 Sheng Yuan1†
Sheng Yuan1† Xin-Yan Yao
Xin-Yan Yao Chun-Yang Lian
Chun-Yang Lian Jian-Wei Shao
Jian-Wei Shao Xue-Lian Zhang
Xue-Lian Zhang- 1School of Life Science and Engineering, Foshan University, Foshan, China
- 2Beijing Biomedical Technology Center of Jofunhwa Biotechnology (Nanjing) Co., Ltd., Beijing, China
Bovine hepacivirus (BovHepV) is a member of the genus Hepacivirus of the family Flaviviridae, which can cause acute or persistent infections in cattle. Currently, BovHepV strains identified in cattle populations worldwide can be classified into two genotypes with eight subtypes in genotype 1. BovHepV has been identified in a wide geographic area in China. Interestingly, the viral RNA of BovHepV has also been detected in ticks in Guangdong province, China. In this study, Rhipicephalus microplus tick samples were collected in Heilongjiang province, northeastern China, and BovHepV was screened with an overall positive rate of 10.9%. Sequence comparison and phylogenetic analysis showed that the BovHepV strains detected in this study belong to the subtype G. This is the first report about the detection of BovHepV in ticks in Heilongjiang province, China, which expands our knowledge that ticks may be a transmission vector of BovHepV.
1. Introduction
The genus Hepacivirus, which belongs to the family Flaviviridae, comprises a genetically diverse group of human and animal pathogens (1). The genome of the hepacivirus is about 10 kb in length, which contains the 5′ untranslated regions (UTR) and 3′ UTR, and a single large open reading frame (ORF) that encodes a single polyprotein (2). The polyprotein is cleaved by signal peptidase, NS2/NS3 protease and NS3 protease enzymes into three structural proteins (Core, E1, and E2) and seven non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (3, 4).
Hepaciviruses have been identified from a wide variety of mammalian hosts and non-mammalian hosts, such as bats (5), rodents (6, 7), monkeys (8), horses (9), dogs (10), donkeys (11), catshark (12), duck (13), fish and vertebrates (14–16). Currently, members of the genus Hepacivirus have been divided into Hepacivirus A–N based on their phylogenetic relationships and host range (17). In addition, more hepaciviruses have been identified in non-vertebrate hosts, such as mosquitos and ticks (18, 19), although their route of infection and transmission is uncertain.
Bovine hepacivirus (BovHepV) is the only member of the species Hepacivirus N and likely only infects cattle (20). It was first identified in cattle in Germany and Ghana in 2015 (21, 22). Thereafter, BovHepV has been detected in China (23–25), Brazil (26, 27), Turkey (28), the USA (29), and Italy (30), suggesting the worldwide geographic distribution of BovHepV. Moreover, BovHepV presented highly genetic diversity. As per the genotyping and subtype classification criteria for the hepatitis C virus, the BovHepV strains can be divided into two genotypes, and genotype 1 could be divided into eight subtypes (A to H) (24). Recently, BovHepV identified in Inner Mongolia, northeastern China further divided subtype G into subtypes G1 and G2, indicating the extensive genetic diversity of BovHepV (31).
In China, BovHepV have been determined in cattle herds in Guangdong, Jiangsu, Yunnan, Sichuan, Heilongjiang, Shandong, Henan, Inner Mongolia and Chongqing, with the positive rate of viral RNA ranging from 2.78 to 13.3% (25, 31–34), indicating that BovHepV was circulating in cattle herds in a wide geographic in China. Notably, the viral RNA of BovHepV has also been detected in ticks collected from cattle in Guangdong province, suggesting that tick maybe play an important role in the transmission of BovHepV among cattle (24). A previous study has shown that the prevalence of BovHepV in cattle herds in Heilongjiang province was 6.0% (25). However, no information about the epidemiology of BovHepV in ticks in Heilongjiang province is available. Therefore, in this study, blood-sucking ticks were collected from the body surface of cattle in Heilongjiang province to investigate the presence of BovHepV.
2. Materials and methods
2.1. Tick sample collection and RNA extraction
During June to August in 2021, a total of 400 blood-sucking adult tick were simple random collected from 18 cattle herds in Harbin, Heilongjiang, northeastern China (Figure 1). The tick species were identified following morphological criteria and further confirmed by sequencing and analyzing the 16S ribosomal RNA (rrs) gene of ticks (35). Five ticks were collected from one cattle individual and merged as one pool and stored at −80°C for further use.
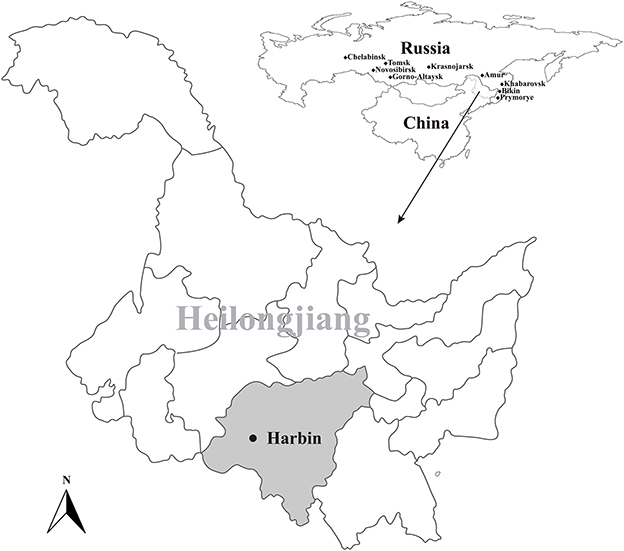
Figure 1. Geographic maps showing the location of sampling sites from where the rodents were captured in this study. This map was plotted using a combination of Surfer software, Version-4 (Golden Software, Golden, CO, USA), and Adobe illustrator, Version CC2017 (Adobe, San Jose, CA, USA). The black dots indicate the sampling regions in this study.
Each pooled tick sample was soaked in 70% ethanol for 30 min, and washed with double distilled water three times. The samples were homogenized in 500 μL sterile phosphate buffered saline (PBS), and their total RNA was extracted from 200 μL homogenates using the TRIzol LS reagent (Invitrogen, Carlsbad, CA, USA) and subsequently purified using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). The extracted RNA was stored at −80°C until further use.
The prevalence of BovHepV in this study was estimated assuming perfect sensitivity and specificity of molecular detection using the EPITOOLS online statistical program “Pooled prevalence for fixed pool size and perfect test” that based on the model established by Cowling et al. (36).
2.2. BovHepV screening and genome sequencing
Primer pairs designed based on the alignment results of the NS3 region of all BovHepV available in GenBank database were used to detect the presence of BovHepV RNA in blood-sucking ticks. Using nested RT-PCR with the primer pair BovHepV-F1/BovHepV-R1 and BovHepV-F2/BovHepV-R2, the PCR product with 493 bp was confirmed by Sanger sequencing. In addition, over-lapping primers were designed to obtain the complete genome sequence of the BovHepV identified in this study. All primers used in this study were listed in Table 1.
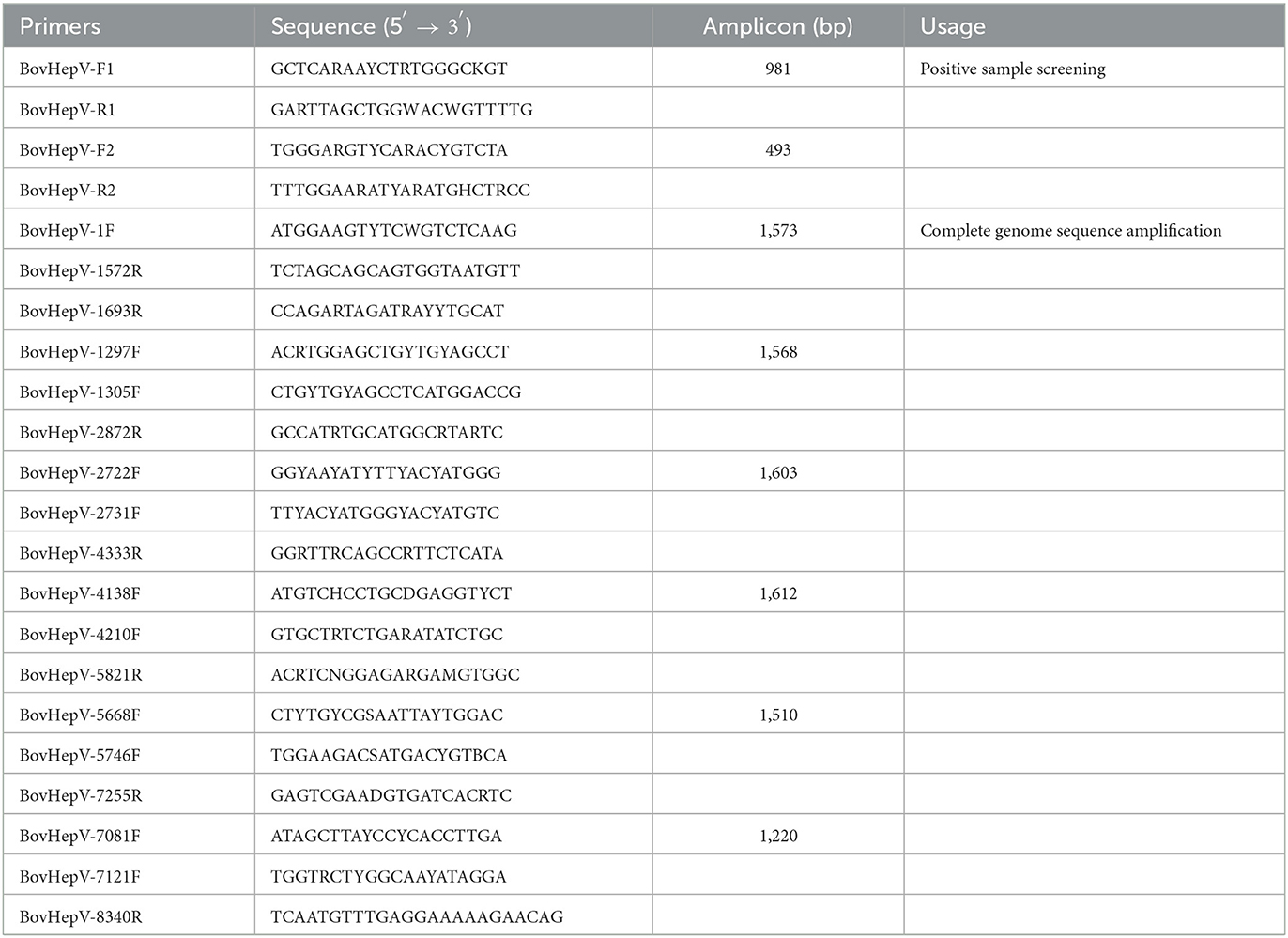
Table 1. Primers used in this study.
The PCR products of the expected size, according to each set of primers, were purified using Gel Extraction kit (TaKaRa, Dalian, China) after electrophoresis. The purified DNA was cloned into pMD19-T vector (TaKaRa, China), and the resulting plasmid was used to transform competent E. coli cells. Positive inserts were confirmed by PCR, and further sequenced by Sangon Biotechnology Company (Shanghai, China). To prevent contamination, the preparation of the PCR mix and the addition of the template DNA were performed in separate rooms using dedicated pipets and filtered tips.
2.3. Sequence comparison and phylogenetic analysis
Sequence assembly and manually editing was performed using the SeqMan program (DNASTAR, Madison, WI), and the nucleotide (nt) and sequence identity were calculated by MegAlign program available within the Lasergene software package (version 7.1, DNAstar). Maximum-likelihood (ML) trees were reconstructed using MEGA version 7.0 (37), based on the best-fit nucleotide substitution model General Time Re-versible (GTR) nucleotide substitution model and optimized parameters of gamma (G)-distribution and proportion of invariable sites (i.e., GTR+G+I) determined by jModel Test (38). Bootstrap values were calculated from 100 replicates, and the phylogenetic trees were mid-point rooted for purposes of clarity only.
2.4. Recombination analysis
The seven methods (RDP, GENECONV, bootscan, maximum chi square, Chimera, SISCAN, and distance plot) within RDP4 program (39) were used to determine the potential recombination events that occurred in the evolutionary history of BovHepV. The analyses were performed based on the complete genome sequences with default settings for the different test methods and a Bonferroni corrected p-value cutoff of 0.05. Only sequences with significant evidence (p < 0.05) of recombination detected by at least two methods and confirmed by phylogenetic analysis were taken to represent strong evidence for recombination. Additionally, similarity plot analyses were inferred to further characterize potential recombination events, including the location of possible breakpoints, as implemented in Simplot version 3.5.1 (40).
2.5. Ethics statement
The study involving animals were reviewed and approved by the ethics committee of College of Life Science and Engineering, Foshan University. Written informed consent was obtained from the owners for the participation of their animals in this study.
3. Results
3.1. Detection of BovHepV in tick sample
A total of 400 ticks, which were identified as Rhipicephalus microplus, were collected from 18 cattle herds in Harbin, Heilongjiang province, from June to August in 2021 (Figure 1). After PCR screening, sequencing and BLAST analysis, 35 out of the 80 tick pools were determined as positive for BovHepV. Among the 18 cattle herds, 17 were detected as positive for BovHepV with the positive rate ranging from 0 to 7.8% and the overall positive rate of BovHepV in this study was 10.9% (95% CI: 7.6–14.9%) (Table 2).
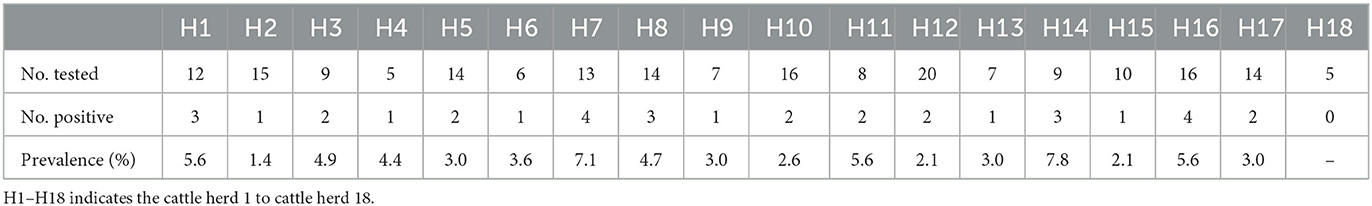
Table 2. Prevalence of BovHepV detected in ticks collected from different cattle herds in Harbin.
3.2. Sequences comparison of BovHepV
The nearly complete genome sequence of the BovHepV strains amplified from six representative samples showed 99.2–99.7% nucleotide identity and 99.0–100% amino acid identity with each other (Table 3). Furthermore, they shared 67.3–93.2% nucleotide identity and 76.4–98.1% amino acid identity with the BovHepV strains identified world-wide, while they shared the highest identity with the subtype G strains determined from cattle in China (Table 3). Moreover, they shared 81.5–81.8% nucleotide identity and 93.5–94.4% amino acid identity with the subtype H strains (Accession Numbers MZ221927, MZ540979, and MZ540980), which were also identified in blood-sucking tick in China (Table 3).
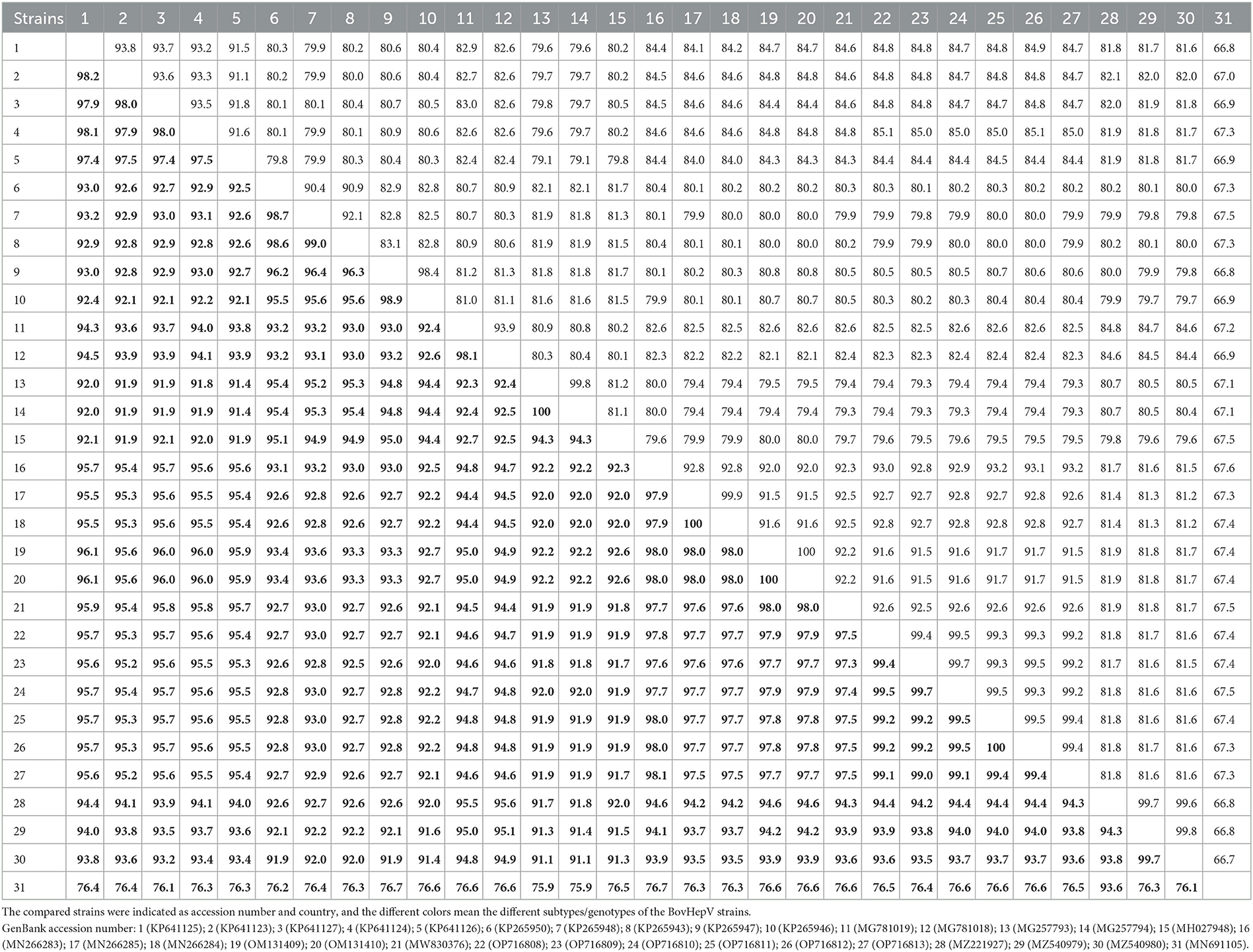
Table 3. The sequence identity within the BovHepV polyprotein at the nucleotide (upper right) and amino acid (lower left, boldface) levels calculated using ClustalW method implemented in MegAlign.
3.3. Recombination and phylogenetic analysis of BovHepV
No statistically supported recombination event was detected within BovHepV strains after systematic analyzes. Phylogenetic analysis reconstructed based on the complete polyprotein coding sequence showed that all BovHepV strains were divided into two genotypes (genotype 1 and genotype 2), and genotype 1 strains are clearly divided into eight well-separated subtypes (subtype A–G). The six viruses identified in this study are more closely related to subtype G viruses that identified in bovine samples collected in Jiangsu, Chongqing, and Inner Mongolia, China, while was distinguished from the subtype H viruses identified in ticks in Guangdong province, China. Notably, virus strains identified in this study showed a closer phylogenetic relationship with those viruses identified in Jiangsu than the Inner Mongolia ones (OM131409–OM131410), although Inner Mongolia is geographically closer to Heilongjiang province (Figure 2).
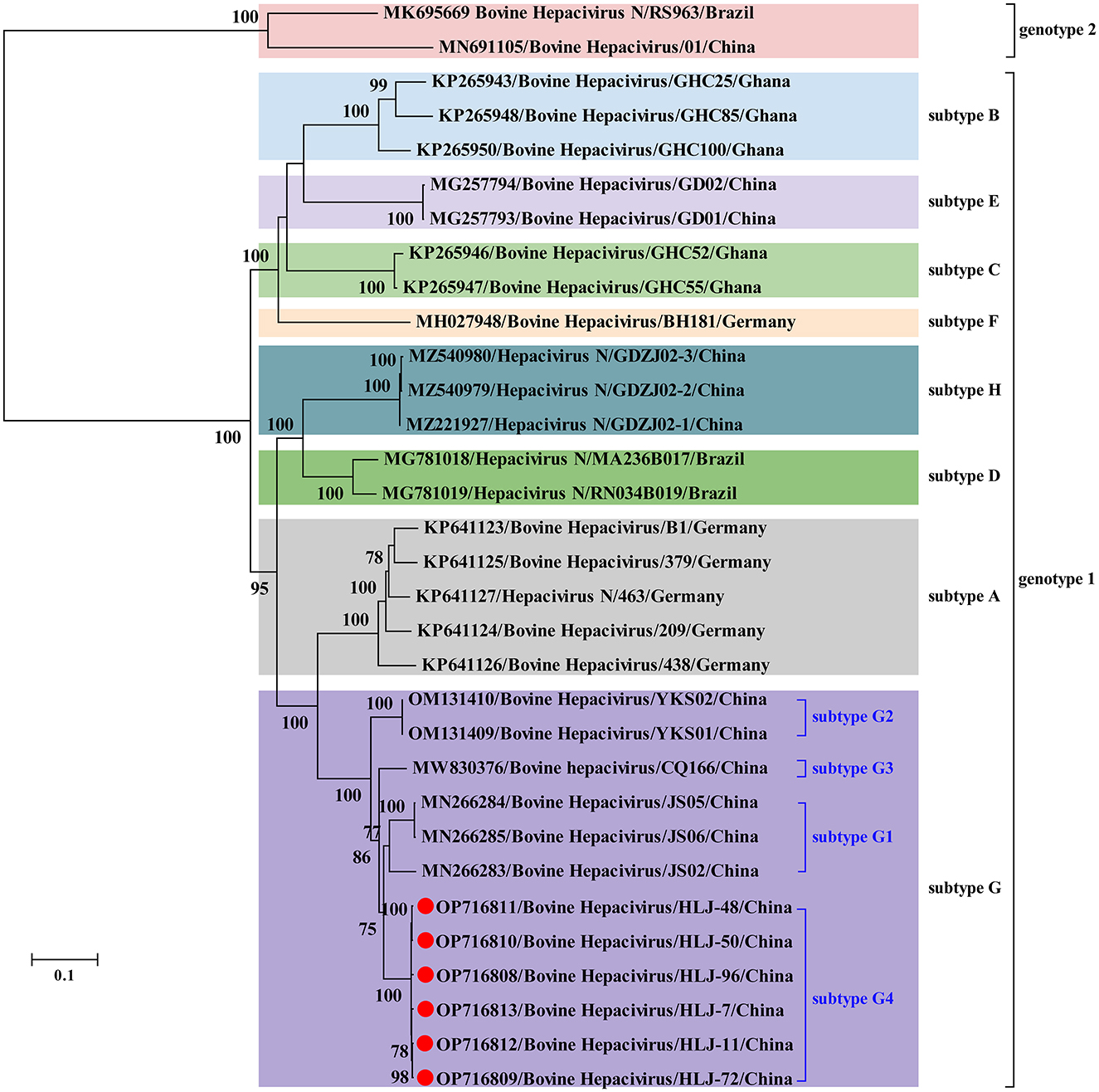
Figure 2. Phylogenetic analysis based on the nucleotide sequences of complete polyprotein-coding region of BovHepV including the newly identified sequences and other reference sequences retrieved from GenBank. The trees were constructed based on the maximum likelihood method implemented in MEGA 7.0, and mid-point rooted for clarity and the scale bar represents the number of nucleotide substitutions per site. Bootstrap values were calculated with 100 replicates of the alignment, and only bootstrap values > 70% are shown at relevant nodes. GenBank accession numbers are followed by the name of hepacivirus strains. Red dots indicate the BovHepV determined in this study.
4. Discussion
Since its first discovery in cattle in Germany and Ghana in 2015, BovHepV has been identified in seven continents (21, 22, 24, 27–30), indicating the worldwide geographical distribution of BovHepV. Moreover, the currently identified BovHepV could be classified into two genotypes, and genotype 1 could be further divided into eight subtypes, suggesting the extensive genetic diversity of BovHepV (24). Interestingly, in this study, the viral RNA of BovHepV was detected in blood-sucking ticks, suggesting that ticks may be serve as an arthropod vector for hepacivirus. However, no information about bovine hepacivirus in ticks collected from vegetation. Moreover, although bovine hepacivirus have been detected in blood-sucking ticks in this study and our previous study (24), we did not simultaneously analyze the active infection in the cattle that the questing tick removed. Together, these results suggest that ticks may be a transmission vector of BovHepV, although it needs further investigation.
In China, BovHepV has been detected in Guangdong, Jiangsu, Yunnan, Sichuan, Heilongjiang, Shandong, Henan, Inner Mongolia and Chongqing (25, 31–34), suggesting that BovHepV was circulating in cattle herds in a wide geographic in China. Notably, BovHepV strains identified in China were segregated into two genotypes and thee subtypes in subtype 1 in the phylogenetic tree. In addition, previous study performed in Inner Mongolia further divided the subtype G into subtype G1–G2 (31). In this study, BovHepV strains in subtype G were divided into four clades (subtype G1–G4), and the BovHepV strains identified in this study were classified as the subtype G4. These results imply the extensive genetic diversity of BovHepV that circulates in China. Additionally, among all genotypes or subtypes of BovHepV identified in China, the viruses belonging to the subtype G were widely detected in Jiangsu, Chongqing, Inner Mongolia, and Heilongjiang, China, indicating that BovHepV in subtype G may be the primary circulating subtype in China.
Previous studies have shown that BovHepV subtypes are associated with their geographic origins based on the limited number of BovHepV sequences (21, 22, 26, 28), and our results were generally consistent with this conclusion. However, the results in this study also show the complex geographic distribution of BovHepV genotypes or subtypes. For example, viruses in subtypes E, H, and G that were identified in China showed a closer phylogenetic relationship with those in subtypes B, D, and A, which were identified in Ghana, Brazil, and Germany, respectively. This could have resulted from frequent international trade of live cattle, which can facilitate transboundary transmission of BovHepV. In addition, the BovHepV strains detected in this study showed a closer phylogenetic relationship with those identified in Jiangsu than the Inner Mongolia strains, although Inner Mongolia is geographically closer to Heilongjiang province. These results indicating an intriguing evolutionary route of BovHepV.
In conclusion, BovHepV belong to the subtype G was detected in Rhipicephalus microplus ticks collected from cattle in Heilongjiang province, northeastern China with an overall prevalence of 10.9%. This is the first reports about the detection of BovHepV in ticks in Heilongjiang province, which expands our knowledge that ticks may be a transmission vector of BovHepV.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OP716808–OP716813.
Ethics statement
The animal study was reviewed and approved by the Ethics Committee of College of Life Science and Engineering, Foshan University. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
X-LZ and J-WS: conceived, designed the experiments, and writing—review and editing. SY, X-YY, C-YL, and SK: collect the samples, performed the experiments, and analyzed the data. SK: help to collect the samples. X-YY: writing—original draft preparation. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by Guangdong Basic and Applied Basic Research Foundation, Grant Nos. 2021A1515110450 and 2022A1515012194.
Conflict of interest
SK was employed by the company Beijing Biomedical Technology Center of Jofunhwa Biotechnology (Nanjing) Co., Ltd., China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Smith DB, Becher P, Bukh J, Gould EA, Meyers G, Monath T, et al. Proposed update to the taxonomy of the genera Hepacivirus and Pegivirus within the Flaviviridae family. J Gen Virol. (2016) 97:2894–907. doi: 10.1099/jgv.0.000612
2. Scheel TK, Simmonds P, Kapoor A. Surveying the global virome: identification and characterization of HCV-related animal hepaciviruses. Antiviral Res. (2015) 115:83–93. doi: 10.1016/j.antiviral.2014.12.014
3. Bartenschlager R, Lohmann V. Replication of the hepatitis C virus. Best practice and research clinical. Gastroenterology. (2000) 14:241–54. doi: 10.1053/bega.1999.0073
4. Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology. (2004) 39:5–19. doi: 10.1002/hep.20032
5. Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, et al. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc Natl Acad Sci U S A. (2013) 110:8194–9. doi: 10.1073/pnas.1303037110
6. Drexler JF, Corman VM, Muller MA, Lukashev AN, Gmyl A, Coutard B, et al. Evidence for novel hepaciviruses in rodents. PLoS Pathog. (2013) 9:e1003438. doi: 10.1371/journal.ppat.1003438
7. Wu Z, Lu L, Du J, Yang L, Ren X, Liu B, et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome. (2018) 6:178. doi: 10.1186/s40168-018-0554-9
8. Lauck M, Sibley SD, Lara J, Purdy MA, Khudyakov Y, Hyeroba D, et al. A novel hepacivirus with an unusually long and intrinsically disordered NS5A protein in a wild Old World primate. J Virol. (2013) 87:8971–81. doi: 10.1128/JVI.00888-13
9. Lyons S, Kapoor A, Sharp C, Schneider BS, Wolfe ND, Culshaw G, et al. Nonprimate hepaciviruses in domestic horses, United kingdom. Emerg Infect Dis. (2012) 18:1976–82. doi: 10.3201/eid1812.120498
10. Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, et al. Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci U S A. (2011) 108:11608–13. doi: 10.1073/pnas.1101794108
11. Walter S, Rasche A, Moreira-Soto A, Pfaender S, Bletsa M, Corman VM, et al. Differential infection patterns and recent evolutionary origins of equine hepaciviruses in donkeys. J Virol. (2017) 91:e01711–6. doi: 10.1128/JVI.01711-16
12. Shi M, Lin XD, Vasilakis N, Tian JH, Li CX, Chen LJ, et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the flaviviridae and related viruses. J Virol. (2016) 90:659–69. doi: 10.1128/JVI.02036-15
13. Zhang XL, Yao XY, Zhang YQ, Lv ZH, Liu H, Sun J, et al. A highly divergent hepacivirus identified in domestic ducks further reveals the genetic diversity of hepaciviruses. Viruses. (2022) 14:371. doi: 10.3390/v14020371
14. Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, et al. The evolutionary history of vertebrate RNA viruses. Nature. (2018) 556:197–202. doi: 10.1038/s41586-018-0012-7
15. Chang WS, Eden JS, Hartley WJ, Shi M, Rose K, Holmes EC. Metagenomic discovery and co-infection of diverse wobbly possum disease viruses and a novel hepacivirus in Australian brushtail possums. One Health Outlook. (2019) 1:5. doi: 10.1186/s42522-019-0006-x
16. Porter AF, Pettersson JH, Chang WS, Harvey E, Rose K, Shi M, et al. Novel hepaci- and pegi-like viruses in native Australian wildlife and non-human primates. Virus Evol. (2020) 6:veaa064. doi: 10.1093/ve/veaa064
17. Simmonds P, Becher P, Bukh J, Gould EA, Meyers G, Monath T, et al. ICTV virus taxonomy profile: flaviviridae. J Gen Virol. (2017) 98:2–3. doi: 10.1099/jgv.0.000672
18. Harvey E, Rose K, Eden JS, Lo N, Abeyasuriya T, Shi M, et al. Extensive diversity of RNA viruses in Australian ticks. J Virol. (2019) 93:e01358–e01318. doi: 10.1128/JVI.01358-18
19. Williams SH, Levy A, Yates RA, Somaweera N, Neville PJ, Nicholson J, et al. Discovery of Jogalong virus, a novel hepacivirus identified in a Culex annulirostris (Skuse) mosquito from the Kimberley region of Western Australia. PLoS ONE. (2020) 15:e0227114. doi: 10.1371/journal.pone.0227114
20. Da Silva MS, Junqueira DM, Baumbach LF, Cibulski SP, Mosena ACS, Weber MN, et al. Comprehensive evolutionary and phylogenetic analysis of Hepacivirus N (HNV). J Gen Virol. (2018) 99:890–6. doi: 10.1099/jgv.0.001082
21. Baechlein C, Fischer N, Grundhoff A, Alawi M, Indenbirken D, Postel A, et al. Identification of a novel hepacivirus in domestic cattle from Germany. J Virol. (2015) 89:7007–15. doi: 10.1128/JVI.00534-15
22. Corman VM, Grundhoff A, Baechlein C, Fischer N, Gmyl A, Wollny R, et al. Highly divergent hepaciviruses from African cattle. J Virol. (2015) 89:5876–82. doi: 10.1128/JVI.00393-15
23. Lu G, Ou J, Zhao J, Li S. Presence of a novel Subtype of bovine hepacivirus in China and expanded classification of bovine hepacivirus strains worldwide into 7 subtypes. Viruses. (2019) 11:843. doi: 10.3390/v11090843
24. Shao JW, Guo LY, Yuan YX, Ma J, Chen JM, Liu Q. A novel subtype of bovine hepacivirus identified in ticks reveals the genetic diversity and evolution of bovine hepacivirus. Viruses. (2021) 13:2206. doi: 10.3390/v13112206
25. Lu G, Chen C, Shao R, Zhang J, Li J, Cai S, et al. Identification and genetic characterization of bovine hepacivirus in China: a large scale epidemiological study. Virol Sin. (2022) 37:223–8. doi: 10.1016/j.virs.2022.02.003
26. Canal CW, Weber MN, Cibulski SP, Silva MS, Puhl DE, Stalder H, et al. A novel genetic group of bovine hepacivirus in archival serum samples from Brazilian cattle. Biomed Res Int. (2017) 2017:4732520. doi: 10.1155/2017/4732520
27. Da Silva MS, Weber MN, Baumbach LF, Cibulski SP, Budaszewski RF, Mosena ACS, et al. Highly divergent cattle hepacivirus N in Southern Brazil. Arch Virol. (2019) 164:3133–6. doi: 10.1007/s00705-019-04419-2
28. Yesilbag K, Baechlein C, Kadiroglu B, Baldan Toker E, Alpay G, Becher P. Presence of bovine hepacivirus in Turkish cattle. Vet Microbiol. (2018) 225:1–5. doi: 10.1016/j.vetmic.2018.09.001
29. Sadeghi M, Kapusinszky B, Yugo DM, Phan TG, Deng X, Kanevsky I, et al. Virome of US bovine calf serum. Biologicals. (2017) 46:64–7. doi: 10.1016/j.biologicals.2016.12.009
30. Elia G, Caringella F, Lanave G, Martella V, Losurdo M, Tittarelli M, et al. Genetic heterogeneity of bovine hepacivirus in Italy. Transbound Emerg Dis. (2020) 67:2731–40. doi: 10.1111/tbed.13628
31. Liu Z, Li L, Guo Y, Xu W, Yuan Y, Liang X, et al. Genome characterization and phylogenetic analysis of bovine hepacivirus in Inner Mongolia, Northeastern China. Zoonoses. (2022) 2:13. doi: 10.15212/ZOONOSES-2022-0003
32. Deng Y, Guan SH, Wang S, Hao G, Rasmussen TB. The detection and phylogenetic analysis of bovine hepacivirus in China. Biomed Res Int. (2018) 2018:6216853. doi: 10.1155/2018/6216853
33. Lu G, Jia K, Ping X, Huang J, Luo A, Wu P, et al. Novel bovine hepacivirus in dairy cattle, China. Emerg Microbes Infect. (2018) 7:54. doi: 10.1038/s41426-018-0055-8
34. Qiang X, Shen X, Peng H, Guo X, He Z, Yao M, et al. Complete genome sequence of a novel bovine hepacivirus from Yunnan, China. Arch Virol. (2020) 165:1489–94. doi: 10.1007/s00705-020-04611-9
35. Liu H, Li Q, Zhang X, Li Z, Wang Z, Song M, et al. Characterization of rickettsiae in ticks in northeastern China. Parasit Vectors. (2016) 9:498. doi: 10.1186/s13071-016-1764-2
36. Cowling DW, Gardner IA, Johnson WO. Comparison of methods for estimation of individual-level prevalence based on pooled samples. Prev Vet Med. (1999) 39:211–25. doi: 10.1016/S0167-5877(98)00131-7
37. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
38. Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. (2008) 25:1253–6. doi: 10.1093/molbev/msn083
39. Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. (2015) 1:vev003. doi: 10.1093/ve/vev003
Keywords: bovine hepacivirus, subtype G, genetic diversity, tick, Northeastern China
Citation: Yuan S, Yao X-Y, Lian C-Y, Kong S, Shao J-W and Zhang X-L (2023) Molecular detection and genetic characterization of bovine hepacivirus identified in ticks collected from cattle in Harbin, northeastern China. Front. Vet. Sci. 10:1093898. doi: 10.3389/fvets.2023.1093898
Received: 09 November 2022; Accepted: 09 February 2023;
Published: 01 March 2023.
Edited by:
Dasiel Obregon, University of Guelph, CanadaReviewed by:
Mario Frías, Maimonides Biomedical Research Institute of Cordoba (IMIBIC), SpainGianvito Lanave, University of Bari Aldo Moro, Italy
Matheus Nunes Weber, Feevale University, Brazil
Copyright © 2023 Yuan, Yao, Lian, Kong, Shao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-Lian Zhang, enhsc2p3MDMxMkAxNjMuY29t
†These authors have contributed equally to this work