compatible with quantum circuits.
5.3 Quantum feature mapping
Encoded data is embedded into a high-dimensional Hilbert space using parameterized quantum circuits. This transformation allows weak or nonlinear patterns, such as subtle gene flow or cryptic structure, to become more separable, much like kernel methods in classical ML but with exponentially expanded feature space.
5.4 Quantum modeling
With data now embedded, quantum machine learning algorithms (e.g., quantum support vector machines, quantum k-means, quantum neural networks) are applied to detect population differentiation, cluster individuals, or identify outliers. These models operate in the quantum feature space and may outperform classical approaches in noisy or weakly structured datasets.
5.5 Measurement and interpretation
Quantum measurement collapses the qubit state into classical outcomes, typically as bitstrings or probability distributions. These outputs are interpreted as cluster memberships, structure probabilities, or other biologically relevant predictions.
5.6 Classical post-processing
Finally, results are validated, visualized, and interpreted using traditional statistical and genetic tools (e.g., STRUCTURE, ADMIXTURE, AMOVA, or DAPC). This hybrid approach leverages the strengths of both quantum and classical systems.
Together, these steps provide a flexible but actionable roadmap for bringing quantum computing into real-world population genetic research.
6 Caveats
The potentially enormous gains in data analysis of population-level genetic data are contingent on several hardware and algorithmic limitations. Current quantum devices (termed Noisy Intermediate-Scale Quantum or ‘NISQ’ systems), have relatively few qubits (often fewer than 100 usable ones) and are prone to error and decoherence (Lau et al., 2022; Chen et al., 2023). These constraints limit the size and depth of quantum circuits, which in turn restricts the scale of biological datasets that can be analyzed directly on hardware. Moreover, data encoding itself presents a bottleneck in the proposed pipeline. While angle encoding is conceptually straightforward and compatible with small-scale data (e.g., mtDNA haplotypes or select SNP panels), amplitude encoding – which enables exponentially efficient processing, requires complex normalization and may be impractical on current devices. Encoding hundreds or thousands of SNPs across many individuals is still computationally expensive, even on simulated backends.
Given the early stage of development, many proposed quantum applications in biology remain speculative. Empirical benchmarking, especially using small, tractable datasets such as mtDNA haplotypes, SNP panels, or synthetic admixed population, is critical for validating the feasibility and value of QML in real-world scenarios. Such preliminary studies can generate essential performance metrics, guide algorithm refinement, and justify further investment in scaling up applications. Finally, one significant challenge is the fact that many quantum algorithms lack biological grounding, meaning that they were not designed with population genetics principles in mind, and few benchmarks exist to compare quantum outputs to traditional methods like DAPC or ADMIXTURE. As such, interpretability and validation will remain critical challenges, and early applications will likely depend on hybrid quantum-classical workflows that combine quantum pattern recognition with classical biological inference.
In short, quantum tools may offer qualitatively different insights, especially in difficult datasets where classical tools struggle. But these gains must be evaluated against current limitations in scale, stability and interpretability–challenges that are likely to be overcome only through cross-disciplinary innovation and algorithm refinement.
7 Conclusion and a call to action
Quantum computing is not a distant future, it is already reshaping fields from chemistry to bioinformatics. For population genetics and molecular ecology, the moment is ripe to explore how quantum tools might uncover the hidden structure that classical methods miss. This transformation will not happen passively though. It will require intentional collaboration between quantum physicists, data scientists, and biologists. Pilot studies on small datasets for e.g., mitochondrial haplotypes, reduced SNP panels, or simulated admixed populations, can provide early proof-of-concept and establish benchmarks for quantum vs. classical performance. Data challenges, benchmarking competitions, and shared datasets can accelerate this process, especially when supported by institutions that span both fields. Equally important is education: graduate programs and workshops should begin introducing ecologists to quantum concepts, particularly in encoding, modeling, and quantum-enhanced optimization. National and international funding agencies are already prioritizing quantum innovation, including through interdisciplinary research initiatives. Molecular ecologists should seize this opportunity to lead, not follow, in applying quantum tools to biological questions. The tools are available. The theory is sound. What’s needed now is action. By embracing quantum methods today, we can reimagine how we study population structure, biodiversity, and evolution tomorrow.
Author contributions
AD: Writing – review and editing, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G., and Hohenlohe, P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 17, 81–92. doi:10.1038/nrg.2015.28
Avise, J. C. (1991). Ten unorthodox perspectives on evolution prompted by comparative population genetics findings on mitochondrial DNA. Annu. Rev. Genet. 25, 45–69. doi:10.1146/annurev.ge.25.120191.000401
Avise, J. C. (2009). Phylogeography: retrospect and prospect. J. Biogeogr. 36, 3–15. doi:10.1111/j.1365-2699.2008.02032.x
Avise, J. C., Arnold, J., Ball, R. M., Bermingham, E., Lamb, T., Neigel, J. E., et al. (1987). Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 18, 489–522. doi:10.1146/annurev.es.18.110187.002421
Boev, A. S., Rakitko, A. S., Usmanov, S. R., Kobzeva, A. N., Popov, I. V., Ilinsky, V. V., et al. (2021). Genome assembly using quantum and quantum-inspired annealing. Sci. Rep. 11, 13183. doi:10.1038/s41598-021-88321-5
Chen, S., Cotler, J., Huang, H. -Y., and Li, J. (2023). The complexity of NISQ. Nature Commun. 14, 6001. doi:10.1038/s41467-023-41217-6
Davey, J. W., and Blaxter, M. L. (2010). RADSeq: next-generation population genetics. Briefings Funct. Genomics 9, 416–423. doi:10.1093/bfgp/elq031
David, A. A. (2018). Reconsidering panmixia: the erosion of phylogeographic barriers due to anthropogenic transport and the incorporation of biophysical models as a solution. Front. Mar. Sci. 5, 280. doi:10.3389/fmars.2018.00280
David, A. A., and Loveday, B. R. (2018). The role of cryptic dispersal in shaping connectivity patterns of marine populations in a changing world. J. Mar. Biol. Assoc. U. K. 98, 647–655. doi:10.1017/s0025315417000236
Dhoriyani, J., Bergman, M. T., Hall, C. K., and You, F. (2025). Integrating biophysical modeling, quantum computing and AI to discover plastic-binding peptides that combat microplastic pollution. PNAS Nexus 4, pgae572. doi:10.1093/pnasnexus/pgae572
Hodel, R. G. J., Chen, S., Payton, A. C., McDaniel, S. F., Soltis, P., and Soltis, D. E. (2017). Adding loci improves phylogeographic resolution in red mangroves despite increased missing data: comparing microsatellites and RAD-Seq and investigating loci filtering. Sci. Rep. 7, 17598. doi:10.1038/s41598-017-16810-7
Hohenlohe, P. A., Funk, W. C., and Rajora, O. P. (2020). Population genomics for wildlife conservation and management. Mol. Ecol. 30, 62–82. doi:10.1111/mec.15720
Huang, X., Rymbekova, A., Dolgova, O., Lao, O., and Kuhlwilm, M. (2024). Harnessing deep learning for population genetic inference. Nat. Rev. Genet. 25, 61–78. doi:10.1038/s41576-023-00636-3
Kelly, R. P., and Palumbi, S. R. (2010). Genetic structure among 50 species of the northeastern Pacific rocky intertidal community. PLoS ONE 5, e8594. doi:10.1371/journal.pone.0008594
Lau, J. W. Z., Lim, K. H., Shrotriya , H., and Kwek, L. C. (2022). NISQ computing: where are we and where do we go?. AAPPS Bulletin. 32, 27. doi:10.1007/s43673-022-00058-z
Li, H., and Ralph, P. (2019). Local PCA shows how the effect of population structure differs along the genome. Genetics 211, 289–304. doi:10.1534/genetics.118.301747
Lowe, W. H., and Allendorf, F. W. (2010). What can genetics tell us about population connectivity. Mol. Ecol. 19, 3038–3051. doi:10.1111/j.1365-294x.2010.04688.x
Lowry, D. B., Hoban, S., Kelley, J. L., Lotterhos, K. E., Reed, L. K., Antolin, M. F., et al. (2016). Breaking RAD: an evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol. Ecol. Resour. 17, 142–152. doi:10.1111/1755-0998.12635
Meirmans, P. G. (2015). Seven common mistakes in population genetics and how to avoid them. Mol. Ecol. 24, 3223–3231. doi:10.1111/mec.13243
Schrider, D. R., and Kern, A. D. (2018). Supervised machine learning for population genetics: a new paradigm. Trends Genet. 34, P301–P312. doi:10.1016/j.tig.2017.12.005
Sheehan, S., and Song , Y. S. (2016). Deep learning for population genetics. PLOS Comput. Biol. 12, e1004845. doi:10.1371/journal.pcbi.1004845
Vakili, M. G., Gorgulla, C., Snider, J., Nigam, A., Bezrukov, D., Varoli, D., et al. (2025). Quantum-computing enhanced algorithm unveils potential KRAS inhibitors. Nat. Biotechnol. doi:10.1038/s41587-024-02526-3
Keywords: QSVM, neural network, next generating sequencing, bioinformatcs, RAD-seq
Citation: Davinack AA (2025) The promise of quantum computing for population genetics and molecular ecology. Front. Quantum Sci. Technol. 4:1657832. doi: 10.3389/frqst.2025.1657832
Received: 01 July 2025; Accepted: 04 September 2025;
Published: 16 September 2025.
Edited by:
Marcelo Victor Pires de Sousa, D’Or Institute for Research and Education (IDOR), BrazilReviewed by:
Elibio Rech, Embrapa Genetic Resources and Biotechnology, BrazilCopyright © 2025 Davinack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew A. Davinack, ZGF2aW5hY2tfZHJld0B3aGVhdG9uY29sbGVnZS5lZHU=