 Chengzhen Weng1,2,3†
Chengzhen Weng1,2,3† Xinxin Huang1,2,3†Zhian Chen1,2Minjia He1,2,3Beiwen Zhang1,2,3Hongxi Li1,2,3Jingrui Xie1,2Meichun Chen1,2,3Longxin Qiu1,2Xiaobing Li1,2*
Xinxin Huang1,2,3†Zhian Chen1,2Minjia He1,2,3Beiwen Zhang1,2,3Hongxi Li1,2,3Jingrui Xie1,2Meichun Chen1,2,3Longxin Qiu1,2Xiaobing Li1,2* Chong Cao1,2*
Chong Cao1,2* Hongbo Chen1,2*
Hongbo Chen1,2*- 1Longyan College Life Science School, Longyan, China
- 2Fujian Provincial Key Laboratory of Preventive Veterinary Medicine and Veterinary Biotechnology, Longyan, China
- 3College of Animal Science, Fujian Agriculture and Forestry University, Fuzhou, China
The persistent threat of porcine reproductive and respiratory syndrome virus (PRRSV) to the global swine industry is exacerbated by the virus’s high mutation rate and frequent recombination events. In China, the emergence of new PRRSV-1 strains in recent years has posed a significant challenge to the sustainability of pork production. This study systematically investigated the epidemiological patterns, genetic evolution, recombination dynamics, GP5 genetic diversity, and N-glycosylation variants of PRRSV-1 strains circulating in China. Whole-genome analysis demonstrated that Chinese PRRSV-1 isolates clustered within subtype 1, with BJEU06-1-like as the predominant subgroup and NMEU09-1-like as the secondary subgroup. Novel subgroups (new subgroups 1, 2, and 3), a new strain, GD2022, and an independent branch represented by strain GXFS20220129 were concurrently identified. High genetic diversity existed both within and between subgroups of Chinese PRRSV-1 strains. Whole-genome recombination has predominantly occurred through inter-subgroup exchange, primarily involving the BJEU06-1-like and Amervac-like lineages. Additionally, recombination events were identified between the field strain NVDC-FJ and the vaccine strain PRRSV1-CN-FJFQ-1-2023. Interestingly, the diversity of the ORF5 gene was consistent with that of the whole genome; however, there is a deviation in the phylogenetic tree position (BJEU06-1-like: 22 vs. 16). To understand the differences between ORF5 and whole-genome variations, we analyzed amino acid and glycosylation sites of the GP5 protein encoded by ORF5. The results indicated that mutations had occurred at amino acid sites within the antigenic epitopes and functional domains of GP5. Additionally, the prediction of potential N-glycosylation sites identified five locations in GP5: positions 35, 37, 38, 46, and 53. Alterations at these sites could facilitate immune evasion. Our analysis of the ORF5 gene suggests that PRRSV-1 research should not focus solely on ORF5 but rather must consider whole-genome variation, as this may provide insights for vaccine development. In summary, whole-genome studies of PRRSV-1 demonstrated that major recombinant subgroups and genetic evolution align with the current prevalence of BJEU06-1-like strains in China. Analysis of GP5 encoded by ORF5 confirmed the presence of differences between whole-genome and ORF5 data, exhibiting minor discrepancies in both the phylogenetic trees and the level of genetic diversity. Thus, instead of focusing solely on specific regions, whole-genome studies are needed to effectively track variation in PRRSV. This study fills a knowledge gap in our understanding of the prevalence and genetic variation of PRRSV-1 in China, providing crucial insights for developing PRRS control strategies and offering theoretical support for vaccine development.
Introduction
Porcine reproductive and respiratory syndrome (PRRS), caused by the PRRS virus (PRRSV), is a natural pathogen affecting domestic pigs and wild boar. First identified in the United States in 1987, the virus has persisted as a major threat to the global swine industry for over 3 decades (1). Characteristic clinical manifestations include severe reproductive disorders in pregnant sows (e.g., fetal mummification, stillbirth, and abortion) and respiratory symptoms in piglets, with rapid transmission having been documented in nearly all pig-producing countries (2). PRRSV is an enveloped, single-stranded positive-sense RNA virus featuring a ~ 15 kb genome and spherical virions (3). Its genomic architecture includes a 5′-untranslated region (UTR), a 3′-terminal poly (A) tail, and at least 11 open reading frames (ORFs)– ORF1a and ORF1b encode 16 non-structural proteins (Nsps) spanning ~80% of the genome, while ORF2–ORF7 encode eight structural proteins (4). ORF5 serves as the primary target for molecular surveillance and phylogenetic analysis (5, 6), with its encoded GP5 protein (a major immunogenic structural protein) representing the focal point for vaccine development (7–10). Phylogenetic analysis based on ORF5 gene sequences and global PRRSV classification systems assigns PRRSV-1 into three or four subtypes [subtype 1 (global), subtype 1 (Russian), subtype 2, and subtype 3] (11, 12), while PRRSV-2 comprises 11 lineages (L1–L11) (13). Despite sharing only ~60% nucleotide sequence homology, PRRSV-1 and PRRSV-2 exhibit nearly identical pathogenic mechanisms and transmission patterns (14, 15). PRRSV’s high rates of mutation and recombination are the primary obstacles to disease control, creating an urgent global public health challenge that demands effective prevention strategies. In China, PRRSV-2 predominates through widespread circulation of lineages L8 (HP-PRRSV), L5 (VR-2332), L3 (QYYZ), and L1 (NADC30-like, NADC34-like) (16–21). Concurrently, PRRSV-1 has persisted for decades, with contemporary Chinese isolates clustering within Subtype 1 and forming four principal subgroups: Amervac-like, BJEU06-1-like, HKEU16-like, and NMEU09-like (22, 23). The Chinese strains have historically been understudied due to their low pathogenicity and low detection rates. Recent evidence indicates divergence in virulence among the Chinese PRRSV-1 strains (24). For example, the domestically isolated ZD-1 strain induces fever, pulmonary lesions, and mortality in piglets, suggesting a shift in virulence toward low-to-moderately virulent phenotypes (25). Notably, PRRSV-1 detection has surged, with genetically diverse strains now reported across more than 23 provinces. The southward expansion of BJEU06-1-like strains from northern regions is an emerging concern (26). Persistent PRRSV-1 detection, coupled with high mutation rates and genetic diversity, poses a substantial latent threat to China’s swine industry (19). Critical research gaps persist regarding the pathogenesis and immune mechanisms, as well as vaccine development, resulting in inadequate control strategies and surveillance systems. Consequently, commercial PRRSV-1 vaccines remain rare. China prohibits modified live vaccines (MLVs) due to recombination risks: MLV strains may recombine with endemic field variants (e.g., BJEU06-1-like strains), potentially generating novel variants with enhanced virulence or transmissibility (11, 27). Given these findings, there is an urgent need for enhanced surveillance of the understudied PRRSV-1 strains. A comprehensive analysis of Chinese PRRSV-1 whole-genome sequences, including genetic divergence, diversity patterns, recombination events, and GP5 protein characteristics (amino acid substitutions and N-glycosylation sites), will enhance epidemic monitoring and provide foundational insights for PRRS control strategies.
Materials and methods
Genome sequence retrieval and processing
All available Chinese PRRSV-1 complete genomes (n = 46; Supplementary Table S2) were retrieved from GenBank (Release 265). The sequences were aligned with MAFFT along with two reference strains (PRRSV-1: M96262.2; PRRSV-2: AY150564.1; Supplementary Table S2), followed by manual trimming of the terminal non-coding regions (28).
Genotyping and genetic distance analysis
The trimmed sequences were used to construct phylogenetic trees in MEGA7.0.26 using the Kimura two-parameter model and the neighbor-joining method, with 1,000 bootstrap replicates. Genotypic classification was determined based on topological clustering. Pairwise nucleotide p-distances were calculated genome-wide using the same K2P model (29, 30).
Detection of recombination
Putative recombination events were identified using RDP4, with eight detection algorithms (3seq, BootScan, Chimera, GENECONV, LARD, MaxChi, RDP, and SiScan). Events supported by three or more methods were considered validated. Recombination breakpoints within the ORF1–ORF7 regions were confirmed via Simplot 3.5.1 (reference strain: M96262.2; Supplementary Figure S2). Recombination frequency statistics were computed in Excel 365 (31).
GP5 protein characterization
ORF5-encoded GP5 protein sequences were analyzed for the following. Sequence homology and variation: alignment via MegAlign (DNASTAR) with the identification of substitutions/deletions (32). N-glycosylation sites: prediction using NetNGlyc 1.0 (threshold: > 0.5) at the Asn-X-Ser/Thr motifs (33).
Results
Phylogenetic analysis of whole genomes of Chinese PRRSV-1
A phylogenetic analysis of 46 complete genomes of Chinese PRRSV-1 and representative strains (PRRSV-1: LV; PRRSV-2: VR-2332) demonstrated that all Chinese isolates belonged to subtype 1, forming four subgroups: BJEU06-1-like, Amervac-like, HKEU16-like, and NMEU09-1-like (Figure 1). Among these, BJEU06-1-like was the predominant subgroup (16 strains in the whole-genome vs. 22 in the ORF5 analysis). Additionally, we identified several small novel subgroups designated as new subgroups 1, 2, and 3, while also detecting a new strain, GD2022, within these subgroups.
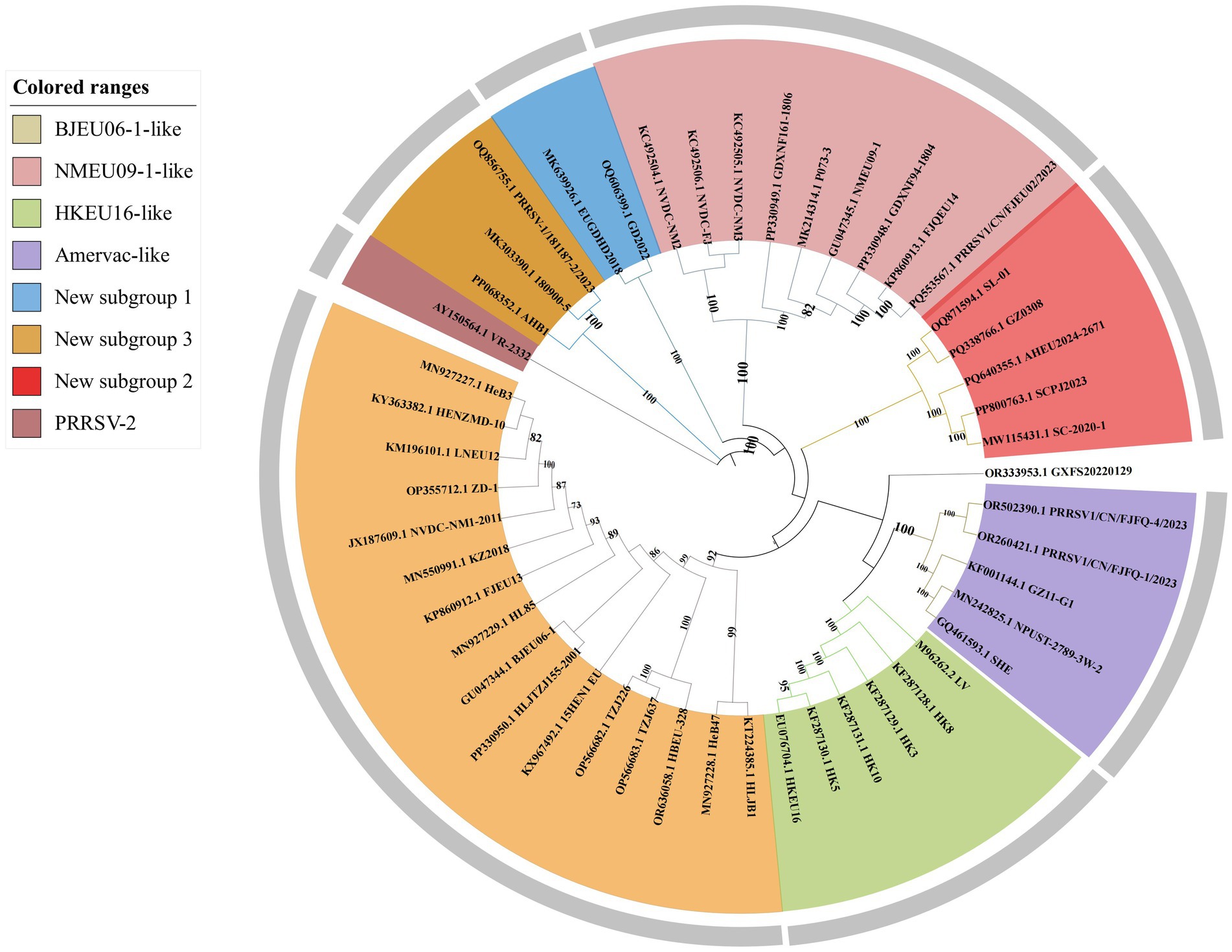
Figure 1. Phylogenetic tree of the complete genome of PRRSV-1 in China.
There were several notable differences between the whole-genome and ORF5 phylogenies (Figure 2). Vaccine strains (SHE, NPUST-2789-3 W-2) clustered within Amervac-like in the whole-genome trees but grouped with field strains (HeB47, LV, HLJB1, and GZ11-G1) in the ORF5-based analysis. New subgroups 2 and 3 formed separate branches in the whole-genome trees, but clustered within BJEU06-1-like in the ORF5 phylogenies. Strain GXFS20220129 comprised an independent branch in both analyses. These differences indicated that whole-genome analysis provides a more detailed characterization of the variation in PRRSV-1. The continuous emergence of new strains and variants necessitates more intensive research on PRRSV-1 in China, and whole-genome analysis would better capture the epidemiological trends of the virus, thereby improving the control of PRRS.
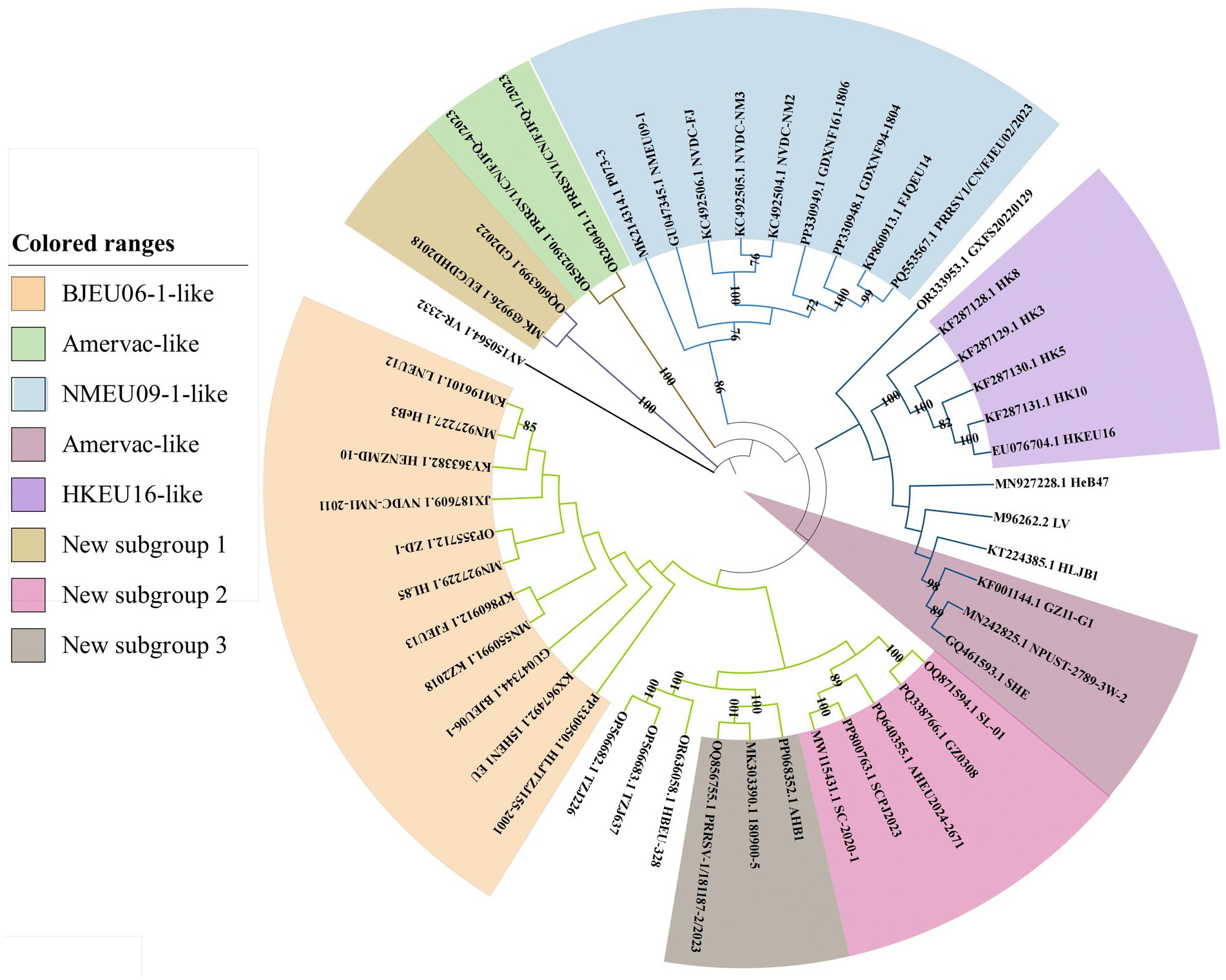
Figure 2. Phylogenetic tree of ORF5 Chinese PRRSV-1.
Genetic diversity of Chinese PRRSV-1 whole genomes
The observed variation in whole genomes of PRRSV-1 reflects inter-subgroup divergence. We calculated nucleotide genetic distances for the complete genome sequences to understand evolutionary relationships among the PRRSV-1 subgroups. Intra-subgroup genetic distances were lowest in new subgroup 1 (0.005), while BJEU06-1-like (0.118), NMEU09-1-like (0.127), and new subgroup 2 (0.139) exhibited greater distances than the other subgroups, consistent with their status as predominant epidemic subgroups (Table 1). Inter-subgroup genetic distances showed greater divergence between BJEU06-1-like and NMEU09-1-like (0.189), new subgroups 1 (0.190), 2 (0.191), and 3 (0.207). BJEU06-1-like had the least distance from the prototype strain LV (0.110). Strain GXFS20220129 was more distant than NMEU09-1-like (0.193), new subgroup 1 (0.191), new subgroup 3 (0.205), and BJEU06-1-like (0.164). New subgroup 3 exhibited significant divergence from all other subgroups (0.173 ~ 0.220). Greater genetic distances are correlated with increased genetic diversity. Considerable genetic variation exists among Chinese PRRSV-1 subgroups, particularly in phylogenetically distinct lineages GXFS20220129 and new subgroup 3, both of which exhibit diversity across subgroups. This variability complicates the control of PRRSV-1 in China and requires attention (Table 2).
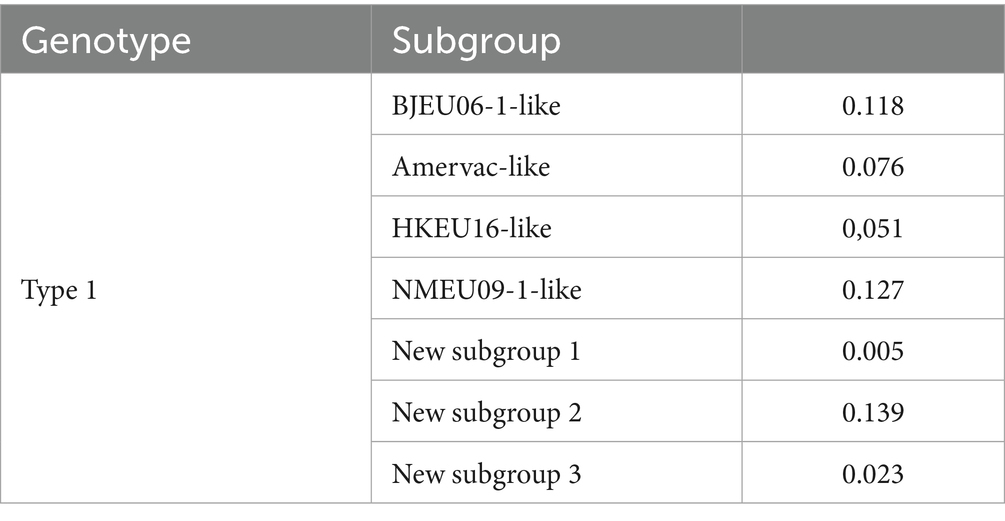
Table 1. Genetic distance of nucleotides within subgroups.
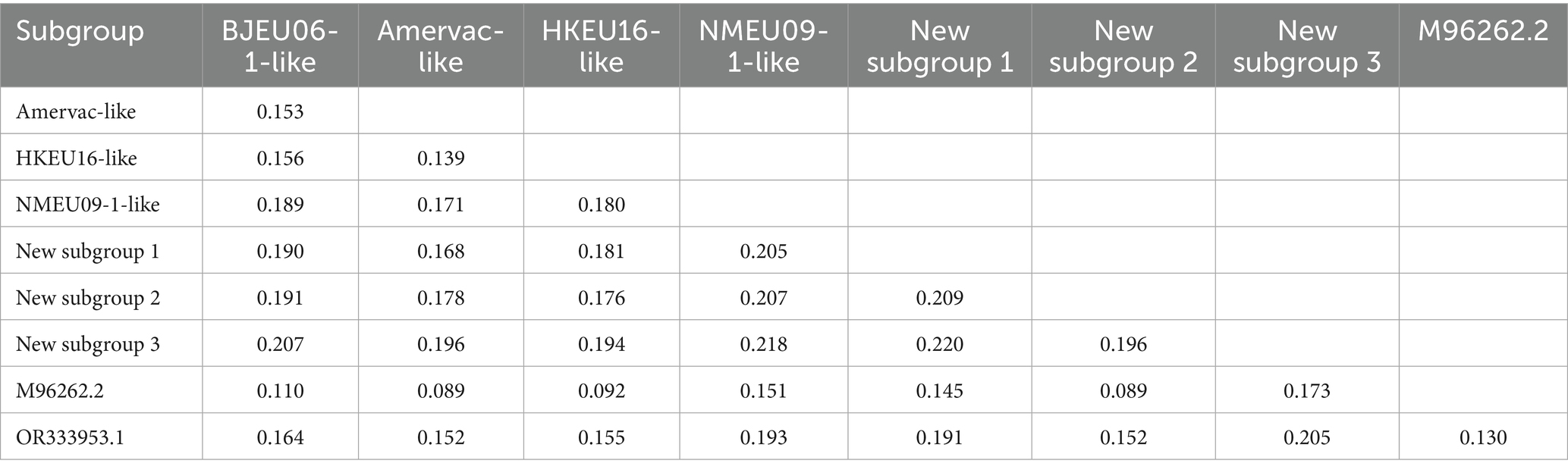
Table 2. Genetic distances of nucleotides between subgroups.
Recombination analysis of Chinese PRRSV-1 whole genomes
Recombination is recognized as a key driving force of high mutation rates and genetic diversity in PRRSV genomes (11, 34–36). We analyzed the recombination events in PRRSV-1 whole genomes, identifying 32 recombination events, of which 9 intra-subgroup events (mainly BJEU06-1-like) and 23 inter-subgroup events (primarily BJEU06-1-like + Amervac-like, n = 7). A recombination analysis of strain HLJB1 using Simplot 3.5.1 and RDP4 confirmed a recombination event with breakpoints at nucleotides 9,966–12,606 nt (Figure 3A). Subsequent segmented phylogenetic analysis of recombinant regions in this strain (HLJB1) identified both major and minor parental lineages belonging to PRRSV-1 (Figure 3B). In addition, inter-subgroup recombination occurred between the vaccine and field strains. For the vaccine strain PRRSV1-CN-FJFQ-1-2023 (Amervac-like), the major parental strain was GZ11-G1 (Amervac-like), and the minor parental strain was NVDC-FJ (NMEU09-1-like). A recombination hotspot analysis showed frequent breakpoints in the ORF1a and ORF1b genes (Supplementary Figure S2).
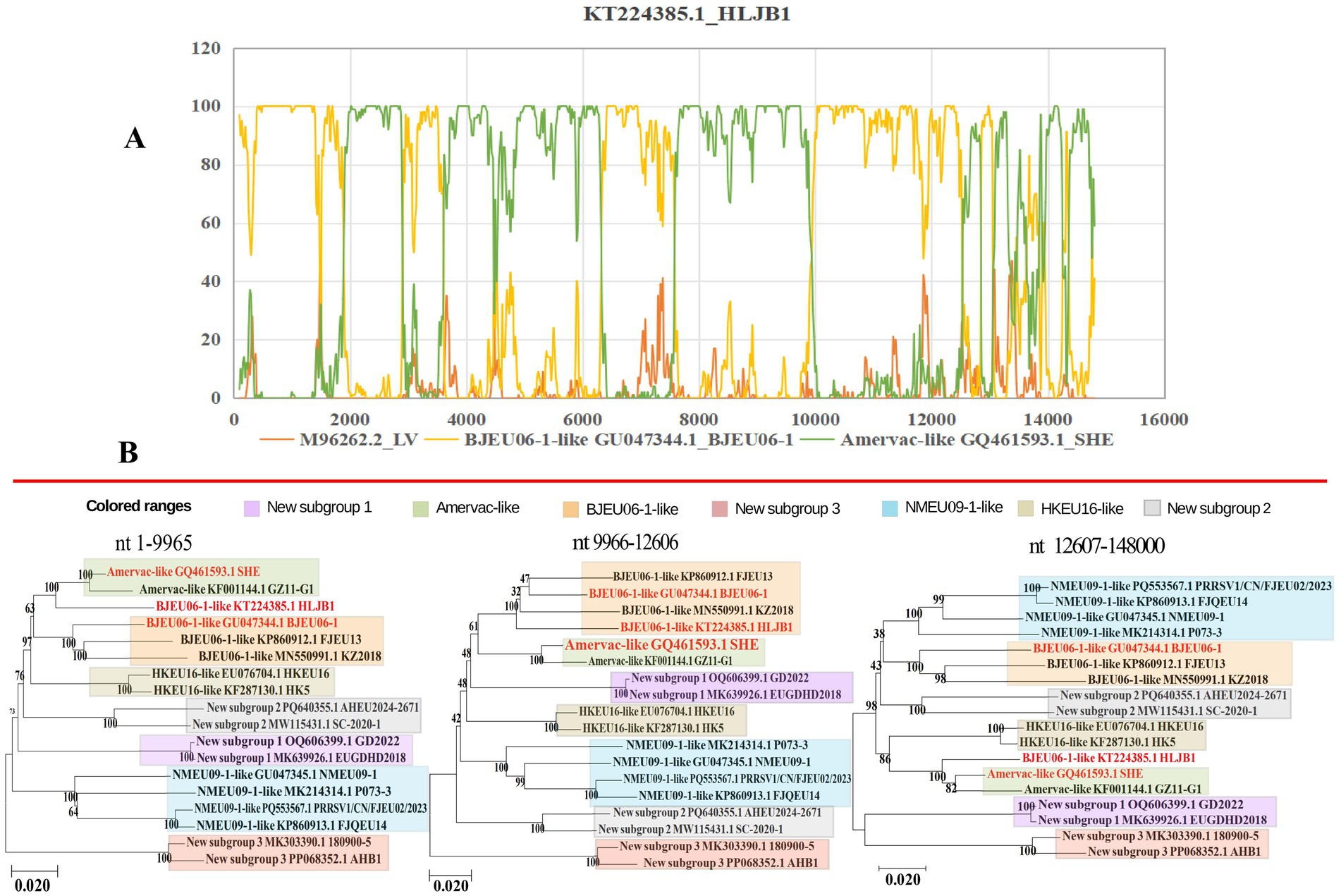
Figure 3. Reorganization analysis results of HLJB1 strain.
Novel subgroups exhibited inter-subgroup recombination: strain MK303390.1 (new subgroup 3) had PRRSV1-CN-FJEU02-2023 (NMEU09-1-like) as the major parent and LV as the minor parent. Recombination occurred between two of the new subgroups: strain SCPJ2023 (new subgroup 2) had EUGDHD2018 (new subgroup 1) as the major parent and SL-01 (new subgroup 2) as the minor parent. These findings indicate that recombination has contributed to the genetic variation and phylogenetic differences among subgroups, thereby explaining, to some extent, the recent spread of PRRSV-1 in China. We observed a shift from simple intra-subgroup recombination to complex inter-subgroup recombination in Chinese PRRSV-1, a factor that complicates the potential control of the disease (Supplementary Table S1).
ORF5 nucleotide and amino acid homology analysis
ORF5 is routinely used for molecular epidemiological surveillance and vaccine development. The genetic diversity analysis of ORF5 has provided insights into the evolutionary trends of PRRSV-1 in China. We therefore examined the nucleotide and amino acid homology of ORF5 genes from Chinese PRRSV-1 whole genomes. The results showed that the nucleotide homology of PRRSV-1 ORF5 genes ranged from 77.7 to 100%, with amino acid homology ranging from 77.2 to 100%. Within HKEU16-like strains, the level of nucleotide homology (92.4–99.7%) indicated higher conservation and lower genetic variation compared to other subgroups. Chinese PRRSV-1 sequences shared 81.8–94.6% homology with European reference strain M96262.2 (LV), suggesting limited genetic diversity between regional and European strains. Notably, intra-subgroup homology varied substantially, with BJEU06-1-like, NMEU09-1-like, and new subgroup 2 exhibiting higher variability. The amino acid homology was elevated in new subgroup 1 (98.5%), new subgroup 3 (92.6–98%), and HKEU16-like (92.1–99.5%), patterns consistent with the nucleotide results. Inter-subgroup comparisons revealed greater genetic divergence, with nucleotide and amino acid homology in the ranges 77.7–100% and 77.2–100%, respectively (Table 3).
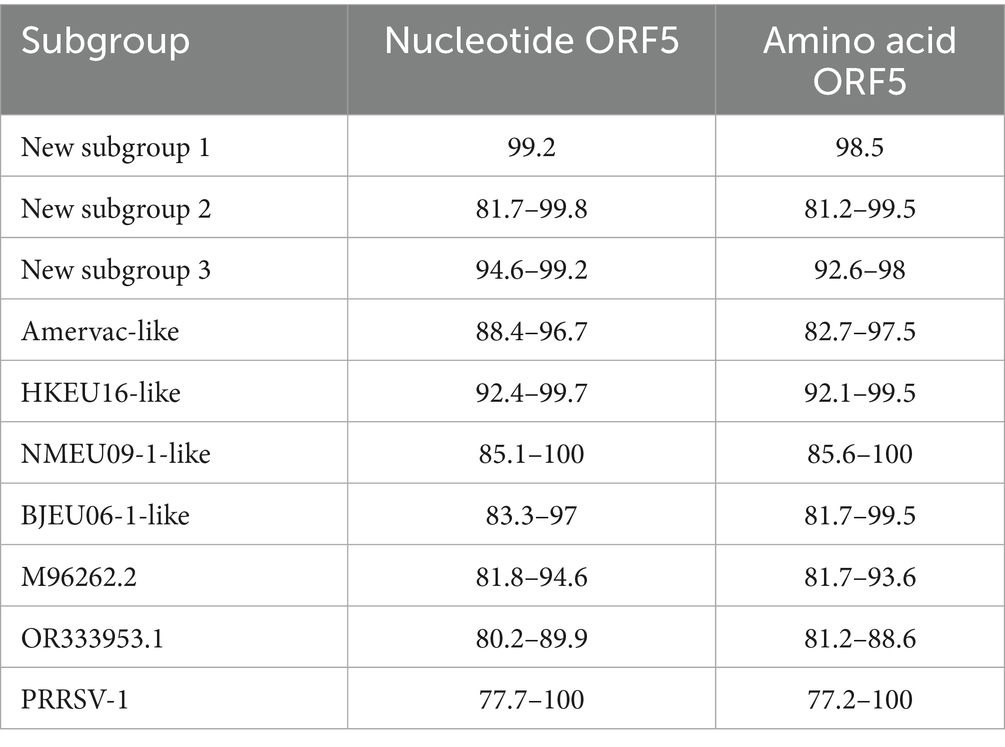
Table 3. ORF5 nucleotide and amino acid homology analysis.
GP5 protein amino acid substitution analysis
Given the high variability of the ORF5-encoded GP5 protein, analyzing GP5 genetic variation has become essential for developing novel PRRSV vaccines and controlling viral transmission. We analyzed the amino acid substitution sites in GP5 from 47 PRRSV-1 ORF5 genes using MegAlign (DNASTAR) to characterize the GP5 composition for improved control of PRRSV-1. The GP5 protein comprises 201 amino acids encoded by 603 nucleotides. Structural prediction revealed a signal peptide, a hypervariable region, and multiple B-cell and T-cell epitopes. In the signal peptide region, position 9 Arg (R) was mutated to His (H) in most strains (Figure 4). Cys24 (C24) and 29WSFADGN35 regions were correlated with neutralizing antibody epitopes, where C24 was highly conserved except for a mutation in GZ11-G1 (Figure 5). Neutralizing epitopes play critical roles in anti-PRRSV responses; we observed substitutions at the 29W-35N epitopes, including Trp29 → Cys (C) in HKEU16. At B-cell epitope position 37 Asn (N), the GD2022 and LV strains exhibited Asn37 → Asp (D) mutations, BJEU06-1 had Asn37 → Thr (T), and SHE/TZJ226/TZJ637/180900–5 exhibited Asn37 → Ser (S). In addition, mutations of Val32 (V) occurred in the Amervac-like strains (SHE, GZ11-G1, NPUST-2789-3 W-2) and the BJEU06-1-like strain LNEU12 (Table 3).
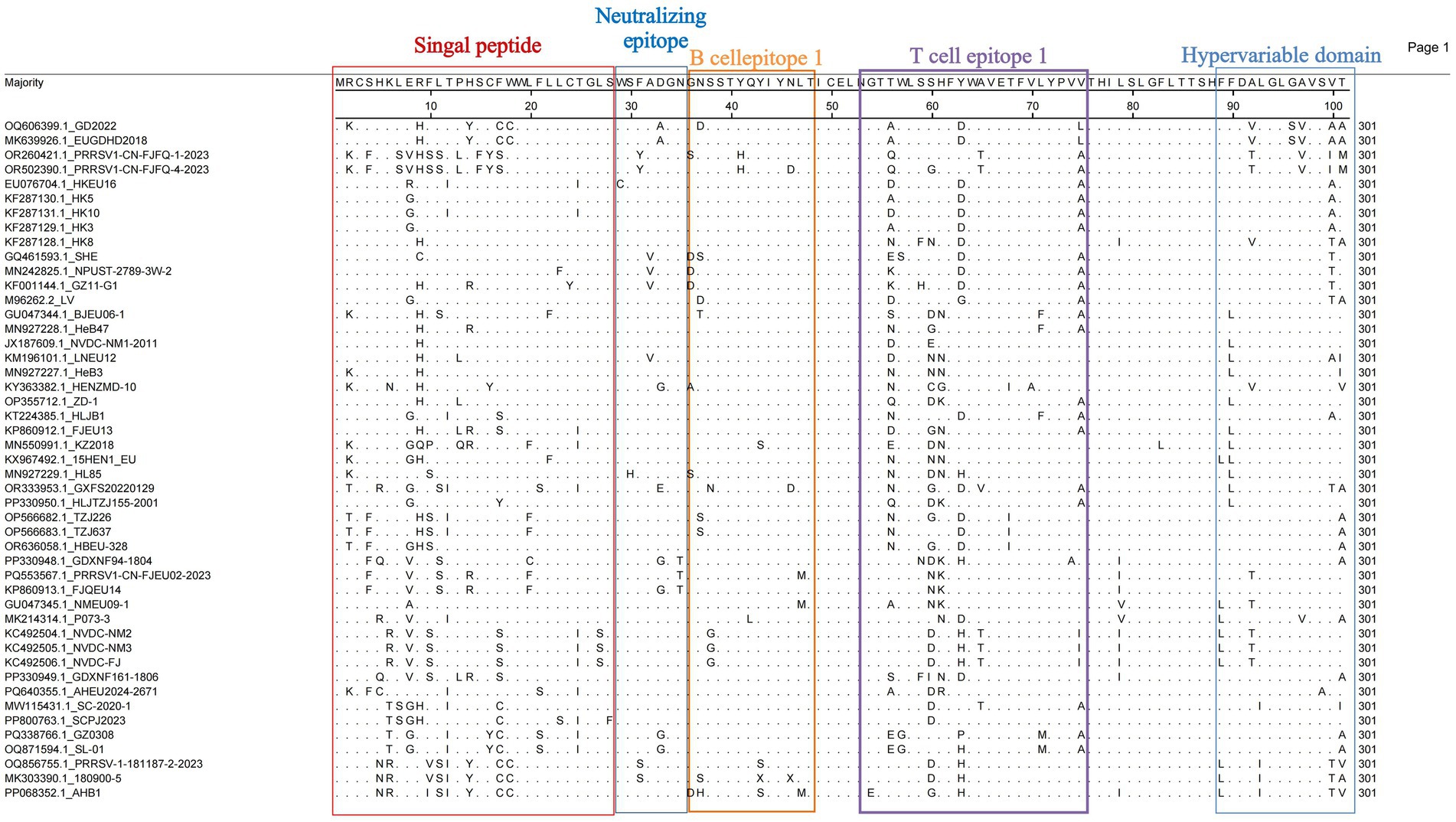
Figure 4. Amino acid site analysis of Chinese PRRSV-1 GP5 protein (Amino acids 1 to 101).
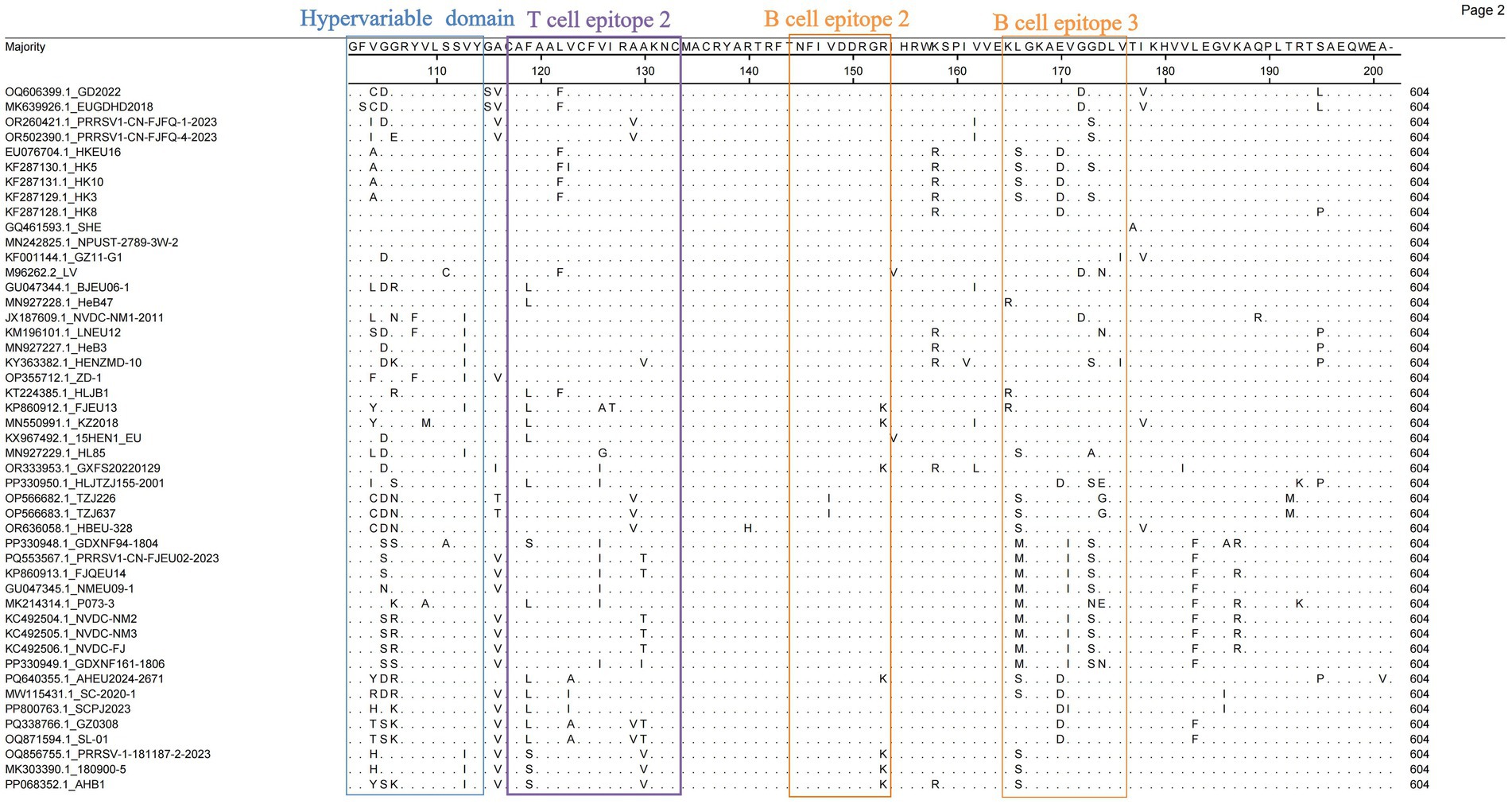
Figure 5. Amino acid site analysis of Chinese PRRSV-1 GP5 protein (Amino acids 102 to 201).
Analysis of potential N-glycosylation sites in the GP5 protein
As a major structural protein inducing protective antibodies, alterations in the glycosylation sites of the PRRSV-1 GP5 protein will impact its immunological efficacy. We thus investigated potential glycosylation sites for the protein. The analysis revealed five potential N-glycosylation sites at amino acid positions 35, 37, 38, 46, and 53 (Table 4). Consistent with the patterns of amino acid variation, mutations occurred at position 37 N in strains GD2022, LV, SHE, BJEU06-1, TZJ226, TZJ637, 180,900–5, and AHB1. At position 35 N—an antigenic epitope site—amino acid additions were observed in SHE, BJEU06-1, TZJ226, TZJ637, and 180,900–5. Concurrently, position 46 N exhibited deletions in GXFS20220129, PRRSV1-CN-FJFQ-4-2023, and 180,900–5 (Table 4).
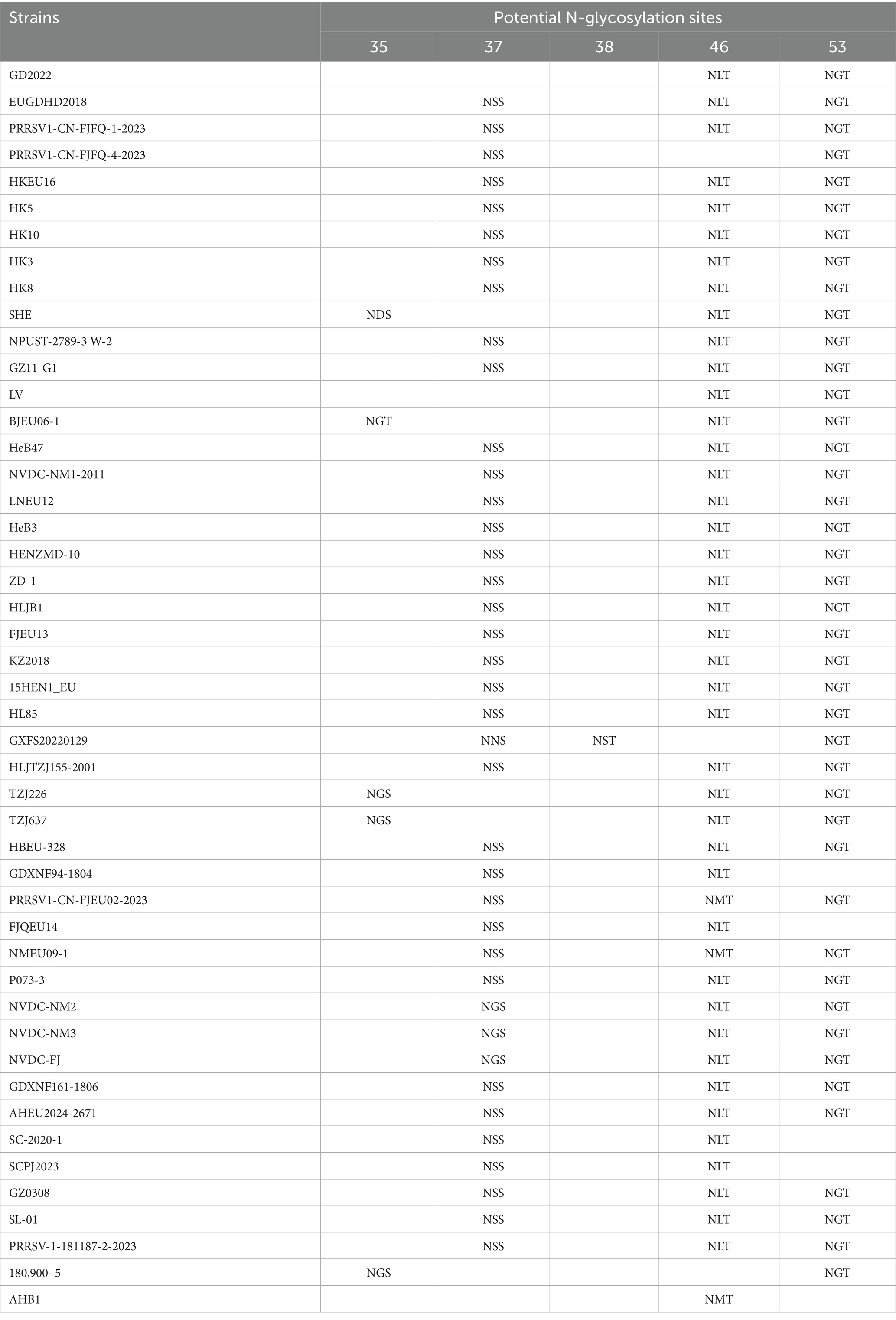
Table 4. Analysis of potential N-glycosylation sites in the GP5 protein.
Discussion
The increasing prevalence of PRRSV, driven by its characteristically high mutation and recombination rates (37, 38), coincides with the rise in PRRSV-1 detection across China. Concurrently, effective prevention and control strategies remain critically underdeveloped. The investigation of epidemiological trends and geographic distribution of PRRSV-1 in China provides essential data to inform the development of vaccines and efforts to contain the epidemic. Genetic differentiation reflects inter-population genetic variation and the dynamics of disease prevalence. Our genetic differentiation analysis of 46 Chinese PRRSV-1 whole-genome strains, together with the reference strain VR-2332 and thorough phylogenetic analysis, revealed that contemporary Chinese PRRSV-1 primarily clustered into four subgroups: BJEU06-1-like, Amervac-like, HKEU16-like, and NMEU09-1-like (23). Among these, BJEU06-1-like represented the dominant subgroup, followed by NMEU09-1-like. The identification of novel branches and emerging strains (e.g., GD2022) (11, 39) indicates increasing complexity in PRRSV-1 epidemiology within China, with substantial genetic variation necessitating heightened vigilance. Since the ORF5 gene serves as the primary diagnostic target for PRRSV detection, we constructed a phylogenetic tree using ORF5 sequences derived from whole genomes. Notably, discrepancies emerged between the ORF5-based and whole-genome phylogenies: strains classified as Amervac-like (SHE, NPUST-2789-3 W-2, GZ11-G1) in the whole-genome analysis clustered with LV and BJEU06-1-like strains (HeB47, HLJB1) in the ORF5 tree. Previous studies have documented recombination between HeB47/HLJB1 and Amervac-like strains (23, 40). We attribute this phylogenetic convergence to high ORF5 sequence homology and inter-strain recombination events. The new subgroups 2 and 3 clustered within BJEU06-1-like in the ORF5 tree, whereas new subgroup 1 formed a distinct branch in both phylogenies, suggesting potential inter-subgroup recombination. The continuous emergence of novel branches and strains demonstrates the persistent evolution of the virus in China, posing a significant risk of viral enhancement that demands increased surveillance. Genetic diversity reflects viral divergence and the potential for mutation. Analysis of whole-genome genetic diversity revealed elevated diversity within the prevalent subgroups BJEU06-1-like, NMEU09-1-like, and Amervac-like, which may explain their epidemiological success. Novel branches also exhibited high diversity, indicating potential for future dominance and warranting close monitoring. Motivated by the observed phylogenetic discrepancies, we conducted nucleotide and amino acid homology analyses of the ORF5 gene. The results confirmed high intra-subgroup diversity within BJEU06-1-like, NMEU09-1-like, and new subgroup 3, consistent with the whole-genome findings and indicative of heightened mutability. The intricate relationship between genetic diversity and variation warrants increased attention. While the ORF5 phylogeny partially diverged from the whole-genome analysis, homology assessment alone cannot establish causality. Frequent recombination of PRRSV-1 drives continuous variation among strains and significantly facilitates the emergence of novel strains. The recombination analysis of 46 whole-genome sequences and reference strain LV identified 32 recombination events, with inter-subgroup recombination exceeding intra-subgroup events. This indicates an epidemiological shift from single-subgroup dominance to co-circulation among multiple subgroups in China. BJEU06-1-like (12 events) and NMEU09-1-like (seven events) were the predominant recombinant subgroups, aligning with their status as major epidemic strains. Recombination is thus a key mechanism driving their dominance, high level of diversity, and adaptability. Beyond confirming the documented recombination between vaccine strains (e.g., Amervac-like) and field strains such as HLJB1/HeB47 (23, 40), we detected additional vaccine-field recombination events. Notably, vaccine strain Amervac-like_GQ461593.1_SHE exhibited frequent recombination with field strains—a finding with potential implications for vaccine design that warrants further vigilance. Frequent recombination between BJEU06-1-like and Amervac-like strains necessitates caution to prevent the emergence of epidemic strains and co-infections involving vaccine and field viruses. Strains showing phylogenetic discordance (Amervac-like: SHE, NPUST-2789-3 W-2, GZ11-G1; BJEU06-1-like: HeB47, HLJB1) demonstrated mutual recombination, which explains their ORF5 clustering. The novel branch (11), arising exclusively from inter-subgroup recombination, exhibits high variability and potential epidemic significance, thereby demanding prioritized surveillance. Collectively, these findings reveal increasingly complex transmission dynamics and growing challenges to controlling PRRSV-1 in China. There was partial discordance between whole-genome and ORF5 phylogenies; GD2022 formed a distinct branch in both trees without evidence of recombination, suggesting an emerging lineage (39). The disparity in detected recombination events (32 whole-genome vs. one ORF5) highlights the inadequacy of ORF5 as a full-genome surrogate. Nevertheless, ORF5 remains crucial for surveillance due to its key roles in neutralizing antibody responses, receptor binding, immune evasion (41), and as a primary subunit vaccine target. Substitutions, deletions, and insertions drive PRRSV evolution (42), with variation in the GP5 protein concentrated in the signal peptide as well as neutralizing and non-neutralizing epitope regions (43). Further GP5 amino acid analysis revealed that GD2022’s B-cell epitope mutation (33A) significantly reduces neutralizing antibody titers against vaccine strains and compromises cross-protection (39). Position C24 is highly conserved in PRRSV-1 genomes, with mutation observed only in GZ11-G1 (44), demonstrating intra-subgroup amino acid heterogeneity and highlighting viral complexity. Detailed characterization of the GP5 amino acid distribution is vital for vaccine design (45) and understanding the patterns of viral pathogenicity and transmission (46). Predicting N-glycosylation sites offers insights into the evolution and regulation of PRRSV-1, as glycosylation modulates immune recognition (47). The amino acid acquisitions at position 35 N in strains SHE, BJEU06-1, TZJ226, TZJ637, and 180,900–5 may have contributed to the prevalence of BJEU06-1-like/NMEU09-1-like. Interestingly, GXFS20220129 possesses a unique acquisition at 38 N, potentially altering the virulence of the strain, explaining its phylogenetic divergence, and signaling an emerging risk that demands specific attention. In summary, these findings have advanced our understanding of the prevalence, genetic diversity, recombination patterns, and divergent amino acid sites of PRRSV-1 in China. Future research must prioritize whole-genome sequencing over exclusive reliance on ORF5, as single-gene analysis cannot fully capture epidemiological complexity. This study provides critical insights for developing effective PRRS prevention and control strategies to mitigate viral transmission.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
CW: Methodology, Formal analysis, Data curation, Validation, Conceptualization, Visualization, Software, Writing – original draft, Investigation. XH: Methodology, Writing – original draft, Conceptualization, Formal analysis, Data curation. ZC: Writing – original draft, Methodology, Investigation. MH: Writing – original draft, Software, Investigation, Conceptualization. BZ: Software, Writing – original draft, Methodology, Supervision. HL: Writing – original draft, Methodology, Investigation. JX: Investigation, Methodology, Writing – original draft. MC: Investigation, Writing – original draft. LQ: Investigation, Supervision, Writing – original draft, Project administration. XL: Resources, Project administration, Writing – review & editing. CC: Writing – review & editing, Resources, Supervision. HC: Funding acquisition, Project administration, Supervision, Writing – review & editing, Resources, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (no. 32102628) and the Doctoral Start-up Project of Longyan University, Fujian Province (no. LB2022009).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1632917/full#supplementary-material
References
2. Snijder, EJ, and Kikkert, M. Arteriviruses. Encycl Virol. (2008) 1:176–86. doi: 10.1016/B978-012374410-4.00537-9
3. Chen, Z, Zhou, X, Lunney, JK, Lawson, S, Sun, Z, Brown, E, et al. Immunodominant epitopes in nsp2 of porcine reproductive and respiratory syndrome virus are dispensable for replication, but play an important role in modulation of the host immune response. J Gen Virol. (2010) 91:1047–57. doi: 10.1099/vir.0.016212-0
4. Ruedas-Torres, I, Rodríguez-Gómez, IM, Sánchez-Carvajal, JM, Larenas-Muñoz, F, Pallarés, FJ, Carrasco, L, et al. The jigsaw of Prrsv virulence. Vet Microbiol. (2021) 260:109168. doi: 10.1016/j.vetmic.2021.109168
5. Martín-Valls, GE, Kvisgaard, LK, Tello, M, Darwich, L, Cortey, M, Burgara-Estrella, AJ, et al. Analysis of Orf5 and full-length genome sequences of porcine reproductive and respiratory syndrome virus isolates of genotypes 1 and 2 retrieved worldwide provides evidence that recombination is a common phenomenon and may produce mosaic isolates. J Virol. (2014) 88:3170–81. doi: 10.1128/JVI.02858-13
6. Guzmán, M, Meléndez, R, Jiménez, C, Piche, M, Jiménez, E, León, B, et al. Analysis of Orf5 sequences of porcine reproductive and respiratory syndrome virus (Prrsv) circulating within swine farms in Costa Rica. BMC Vet Res. (2021) 17:217. doi: 10.1186/s12917-021-02925-7
7. Zhang, H, Luo, Q, He, Y, Zheng, Y, Sha, H, Li, G, et al. Research progress on the development of porcine reproductive and respiratory syndrome vaccines. Vet Sci. (2023) 10:491. doi: 10.3390/vetsci10080491
8. Luo, Q, Zheng, Y, Zhang, H, Yang, Z, Sha, H, Kong, W, et al. Research Progress on glycoprotein 5 of porcine reproductive and respiratory syndrome virus. Animals (Basel). (2023) 13:813. doi: 10.3390/ani13050813
9. Zhao, G, Zhang, J, Sun, W, Xie, C, Zhang, H, Gao, Y, et al. Immunological evaluation of recombination Prrsv Gp3 and Gp5 Dna vaccines in vivo. Front Cell Infect Microbiol. (2022) 12:1016897. doi: 10.3389/fcimb.2022.1016897
10. Liu, D, and Chen, Y. Epitope screening and vaccine molecule design of Prrsv Gp3 and Gp5 protein based on immunoinformatics. J Cell Mol Med. (2024) 28:e18103. doi: 10.1111/jcmm.18103
11. Sun, Q, Xu, H, An, T, Cai, X, Tian, Z, and Zhang, H. Recent Progress in studies of porcine reproductive and respiratory syndrome virus 1 in China. Viruses. (2023) 15:1528. doi: 10.3390/v15071528
12. Li, Y, Tas, A, Sun, Z, Snijder, EJ, and Fang, Y. Proteolytic processing of the porcine reproductive and respiratory syndrome virus replicase. Virus Res. (2015) 202:48–59. doi: 10.1016/j.virusres.2014.12.027
13. Yim-Im, W, Anderson, TK, Paploski, I, VanderWaal, K, Gauger, P, Krueger, K, et al. Refining Prrsv-2 genetic classification based on global Orf5 sequences and investigation of their geographic distributions and temporal changes. Microbiol Spectr. (2023) 11:e291623. doi: 10.1128/spectrum.02916-23
14. Guo, Z, Chen, XX, Li, R, Qiao, S, and Zhang, G. The prevalent status and genetic diversity of porcine reproductive and respiratory syndrome virus in China: a molecular epidemiological perspective. Virol J. (2018) 15:2. doi: 10.1186/s12985-017-0910-6
15. Lunney, JK, Fang, Y, Ladinig, A, Chen, N, Li, Y, Rowland, B, et al. Porcine reproductive and respiratory syndrome virus (Prrsv): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci. (2016) 4:129–54. doi: 10.1146/annurev-animal-022114-111025
16. Zhang, H, Luo, Q, Zheng, Y, Sha, H, Li, G, Kong, W, et al. Genetic variability and recombination of the Nsp2 gene of Prrsv-2 strains in China from 1996 to 2021. Vet Sci. (2023) 10:325. doi: 10.3390/vetsci10050325
17. Zhou, L, and Yang, H. Porcine reproductive and respiratory syndrome in China. Virus Res. (2010) 154:31–7. doi: 10.1016/j.virusres.2010.07.016
18. Han, J, Zhou, L, Ge, X, Guo, X, and Yang, H. Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet Microbiol. (2017) 209:30–47. doi: 10.1016/j.vetmic.2017.02.020
19. Liu, J, Zhou, X, Zhai, J, Wei, C, Dai, A, Yang, X, et al. Recombination in Jxa1-R vaccine and Nadc30-like strain of porcine reproductive and respiratory syndrome viruses. Vet Microbiol. (2017) 204:110–20. doi: 10.1016/j.vetmic.2017.04.017
20. Su, Z, Wang, X, Liu, K, Chen, G, Zhang, K, Liu, J, et al. Recombination and pathogenicity analysis of Nadc30-like and Qyyz-like Prrsv strains in South China. Microb Pathog. (2025) 200:107351. doi: 10.1016/j.micpath.2025.107351
21. Jiao, S, Zhang, J, Wang, J, Ma, X, Li, G, Li, J, et al. Whole-genome analysis of the recombination and evolution of newly identified Nadc30-like porcine reproductive and respiratory syndrome virus strains circulated in Gansu province of China in 2023. Front Vet Sci. (2024) 11:1372032. doi: 10.3389/fvets.2024.1372032
22. Li, C, Xu, H, Zhao, J, Gong, B, Sun, Q, Xiang, L, et al. Epidemiological investigation and genetic evolutionary analysis of Prrsv-1 on a pig farm in China. Front Microbiol. (2022) 13:1067173. doi: 10.3389/fmicb.2022.1067173
23. Chen, N, Liu, Q, Qiao, M, Deng, X, Chen, X, and Sun, M. Whole genome characterization of a novel porcine reproductive and respiratory syndrome virus 1 isolate: genetic evidence for recombination between Amervac vaccine and circulating strains in mainland China. Infect Genet Evol. (2017) 54:308–13. doi: 10.1016/j.meegid.2017.07.024
24. Zhai, SL, Lin, T, Zhou, X, Pei, ZF, Wei, ZZ, Zhang, H, et al. Phylogeographic analysis of porcine reproductive and respiratory syndrome virus 1 in Guangdong province, southern China. Arch Virol. (2018) 163:2443–9. doi: 10.1007/s00705-018-3873-z
25. Xu, H, Gong, B, Sun, Q, Li, C, Zhao, J, Xiang, L, et al. Genomic characterization and pathogenicity of Bjeu06-1-like Prrsv-1 Zd-1 isolated in China. Transbound Emerg Dis. (2023) 2023:6793604. doi: 10.1155/2023/6793604
26. Gong, B, Xu, H, Sun, Q, Li, C, Xiang, L, Zhao, J, et al. Dissecting genetic diversity and evolutionary trends of Chinese Prrsv-1 based on whole-genome analysis. Transbound Emerg Dis. (2024) 2024:9705539. doi: 10.1155/2024/9705539
27. Kimman, TG, Cornelissen, LA, Moormann, RJ, Rebel, JM, and Stockhofe-Zurwieden, N. Challenges for porcine reproductive and respiratory syndrome virus (Prrsv) vaccinology. Vaccine. (2009) 27:3704–18. doi: 10.1016/j.vaccine.2009.04.022
28. Toh, H. Parallelization of the Mafft multiple sequence alignment program. Bioinformatics. (2010) 26:1899. doi: 10.1093/bioinformatics/btq224
29. Sudhir, K, Glen, S, and Koichiro, T. Mega7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 7:1870–4. doi: 10.1093/molbev/msw054
30. Nakamura, T, Yamada, KD, Tomii, K, and Katoh, K. Parallelization of Mafft for large-scale multiple sequence alignments. Bioinformatics. (2018) 34:2490–2. doi: 10.1093/bioinformatics/bty121
31. Martin, DP, Murrell, B, Khoosal, A, and Muhire, B. Detecting and analyzing genetic recombination using Rdp 4. Methods Mol Biol. (2017) 1525:433–60. doi: 10.1007/978-1-4939-6622-6_17
32. Irum, B, Kabir, F, Shoshany, N, Khan, SY, Rauf, B, Naeem, MA, et al. A genomic deletion encompassing Crybb2-Crybb2P1 is responsible for autosomal recessive congenital cataracts. Hum Genome Var. (2022) 9:31. doi: 10.1038/s41439-022-00208-7
33. Sukreet, S, Zhang, H, Adamec, J, Cui, J, and Zempleni, J. Identification of glycoproteins on the surface of bovine milk exosomes and intestinal cells that facilitate exosome uptake in human colon carcinoma Caco-2 cells. FASEB J. (2018) 31:646. doi: 10.1096/fasebj.31.1_supplement.646.25
34. Cui, XY, Xia, DS, Luo, LZ, and An, TQ. Recombination of porcine reproductive and respiratory syndrome virus: features, possible mechanisms, and future directions. Viruses. (2024) 16:929. doi: 10.3390/v16060929
35. Liu, D, Zhou, R, Zhang, J, Zhou, L, Jiang, Q, Guo, X, et al. Recombination analyses between two strains of porcine reproductive and respiratory syndrome virus in vivo. Virus Res. (2011) 155:473–86. doi: 10.1016/j.virusres.2010.12.003
36. Wang, X, Zhang, K, Mo, Q, Chen, G, Lv, J, Huang, J, et al. The emergence and pathogenesis of recombinant viruses associated with Nadc34-like strains and the predominant circulating strains of porcine reproductive and respiratory syndrome virus in southern China. Viruses. (2022) 14:1695. doi: 10.3390/v14081695
37. Zhou, L, Han, J, and Yang, H. The evolution and diversity of porcine reproductive and respiratory syndrome virus in China. Vet Microbiol. (2024) 298:110252. doi: 10.1016/j.vetmic.2024.110252
38. Wang, HM, Liu, YG, Tang, YD, Liu, TX, Zheng, LL, Wang, TY, et al. A natural recombinant Prrsv between Hp-Prrsv Jxa1-like and Nadc30-like strains. Transbound Emerg Dis. (2018) 65:1078–86. doi: 10.1111/tbed.12852
39. Xu, H, Xie, Y, Deng, K, and He, D. Isolation and identification, genome-wide analysis and pathogenicity study of a novel Prrsv-1 in southern China. Front Microbiol. (2024) 15:1465449. doi: 10.3389/fmicb.2024.1465449
40. Yu, F, Liu, L, Tian, X, Chen, L, Huang, X, Sun, Y, et al. Genomic analysis of porcine reproductive and respiratory syndrome virus 1 revealed extensive recombination and potential introduction events in China. Vet Sci. (2022) 9:450. doi: 10.3390/vetsci9090450
41. Kim, WI, Kim, JJ, Cha, SH, Wu, WH, Cooper, V, Evans, R, et al. Significance of genetic variation of Prrsv Orf5 in virus neutralization and molecular determinants corresponding to cross neutralization among Prrs viruses. Vet Microbiol. (2013) 162:10–22. doi: 10.1016/j.vetmic.2012.08.005
42. Music, N, and Gagnon, CA. The role of porcine reproductive and respiratory syndrome (Prrs) virus structural and non-structural proteins in virus pathogenesis. Anim Health Res Rev. (2010) 11:135–63. doi: 10.1017/S1466252310000034
43. Chen, W, Gao, Y, Xie, W, Gong, L, Lu, K, Wang, W, et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet. (2014) 46:714–21. doi: 10.1038/ng.3007
44. Wang, X, Yang, X, Zhou, R, Zhou, L, Ge, X, Guo, X, et al. Genomic characterization and pathogenicity of a strain of type 1 porcine reproductive and respiratory syndrome virus. Virus Res. (2016) 225:40–9. doi: 10.1016/j.virusres.2016.09.006
45. Xu, W, Du, S, Li, T, Wu, S, Jin, N, Ren, L, et al. Generation and evaluation of recombinant Baculovirus Coexpressing Gp5 and M proteins of porcine reproductive and respiratory syndrome virus type 1. Viral Immunol. (2021) 34:697–707. doi: 10.1089/vim.2021.0018
46. Wissink, EH, Kroese, MV, van Wijk, HA, Rijsewijk, FA, Meulenberg, JJ, and Rottier, PJ. Envelope protein requirements for the assembly of infectious virions of porcine reproductive and respiratory syndrome virus. J Virol. (2005) 79:12495–506. doi: 10.1128/JVI.79.19.12495-12506.2005
Keywords: PRRSV-1, whole genome, amino acid site, genetic evolution, GP5, mutation
Citation: Weng C, Huang X, Chen Z, He M, Zhang B, Li H, Xie J, Chen M, Qiu L, Li X, Cao C and Chen H (2025) Genetic evolution, epidemic trends, and recombination dynamics of PRRSV-1 in China. Front. Vet. Sci. 12:1632917. doi: 10.3389/fvets.2025.1632917
Edited by:
Mengmeng Zhao, Foshan University, ChinaReviewed by:
Huixing Lin, Nanjing Agricultural University, ChinaFeipeng Zhao, Mudanjiang Medical University, China
Copyright © 2025 Weng, Huang, Chen, He, Zhang, Li, Xie, Chen, Qiu, Li, Cao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaobing Li, OTI0NDY0MjcwQHFxLmNvbQ==; Chong Cao, ODk1MjU1ODk0QHFxLmNvbQ==; Hongbo Chen, bHl4eV92ZXRAMTYzLmNvbQ==
†These authors have contributed equally to this work