 Dehong Tian
Dehong Tian Buying Han1,2,3
Buying Han1,2,3- 1Key Laboratory of Adaptation and Evolution of Plateau Biota, Qinghai Provincial Key Laboratory of Animal Ecological Genomics, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining, China
- 2College of Life Science, University of Chinese Academy of Sciences, Beijing, China
- 3Key Laboratory of Adaptation and Evolution of Plateau Biota, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining, China
- 4Qinghai Sheep Breeding and Promotion Service Center, Gangcha, China
- 5Qinghai Yak Breeding and Promotion Service Center, Datong, China
Under the combined effects of long-term natural selection and artificial domestication, Tibetan sheep on the Qinghai-Tibet Plateau have evolved distinct ecotypes to survive extreme high-altitude conditions, including hypoxia, cold, and low oxygen levels. These ecotypic variations not only serve as an ideal model for studying plateau livestock adaptation but also harbor valuable genetic diversity. However, the lack of comprehensive genetic analyses on their adaptive and phenotypic traits has hindered the effective conservation and utilization of these resources. Using whole-genome resequencing, we systematically studied seven Tibetan sheep breeds, uncovering their genetic structure and diversity. Population analyses, including NJ and maximum likelihood trees, revealed clear genetic differentiation and migration patterns. Selective sweep analyses (Fst and θπ) identified hypoxia-related genes (DOCK8, IGF1R, JAK1, SLC47, TMTC2, and VPS13A) and wool color genes (TCF25, MITF, and MC1R). GWAS further detected candidate genes for body size traits (height, length, weight), enriched in cGMP-PKG, cAMP, and Hedgehog signaling pathways. Integrating GWAS and transcriptomics, we pinpointed key wool trait genes, including WNT16 (non-synonymous mutations), PRKCA, MAP3K8, MMP7, OVOL2 (intergenic SNPs), and COL7A1, KDM8, ZNF385D (intronic SNPs). Notably, HOX family transcription factors were found to critically regulate hair follicle development. These genetic markers offer promising targets for molecular breeding to enhance wool quality and adaptive traits. Our findings provide a genetic basis for understanding Tibetan sheep’s unique adaptations and production traits, supporting future breeding strategies and sustainable utilization of their genetic resources.
Introduction
With the migration of human populations, particularly nomadic groups, domesticated sheep (Ovis aries) were first domesticated in the Fertile Crescent approximately 9,500 to 10,000 years ago (BCE, BP). Genetic analyses have revealed multiple distinct domestication lineages that have subsequently dispersed nearly worldwide (1). Tibetan sheep gradually disseminated from the northeastern region of the Tibetan Plateau to its central area as the Di-Qiang people expanded 3,100 years ago, and subsequently from the southwestern region to the central area by 1,300 years ago (2, 3). Tibetan sheep, inhabiting the relatively isolated Tibetan Plateau, have developed rich genetic resources and distinct local varieties due to natural geographical barriers and limited external species intrusion. Over time, these sheep have undergone both natural and artificial selection, resulting in significant phenotypic diversity, including variations in hair type, hair color, horn morphology, and tail structure, etc. The diverse phenotypes of Tibetan sheep, along with the extensive variation in economically important traits, offer researchers a wealth of genetic resources.
Previous research has identified approximately 17 local Tibetan sheep populations on the Qinghai-Tibet Plateau (4). As the exploration of germplasm resources continues, an increasing number of new germplasms have been discovered, including the Zashgar, Zeku, and Maduo sheep. These breeds, belonging to distinct groups within the Tibetan sheep population, have since evolved into independent breeds. Tibetan sheep breeds on the Qinghai-Tibet Plateau exhibit remarkable traits including drought tolerance, cold resistance, roughage adaptability, disease resilience, high-altitude acclimatization, foraging capability, robust physique, and strong genetic adaptability (5). Furthermore, Tibetan sheep hold significant agricultural, economic, cultural, and religious value in the Tibetan Plateau region of China, contributing substantially to the economic development of pastoral areas (6).
Artificial selection has significantly influenced the genetic diversity of sheep during domestication and production-oriented breeding, resulting in populations with distinctive characteristics and valuable genetic resources. The genome of Tibetan sheep offers a unique opportunity to identify traits associated with this selection. Under artificial intervention, the genetic variation affecting the fertility of consecutive multiple births in Tibetan sheep was investigated. It was discovered that PAPPA is a key gene responsible for stimulating the growth and development of ovarian follicles and enhancing steroid production, thereby improving reproductive success (7). A study examining genomic variation in 986 Tibetan sheep samples across their range identified strong selection signals in genes related to hypoxia and ultraviolet signaling pathways, including the HIF-1 pathway and the HBB and MITF genes. Additionally, strong selection was observed in genes associated with morphological traits, such as horn size and shape, particularly the RXFP2 gene. Furthermore, the study detected 5.23–5.79% gene introgression from argali (Ovis ammon) into Tibetan sheep (2). Genome-wide association analysis was conducted on 103 subtypes, including normal large horns, scurs, and polled, derived from the second generation (G2) of a Tibetan sheep polled core herd. Six SNPs located on chromosome 10 within the RXFP2 gene showed significant positive correlations with horn length, horn base circumference, and horn base interval (8). High-frequency structural variant genes, including EPAS1, PAPSS2, and PTPRD, represent significant sources of genetic variation in the gene expression of Tibetan sheep and play a crucial role in their adaptation to high-altitude environments (9). Positive selection genes related to meat production, coat color, wool traits, horn type or adaptability of different ecological types of Tibetan sheep have also been explored (10). During the successive generations of Tibetan sheep domestication, varying degrees of genetic imprints have been left on the genome, ultimately becoming fixed in certain domesticated breeds of Tibetan sheep. These valuable genetic resources delineate the genomic landscape of various ecological types of Tibetan sheep across their distribution range. However, each species may possess distinct candidate genes that are responsible for their unique traits, and there is a relative paucity of research on the selection markers for various economic traits across different ecological types.
In this study, we conducted whole-genome resequencing of Tibetan sheep populations from diverse ecological environments. Each sample possesses distinct traits, including hair length, high-altitude adaptation, body size, and coat color. Through the application of signal analysis methods for whole-genome scanning, we identified the specific genetic markers responsible for these traits in Tibetan sheep. This study enhances our understanding of the underlying genetic mechanisms and contributes to the broader field of fundamental biological research.
Materials and methods
Animal care
The study was conducted according to the guidelines of the Institutional Animal Care and Use Committee of Institute of Animal Science and Veterinary Medicine, Chinese Academy of Sciences (IACUC2021311).
Animals and phenotypic measurement
Seven Tibetan sheep breeds exhibiting significant geographical and phenotypic variations were selected from Qinghai Province (plateau-type Tibetan sheep, valley-type Tibetan sheep, Black Tibetan sheep, Qumaari Speckled sheep, Zeku sheep, Oula sheep, and Zashgar sheep). A total of 140 blood samples were collected from unrelated adult ewes (n = 20 per breed). Each sample was meticulously documented with the species name, codes, geographical coordinates of the sampling site (longitude, latitude, and altitude), and phenotypic characteristics (see Figure 1a and Supplementary Table 1 for further details). All tissue samples were preserved in 95% ethanol and stored at −80°C for subsequent genomic analysis. The phenotypes of five traits were measured in 20 different breeds of Tibetan sheep, including three body size traits (body weight, body length, and body height) and two wool quality traits (wool fiber length and fiber fineness).
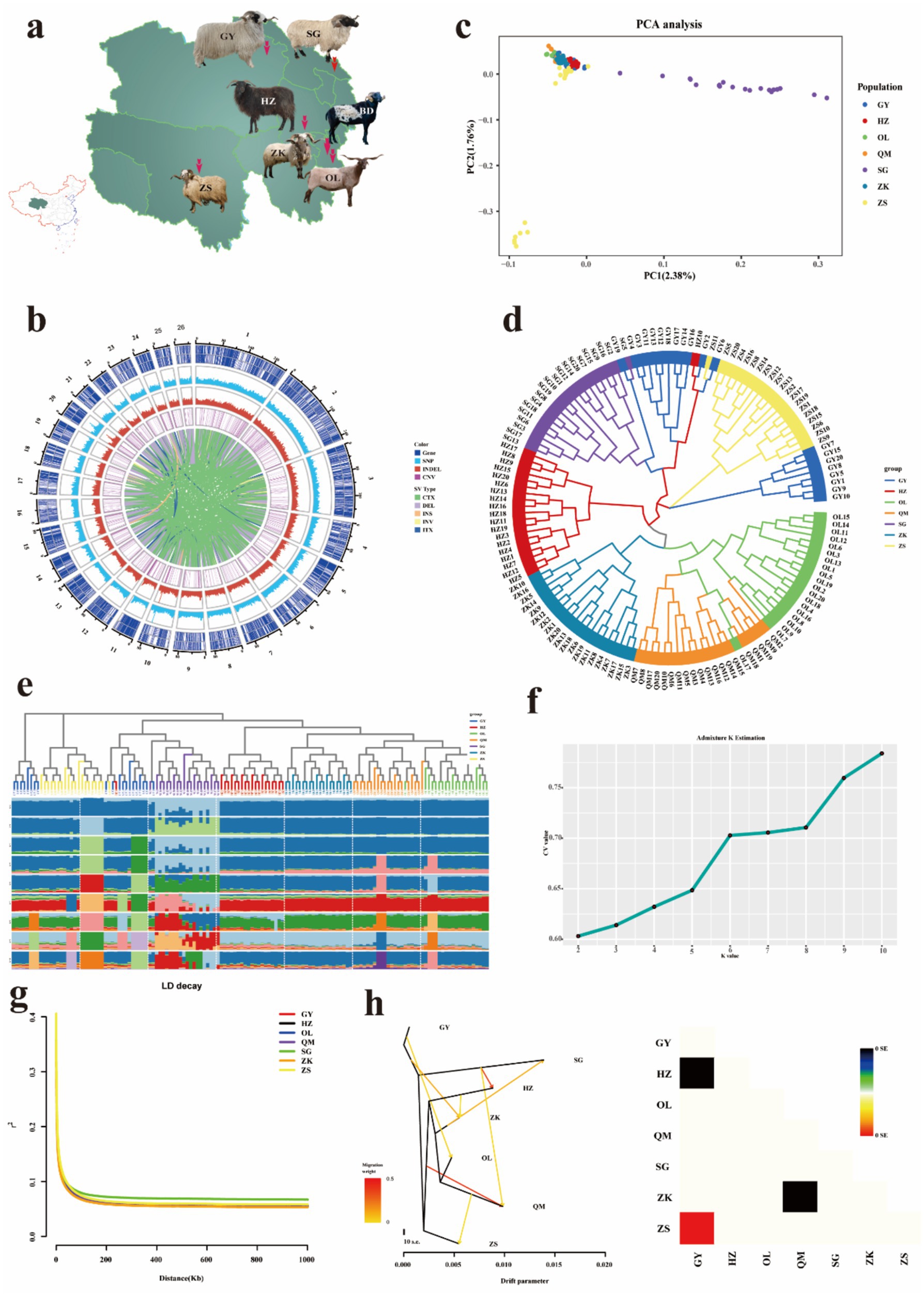
Figure 1. Geographic distribution and genetic diversity analyses. (a) Geographic distribution of seven Tibetan sheep breeds in Qinghai. The map was generated using Adobe Illustrator software. (b) SNPs and InDels density distribution circle. (c) Plots of principal components. (d) Neighbor-joining tree constructed from SNP data among six sheep populations. (e) Genetic structure analysis of samples using admixture, with changing ancestral populations from K = 2 to K = 10. (f) Distribution of cross-validation error values associated with varying K values. (g) The linkage disequilibrium (LD) decay analysis. (h) Gene flow diagram and residual fitting heat map.
Whole-genome resequencing
Total genomic DNA was extracted from the samples, and at least 3 μg of genomic DNA was used to construct paired-end libraries with a read length of 2 × 150 bp for paired-end sequencing. Simultaneously, we obtained the farmer’s consent to collect skin samples. We randomly selected three unrelated plateau Tibetan sheep and three unrelated Oula sheep, and collected 2 square centimeters of skin tissue from the shoulder region using sterile surgical blades. The samples were promptly immersed in liquid nitrogen (−80°C) for subsequent transcriptome sequencing and real-time quantitative polymerase chain reaction (RT-qPCR) analysis. These libraries were sequenced utilizing the Illumina NovaSeq 6000 platform located in Shanghai, China.
Read processing and variant calling
ASTP Toolkit v0.18.0 was utilized for stringent quality control of the raw reads based on the following criteria: (1) removal of reads containing ≥ 10% unidentified nucleotides (N); (2) elimination of reads with > 50% bases having Phred quality scores ≤ 20; and (3) discarding reads that aligned to the barcode adapter. The Burrows–Wheeler Aligner (BWA) was employed to map the cleaned reads from each sample to the CAU O.aries_1.0 reference genome1 using the parameters “mem 4 -k 32 -M”. Here, -k specifies the minimum seed length, and -M marks shorter split alignment hits as secondary alignments (11). Variant calling was conducted across all samples using GATK’s Unified Genotyper (12). Subsequently, SNPs and InDels were filtered through GATK’s Variant Filtration tool, applying specific thresholds (-Window 4, -filter “QD <2.0||FS >60.0||MQ <40.0,” -G_filter “GQ <20”), and variants showing segregation distortion or indicative of sequencing errors were excluded (13). For the determination of the physical positions of each SNP, the ANNOVAR software (14) was applied for alignment and annotation of SNPs or InDels. Structural variations (SVs), encompassing translocations, inversions, and insertions, were identified using the breakdancer software (Max1.1.2.) (15). Copy number variants (CNVs) were classified using CNVnator (0.3.2) (16).
Population genetic analyses
The SNP-only dataset was analyzed utilizing a maximum likelihood algorithm. A phylogenetic tree was constructed using PHYML 3.0 (17) and FastTree, based on the selected optimal nucleotide substitution model, generalized time reversible (GTR). Node support was assessed through 1,000 bootstrap replicates. Principal component analysis (PCA) was conducted using SNP data [excluding SNPs with a minor allele frequency (MAF) less than 0.05] via the GCTA software. Individuals were subsequently clustered into distinct subgroups based on the principal components derived from the analysis. The ADMIXTURE software2 was utilized to analyze the genetic structure of the population using SNP data. A mixed model was selected, with K ranging from 2 to 10 (assuming 2 to 10 ancestral populations). All other parameters were set to the software’s default values. The optimal K value, which is closest to the true number of ancestral populations, was determined based on the cross-validation (CV) error values.
Selected sweep
Select regions with extremely low or high θ, where the π ratio is in the 5% left and right tails, and those with significantly high Fst values (i.e., the top 5% of Fst), as these are identified as regions that have undergone strong selective sweeps.
Enrichment analysis of selected candidate genes
Use top GO/KEGG to conduct enrichment analysis. In this process, the gene list and gene count for each pathways are derived from the candidate region genes annotated. The p-value is then calculated using the hypergeometric distribution method, with a threshold of p-value <0.05 indicating significant enrichment. This approach identifies the pathways that are significantly enriched in the candidate region genes relative to the whole-genome background, thereby elucidating the primary biological functions of these genes.
GWAS analysis
Based on inter-population SNPs and linkage disequilibrium, the EMMAX software was utilized to perform association analysis between molecular markers and trait phenotypes,3 identifying markers or candidate genes closely associated with the target traits. These findings were visualized using Manhattan plots and QQ plots.
RNA extraction, library construction
Total RNA was extracted using the Trizol Reagent (Invitrogen Life Technologies). Subsequently, the concentration, quality, and integrity of the RNA were assessed using a NanoDrop spectrophotometer (Thermo Scientific). To isolate cDNA fragments of the desired 400–500 base pairs in length, the library fragments were purified utilizing the AMPure XP system (Beckman Coulter, Beverly, CA, United States). Products were purified using the AMPure XP system and quantified with the Agilent High Sensitivity DNA Assay on a Bioanalyzer 2100 (Agilent Technologies). Subsequently, the sequencing library was subjected to sequencing on the NovaSeq X Plus platform (Illumina).
Library construction and sequencing
Fastp (18) (version 0.18.0) was used to filter adapters or low quality bases from raw reads. The reference genome and gene annotation files were retrieved from the genome database. The filtered reads were aligned to the reference genome using HISAT2 version 2.0.5.
Differential expression analysis
We utilized HTSeq (version 0.9.1) to compare the read count values for each gene as a measure of its original expression, followed by normalization using FPKM. Differential gene expression was analyzed using DESeq (version 1.30.0) with the following screening criteria: an absolute log2 fold change greater than 1 and a significant p-value less than 0.05. Additionally, we employed the R language Pheatmap (version 1.0.8) package to conduct bi-directional clustering analysis of all differentially expressed genes across samples. The heatmap was generated based on the expression levels of the same gene in different samples and the expression patterns of different genes within the same sample, using the Euclidean method to calculate distances and the complete linkage method for clustering.
Analysis integrating GWAS and transcriptomic data
The SNPs identified through GWAS were mapped to the corresponding genes in the expression dataset to evaluate their impact on gene expression. The significant genes obtained from GWAS were intersected with differentially expressed genes (DEGs), and KEGG/GO enrichment analysis was performed on the overlapping genes. Cluster analysis of the commonly differentially expressed genes was carried out using the Pheatmap package (version 1.0.8) in the R programming language.
RT-qPCR analysis
Seven DEGs were selected and confirmed by RT-qPCR. The housekeeping gene actin served as an internal control for normalizing mRNA expression levels. Primers were designed using Oligo 6.0 software (Supplementary Table 30). Quantitative real-time PCR was performed in a 20 μL reaction volume containing 10 μL of 2 × Top Green EX-Taq Mix, 2 μL of cDNA, 7 μL of ddH2O, and 0.5 μL each of forward and reverse primers. The thermocycling conditions were as follows: initial denaturation at 94°C for 30 s; followed by 42 cycles of denaturation at 94°C for 5 s, annealing at 61°C for 35 s; a final extension at 97°C for 10 s, and a dissociation curve analysis stage consisting of 65°C for 60 s and 97°C for 1 s. Relative mRNA expression levels were determined using the 2−ΔΔCT method. All experiments were conducted with six biological replicates.
Results
Sequencing, mapping and SNP/InDel detection
Whole-genome resequencing of 140 samples generated 36.4 billion paired-end raw reads with an insert size of 400 bp, and 35.98 billion high-quality reads, achieving an average depth of 14.44 × per sample and an average genome coverage of 99.33%. The GC content varied between 42.77 and 44.30%, the Q20 value was no less than 97%, the Q30 value was at least 93.69%, and the mapping rate against the reference genome surpassed 99.84% (Supplementary Table 2). Furthermore, a total of 46,681,283 SNP sites were identified for purine-pyrimidine transitions and transversions, with a Ts/Tv ratio of 1.86 (Supplementary Table 3). A total of 46.69 million SNPs were identified and utilized for subsequent analyses. The majority of these high-quality SNPs (64.35%) were found in intergenic regions, characterized by T/C and A/G transitions, while only 0.67% were located within exonic regions. The remaining SNPs were distributed as follows: 0.58% upstream, 0.56% downstream, and 33.82% within introns. In total, 314,431 SNPs were detected in exons, of which 46.63% were non-synonymous and 51.09% were synonymous, yielding a non-synonymous to synonymous ratio of 0.913 (Supplementary Table 4). In addition, a total of 1,880,322 insertions and 2,680,046 deletions were identified across seven indigenous sheep breeds (Supplementary Table 5).
The majority of insertions and deletions were situated in intergenic regions (63.71%). Additionally, exon InDels predominantly comprised in-frame deletions or insertions (56.62%), leading to alterations in the reading frame of protein-coding genes, which often exhibited a multiple of the triplet codon length (Supplementary Table 6). After quality control, the following copy number variants (CNVs) were detected: 24,803 in GY, 238,655 in HZ, 24,984 in OL, 24,960 in QM, 23,435 in SG, 22,029 in ZK, and 24,868 in ZS (Supplementary Table 7). A total of 337,491 reliable structural variants (SVs) were identified. Specifically, this included 3,469 insertions, 186,018 deletions, 15,204 inversions, 46,226 intrachromosomal translocations, and 86,574 interchromosomal translocations (Supplementary Table 8).
Genetic diversity analysis and population genetic structure
The observed heterozygosity (Ho) of seven Tibetan sheep populations ranged from 0.358 to 0.372, while the expected heterozygosity (He) ranged from 0.349 to 0.358 (Supplementary Table 9). The observed heterozygosity was consistently higher than the expected heterozygosity, Fis values for ZS, SG, and GY were all less than 0. To gain a more comprehensive understanding of the distribution of chromosomal variations in Tibetan sheep, we established a 1 Mb sliding window and generated density maps for genes, SNPs, CNVs, and InDels within each window (Figure 1b). Notably, the genomic variation exhibits a high degree of homogeneity. The constructed weighted phylogenetic tree elucidates the relationships among seven Tibetan sheep breeds, and the resulting neighbor-joining (NJ) tree provides evidence of distinct separations between breeds, with each breed forming its own branch, although some overlap is observed in Tibetan sheep populations (Figure 1d). On the other hand, PCA cluster analysis offers further empirical support for the delineation of these groups, thereby demonstrating the robust consistency of the selected samples (Figure 1c). The mixed model was employed to assess the genetic structure of the population. At K = 2, the cross-validation error reached its minimum, resulting in the division of 140 germplasm resources into two distinct groups. When K = 3, SG tends to diverge in a different direction from the main group, while the remaining breeds are distributed across the other two clusters. When K = 7, the majority of the genetic information of GY, HZ, OL, QM, ZK, and ZS appears to stem from a shared ancestral population (red). When K = 10, the Tibetan sheep population exhibits extremely rich genetic diversity, which may be attributed to its highly complex population history, extensive geographical distribution, or diverse adaptive traits (Figures 1e,f). Linkage disequilibrium (LD, r2) decreases to half of its maximum value within less than 10 kb, with ZK populations exhibiting the fastest decline and SG populations showing the slowest decline. These findings indicate that SG species exhibit strong LD and are more prone to inbreeding, which may be a result of long-term artificial selection (Figure 1g). The maximum likelihood tree, based on the number of optimal migration events, provided evidence of gene flow between distinct Tibetan sheep populations (Figure 1h).
Genome-wide study of selective sweeps for hypoxic adaptability
Through the analysis of genomic regions, we identified loci with high levels of Fst and nucleotide diversity ratio (θπ), which are associated with hypoxic adaptability. Specifically, by comparing varieties below 3,000 meters and those above 4,000 meters, we identified 2,900 and 2,618 candidate positively selected regions in the SG (Fst >0.082, θπ >0.496) (Supplementary Table 9) and HZ (Fst >0.044, θπ >0.365) (Supplementary Table 10) varieties, respectively. These two varieties exhibited 607 and 586 candidate genes, respectively, during variety-specific selection events, with a total of 218 co-selected genes (Figure 2a). These shared genes were selected through critical adaptive responses to hypoxic conditions in high-altitude environments, exhibiting higher levels of differentiation. For instance, several positively selected genes associated with hypoxic adaptation were identified in one or more of the studied populations, including DOCK8, IGF1R, JAK1, SLC4A7, TMTC2, and VPS13A. KEGG enrichment analysis revealed that these genes were predominantly enriched in signal transduction, development and regeneration, and endocrine system (Figure 2b and Supplementary Table 11).
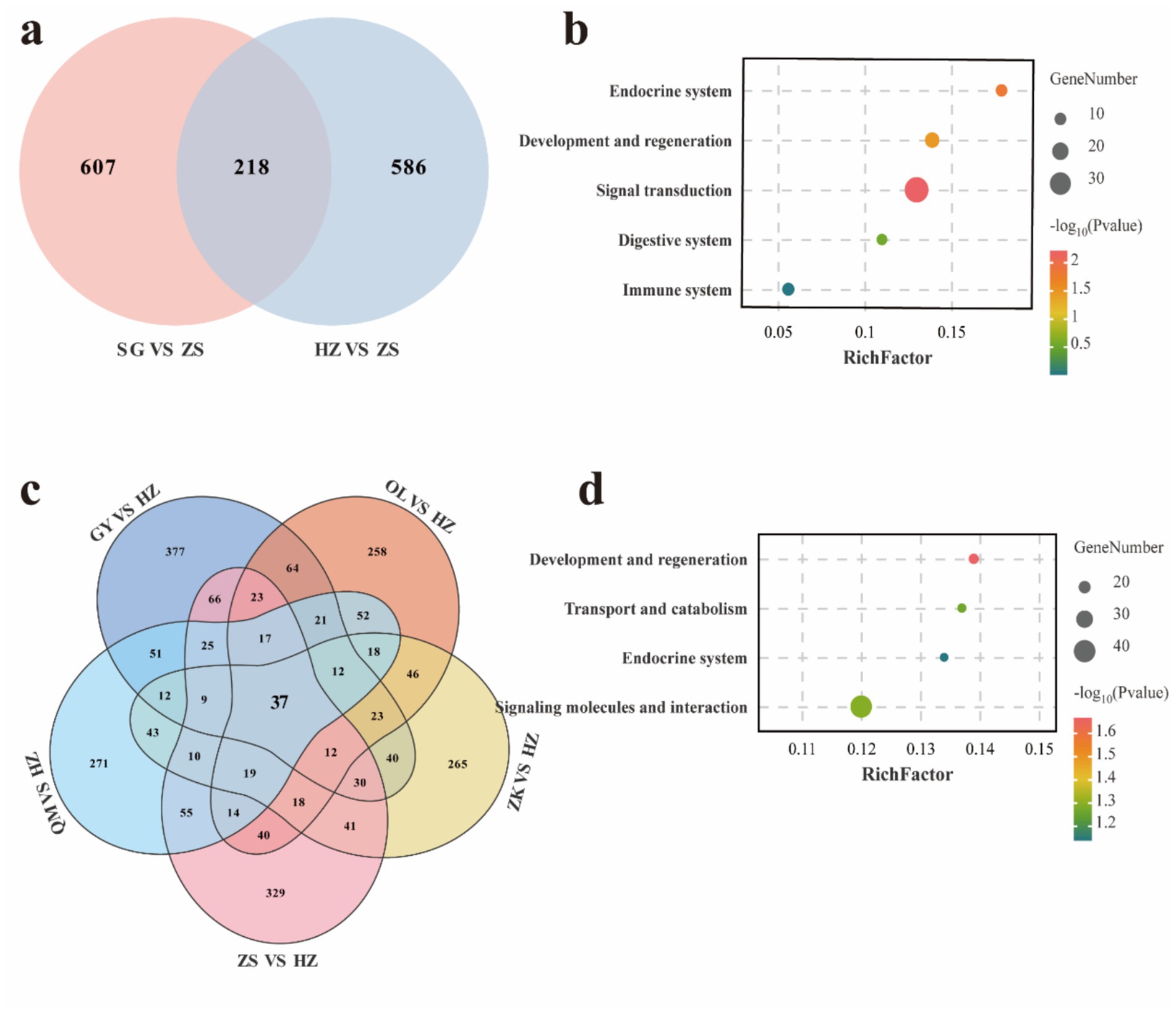
Figure 2. Venn diagrams of co-selected genes for hypoxic adaptability (a) and KEGG enrichment of the corresponding genes (b) among different comparisons. Venn diagrams of common selected genes for coat color (c) and KEGG enrichment of the corresponding genes (d) among different comparisons.
Genome-wide study of selective sweeps for coat color
Considering the physical properties of coat color, we employed a comprehensive comparative analysis, evaluating the OL, GY, HZ, ZS, and ZK varieties against the black-coated HZ variety. For OL (Fst >0.037, θπ <−0.305), GY (Fst >0.039, θπ >0.273), QM (Fst >0.039, θπ <−0.250), and ZS (Fst >0.044, θπ <−0.257) and ZK (Fst >0.039, θπ <−0.363), a total of 2,136, 2,625, 2,193, 2,376, and 2,034 selection regions were identified (Supplementary Tables 12–16), encompassing 37 co-selection genes (Figure 2c). Among the shared genes, including TCF25, MITF, and MC1R, which are associated with hair color, these genes are involved in transport and catabolism, regulation of the endocrine system, development and regeneration processes, as well as signaling molecules and interaction pathways (Figure 2d and Supplementary Table 17).
Genome-wide association studies of body size and wool traits
GWAS serve as a crucial tool for elucidating the genetic underpinnings of traits, revealing the relationship between genetic variation and these traits. To account for known confounding factors, we incorporated gender and age as fixed effects into the mixed linear model using a general linear modeling approach. Based on the significance criterion of a p-value with a −log10 value exceeding 4, we identified several significant SNP loci associated with body height (BH), body length (BL), and body weight (BW) at the genomic level (Supplementary Tables 18–20). The p-value Manhattan plot revealed significant associations between SNPs and BH, BL, and BW. Through gene annotation, 30 significant loci related to body shape traits were identified. For the BH trait, five significant SNPs were located on OAR1, while one SNP was found on OAR2, OAR5, OAR21, and OAR6 (Figures 3a,d). For BL, five SNPs showed significant associations, with candidate regions identified on OAR1, OAR3, and OAR23. Notably, the five most significant SNPs were located on OAR2 (Figures 3b, e). For BW, 10 significant SNPs were identified, with the most prominent associations occurring in the EVC and EVC2 regions on OAR6 (Figures 3c,f). These SNPs demonstrated a strong positive correlation with body size traits (Table 1), exceeding the predefined threshold for significance.
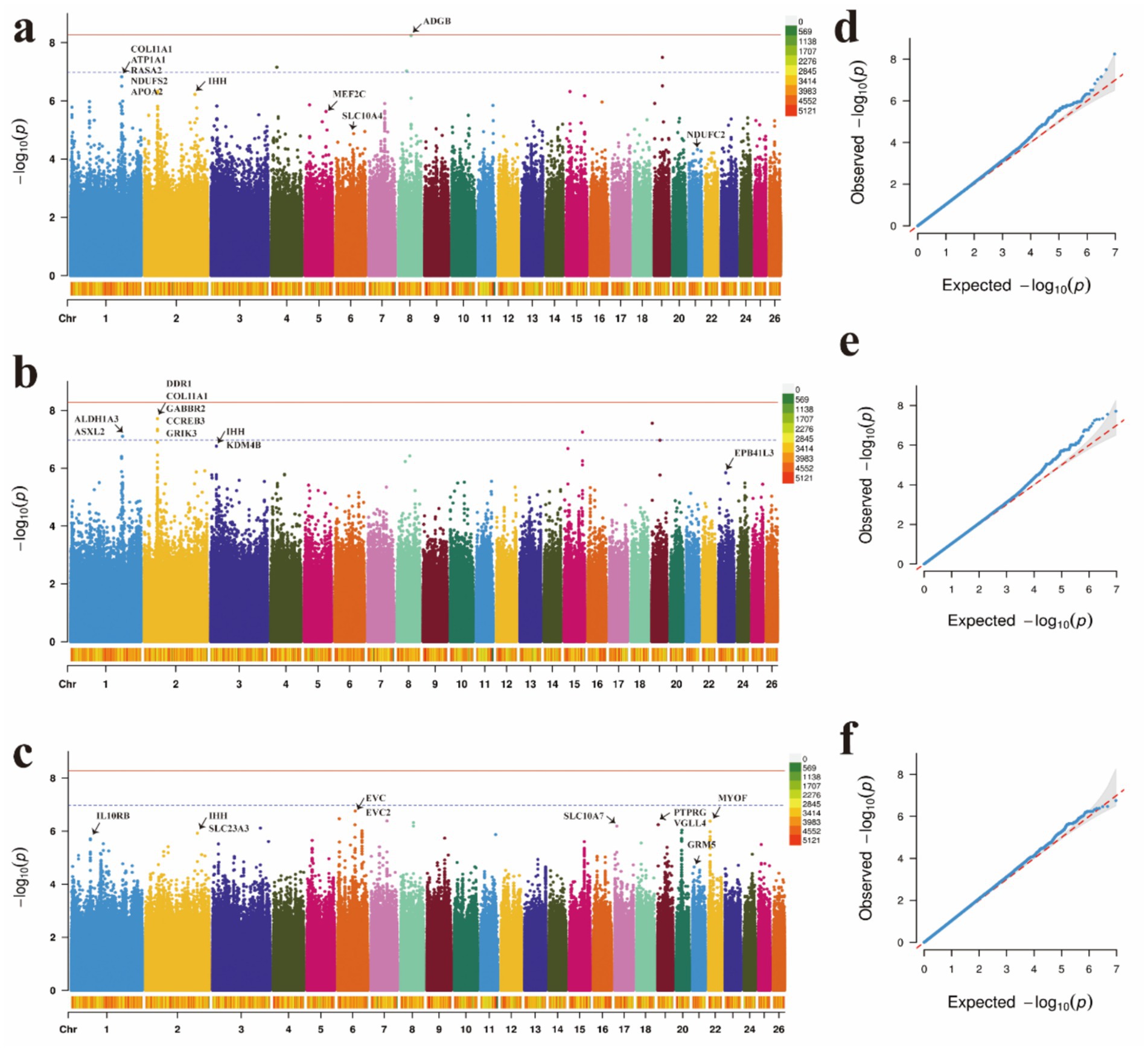
Figure 3. Manhattan plots and QQ plots of (a) body height, (b) body length, and (c) body weight traits. (d) QQ plots of the body height, (e) QQ plots of the body length, (f) QQ plots of the body weight.
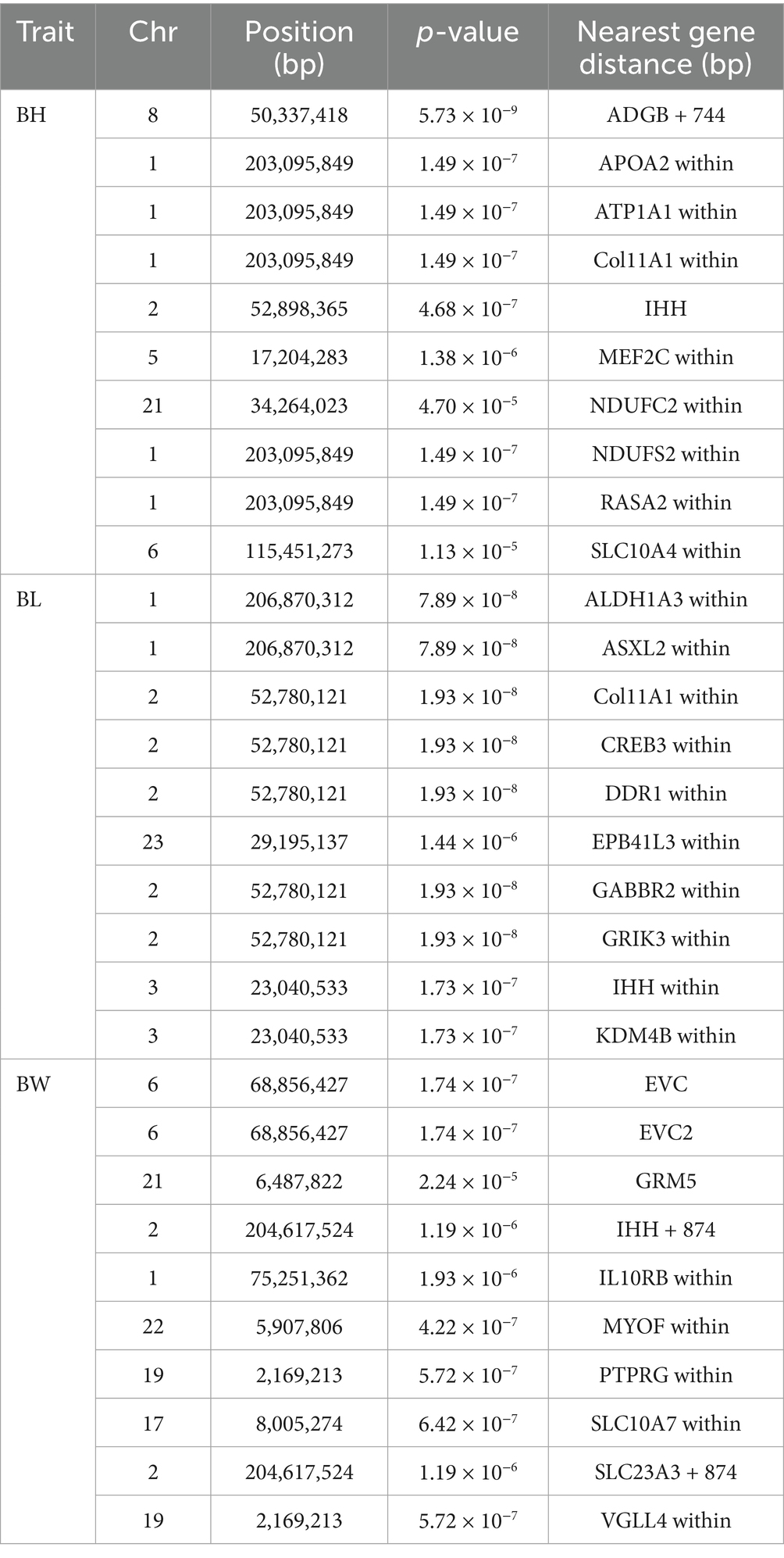
Table 1. Significant SNPs associated with body traits.
Similarly, through GWAS analysis, several key genes associated with wool traits and their genetic variations were identified (Supplementary Tables 21, 22). Further annotation analysis revealed 19 significant loci linked to these traits. For wool fiber length (WL), the two most significant SNPs were located on OAR1, while one SNP was identified on each of OAR26, OAR11, OAR15, OAR24, OAR19, and OAR4, and two SNPs were found on OAR13 (Figures 4a,c). Regarding wool fiber fineness (WF), five SNPs were significantly associated with candidate regions on OAR8, OAR11, and OAR2, with the two most significant SNPs located on OAR6 (Figures 4b,d). We identified a set of significantly correlated SNP loci, with detailed information regarding these significant SNPs presented in Table 2.
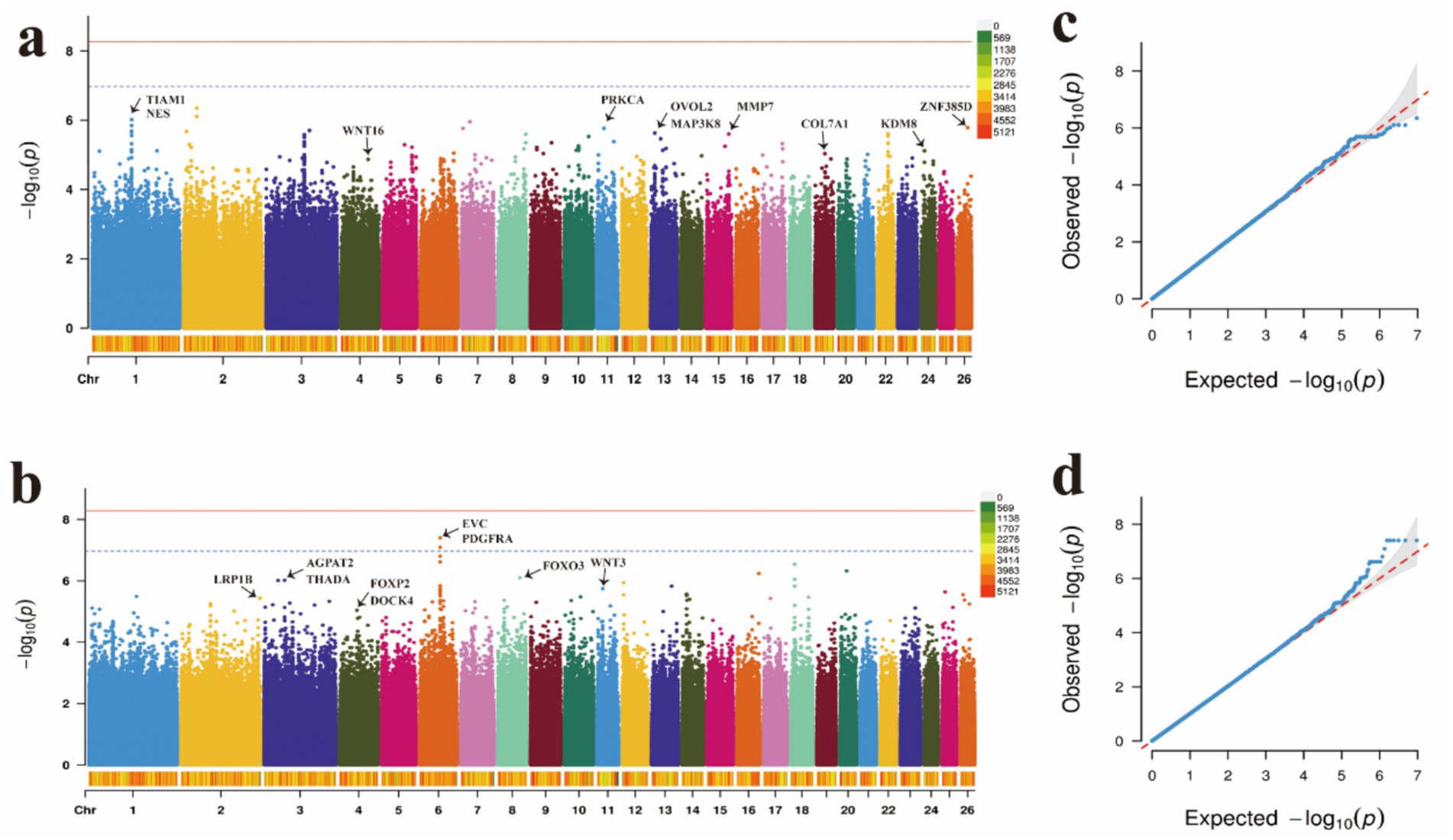
Figure 4. Manhattan plots and QQ plots of (a) wool fiber length, (b) wool fiber fineness traits, and (c) wool fiber length. (d) QQ plots of the wool fiber fineness.
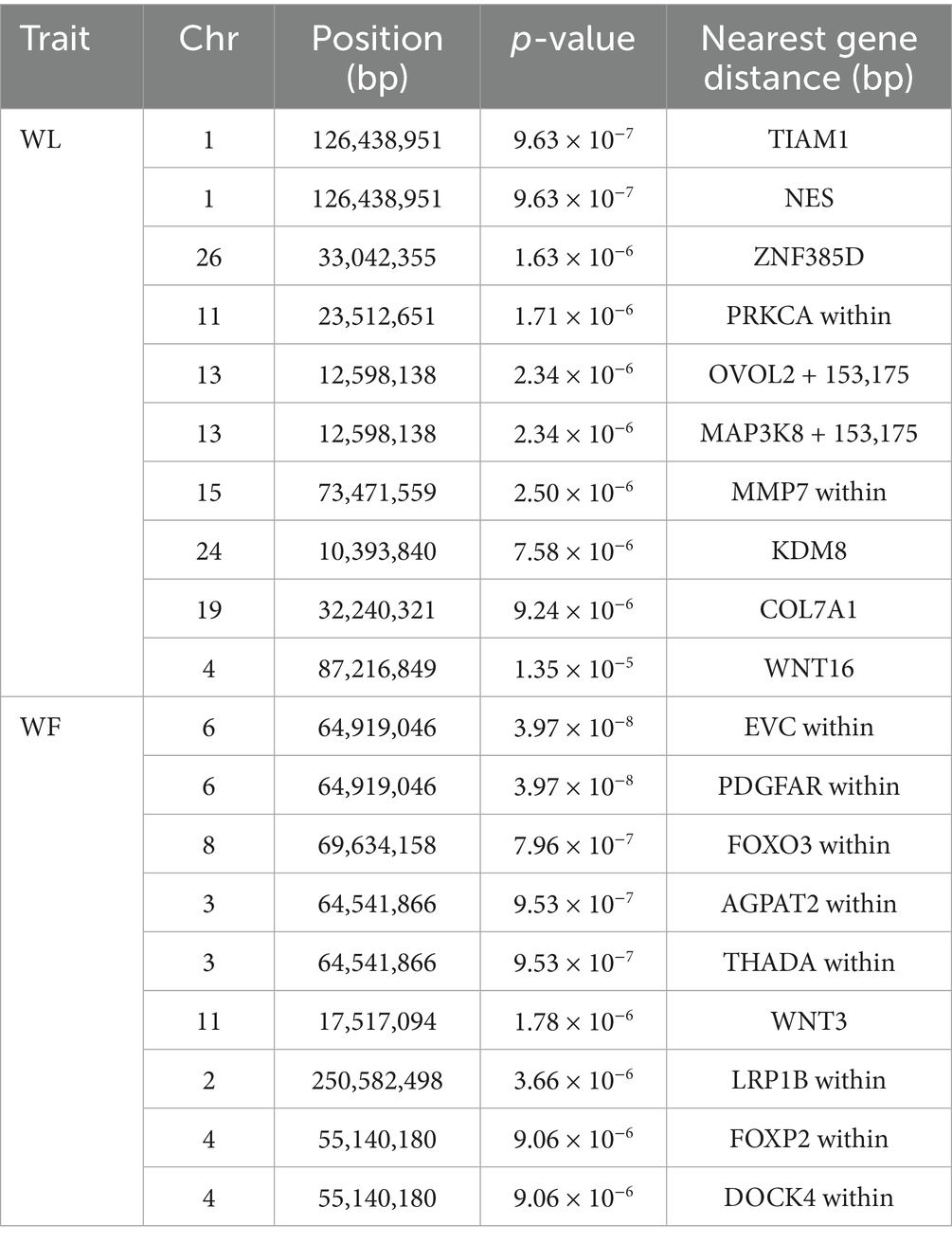
Table 2. Significant SNPs associated with body traits.
To comprehensively assess the characteristics of the candidate genes annotated by SNPs, we conducted enrichment analyses to identify the functional categories associated with these genes. In terms of body size traits, the enriched KEGG pathways are primarily associated with the cytoskeleton in muscle cells, cGMP-PKG signaling pathway, cAMP signaling pathway and hedgehog signaling pathway (Figures 5a–c and Supplementary Table 23). For wool fiber traits, the focus is mainly on the melanogenesis, TNF signaling pathway, Wnt signaling pathway, EGFR tyrosine kinase inhibitor resistance, hedgehog signaling pathway and Rap1 signaling pathway (Figures 5d,e and Supplementary Table 24).
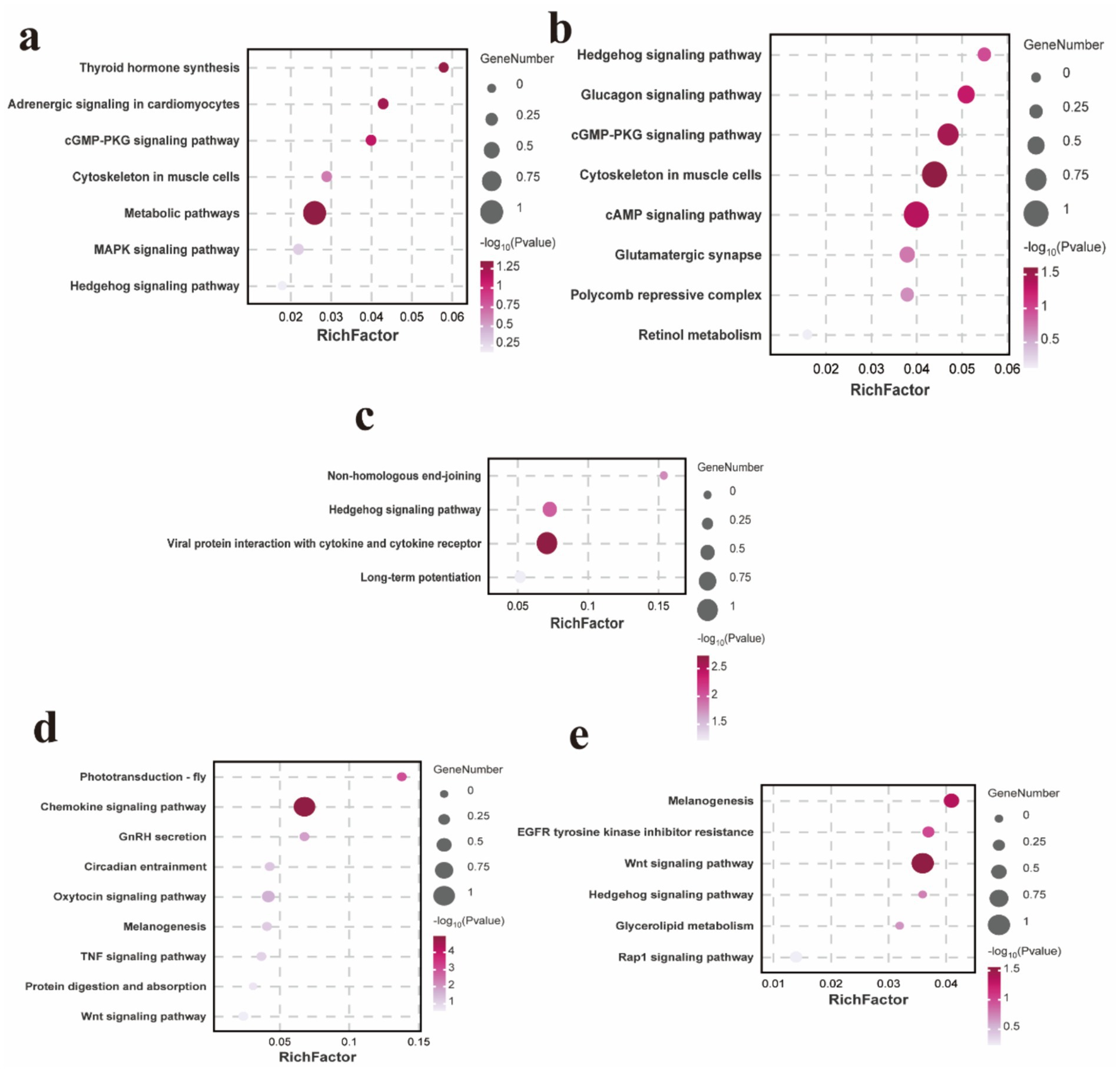
Figure 5. KEGG enrichment analysis pathway map of candidate genes associated with body height (a). KEGG enrichment analysis pathway map of candidate genes associated with body length (b). KEGG enrichment analysis pathway map of candidate genes associated with body weight (c). KEGG enrichment analysis pathway map of candidate genes associated with wool fiber length (d). KEGG enrichment analysis pathway map of candidate genes associated with wool fiber fineness (e).
Analysis of RNA-seq data and functional annotation of DEGs associated with wool traits
To elucidate the molecular mechanisms underlying the distinctive long hair traits of Tibetan sheep, we conducted transcriptomic analyses on skin samples from two Tibetan sheep breeds, ZY and OL, which exhibit significant differences in wool length. On average, 143,236,464 and 126,767,572 original reads were produced in the ZY and OL cDNA libraries, respectively. The GC contents of ZY and OL were 47.09 to 48.30% and 48.05 to 48.25%, respectively (Supplementary Table 25). Based on the significance threshold (|FC| >2 and p-value <0.05), a total of 1,065 DEGs were identified, including 434 up-regulated and 631 down-regulated genes (Figure 6a). The hierarchical clustering showed that there were significant differences in the expression profiles of DEGs between the treat group (GY) and the control group (OL) (Figure 6b). GO analysis revealed significant enrichment in several biological processes, including fibrillar collagen trimer, melanin metabolic process, peptidase regulator activity, and cellular lipid metabolism process (Figure 6c and Supplementary Table 26). A total of 39 significant pathways were identified through KEGG pathway analysis. The top 20 enriched pathways are presented in Figure 6d, indicating that DEGs (73 up-regulated and 155 down-regulated) were predominantly enriched in ECM-receptor interaction, PPAR signaling pathway, focal adhesion, fatty acid elongation and biosynthesis of unsaturated fatty acids these pathways (Supplementary Table 27). Based on the threshold for DEGs analysis, we identified and screened protein pairs with a node score greater than 0.95 in the STRING database to construct the PPI network (Figure 6e). According to the interaction results, there are four up-regulated gene modules (HOXC6, HOXA6, HOXA7, and HOXB6) and four down-regulated gene modules (COL1A1, COL1A2, COL3A1, and SPARC). Additionally, some modules contain both up-regulated genes (such as ATP6, COX2) and down-regulated genes (such as CYTB). Notably, the HOX homeobox genes, which encodes transcription factors (TFs), exhibited a significantly high degree of connectivity within this network. To further investigate the distribution characteristics of differentially expressed TFs, we conducted a statistical analysis of the number of differentially expressed TFs in each family within the control group. The results indicate that the homeobox family exhibits the most significant differential expression, with 10 upregulated TFs identified, including HOXC6, HOXC8, HOXA6, HOXA10, HOXC9, HOXA1, HOXB6, and HOXC10, as well as six downregulated factors (Figure 6f and Supplementary Table 28). Notably, the Hox subfamily demonstrates the most extensive distribution pattern within this family. This finding highlights the critical role of the HOX homeobox family, particularly its Hox subfamily, in gene regulatory networks.
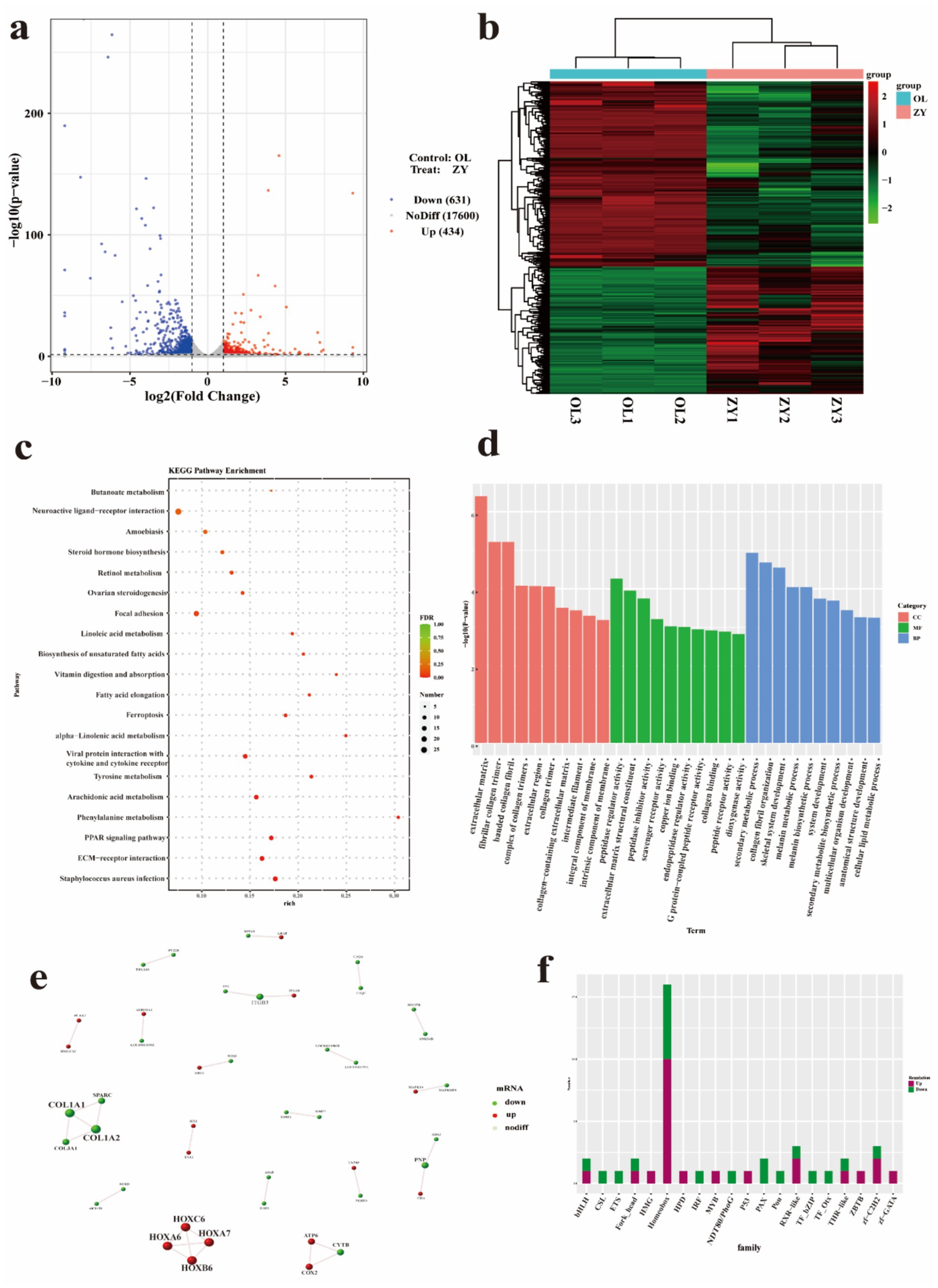
Figure 6. Skin transcriptome analysis. (a) Volcano plot of DEGs. (b) Clustered expression heatmaps of all mRNAs. (c) The top 20 KEGG pathways for DE mRNAs. (d) GO enrichment pathways for DE mRNAs. (e) PPI network of DEGs. (f) Histogram of differentially expressed transcription factors.
Identification of functional mutations in DEGs supported by whole-genome resequencing data
To further evaluate the expression levels of candidate genes identified through GWAS, we examined the intersection between these candidate genes and DEGs associated with two distinct wool traits. SNPs were identified in critical regions of 10 key overlapping DEGs. For instance, a non-synonymous SNP in the coding region of WNT16 was predicted to alter the protein sequence, whereas synonymous mutations were observed in the coding regions of NES and TIAM1. SNPs were also detected in intergenic regions of PRKCA, MAP3K8, MMP7, and OVOL2, as well as within introns of COL7A1, KDM8, and ZNF385D (Table 3). The expression levels of 10 overlapping DEGs exhibited differential expression and distinct clustering patterns, with 8 genes up-regulated and 2 genes down-regulated (Figure 7a). Subsequently, KEGG enrichment analysis was conducted on these overlapping genes to elucidate their biological functions. The results indicate that these genes are involved in multiple metabolic pathways, including the Wnt signaling pathway, Ras signaling pathway, protein digestion and absorption, TNF signaling pathway, melanogenesis and chemokine signaling pathway (Figure 7b and Supplementary Table 29). Heat maps were utilized to visualize the genotypes of overlapping genes, and the results demonstrated that the polymorphism of the GY genotype was significantly higher compared to that of the OL genotype (Figure 7c).
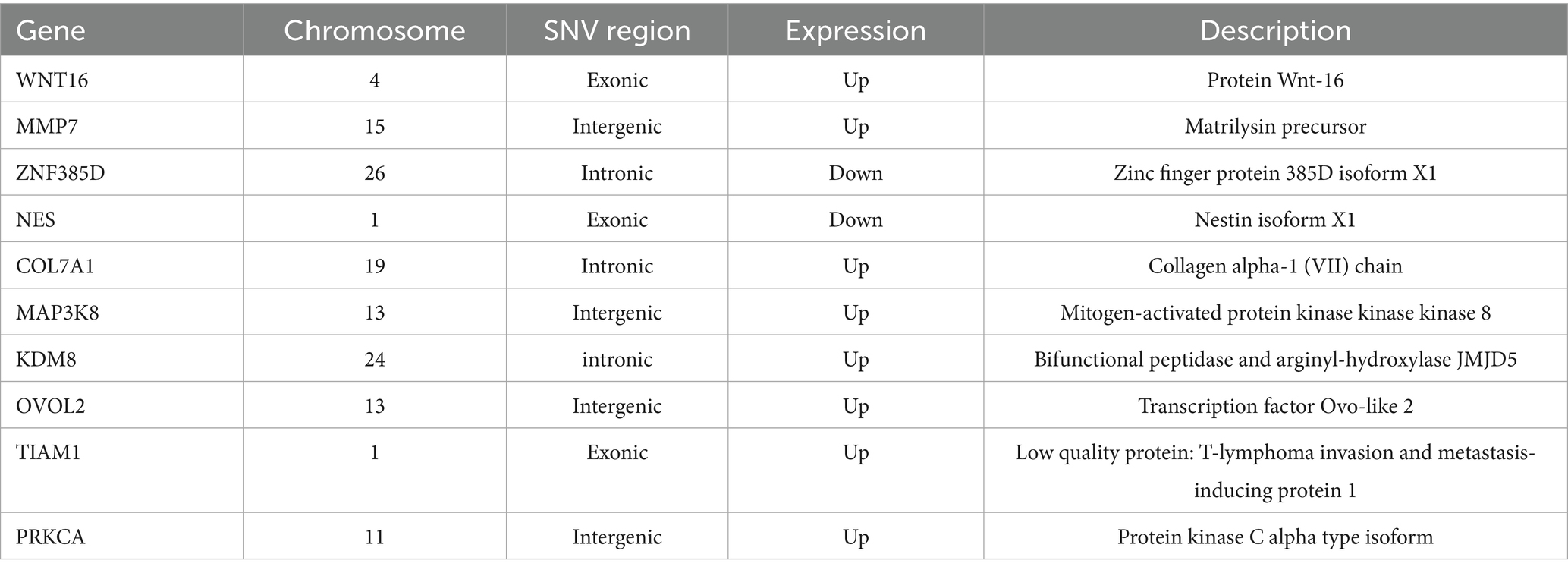
Table 3. Shared genes exhibiting similar expression patterns and functional annotations.
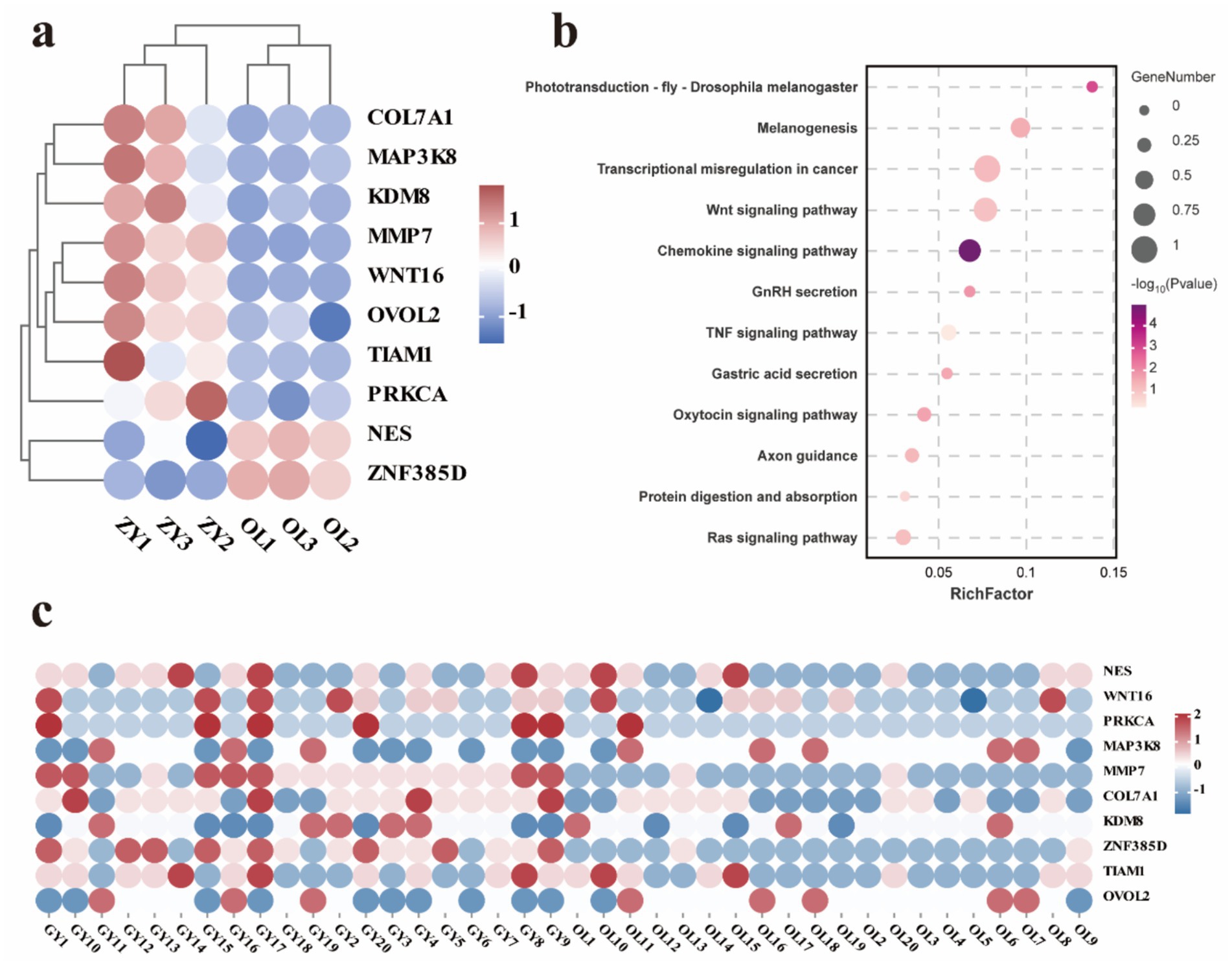
Figure 7. Analysis of key gene expression and variation. (a) Clustered expression heatmap of 10 key genes. (b) KEGG enrichment analysis of key expression genes. (c) Heatmap of variation of 10 key expressed genes.
RT-qPCR validation of DEGs
To validate the accuracy of the DEGs assay at the transcriptome level, we selected seven genes and evaluated their expression levels using six replicates for each sample. The results demonstrate a high degree of consistency between the two methods at the expression level, with a Pearson correlation coefficient of 0.75 (Figure 8). This indicates that the RNA-seq data exhibit strong reliability. The selected candidate genes exhibited strong concordance at both the transcriptional and actual expression levels, further substantiating their potential significance in subsequent functional analyses.
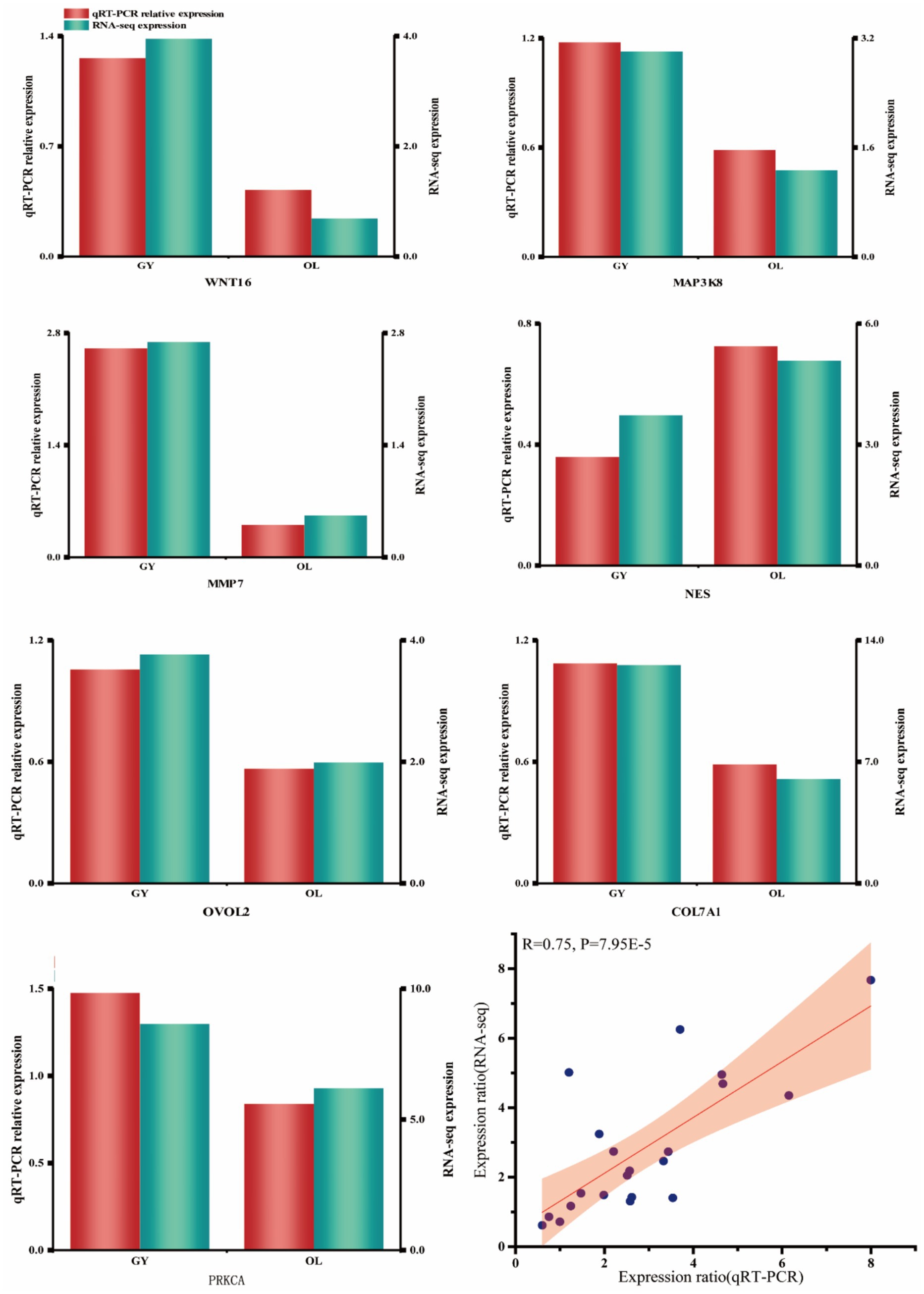
Figure 8. RT-qPCR validation of DEGs and correlation scatter plot of DEGs.
Discussion
The genetic diversity of sheep constitutes the foundation of their evolutionary success. This diversity arises from both natural selection in response to diverse environmental pressures and artificial selection driven by human breeding programs designed to enhance economically valuable traits. There is compelling evidence indicating accelerated changes in specific genomic regions under artificial selection (19). A lower level of genomic diversity in domestic breeds compared to their wild ancestors, suggesting that a significant amount of genetic variation has been lost during and after the domestication process. While the genomic diversity of local breeds has largely been retained in improved varieties, contemporary breeding practices that focus on a limited range of commercial varieties have led to genetic homogenization. This increases the risk of adaptive allelic loss in native populations (20). Despite significant advancements in genomics, systematic studies comparing genetic divergence among geographically distinct sheep populations are still limited, especially for local breeds that possess unique ecological adaptations. In this study, we performed a comprehensive analysis of seven indigenous Chinese sheep breeds, including several Tibetan sheep ecotypes, to elucidate the genetic basis underlying their divergent traits such as hypoxic adaptation, growth performance, and wool characteristics, and to investigate their population structure. Our results revealed significant genetic differentiation among Tibetan sheep subpopulations, driven by both natural selection and historical pastoral management practices. Notably, genome-wide comparisons of local breeds have been limited, and our study addresses this gap by providing high-resolution insights into the genetic variation within these understudied populations. These findings underscore the indispensable role of the distinctive phenotypic traits of local sheep breeds in preserving the genetic diversity of sheep within our country. Additionally, the molecular markers identified through whole-genome resequencing that are significantly associated with key economic traits offer a robust foundation for developing a marker-assisted selection system. The seven native sheep breeds examined in this study have developed distinct adaptive phenotypic differentiation across varying elevations on the Tibetan Plateau. Among these, GY, ZK, HZ, and ZS, as typical high-altitude adapted breeds (≥3,000 m), exhibit longer wool fiber lengths and higher hair follicle densities. These traits likely represent a low-temperature adaptive strategy achieved through positive selection of genes associated with hair follicle development (21). In contrast, QM and OL display a “short coarse hair” phenotype, which, while reducing textile suitability, is significantly positively correlated with muscle growth efficiency, reflecting the selection pressure driven by the economic needs of herders (22, 23). Domestication, adaptation, and artificial selection have led to a diverse array of coat colors, which are the most distinctive characteristics among different breeds (24). The ancestral coat color of sheep was predominantly brown; however, domesticated breeds now exhibit a wide variety of colors and patterns (25). Notably, the QM breed primarily displays black-brown and yellow-brown coats, often with white spotting on the back, sides, and hips. Some studies suggest that this coloration may be influenced by upstream regions of the MITF gene and strong linkage disequilibrium with other loci (26), providing a novel model for studying the formation of body surface patterns in mammals.
Hypoxic adaptation in plateau animals involves a complex, multi-layered molecular regulatory network wherein key genes enhance oxygen utilization efficiency through synergistic interactions. IGF1R is a tyrosine kinase receptor located on the cell surface. Its binding to its ligands, IGF1 or IGF2, activates downstream PI3K/Akt and RAS/MAPK signaling pathways, thereby promoting cell proliferation, differentiation, migration, and survival while inhibiting apoptosis (27). Hypoxia can induce alterations in the IGF system, potentially due to diminished anabolic effects of IGFs. Additionally, increased expression of IGF1R may reflect a tissue-protective mechanism that compensates for changes in the IGF system, such as reduced serum levels of IGF-I and -II and elevated IGFBP-1 (28). Decreased expression of SLC4A7 under hypoxic conditions may compromise critical intracellular pH regulatory functions that cells normally maintain through substantial resource allocation. This adverse effect is particularly pronounced in the mammalian central nervous system, where hypoxia induces a spectrum of physiological responses that vary depending on the developmental stage (29). This upregulation suggests that enhanced fluid reabsorption may serve to prevent edema and inhibit T cell proliferation, thereby mitigating acute lung inflammation and facilitating adaptation to hypoxic conditions (30). DOCK8 functions as a negative regulator of HIF2α nuclear translocation in CD4+ T cells (31). VPS13A, JAK1, TMTC2, and other genes are implicated in multi-dimensional biological regulation, collectively mediating the complex, multi-level response mechanisms involved in hypoxia adaptation (32–34). The stable genetic adaptability of Tibetan sheep facilitates dynamic multilevel adaptive variations at the genomic level, thereby enhancing the species’ systemic ecological response efficiency to extreme high-altitude hypoxia stress.
The high diversity of domestic animal coat color results from a combination of artificial selection and adaptive evolution. This diversity largely reflects variations in human preferences or the fixation of certain colors associated with desirable domestication traits such as docility, reproductive rate, and growth rate. In the local Tibetan sheep population, coat colors include black, white, brown, and variegated phenotypes, with white being the most common. MITF serves as a critical regulator in the development, proliferation, and survival of melanocytes. Mutations in MITF can result in metabolic dysfunction of pigment cells, thereby impacting fur pigmentation (35). A synonymous MITF g.1548 C/T mutation was identified, and the frequency of the C allele was strongly associated with the pure white coat in Tibetan sheep, indicating that the C allele is likely the dominant allele for white coat color (36). Loss-of-function mutations in the MC1R gene result in a shift towards phaeomelanin synthesis, while SNPs at position 901C/T within the MC1R coding region are associated with white coat color (37). In mouse embryos, TCF25 is robustly expressed in the dorsal root ganglion. Based on this observation, we hypothesize that the regional differential expression of TCF25 may influence subsequent melanocyte migration, potentially leading to the formation of dark streaks along the midline of the back in goats (38).
Body size traits, including BH, BL, and BW, are critical indicators of production performance in livestock. Their genetic architecture is shaped by both natural selection (e.g., optimization of the body surface area-to-volume ratio for cold adaptation) and artificial selection (targeted breeding for economically important traits such as meat yield and feed conversion ratio). Variance estimation for sheep body size traits revealed heritability (h2) values of 0.22 ± 0.08 for BW, 0.11 ± 0.06 for BL, and 0.17 ± 0.06 for BH, with significant genetic correlations among these traits. The observed positive genetic and phenotypic correlations indicate that selection for body size traits can improve both genetic and phenotypic body size, making it a key target for modern molecular breeding (39). As a crucial approach in modern quantitative genetics research, GWA studies have showcased substantial advantages in the genetic analysis of body shape traits in sheep. In this study, by employing the mixed linear model analysis method, we systematically identified genetic variants significantly associated with body height, body length, and body weight in Tibetan sheep. These variants may represent key genetic factors influencing the development of body shape traits in Tibetan sheep. RASA2 is a compelling candidate gene for regulating height, as copy number variants (CNVs) and loss-of-function mutations in RASA2 have been associated with short stature (40). NDUFS2 is considered a functional candidate gene influencing meat quality, primarily governing phenotypes such as fat metabolism and muscle development, and playing a crucial role in energy metabolism synthesis (41). IHH, as a critical signal for the local growth of endochondral bone, regulates bone development through multiple parallel pathways. Disruption of IHH signaling leads to a progressive reduction in embryonic bone size (42). Inactivation mutations in Evc or Evc2 within the perichondrium result in markedly elevated FGF signaling, leading to severe dwarfism characteristic of Evc syndrome (43). COL11A1 plays an essential role in bone morphogenesis, and variations in this gene are linked to human height (44). ALDH1A3, ATP1A1, MEF2C, NDUFC2, EPB41L3, IL10RB, PTPRG, and GRM5 are likely to play a significant role in growth traits (45–52). The effects of the aforementioned diverse genes highlight the intricate genetic regulatory mechanisms underlying body size, a quintessential quantitative trait. The development of body size is a dynamic and multi-tiered regulatory process. Future studies can achieve a more comprehensive understanding of the genetic basis of body size by constructing gene regulatory networks and identifying functional modules.
In an integrated analysis of GWAS and transcriptome data, we combined genomic variation with gene expression profiles to identify a set of genes whose expression levels are significantly influenced by genetic variation. These candidate genes may contribute to phenotypic variation through the regulation of key biological pathways or molecular mechanisms. Long wool is a crucial genetic resource in Qinghai Tibetan sheep, characterized by its high content of medullated fibers. Consequently, Tibetan wool is extensively utilized in carpet manufacturing and renowned for its superior quality (53, 54). CircRNAs may play a crucial role in the development of hair follicles and the growth of cashmere by forming a balanced regulatory relationship with their host gene, TIAM1 (55). The long non-coding RNA (lncRNA) MSTRG.20890.1, transcribed from the intronic region of the ZNF385D gene, inhibits the proliferation and migration of dermal fibroblasts by competitively binding to chi-miR-24-3p with ADAMTS3. Consequently, this interaction leads to the inhibition of dermal papillary structure formation and secondary hair follicle morphogenesis (56). OVOL1-OVOL2 axis serves as a positive regulator in normal hair development and differentiation, facilitating the proliferation and differentiation of hair follicle cells. Therefore, OVOL1 and OVOL2 may represent potential therapeutic targets for the treatment of hair loss (57). Nestin (NES)-containing cells constitute the predominant cell population in the hair follicle throughout each follicle cycle, and nestin-expressing cells serve as stem cells for the entire hair follicle (58). The down-regulated expression of MAP3K8 inhibits the proliferation and melanin synthesis in sheep melanocytes (59). PKC plays a pivotal role in cellular signal transduction, modulating the proliferation and differentiation of hair follicle cells (60). It is noteworthy that in our study, the HOX homeobox genes, which encode TFs, exhibited exceptionally high connectivity within the protein–protein interaction (PPI) network and demonstrated the most significant differential expression. Previous research has found that HOX family TFs play a pivotal role in regulating cell differentiation, function, proliferation, embryonic development, and tissue homeostasis by modulating the promoter regions of multiple target genes (61). Specifically, in hair follicle biology, HOX homeobox genes are crucial for establishing the topological specificity of hair follicles and are integral to their development, cycle regulation, and regeneration (62–64). These genes may significantly influence hair follicle formation and function through the regulation of hair follicle stem cell activity, interaction with signaling pathways, and region-specific expression patterns. These findings underscore the important role of HOX genes in hair follicle biology and provide valuable insights for further research into the mechanisms of hair follicle development and regeneration. These results established a critical foundation for the subsequent validation studies and provided novel insights into the genetic analysis of sheep wool traits. Moving forward, we intend to conduct in-depth investigations of the identified candidate genes, encompassing gene function elucidation, regulatory network construction, and both in vitro and in vivo functional validations. This will further uncover the causal mutations influencing sheep wool traits and their associated molecular regulatory pathways, thereby offering alternative molecular targets and a robust theoretical basis for sheep breeding.
Conclusion
Our experimental strategy is based on selective clearance analysis of whole genome resequencing and integrated analysis of GWAS and transcriptome data to identify key genomic regions and genes under selection in the Tibetan sheep genome. By combining genomic variation and gene expression profiles, we have successfully pinpointed candidate genes associated with wool traits and their regulatory pathways. These findings not only elucidate the genetic mechanisms driving the unique adaptability of Tibetan sheep to extreme environments but also offer novel insights into the molecular basis of economically important traits. Furthermore, our results provide a robust theoretical foundation for the conservation and utilization of sheep genetic resources, significantly advancing molecular breeding and genetic improvement efforts for Tibetan sheep.
Data availability statement
The raw reads produced in this study were deposited in the NCBI SRA with the accession number SRA SUB15389572 under Bio-project PRJNA1280483. Additional data can be found in Supplementary files.
Ethics statement
The animal studies followed the recommendations of the “Regulations for the Management of Affairs Concerning Experimental Animals” (Ministry of Science and Technology, China, revised in June 2004). The study was approved by the Animal Care and Use Committees of the Northwest Institute of Plateau Biology, Chinese Academy of Sciences. The animals are not harmed during sample collection. The study was conducted in accordance with the ARRIVE Guidelines. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
DT: Writing – review & editing, Data curation, Funding acquisition, Visualization, Writing – original draft, Conceptualization, Formal analysis. BH: Formal analysis, Data curation, Investigation, Writing – review & editing, Software. XL: Writing – review & editing, Software, Methodology, Data curation. QP: Writing – review & editing, Resources, Project administration, Data curation. BZ: Investigation, Resources, Data curation, Writing – review & editing. KZ: Validation, Supervision, Investigation, Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Qinghai Science and Technology Major Program (2021-NK-A5) and National Breeding Joint Research Project.
Acknowledgments
We greatly appreciate our collaborators for their assistance in sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1605252/full#supplementary-material
Footnotes
1. ^https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_017524585.1/
References
1. Zeder, MA. Domestication and early agriculture in the Mediterranean basin: origins, diffusion, and impact. Proc Natl Acad Sci USA. (2008) 105:11597–604. doi: 10.1073/pnas.0801317105
2. Hu, X-J, Yang, J, Xie, X-L, Lv, F-H, Cao, Y-H, Li, W-R, et al. The genome landscape of Tibetan sheep reveals adaptive introgression from argali and the history of early human settlements on the Qinghai-Tibetan plateau. Mol Biol Evol. (2019) 36:283–303. doi: 10.1093/molbev/msy208
3. Zhao, Y-X, Yang, J, Lv, F-H, Hu, X-J, Xie, X-L, Zhang, M, et al. Genomic reconstruction of the history of native sheep reveals the peopling patterns of nomads and the expansion of early pastoralism in East Asia. Mol Biol Evol. (2017) 34:2380–95. doi: 10.1093/molbev/msx181
5. Zhao, Y, Zhao, E, Zhang, N, and Duan, C. Mitochondrial DNA diversity, origin, and phylogenic relationships of three Chinese large-fat-tailed sheep breeds. Trop Anim Health Prod. (2011) 43:1405–10. doi: 10.1007/s11250-011-9869-2
6. Zhang, Z, Xu, D, Wang, LI, Hao, J, Wang, J, Zhou, X, et al. Convergent evolution of rumen microbiomes in high-altitude mammals. Curr Biol. (2016) 26:1873–9. doi: 10.1016/j.cub.2016.05.012
7. Han, B, Tian, D, Li, X, Liu, S, Tian, F, Liu, D, et al. Multiomics analyses provide new insight into genetic variation of reproductive adaptability in Tibetan sheep. Mol Biol Evol. (2024) 41:msae058. doi: 10.1093/molbev/msae058
8. Tian, D, Zhang, Z, Huang, B, Han, B, Li, X, and Zhao, K. Genome-wide association analyses and population verification highlight the potential genetic basis of horned morphology during polled selection in Tibetan sheep. Animals. (2024) 14:2152. doi: 10.3390/ani14152152
9. Liang, X, Duan, Q, Li, B, Wang, Y, Bu, Y, Zhang, Y, et al. Genomic structural variation contributes to evolved changes in gene expression in high-altitude Tibetan sheep. Proc Natl Acad Sci USA. (2024) 121:e2322291121. doi: 10.1073/pnas.2322291121
10. Tian, D, Han, B, Li, X, Liu, D, Zhou, B, Zhao, C, et al. Genetic diversity and selection of Tibetan sheep breeds revealed by whole-genome resequencing. Anim Biosci. (2023) 36:991–1002. doi: 10.5713/ab.22.0432
11. Li, H, and Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
12. Van der Auwera, GA, Carneiro, MO, Hartl, C, Poplin, R, Del Angel, G, Levy-Moonshine, A, et al. From fastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. (2013) 43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
13. Yu, Q, Ling, Y, Xiong, Y, Zhao, W, Xiong, Y, Dong, Z, et al. RAD-seq as an effective strategy for heterogenous variety identification in plants—a case study in Italian ryegrass (Lolium multiflorum). BMC Plant Biol. (2022) 22:231. doi: 10.1186/s12870-022-03617-6
14. Wang, K, Li, M, and Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164–4. doi: 10.1093/nar/gkq603
15. Chen, K, Wallis, JW, McLellan, MD, Larson, DE, Kalicki, JM, Pohl, CS, et al. Breakdancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. (2009) 6:677–81. doi: 10.1038/nmeth.1363
16. Abyzov, A, Urban, AE, Snyder, M, and Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. (2011) 21:974–84. doi: 10.1101/gr.114876.110
17. Guindon, S, Dufayard, J-F, Lefort, V, Anisimova, M, Hordijk, W, and Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. (2010) 59:307–21. doi: 10.1093/sysbio/syq010
18. Chen, S, Zhou, Y, Chen, Y, and Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
19. Kijas, JW, Lenstra, JA, Hayes, B, Boitard, S, Porto Neto, LR, San Cristobal, M, et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. (2012) 10:e1001258. doi: 10.1371/journal.pbio.1001258
20. Li, X, Yang, J, Shen, M, Xie, XL, Liu, GJ, Xu, YX, et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat Commun. (2020) 11:2815. doi: 10.1038/s41467-020-16485-1
21. Wu, C, Ma, S, Zhao, B, Qin, C, Wu, Y, Di, J, et al. Drivers of plateau adaptability in cashmere goats revealed by genomic and transcriptomic analyses. BMC Genomics. (2023) 24:428. doi: 10.1186/s12864-023-09333-1
22. Zhang, Y, Xue, X, Liu, Y, Abied, A, Ding, Y, Zhao, S, et al. Genome-wide comparative analyses reveal selection signatures underlying adaptation and production in Tibetan and Poll Dorset sheep. Sci Rep. (2021) 11:2466. doi: 10.1038/s41598-021-81932-y
23. Mortimer, SI, Hatcher, S, Fogarty, NM, van der Werf, JHJ, Brown, DJ, Swan, AA, et al. Genetic correlations between wool traits and carcass traits in merino sheep. J Anim Sci. (2017) 95:2385–98. doi: 10.2527/jas.2017.1385
24. Darwin, C, and Charles, D. The variation of animals and plants under domestication. Cambridge: Cambridge University Press (2010).
25. Lundie, RS. The genetics of colour in fat-tailed sheep: a review. Trop Anim Health Prod. (2011) 43:1245–65. doi: 10.1007/s11250-011-9850-0
26. Edea, Z, Dadi, H, Dessie, T, Kim, I-H, and Kim, K-S. Association of MITF loci with coat color spotting patterns in Ethiopian cattle. Genes Genomics. (2017) 39:285–93. doi: 10.1007/s13258-016-0493-4
27. Griffiths, CD, Bilawchuk, LM, McDonough, JE, Jamieson, KC, Elawar, F, Cen, Y, et al. IGF1R is an entry receptor for respiratory syncytial virus. Nature. (2020) 583:615–9. doi: 10.1038/s41586-020-2369-7
28. Custodio, RJ, Do Carmo Custodio, VI, Scrideli, CA, SLS, M, Cervi, MC, Cupo, P, et al. Impact of hypoxia on IGF-I, IGF-II, IGFBP-3, ALS and IGFBP-1 regulation and on IGF1R gene expression in children. Growth Hormon IGF Res. (2012) 22:186–91. doi: 10.1016/j.ghir.2012.07.001
29. Chen, L-M, Choi, I, Haddad, GG, and Boron, WF. Chronic continuous hypoxia decreases the expression of SLC4A7 (NBCn1) and SLC4A10 (NCBE) in mouse brain. Am J Phys Regul Integr Comp Phys. (2007) 293:R2412–20. doi: 10.1152/ajpregu.00497.2007
30. Sharma, P, Bansal, A, and Sharma, PC. RNA-seq-based transcriptome profiling reveals differential gene expression in the lungs of Sprague–Dawley rats during early-phase acute hypobaric hypoxia. Mol Gen Genomics. (2015) 290:2225–40. doi: 10.1007/s00438-015-1064-0
31. Yamamura, K, Uruno, T, Shiraishi, A, Tanaka, Y, Ushijima, M, Nakahara, T, et al. The transcription factor EPAS1 links DOCK8 deficiency to atopic skin inflammation via IL-31 induction. Nat Commun. (2017) 8:13946. doi: 10.1038/ncomms13946
32. Dong, K, Yao, N, Pu, Y, He, X, Zhao, Q, Luan, Y, et al. Genomic scan reveals loci under altitude adaptation in Tibetan and Dahe pigs. PLoS One. (2014) 9:e110520. doi: 10.1371/journal.pone.0110520
33. Lin, Q, Chen, Z, Shi, W, Lv, Z, Wan, X, and Gao, K. JAK1 inactivation promotes proliferation and migration of endometrial cancer cells via upregulating the hypoxia-inducible factor signaling pathway. Cell Commun Signal. (2022) 20:177. doi: 10.1186/s12964-022-00990-5
34. Han, Z, Yang, R, Zhou, W, Zhang, L, Wang, J, Liu, C, et al. Population structure and selection signal analysis of indigenous sheep from the southern edge of the Taklamakan Desert. BMC Genomics. (2024) 25:681. doi: 10.1186/s12864-024-10581-y
35. Fontanesi, L, Scotti, E, and Russo, V. Haplotype variability in the bovine MITF gene and association with piebaldism in Holstein and Simmental cattle breeds. Anim Genet. (2012) 43:250–6. doi: 10.1111/j.1365-2052.2011.02242.x
36. Han, J, Min, Y, Guo, T, Yue, Y, Liu, J, Niu, C, et al. Molecular characterization of two candidate genes associated with coat color in Tibetan sheep (Ovis arise). J Integr Agric. (2015) 14:1390–7. doi: 10.1016/S2095-3119(14)60928-X
37. Almathen, F, Elbir, H, Bahbahani, H, Mwacharo, J, and Hanotte, O. Polymorphisms in MC1R and ASIP genes are associated with coat color variation in the Arabian camel. J Hered. (2018) 109:700–6. doi: 10.1093/jhered/esy024
38. Sun, X, Jiang, J, Wang, G, Zhou, P, Li, J, Chen, C, et al. Genome-wide association analysis of nine reproduction and morphological traits in three goat breeds from southern China. Anim Biosci. (2022) 36:191–9. doi: 10.5713/ab.21.0577
39. Abbasi, M-A, and Ghafouri-Kesbi, F. Genetic (co) variance components for body weight and body measurements in Makooei sheep. Asian Australas J Anim Sci. (2011) 24:739–43. doi: 10.5713/ajas.2011.10277
40. Lin, E, Tsai, S-J, Kuo, P-H, Liu, Y-L, Yang, AC, Conomos, MP, et al. Genome-wide association study in the Taiwan biobank identifies four novel genes for human height: NABP2, RA SA2, RNF41 and SLC39A5. Hum Mol Genet. (2021) 30:2362–9. doi: 10.1093/hmg/ddab202
41. Shen, Y, Wang, H, Xie, J, Wang, Z, and Ma, Y. Trait-specific selection signature detection reveals novel loci of meat quality in large white pigs. Front Genet. (2021) 12:761252. doi: 10.3389/fgene.2021.761252
42. Long, F, Joeng, K-S, Xuan, S, Efstratiadis, A, and McMahon, AP. Independent regulation of skeletal growth by Ihh and IGF signaling. Dev Biol. (2006) 298:327–33. doi: 10.1016/j.ydbio.2006.06.042
43. Zhang, H, Kamiya, N, Tsuji, T, Takeda, H, Scott, G, Rajderkar, S, et al. Elevated fibroblast growth factor signaling is critical for the pathogenesis of the dwarfism in Evc2/Limbin mutant mice. PLoS Genet. (2016) 12:e1006510. doi: 10.1371/journal.pgen.1006510
44. Li, Y, Lacerda, DA, Warman, ML, Beier, DR, Yoshioka, H, Ninomiya, Y, et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. (1995) 80:423–30. doi: 10.1016/0092-8674(95)90492-1
45. Yu, H, Yu, S, Guo, J, Cheng, G, Mei, C, and Zan, L. Genome-wide association study reveals novel loci associated with body conformation traits in Qinchuan cattle. Animals. (2023) 13:3628. doi: 10.3390/ani13233628
46. Zhang, X, Chu, Q, Guo, G, Dong, G, Li, X, Zhang, Q, et al. Genome-wide association studies identified multiple genetic loci for body size at four growth stages in Chinese Holstein cattle. PLoS One. (2017) 12:e0175971. doi: 10.1371/journal.pone.0175971
47. Li, L, Shen, B, Dai, S, Tang, Z, Zhao, W, Zhan, S, et al. The novel mutations in the MEF2C gene associate with growth of Nanjiang Yellow goats. Gene Rep. (2019) 14:100–9. doi: 10.1016/j.genrep.2018.12.003
48. Cai, W, Hu, J, Zhang, Y, Guo, Z, Zhou, Z, and Hou, S. Cis-eQTLs in seven duck tissues identify novel candidate genes for growth and carcass traits. BMC Genomics. (2024) 25:429. doi: 10.1186/s12864-024-10338-7
49. Zhang, L, Liu, J, Zhao, F, Ren, H, Xu, L, Lu, J, et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS One. (2013) 8:e66569. doi: 10.1371/journal.pone.0066569
50. Maculewicz, E, Dzitkowska-Zabielska, M, Antkowiak, B, Antkowiak, O, Mastalerz, A, Garbacz, A, et al. Association of interleukin genes IL10 and IL10RB with parameters of overweight in military students. Genes. (2022) 13:291. doi: 10.3390/genes13020291
51. Liu, D, Li, X, Wang, L, Pei, Q, Zhao, J, Sun, D, et al. Genome-wide association studies of body size traits in Tibetan sheep. BMC Genomics. (2024) 25:739. doi: 10.1186/s12864-024-10633-3
52. Zhang, H, Zhuang, Z, Yang, M, Ding, R, Quan, J, Zhou, S, et al. Genome-wide detection of genetic loci and candidate genes for body conformation traits in Duroc × Landrace × Yorkshire crossbred pigs. Front Genet. (2021) 12:664343. doi: 10.3389/fgene.2021.664343
53. Tian, D, Pei, Q, Jiang, H, Guo, J, Ma, X, Han, B, et al. Comprehensive analysis of the expression profiles of mRNA, lncRNA, circRNA, and miRNA in primary hair follicles of coarse sheep fetal skin. BMC Genomics. (2024) 25:574. doi: 10.1186/s12864-024-10427-7
54. Tian, D, Zhang, W, Wang, L, Qi, J, Xu, T, Zuo, M, et al. Proteo-transcriptomic profiles reveal genetic mechanisms underlying primary hair follicle development in coarse sheep fetal skin. J Proteome. (2025) 310:105327. doi: 10.1016/j.jprot.2024.105327
55. Shen, J, Wang, Y, Bai, M, Fan, Y, Wang, Z, and Bai, W. Novel circRNAs from cashmere goats: discovery, integrated regulatory network, and their putative roles in the regeneration and growth of secondary hair follicles. Czeh J Anim Sci. (2022) 67:237–51. doi: 10.17221/179/2021-CJAS
56. Wang, M, Ma, R, Ma, Q, Ma, B, Shang, F, Lv, Q, et al. Role of lncRNA MSTRG. 20890.1 in hair follicle development of cashmere goats. Genes. (2024) 15:1392. doi: 10.3390/genes15111392
57. Ito, T, Tsuji, G, Ohno, F, Uchi, H, Nakahara, T, Hashimoto-Hachiya, A, et al. Activation of the OVOL1-OVOL2 axis in the hair bulb and in pilomatricoma. Am J Pathol. (2016) 186:1036–43. doi: 10.1016/j.ajpath.2015.12.013
58. Li, L, Mignone, J, Yang, M, Matic, M, Penman, S, Enikolopov, G, et al. Nestin expression in hair follicle sheath progenitor cells. Proc Natl Acad Sci USA. (2003) 100:9958–61. doi: 10.1073/pnas.1733025100
59. Ji, K-Y, Wen, R-J, Wang, Z-Z, Tian, Q-Q, Zhang, W, and Zhang, Y-H. MicroRNA-370-5p inhibits pigmentation and cell proliferation by downregulating mitogen-activated protein kinase kinase kinase 8 expression in sheep melanocytes. J Integr Agric. (2023) 22:1131–41. doi: 10.1016/j.jia.2023.02.018
60. Li, L, Fiedler, VC, and Kumar, R. The potential role of skin protein kinase C isoforms alpha and delta in mouse hair growth induced by diphencyprone-allergic contact dermatitis. J Dermatol. (1999) 26:98–105. doi: 10.1111/j.1346-8138.1999.tb03518.x
61. Li, B, Huang, Q, and Wei, G-H. The role of HOX transcription factors in cancer predisposition and progression. Cancers. (2019) 11:528. doi: 10.3390/cancers11040528
62. Chuong, C-M, Oliver, G, Ting, SA, Jegalian, BG, Chen, HM, and De Robertis, EM. Gradients of homeoproteins in developing feather buds. Development. (1990) 110:1021–30. doi: 10.1242/dev.110.4.1021
63. Kanzler, B, Viallet, JP, Le Mouellic, H, Boncinelli, E, Duboule, D, and Dhouailly, D. Differential expression of two different homeobox gene families during mouse tegument morphogenesis. Int J Dev Biol. (2004) 38:633–40. doi: 10.1387/ijdb.7779685
Keywords: indigenous breeds, GWAS, hypoxic adaptability, production, transcriptome
Citation: Tian D, Han B, Li X, Pei Q, Zhou B and Zhao K (2025) Whole-genome resequencing to investigate the genetic diversity and the molecular basis underlying key economic traits in indigenous sheep breeds adapted to hypoxic environments. Front. Vet. Sci. 12:1605252. doi: 10.3389/fvets.2025.1605252
Edited by:
Fernanda Marcondes de Rezende, University of Florida, United StatesReviewed by:
Raziye Işık, São Paulo State University, BrazilHua Yang, Xinjiang Academy of Agricultural and Reclamation Sciences (XAARS), China
Copyright © 2025 Tian, Han, Li, Pei, Zhou and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Zhao, emhhb2thaUBud2lwYi5jYXMuY24=