 T. Naveenprasath
T. Naveenprasath Badeer Hassan Ummat
Badeer Hassan Ummat Farha Tarique
Farha Tarique Aakash Chawade
Aakash Chawade Sandeep Kushwaha
Sandeep Kushwaha- 1National Institute of Animal Biotechnology, Hyderabad, India
- 2Regional Centre for Biotechnology (RCB), Faridabad, India
- 3Swedish University of Agricultural Sciences, Alnarp, Sweden
Bovine mastitis, a multi-etiological disease, is driven by complex microbial consortia; however, the transcriptional activity of pathogens and their underlying molecular mechanisms remains insufficiently explored. To the best of our knowledge, no metatranscriptome study on bovine mastitis is available in the public domain that identifies transcriptionally active pathogens and their associated molecular signatures. In this study, an in silico metatranscriptomics approach is employed on publicly available bovine mastitis RNA sequencing (RNA-Seq) datasets to identify transcriptionally active pathogens and their gene expression signatures. The analysis of unmapped reads (those not mapped to the bovine genome) identified 25 transcriptionally active pathogenic genera, accounting for 8,995 sequences, approximately from 500 bacterial strains of different species. Major findings of the study includes: (I) list of emerging pathogens “Pseudomonas, Stenotrophomonas, Comamonas, and Sphingomonas” actively contributing to disease development alongside well-known pathogens; (II) expression profiling of 4,121 virulence proteins, 484 peptidases, 432 secretory proteins, and 74 antimicrobial resistance genes; (III) identification of numerous hypothetical proteins in Staphylococcus (112), Mycoplasma (69), and Escherichia (32), representing potential source for diagnostics and multi-epitope vaccine candidates; and (IV) negative correlations between beneficial bacteria (Blautia, Bacillus, Lactobacillus) and pathogenic species in microbial co-occurrence interaction networks, suggesting opportunities for microbiome-based therapeutic strategies to treat subclinical mastitis. This study demonstrated the advantages of the metatranscriptomics approach and publicly available dual RNA-Seq datasets in unraveling the complexity of polymicrobial infectious diseases.
1 Introduction
Bovine mastitis is a multi-etiological disease in dairy cattle, characterised by inflammation of the mammary gland. It affects animal health, milk production and quality and causes substantial economic losses to dairy farmers (1). Additionally, treating bovine mastitis significantly contributes to the bigger problem of antimicrobial resistance (AMR), particularly through the potential use and misuse of antibiotics in livestock production (2). Various culture-dependent and culture-independent high-throughput research studies have reported the association of hundreds of microbial species, including Streptococcus agalactiae, Streptococcus uberis, Staphylococcus aureus, Streptococcus pyogenes, Streptococcus dysgalactiae, Trueperella pyogenes, Escherichia coli, Klebsiella pneumoniae, Klebsiella oxytoca, Enterobacter aerogenes, and Pasteurella spp. 16S rRNA and whole metagenome shotgun sequencing (WMGS) have significantly contributed to the discovery of microbial species associated with bovine mastitis (3–6). A 16S sequencing-based study reported the top 10 genera of causative pathogens in the milk of mastitic quarters from 65 cows, and also reported less-known bacteria, such as Sneathia sanguinegens and Listeria innocua, which are difficult to identify using culture-based diagnostics (7). Another 16S study highlighted significant variation in taxonomic profiles among different udder quarters (healthy, mastitis-affected, and quarters with undetermined status) at the genus level (8). Similarly, another study investigated the impact of bacteria causing subclinical mastitis on the structure of the cow’s milk microbiome, reporting that Firmicutes and Proteobacteria were predominantly present in subclinical mastitis (9). Burakova et al. (10) investigated the relationship between milk microbiome composition and bovine mastitis before and after antibiotic treatment using 16S rRNA sequencing. This study linked the increased abundance of the genera Hymenobacter and Lachnospiraceae NK4A136 group with the development of subclinical and clinical mastitis. In contrast, a reduced abundance of Ralstonia, Lachnospiraceae NK3A20 group, Acetitomaculum, Massilia, and Atopostipes in mastitic milk was linked to their potential role in maintaining udder health. In the 16S sequencing, specific regions of rDNA are targeted to characterise the community composition. However, it has several major limitations, including low taxonomic resolution (primarily at the genus level), PCR bias, limited functional insight, and a lack of viability assessment of the bacterial community (11–13). To overcome these limitations, the WMGS approach was employed to gain a deeper understanding of the functional aspects of microbial species associated with bovine mastitis. A milk WGS metagenome study on healthy and clinical mastitis (CM) cows reported 363 unique bacterial species and strains in CM samples (4). Another metagenomic analysis was performed on the subclinical mastitis milk samples of Kankrej, Gir (Bos indicus), and crossbred (Bos taurus × B. indicus) animals. A total of 56 different species, with varying abundances, were detected in the subclinical mastitis milk samples (6). Recently, a longitudinal study on the udder microbiome of Norwegian Red dairy cows was conducted using a shotgun metagenomic approach to gain insights into pathogen-driven microbial adaptation and succession. This study revealed that samples with low somatic cell counts were enriched with beneficial genera, such as Corynebacterium, Bradyrhizobium, and Lactococcus, while Staphylococcus predominated in high somatic cell count milk samples (14). In WMGS, the whole stretch of DNA is sequenced to identify the presence of genes and associated bacteria. However, it fails to distinguish between transcriptionally active and dead bacteria, and has low real-time functional resolution at the species and gene levels. These limitations can be addressed through a metatranscriptomics approach.
The accurate identification of molecular weapons used by disease-associated pathogens against their hosts is critical for gaining insight into disease initiation and progression (15). RNA-Seq datasets of diseases contain extensive functional information on disease-related pathogens that are expressed simultaneously (16). However, host-centric studies often overlook the functional importance of microbial communities, such as in bovine mastitis. The proportion of opportunistic and commensal microbial populations in the udder tissue constantly shifts as mastitis progresses, which can be utilised to understand in vivo host-pathogen interactions. Therefore, publicly available bovine mastitis dual RNA-Seq studies have been analysed to identify transcriptionally active bacteria associated with bovine mastitis, elucidating their functional role in pathogenesis. We extracted the non-bovine, non-ribosomal sequenced reads and constructed de novo metatranscriptome assemblies (17, 18). The assembled transcripts were further annotated to reveal the taxonomy and functional profile of transcriptionally active pathogens associated with bovine mastitis.
2 Materials and methods
2.1 Selection of bovine mastitis transcriptome studies
A systematic approach was employed to identify publicly available bovine mastitis transcriptomics datasets from the NCBI Sequence Read Archive (SRA) database. A total of eight bovine mastitis studies combining five field (PRJEB43443, PRJNA544129, PRJNA627642, PRJNA551141, and PRJNA668296) and three cell lines studies (PRJNA778892, PRJNA556769, and PRJNA556769) were selected for this study. The NCBI bio project ID and study details for each project are listed in Supplementary Table S1. The three studies (PRJNA778892, PRJNA556769, and PRJNA591729) utilised the bovine mammary alveolar cell line (MACT), while one study (PRJNA668296) employed blood, two studies (PRJEB43443 and PRJNA544129) used milk, and two studies (PRJNA551141 and PRJNA627642) used mammary gland samples to generate RNA-Seq data. A brief information of each study including experimental conditions, sample sizes, sequencing platforms, and read statistics are provided in Supplementary Tables S2.
2.2 Quality check and control of sequence data
The quality of the RNA-Seq datasets from each project was assessed using FastQC v0.11.9 and pre-processed using FastP v0.23.2. using default parameters (19). The pre-processed RNA-Seq reads were aligned against the Bos Taurus ARS-UCD1.3 genome using STAR version 2.7.3a (20) with a maximum number of multiple alignments set to 10. Further, unmapped reads were extracted, and ribosomal reads were removed from unmapped reads using SortMeRNA version 2.1b (21) with default parameters. High-quality unmapped non-ribosomal reads were used to construct the metatranscriptome assembly.
2.3 Metatranscriptome assembly and annotation
The de novo metatranscriptome was constructed from non-ribosomal unmapped short reads using the Trinity version 2.9.0 software (22). Protein-coding sequences were identified and translated using TransDecoder v5.5.0 for ORF identification with a minimum amino acid length of 60. Only complete ORFs with start and stop codons were retained for analysis to ensure the reliability of the generated protein sequences. The abundance of each translated transcript was calculated by counting the number of mapped reads to each transcript using STAR v2.7.3a and featureCounts v2.0 (23), followed by DESeq2 normalisation. Transcripts longer than 60 amino acids and with an average read count of ≥3 were selected for redundancy removal, which was carried out by clustering the sequences at 90% identity using CD-HIT v4.8.1 (24). Further, clustered transcripts were annotated using NCBI BLAST v2.12.0 (25) against the UniProt UniRef100 database. Only the annotated proteins with sequence identity ≥75% and query coverage ≥60% with the reference sequence were selected for further taxonomy and functional assignment. Microbial species and functional information for each sequence were extracted from the description of hit sequences. Furthermore, species-to-kingdom links were traced through the UniProt taxonomy tree map. The project PRJNA668296 was omitted due to technical incompatibility with the downstream analysis.
A list of 102 genera was compiled from published metagenomics studies on bovine mastitis to identify mastitis pathogens in the assembled metatranscriptome. KEGG Orthology (KO) annotations for sequences were obtained from eggNOG-mapper version 2.1.12 (26) using emapper.py, which assigned KO terms to each protein sequence. KO enrichment was performed using the enrichKO function of MicrobiomeProfiler (version 1.15) R package (24) at a p-value cut-off of 0.05 and q-value (FDR) cut-off of 0.2. Benjamini & Hochberg method was applied for p-value correction. Additionally, gene set filter criteria with a minimum size of 10 and a maximum size of 500 were used to reduce noise and exclude overly broad or narrow categories. The annotation of hypothetical protein sequences was performed using the InterProScan database (27). VirulentPred 2.0 (28), and signalP 5.0 (29) were used to identify virulent and secretory proteins. Peptidase and AMR genes were identified using a BLAST similarity search against the above-mentioned criteria in Merops [27] and the Comprehensive Antibiotic Resistance Database (CARD) (30).
2.4 Microbial diversity analysis of metatranscriptome
Microbial abundance data were pre-processed in the R language (version 4.2.3) using the R packages tidyverse (version 2.0) and RColorBrewer (version 1.1.3) (31). Microbial diversity, species evenness and richness analysis were performed using the R package vegan version 2.7. A linear regression model was fitted between Shannon diversity and species richness using the lm function, with 95% confidence intervals. The ggplot2 version 3.5 was used for visualization. Non-parametric Spearman correlation was calculated using the R cor function from the taxonomic abundance matrix. A correlation threshold of ≥0.6 and a p-value of ≤ 0.05 was applied to reduce noise while capturing biologically meaningful co-occurrence patterns. A microbial co-occurrence interaction network was generated using the R package igraph (version 2.1.4).
3 Results
Host-centric RNA-Seq datasets are a valuable resource for exploring in vivo gene expression profiles of pathogens to develop new diagnostic and therapeutic solutions. In this study, non-ribosomal and unmapped reads from the host genome were extracted from all the selected RNA-Seq datasets and used for metatranscriptome assembly and analysis of pathogens. This study aims to identify transcriptionally active pathogens and their associated molecular signatures for bovine mastitis disease. The schematic workflow of the performed study is detailed in Figure 1.
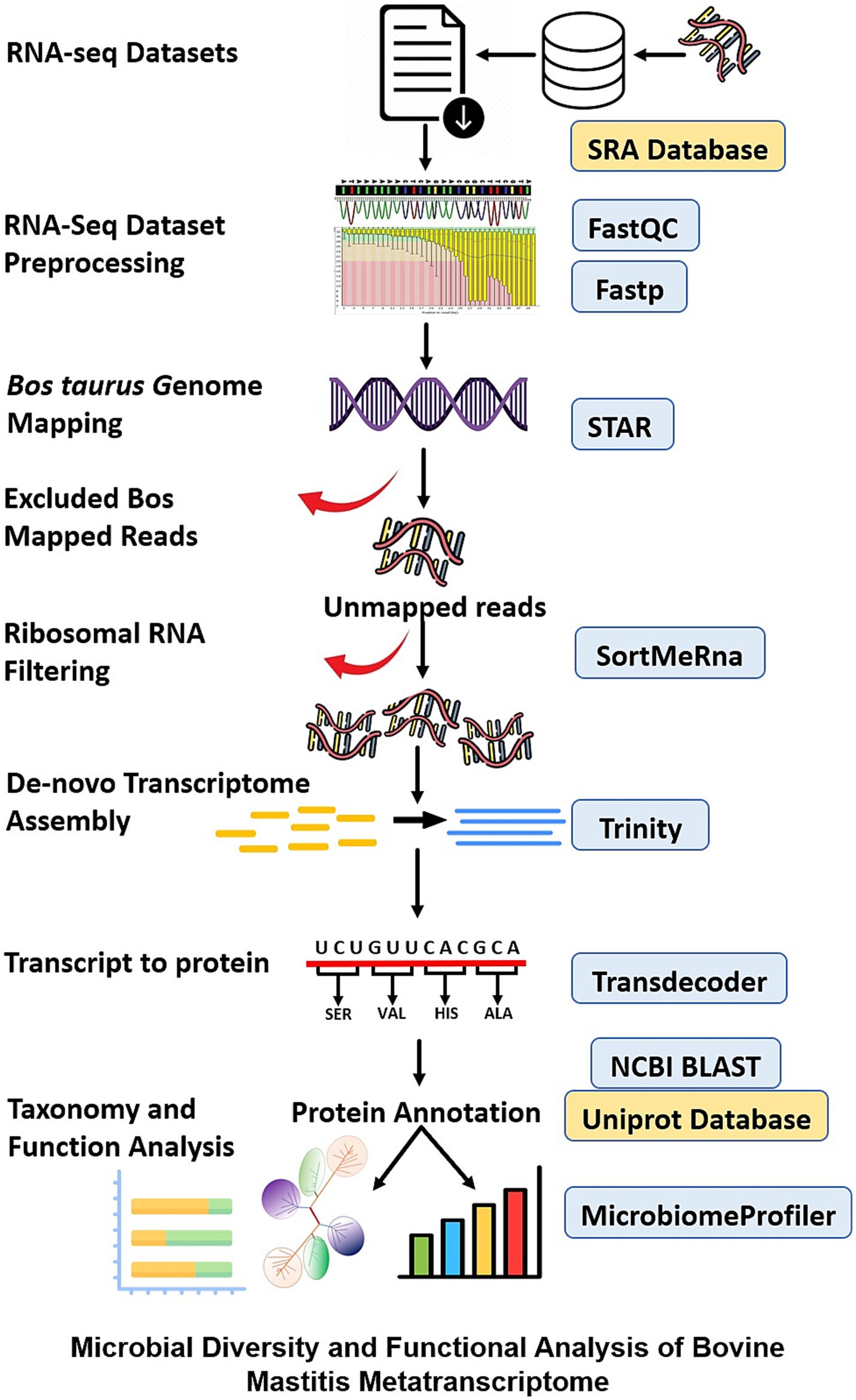
Figure 1. Schematic representation of the performed metatranscriptomics study to identify the transcriptionally active bacterial pathogens associated with bovine mastitis.
3.1 Metatranscriptome assembly and annotation
The assembled metatranscriptomes were translated into protein sequences (Table 1). First, all transcripts were screened for an average read count of ≥3 and coded protein lengths of ≥60 amino acids to exclude chimeric and low-quality partial transcripts. Secondly, all the selected proteins at the first step were clustered at 90% sequence identity to remove redundant and similar proteins generated from different isoforms. In the third step, non-redundant protein sequences were annotated using a BLAST similarity search against the UniProt database. Only sequences that matched the UniProt database with at least 75% sequence identity and 60% query coverage were selected for downstream analysis, aiming to capture the inherent functional diversity of microbial consortia with greater accuracy. Microbial species information for each sequence was extracted from the description of hit sequences. Furthermore, species-to-kingdom links were traced through the UniProt taxonomy tree map. The above-mentioned filtering steps were applied to each project to ensure high-quality assembled transcripts and translated proteins for further taxonomic and functional analysis. The annotated protein sequences were classified into five groups based on their origin, namely bacteria, fungi, bovine, humans, and others (Table 2). This study primarily focused on identifying functionally active bacteria associated with bovine mastitis. Therefore, protein sequences from fungi, bovine, human, and other groups were removed after annotation from further downstream analysis.
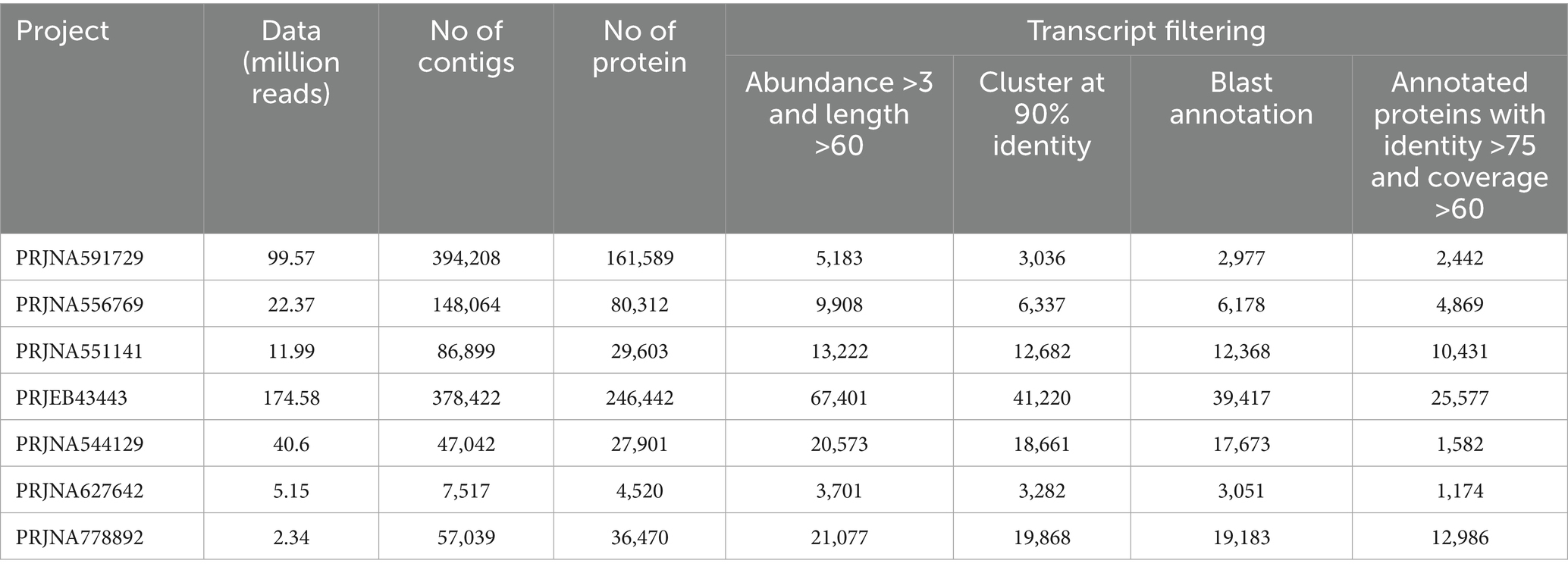
Table 1. Summary of de novo metatranscriptome assembly for each RNA-Seq study.
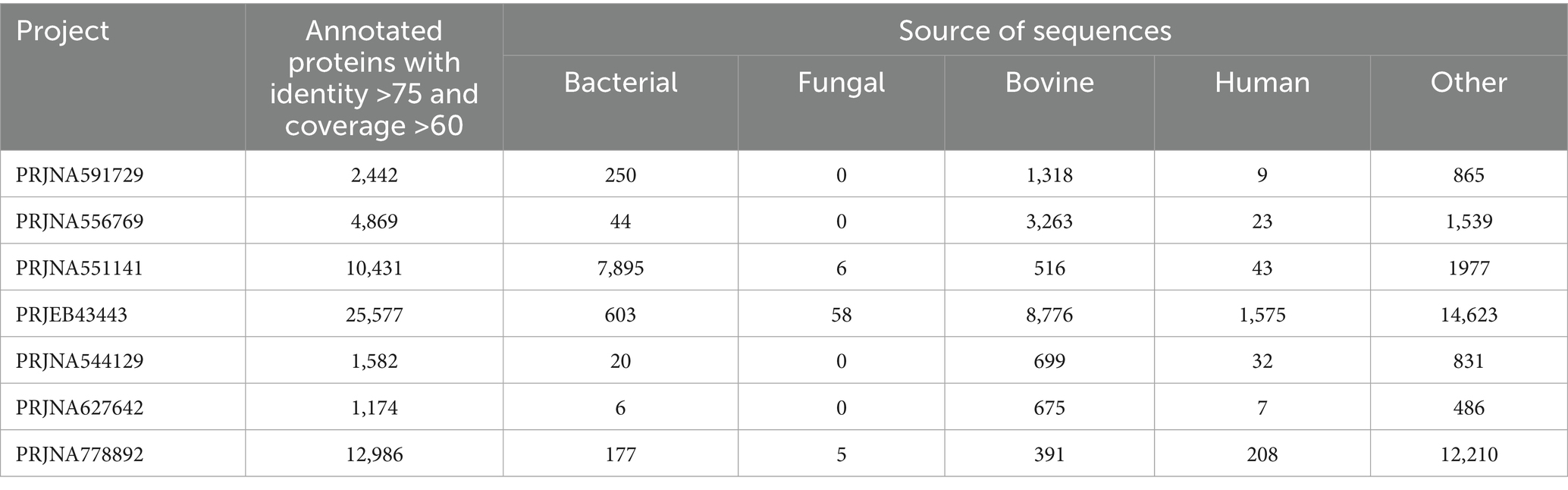
Table 2. Summary of annotated metatranscriptome for each RNA-Seq study with the number of transcripts identified for bacteria, fungi, bovine, human and other categories.
3.2 Metatranscriptome analysis to reveal taxonomic and functional profiling of pathogenic microbial communities associated with bovine mastitis
Protein sequences of bacterial origin were further analysed for each project to determine their taxonomic and functional role. Bacterial sequences were detected in distinct proteins of each project at the phylum, genus and species levels (Figures 2A,B).
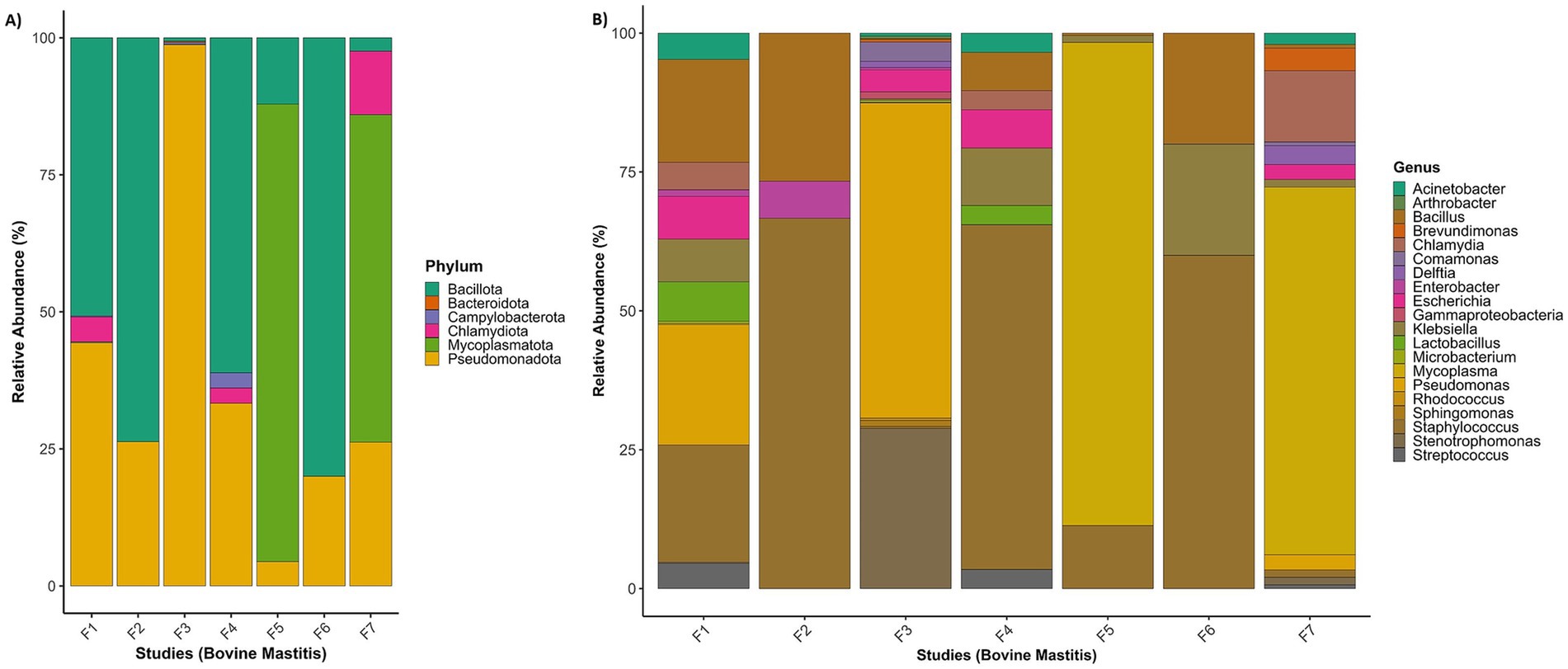
Figure 2. Taxonomic summary of metatranscriptome for each RNA-Seq study: (A) relative abundance of bacterial phyla, and (B) relative abundance of bacterial genera. F1 (PRJNA544129); F2 (PRJEN43443); F3 (PRJNA551141); F4 (PRJNA556769); F5 (PRJNA591729); F6 (PRJNA627642); F7 (PRJNA778892).
However, various sequences were missing species-level information because the UniProt database may not contains species level information for these sequences. The phyla Pseudomonadota and Bacillota were highly prevalent in all projects and had a higher number of distinct sequences. Both phyla cover 16 to 96% of each project, with an average coverage of 70% (Figure 2). Pseudomonas is a highly abundant genus in the phylum Pseudomonadota. P. alcaligenes, P. entomophila, P. marginalis, P. monteilii, P. phage, P. plecoglossicida, P. stutzeri, P. taiwanensis, P. geniculata, P. coronafaciens, P. savastanoi, P. amygdali, P. fluorescens, P. aeruginosa, P. syringae, P. putida were among the most abundant species in the metatranscriptome. In the phylum Bacillota, various Bacillus species (B. amyloliquefaciens, B. cereus, B. obstructivus, B. pumilus, B. subtilis, B. thuringiensis, and B. wiedmannii) were also found in abundance.
In three projects, Staphylococcus aureus was used as a pathogen to study bovine mastitis infection, while the project PRJNA544129 studied naturally infected bovine mastitis. Mycoplasma bovis and Streptococcus uberis are the other two infection agents used in the projects PRJNA551141 and PRJNA627642, respectively. Table 3 shows that Staphylococcus species (S. aureus, S. cohnii, S. sciuri, S. succinus) have a significant number of distinct proteins in four projects. Project-wise identified genera, number of sequences and respective species are provided in Supplementary File 2 (ST1–ST8). Based on the number of sequences (≥5 distinct sequences), 58 transcriptionally active genera were identified. Transcriptionally active pathogenic genera and their top 20 species, with the number of distinct sequences, are provided in Supplementary File 2 (ST9 and ST10).
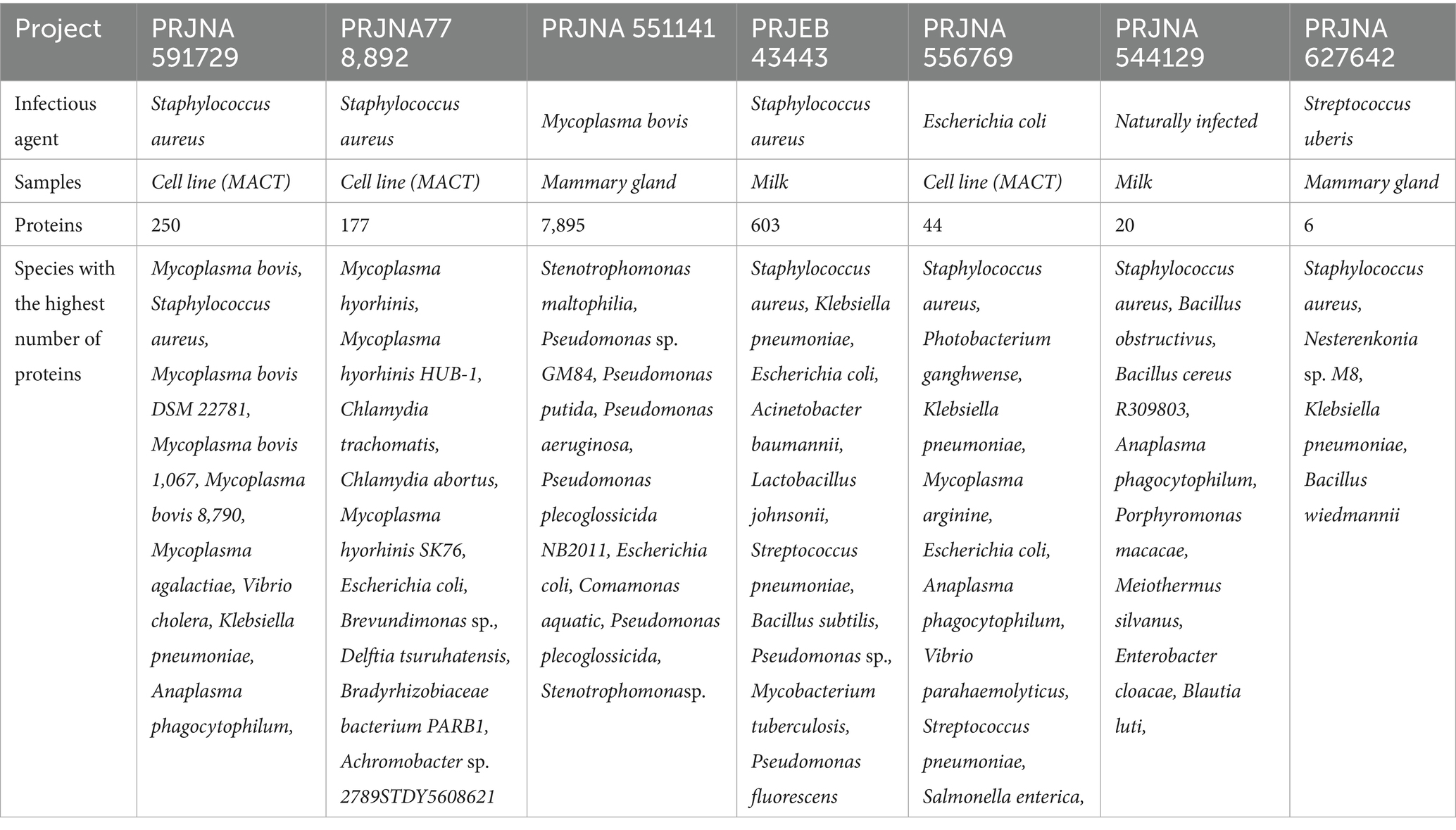
Table 3. Summary of identified bacterial species and number of bacterial proteins associated with species for each study.
The Shannon index of studies (PRJNA544129:1.63 and PRJNA778892: 1.37) exhibits moderate diversity, indicating the dominance of certain species, while others show lower diversity (dominance of a very few species) (Figure 3A). The diversity results are very well supported by the species evenness index. The studies (PRJNA591729, PRJNA627642, and PRJNA551141) have extremely uneven species composition, whereas PRJNA544129, PRJNA778892, and PRJNA556769 have low species evenness (Figure 3B). PRJEN43443 has moderate species evenness (0.5) among all the studies. The Bray-Curtis and Jaccard dissimilarity indices of all the studies are relatively high (>0.6), indicating high dissimilarity in microbial composition and abundance across all the studies (Figure 3C).
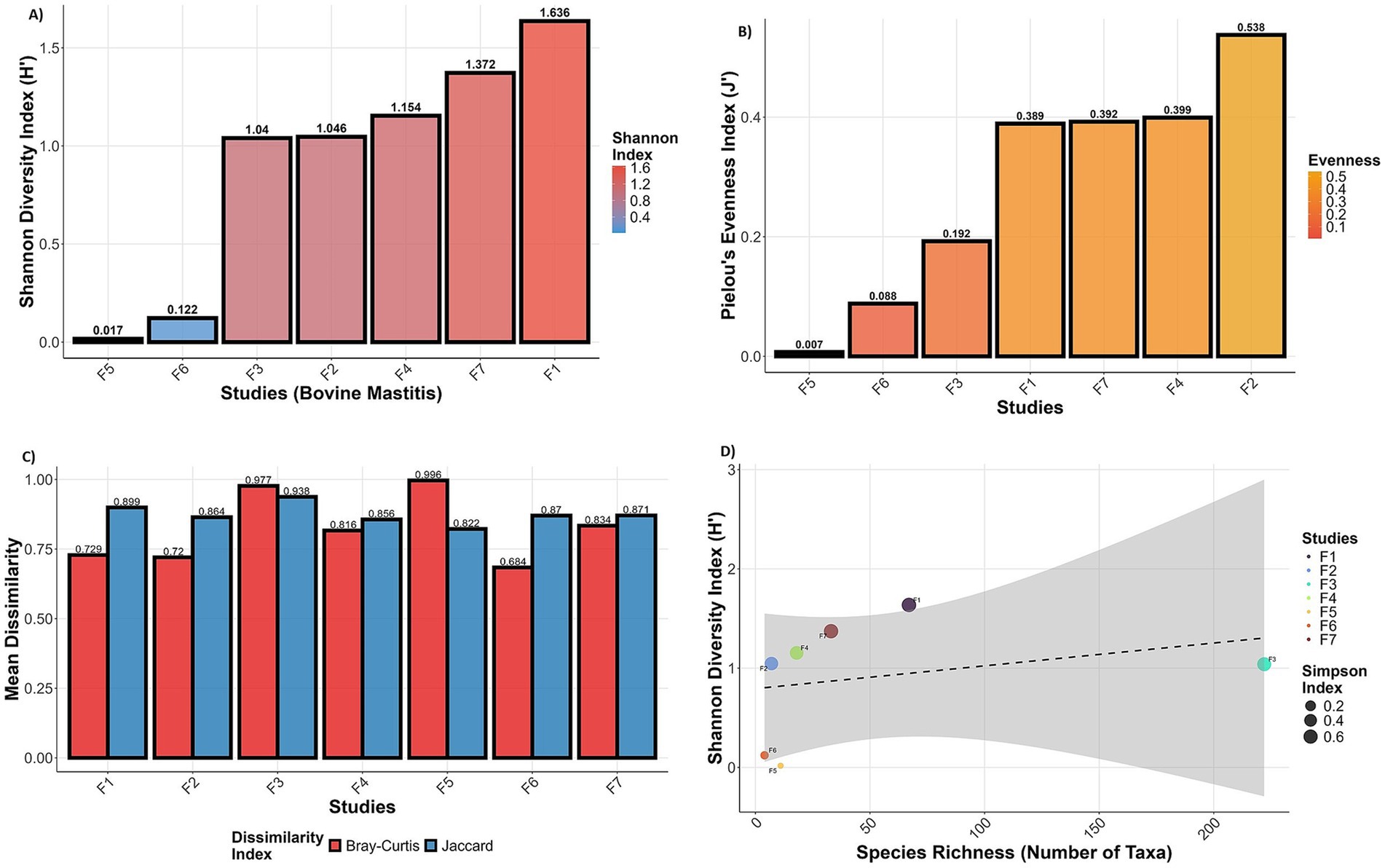
Figure 3. Microbial diversity indices of metatranscriptome for each RNA-Seq study: (A) Shannon diversity, (B) species evenness, (C) Bray–Curtis and Jaccard distance between samples, and (D) relationship between species richness and diversity. Box plots of Shannon, Simpson, Chao1, and Beta diversity for each project are provided in Supplementary Figures S1A–E. F1 (PRJNA544129); F2 (PRJEN43443); F3(PRJNA551141); F4(PRJNA556769); F5(PRJNA591729); F6(PRJNA627642); F7 (PRJNA778892).
Each RNA-Seq study was designed differently according to its objectives. Therefore, we selected only mammary gland and milk samples and grouped them into ‘Mastitic’ and ‘Healthy’ groups to identify the most abundantly expressed proteins by calculating the mean abundance of their transcript sequences for each project separately. In study PRJEB43443, a time-point-based infection study, samples from the zero-hour time point were considered healthy, and all others were grouped as mastitic. Since PRJNA627642 does not have control samples, it was excluded from the abundance calculation. Staphylococcus aureus is the most prevalent pathogen of bovine mastitis reported in studies worldwide, and we also observed that Staphylococcus aureus proteins were among the most abundant proteins in all the projects. In our analysis, other known mastitis pathogens, such as Escherichia, Mycoplasma, Streptococcus and Pseudomonas species, were also present in higher abundance. We have observed that various transcripts exhibit higher average expression levels in mastitis samples. The proteins were sorted according to their abundance in the mastitis samples and provided the top five most abundant proteins across all projects (Table 4), which are primarily associated with cell division and gene regulatory mechanisms. Interestingly, most of the top-abundant proteins were either uncharacterised or hypothetical, with limited information available regarding their function. The project-wise average abundance of pathogens, as determined by identified distinct sequences, and the abundance of all identified sequences, are provided in Supplementary Table S3, Supplementary Files 1 and Supplementary Files 3a,b.
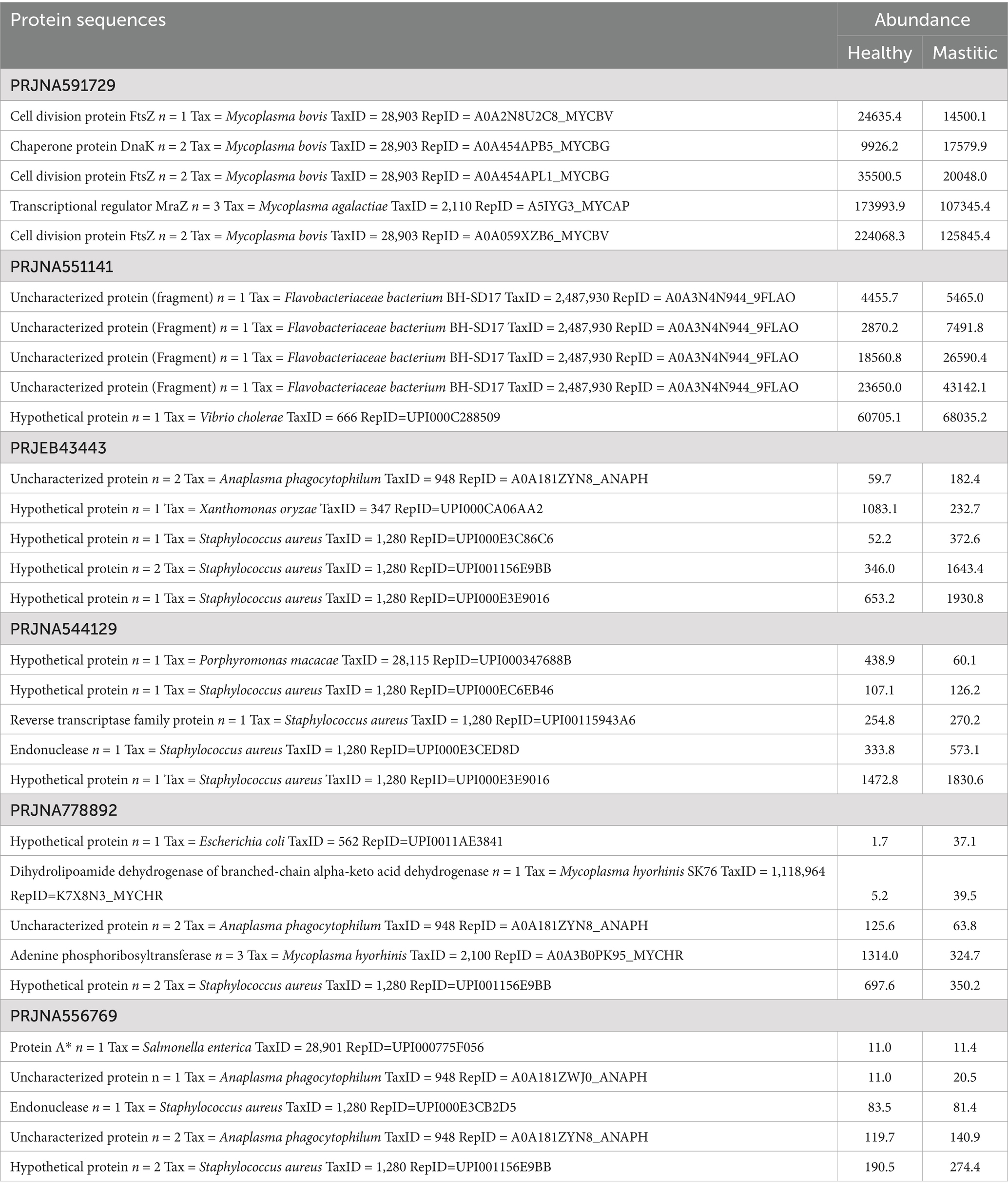
Table 4. List of the five most abundant proteins and their average abundance between two groups for each project.
3.3 KEGG orthology (KO) enrichment analysis of metatranscriptome
KO enrichment analysis of the metatranscriptome is crucial for developing deep insight into bacterial pathogenesis, as enriched pathways can reveal key virulence factors, survival mechanisms, and employed metabolic adaptations in host environments. Overall, 83 pathways were obtained from KO enrichment. The description of pathways and counts of mapped KO terms is provided in Supplementary Table S5, Supplementary Files 1. Our KO analysis shows (Figure 4) that mastitis-associated pathogens are finely tuned to the host environment with a strong representation of signalling and regulation pathways (Two-component systems), nutrient acquisition and metabolism (amino acid and cofactor biosynthesis), motility and colonisation (flagella, chemotaxis), virulence and immune evasion (secretion systems, biofilm formation).
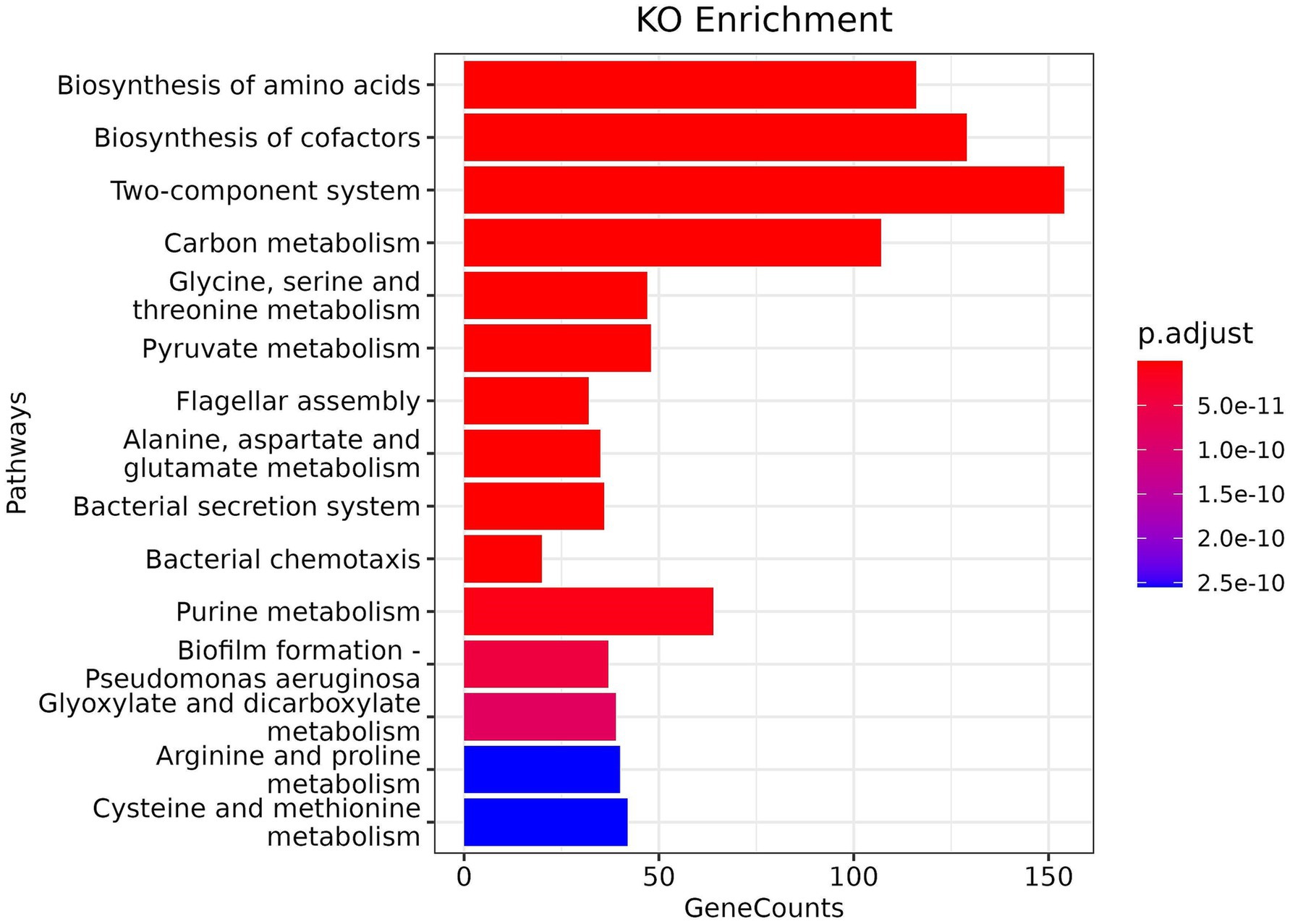
Figure 4. KEGG orthology (KO) enrichment analysis of assembled metatranscriptome.
The two-component system, the most enriched pathway, enables bacteria to sense and respond to environmental changes, such as host defences and antibiotic treatment. The biosynthesis of amino acids and cofactors is another enriched pathway that suggests the survival and growth of pathogens in nutrient-limited host environments, activating pyruvate and amino-acid metabolism (including pyruvate, glycine, serine, and threonine) in response to stress. Flagellar assembly and chemotaxis pathways suggest bacterial motility for initial colonisation and host tissue invasion, and deliver effector proteins into host cells using bacterial secretion systems type III and VI secretion systems, a well-known virulence mechanism of bacteria. Biofilm formation pathways provide shelter against antimicrobials and host immunity, contributing to recurrence, chronic infections, and antimicrobial resistance. The enrichment of arginine, proline, cysteine, and methionine metabolism suggests that the pathogenic bacterial community has developed adaptive mechanisms (redox balance, stress response and polyamine synthesis) against host oxidative stress and immune modulation.
3.4 Identification of virulence factors, peptidase and AMR genes in metatranscriptome assembly
In bacteria, various types of proteins exist which can interact with the host directly or indirectly and may contribute to disease pathogenesis. Therefore, identifying such bacterial proteins in the metatranscriptome is essential for evaluating the pathogenic potential of pathogens. Virulent, peptidase and AMR genes fall under these categories. Therefore, bacterial proteins with virulence features were identified using VirlentPred software. Further, the predicted proteins were also explored in the Virulence Factor Database (VFDB) using BLAST similarity search (Table 5). Detailed descriptions of virulent proteins are provided in Supplementary File 4 (ST2). Predicted virulent proteins from Stenotrophomonas, Mycoplasma, Staphylococcus Species, Comamonas, and Sphingomonas species mostly belong to hypothetical and uncharacterized proteins.
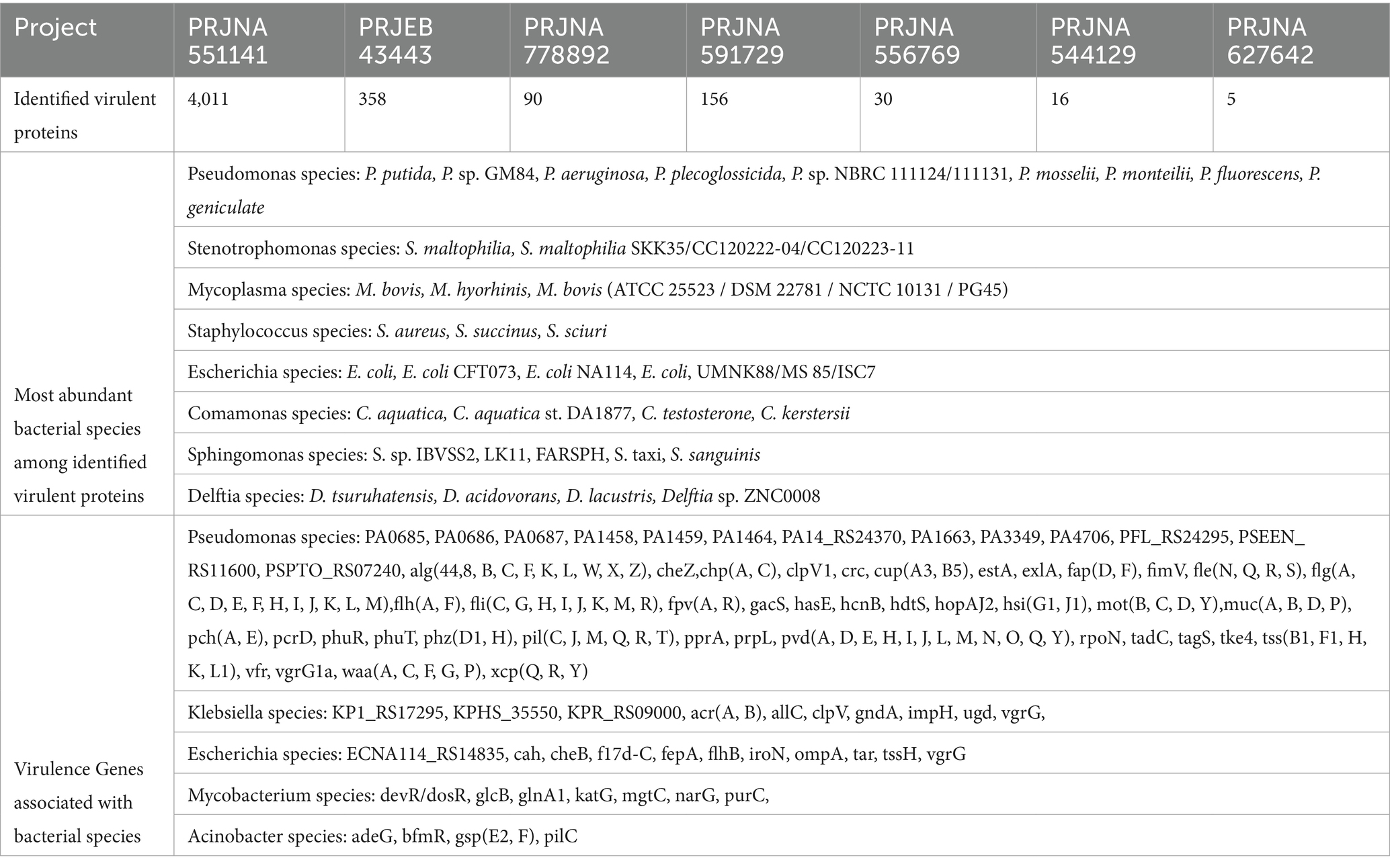
Table 5. Summary of identified virulence protein, most abundant bacterial species, and associated virulence genes.
Proteases are key bacterial proteins that cleave peptide bonds in proteins to facilitate host cell colonisation, defence evasion, and damage. The cross-compatibility of several bacterial proteases with the host system enables them to cleave host proteins and disrupt host cellular functions. MEROPS database was used to identify the peptidase sequences among bacterial proteins (Table 6). The identified peptidases mainly belong to the serine and metalloprotease families, and the PRJNA551141 project has the highest number of peptidases. Proteases were identified for the following projects: PRJNA551141 (625), PRJEB43443 (32), PRJNA778892 (26), PRJNA591729 (9), and PRJNA556769 (4). Project-wise information on identified proteases is provided in Supplementary File 4 (ST3). The prevalence of antibiotic resistance among bacterial pathogens is increasing due to the unsupervised use of antibiotics in treatment, and the treatment of bovine mastitis contributes significantly to antimicrobial resistance. Therefore, the identified bacterial proteins from each project were analysed against the CARD database (Table 7). A total of 85 proteins were predicted to have antibiotic resistance; 82 sequences belonged to study PRJNA551141. Two resistance proteins were identified from PRJEB43443 and PRJNA778892, respectively. The identified proteins from the bovine mastitis metatranscriptome are resistant to major antibiotics and drugs, such as Tetracycline, Penicillin, Macrolides, and Cephalosporins, which are routinely used for bovine mastitis treatments. A project-wise detailed description of antibiotic resistance proteins is provided in Supplementary File 4 (ST4).

Table 6. Summary of identified protease gene families, most abundant bacterial species, and associated protease families in metatranscriptome.
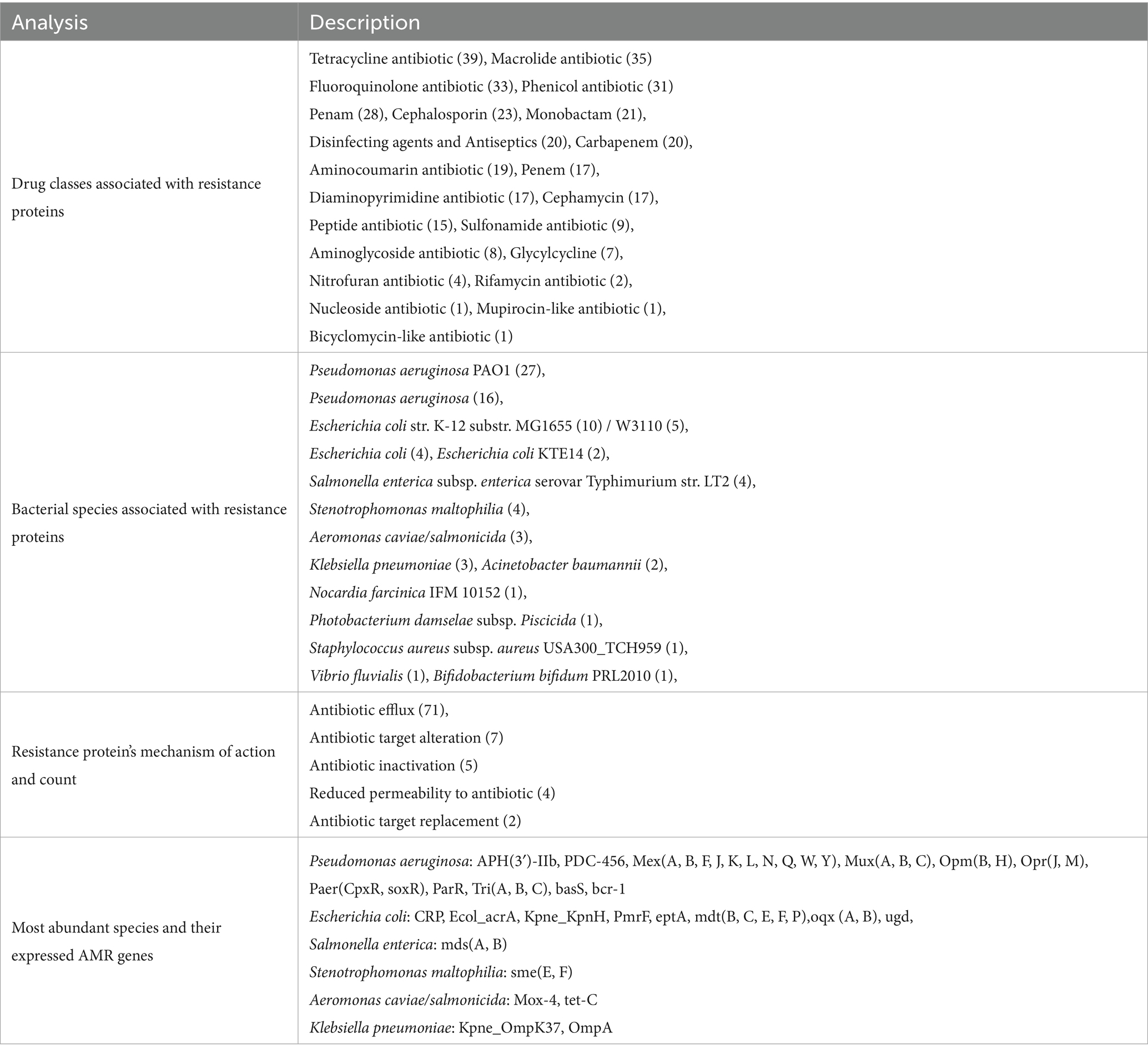
Table 7. Summary of AMR genes and gene families, resistance mechanisms, most abundant bacterial species, and associated AMR genes in metatranscriptome.
Based on metatranscriptome analysis, a list of the 25 most transcriptionally active bacterial genera has been compiled to illustrate the active pathogenic load associated with bovine mastitis, with a particular focus on virulence factors, peptidases, secretory proteins, and antibiotic resistance (AMR) genes. The minimum number of virulent proteins, 10, was considered to select the most transcriptionally active genera from metatranscriptome data (Table 8). A detailed description is provided in Supplementary File 2 (ST11). Microbial co-occurrence interaction network analysis, based on expression abundance, revealed that Blautia, Bacillus, Klebsiella, and Lactobacillus are negatively correlated with other genera of pathogenic consortia. It suggests that the abundance of Blautia and Lactobacillus, a genus with probiotic characteristics, decreases during disease, whereas the genus Bacillus contains both pathogenic and probiotic species; perhaps the abundance of probiotics is more prevalent in healthy conditions. Interestingly, a significant number of virulent (33), peptidase (2), and antibiotic resistance (AMR) (3) proteins were identified in the Klebsiella genus from metatranscriptome data. However, it is negatively correlated to consortium species (Figure 5).
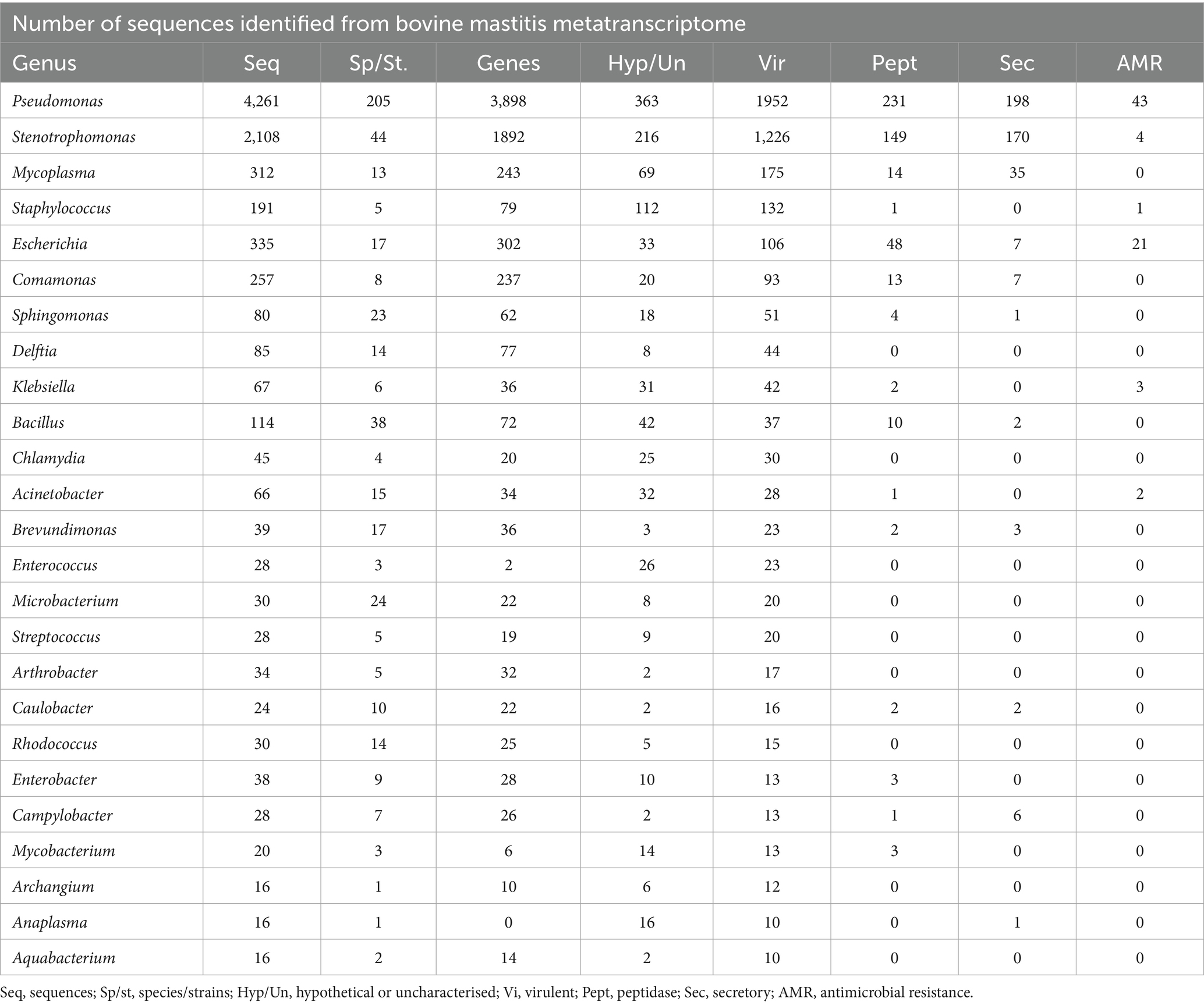
Table 8. List of transcriptionally active bacteria associated with bovine mastitis based on the number of virulent proteins identified for each genus.
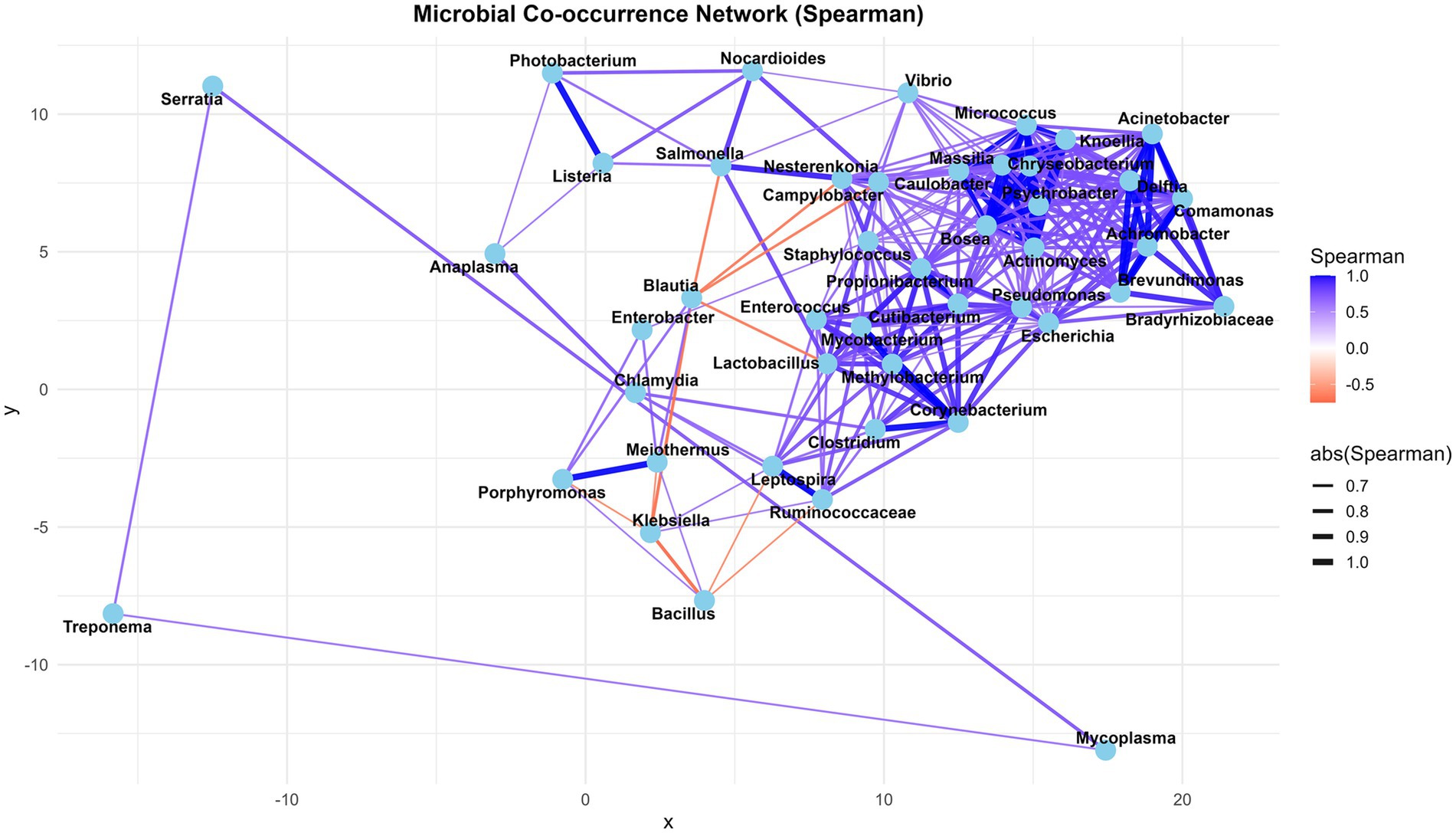
Figure 5. The microbial co-occurrence interaction networks of bovine mastitis-associated microbial communities at the genus level, based on the Spearman correlation coefficient.
4 Discussion
This study primarily focused on identifying transcriptionally active bacteria from publicly available bovine mastitis dual RNA-Seq studies, as these datasets contain extensive functional information on disease-related microbial species within the same biological samples. The microbial content expressed during the host-pathogen interaction, provides in vivo gene expression profile of pathogens, which facilitates the discovery of bacterial virulence factors to develop new diagnostics and therapeutics. Similar attempts have been made to understand the role of microbial communities in human cancer diseases using RNA-Seq data (34, 35).
In this study, rigorous quality control criteria were used to exclude chimeric, low-quality, partial and redundant transcript sequences. A minimum protein length of ≥60 was used to include small proteins from microbial communities that are involved in regulating larger proteins, cell signaling, antibiotic production and toxin regulation, membrane-associated functions, metabolism, and stress response, as well as metal ion homeostasis (36). Moreover, the inclusion of 60 amino acids provided higher confidence in functional gene-to-species assignment compared to shorter lengths and relatively less partial sequences. The average read counts of greater than or equal to 3 for genes across the samples were considered to remove chimeric and rare protein sequences, due to the hypersensitivity of high-throughput sequencing technology. The average read count is a better metric for reducing disparity between small and large numbers of studies. Various isoforms of microbial gene sequences were generated due to the stitching of short k-mers during transcriptome assembly. Therefore, we clustered protein isoforms at 90% sequence identity to reduce functional redundancy and false specificity for cross-species orthology inference. Stringent BLAST similarity search criteria (sequence identity ≥75% and query coverage ≥60%) were used to ensure the high-quality taxonomical and functional assignment (Table 2). This approach is more reliable and accurate than the seed sequence mapping approach, which lacks robustness for assigning gene species links in microbial community analysis.
Even after excluding non-host reads before assembly, a large number of the proteins belonged to the bovine species, which may be due to the presence of conserved domains and motif sequences across the kingdom. In filtered protein sequences, the proportion of bovine sequences varied from 3% to 71%, compared to the low bacterial protein proportion (0.5%–10%) across all projects, except for study PRJNA551141. It has 75% bacterial protein and 5% bovine protein. The enhanced representation of bacterial proteins may be due to the fact that rRNA was removed during sample preparation, as rRNA constitutes 70%–90% of the total RNA in these samples. None of the other projects had used ribo-depletion during their sample preparation.
The microbial diversity, species richness and evenness metric support the functional diversity among the selected RNA-Seq studies. The scatter plot of species richness and Shannon diversity index shows a positive correlation between species richness and Shannon diversity, as indicated by the upward-sloping linear regression line (Figure 3D). This suggests that sample PRJEB43443 demonstrated a relatively high Shannon index with moderate species richness, potentially indicating a more evenly distributed community structure, as supported by its larger point size (Simpson index). In contrast, sample PRJNA551141, while showing the highest richness, exhibited only moderate Shannon diversity, suggesting dominance by a few taxa (Stenotrophomonas and Pseudomonas species) and reduced evenness. Higher or moderate evenness often suggests stable communities, while low evenness may indicate vulnerability to disturbance (PRJNA591729 and PRJNA627642). Low Shannon diversity and high species richness indicate dysbiosis or infection dominance (PRJNA551141). Moderate Shannon diversity and moderate species richness may reflect baseline microbial communities (PRJNA544129), whereas moderate diversity with fewer taxa suggests a balanced microbial environment with minimal colonisation (PRJNA778892), possibly post-antibiotic or in the early stage of infection.
In KO analysis, enriched pathways include the two-component system, amino acid/cofactor biosynthesis, flagellar assembly and secretion systems, and biofilm formation, reflecting bacterial adaptation to host defences, nutrient limitation, and persistence mechanisms. Enrichment of amino acid metabolism further highlights adaptive responses of the pathogenic community to oxidative stress and immune modulation (37–41). The Project-wise KO analysis is provided in Supplementary Figures S2A–E. Various highly abundant sequences were either uncharacterised or hypothetical despite their known genomic sequences (Table 4). Therefore, in silico sequence analysis of hypothetical and uncharacterised proteins was performed to explore their functional information. On average, 62% of the total putative proteins were annotated, which contain domains of catalytic enzymes, transposable elements, retrotransposable elements, reverse transcriptases, prokaryotic membrane lipoproteins, endonucleases, and various other molecular signatures. A summary of hypothetical protein annotation and detailed descriptions of the InterProScan annotation of all hypothetical proteins are provided in Supplementary Table S4 and Supplementary File 4 (ST1).
In our analysis, various abundant hypothetical and uncharacterised proteins contain intrinsically disordered regions, suggesting that these proteins may have roles in host–microbe interactions in vivo conditions, such as bacterial effector protein translocation, evasion of the host immune system, and mimicing host protein functions (42). The presence of intrinsically disordered regions in secreted bacterial effectors is well-reported (43). The biofilm-forming curli protein of E. coli has disordered regions that facilitate its aggregation and contribute to the formation of the extracellular matrix of the biofilm (33). Various intrinsically disordered regions containing hypothetical or uncharacterised proteins exhibit a virulent nature in our analysis. The in silico annotated sequence contains reverse transcriptases, transposable elements and transposon-related domains. Reverse transcriptase converts RNA into double-stranded cDNA, and it is a major component of diversity-generating retroelements (DGRs) in bacteria. Reverse transcriptases in DGRs can create hypervariable regions in target genes, fuelling the faster evolution of bacterial genomes to ensure bacterial survival in harsh environments (44). Transposable elements can also alter the genome by insertion and excision, promoting bacterial evolution. These jumping genes are known to be part of bacterial virulent elements (45). These proteins may have roles in antibiotic resistance and pathogenicity, which need to be explored further.
Based on the number of virulent proteins (≥10), peptidases, secretory proteins, and antibiotic resistance (AMR) genes, a list of 25 transcriptionally active bacterial genera was compiled to highlight pathogenic load associated with bovine mastitis. Pseudomonas species dominated in the constructed metatranscriptome with the highest number of species (205) and expressed genes (3898), including the highest number of virulence (1952), secretory (198), peptidases (231) and antimicrobial resistance genes (44), highlighting a significant role of Pseudomonas species in bovine mastitis pathogenesis. Moreover, P. putida, and P. aeruginosa are emerged as multidrug-resistant pathogens linked with clinical and subclinical bovine mastitis worldwide (46–49). Stenotrophomonas species, a lesser-known pathogen of bovine mastitis, have been shown to express substantial virulence genes (1226) and secretory proteins (170), indicating their active involvement in pathogenesis and potential biofilm formation or host interaction, making them an emerging global opportunistic pathogen (50). Recent research also demonstrated that enterogenic S. maltophilia can migrate from the gut to the mammary gland via the gut-mammary axis to induce mastitis by activating the calcium-ROS-AMPK-mTOR-autophagy pathway (51). Mycoplasma species, despite having a lower virulence gene count (175) out of a total of 243 identified genes, indicate a specialisation in pathogenesis rather than metabolic versatility. Staphylococcus, a known pathogen of mastitis, displayed a lower number of species but a notable number of virulence factors (132). The expression of hypothetical or uncharacterised sequences (112) suggests the presence of novel molecular factors from these species in bovine mastitis pathogenic consortia.
Among all identified virulent genes, 633 were uncharacterized, and 379 were hypothetical by Uniprot definition. The protein sequence (Sequence id: TRINITY_DN43764_c0_g1_i1.p4; Uniprot id of hit: UniRef100_UPI000DEBC0B7) from Pseudomonas aeruginosa is one of the identified virulent hypothetical proteins that also possesses an AMR gene feature (Drug class: disinfecting agents and antiseptics; mode of mechanism: Antibiotic efflux; ARO ID:3003680; Gene name: TriB). In our study, we identified 84 antimicrobial resistance (AMR) genes, some are reported for bovine mastitis, including tetG, tetC, blaZ, sul1, adeB, acrA, APH(3′)-IIb, mexA, mexB, and mexF. These genes are involved in well-known resistance mechanisms, including efflux pumps, antibiotic-modifying enzymes, and target protection proteins. Other genes like adeB, adeF, APH(3′)-IIb, eptA, kpnH, ompA, oqxA, and oqxB were also reported for bovine mastitis. The presence of these AMR genes in this dataset suggests a broader distribution of resistance determinants among mastitis-associated microbiota (52–54). The association of other identified AMR genes with bovine mastitis has not been found in the literature. In the absence of literature support, it is challenging to speculate on the role of these bacteria in pathogenic consortia mastitis, which are predominantly composed of Pseudomonas species. However, the expression of such a large number of AMR genes from Pseudomonas species suggests a potential role in providing protection against antibiotics, which warrants further in-depth investigation. Most Pseudomonas aeruginosa strains possess a Type III Secretion System (T3SS), which plays a crucial role in pathogenesis and has been linked to elevated somatic cell counts in the milk of cows with mastitis (55). Key T3SS-related genes identified in bovine mastitis isolates include exlA, pcrD, and hopAJ2. Additionally, a range of other virulence and secretion system genes—such as clpV1, flgC, flgG, flgK, flgL, fliC, mucA, mucB, algD, algF, algA, pilV, pilY2, pvdF, pvdS, rhlR, rhlI, rhlA, rhlB, rhlC, tssB1, tssF1, tssG1, vgrG1, as well as components of the xcp (PA3095–PA3105) and hxc (PA0677–PA0687) secretion system are regulated by phosphate availability (56).
The negative correlation of Klebsiella with other genera of pathogenic consortia (Bacillus/ Balutia/ Salmonella/ Leptospira/ Porphyromonas/ Ruminococcaceae/ Lactobacillus) in the non-directional microbial co-occurrence network indicates that when Klebsiella abundance increases, Bacillus/ Balutia/ Salmonella/ Leptospira/ Porphyromonas/ Ruminococcaceae/ Lactobacillus tends to decrease, and vice versa. This could be due to competition, niche exclusion, or antagonistic interactions. These species may not co-exist well in the same environment. Some species, such as Bacillus/Lactobacillus, produce bacteriocins that inhibit the growth of Klebsiella (57). Host factors (such as temperature, oxygen level, or pH) may favour the growth of Klebsiella species after environmental E. coli infection compared to other pathogens, due to similar growth conditions to those of other Enterobacteriaceae. The chance of Klebsiella infection after exposure to environmental mastitis conditions is moderate to high in poor bedding conditions (58). Blautia and Lactobacillus, a genus with probiotic characteristics, may decrease during disease, whereas the genus Bacillus contains both pathogenic and probiotic species; perhaps the abundance of probiotic strains is more prevalent in healthy conditions. It is not easy to speculate and interpret until further multi-bacterial culture-based research investigation. Different Bacillus strains have been reported to exhibit antimicrobial, antioxidant, and immune-modulatory activities in the host. A study by Pinchuk et al. (32) has reported the B. subtilis strains’ anti H. pylori activity, which was attributed to the secretion of aninocoumacin A antibiotic. The antagonistic activity of aninocoumacin A was also documented against enteric E. faecium and Shigella flexneri. An interesting communication by Ripert et al. (59) revealed that the probiotic B. clausii O/C strain protected Vero and Caco-2 cells from the cytotoxic effects of Clostridium difficili and B. cereus toxins.
Pseudomonas, Stenotrophomonas, Comamonas, and Sphingomonas are among the most transcriptionally active pathogens. Pseudomonas and Sphingomonas genera are often reported in association with bovine mastitis in various studies (60, 61). The Stenotrophomonas genus is a lesser-known opportunistic pathogen associated with bovine mastitis. However, various Stenotrophomonas isolates were extracted and reported from bovine milk samples for mastitis cases (62). The Prevalence and survival of Stenotrophomonas species in milk and dairy products have also been reported (63, 64). A study has explored the prevalence of genetic relatedness, antimicrobial resistance, biofilm formation, biofilm genes associated with virulence and integron genes among isolates of S. maltophilia recovered from bovine milk with subclinical mastitis (65). Comamonas is another less-known genus associated with bovine mastitis. However, it was isolated from bulk tank milk (66). Therefore, our study emphasises the inclusion of these prominent pathogens in the list of primary bovine mastitis-causing pathogens.
4.1 Limitations
This study utilised different host-centric RNA-Seq studies to identify transcriptionally active bacteria and their molecular signature. However, these studies have their own experimental designs, sequencing methods (single-end/paired-end/different platforms), and data analysis approaches to achieve their objectives. It is very challenging to obtain datasets from the same source with identical technical specifications. Therefore, a uniform data processing approach has been used to extract and assemble the metatranscriptome to minimise confounding risks and technical disparity. Each study was processed and reported separately before being integrated into the overall analysis. A fraction of the non-host reads from host-centric dual RNA-seq studies has been extracted and used to construct a metatranscriptome, rather than a microbial-centric metatranscriptome. Additionally, transcription levels do not always correlate with protein abundance or activity. Therefore, these findings may differ slightly from the actual situation. This study aims to generate research leads for scientists by leveraging computational tools and publicly available datasets. However, in vitro validation of these findings is crucial for a better understanding of the microbial aspects of bovine mastitis, which is beyond the current scope of this work.
5 Conclusion
This is the first in-depth metatranscriptomics study on bovine mastitis, delineating transcriptionally active pathogenic bacterial taxa, their associated molecular signatures, and uncovering a complex pathogenic consortium that goes beyond the well-known causative pathogens. Pseudomonas (P. putida, P. aeruginosa, P. fluorescens, P. monteilii, P. mosselii, and P. plecoglossicida), Stenotrophomonas (S. maltophilia, and S. sp. RIT309, SKA14), Mycoplasma (M. bovis, M. hyorhinis, M. agalactiae, and M. ovipneumoniae), Staphylococcus (S. aureus, S. succinus, and S. sciuri), Escherichia (E. coli CFT073, and NA114), Comamonas (C. aquatica, C. testosteroni) and Sphingomonas (S. sp. IBVSS2, LK11, FARSPH, S. sanguinis, and S. taxi) species were predominant among transcriptionally active pathogens, and should be considered as significant contributors to bovine mastitis pathogenesis alongside established pathogens.
Identified 213 hypothetical and uncharacterized proteins from Staphylococcus, Mycoplasma, and Escherichia, providing a list of unexplored virulence factors that could serve as targets for multi-epitope vaccine development, while the expression of 74 AMR genes, particularly from Pseudomonas and Escherichia species, suggests these pathogens may confer antibiotic protection to the entire pathogenic consortium, necessitating the need to update current treatment strategies. The abundance of uncharacterized proteins presents opportunities for developing novel diagnostics and therapeutics. The negative correlation of beneficial bacteria (Blautia, Bacillus, and Lactobacillus) with pathogenic species indicates that microbiome modulation could be a viable prevention strategy for subclinical mastitis. A balanced microbiome with these antagonistic bacteria could help to reduce pathogen loads and prevent subclinical infections from becoming clinical. However, these findings need to be validated thoughtfully to develop microbiome-assisted prevention strategies for subclinical bovine mastitis. Our study highlights that transcriptionally active bacteria and their expressed genes (AMR, secretory, peptidase and virulence genes) can aid in identifying novel therapeutic targets, early diagnostic biomarkers and expressed candidate genes for developing a multi-epitope vaccine that targets multiple pathogens. Our findings also suggest the potential of microbiome modulation strategies to develop microbiome-assisted prevention approaches for better management of subclinical bovine mastitis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
TN: Data curation, Visualization, Writing – original draft, Formal analysis, Resources, Writing – review & editing. BU: Writing – review & editing, Writing – original draft, Methodology, Formal analysis, Data curation, Visualization. FT: Data curation, Writing – review & editing, Writing – original draft, Visualization. AC: Resources, Visualization, Formal analysis, Writing – original draft, Data curation, Investigation, Writing – review & editing, Software, Methodology. SK: Investigation, Supervision, Validation, Writing – review & editing, Visualization, Project administration, Software, Data curation, Writing – original draft, Formal analysis, Methodology, Resources, Conceptualization, Funding acquisition.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The infrastructural facilities of the National Institute of Animal Biotechnology (NIAB), Hyderabad, are duly acknowledged for the work performed in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1642351/full#supplementary-material
References
1. Seegers, H, Fourichon, C, and Beaudeau, F. Production effects related to mastitis and mastitis economics in dairy cattle herds. Vet Res. (2003) 34:475–91. doi: 10.1051/vetres:2003027
2. Van Boeckel, TP, Brower, C, Gilbert, M, Grenfell, BT, Levin, SA, Robinson, TP, et al. Global trends in antimicrobial use in food animals. Proc Natl Acad Sci. (2015) 112:5649–54. doi: 10.1073/pnas.1503141112
3. Couvillion, SP, Mostoller, KE, Williams, JE, Pace, RM, Stohel, IL, Peterson, HK, et al. Interrogating the role of the milk microbiome in mastitis in the multi-omics era. Front Microbiol. (2023) 14:1105675. doi: 10.3389/fmicb.2023.1105675
4. Hoque, MN, Istiaq, A, Clement, RA, Sultana, M, Crandall, KA, Siddiki, AZ, et al. Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci Rep. (2019) 9:13536. doi: 10.1038/s41598-019-49468-4
5. Porcellato, D, Meisal, R, Bombelli, A, and Narvhus, JA. A core microbiota dominates a rich microbial diversity in the bovine udder and may indicate presence of dysbiosis. Sci Rep. (2020) 10:21608. doi: 10.1038/s41598-020-77054-6
6. Bhatt, VD, Ahir, VB, Koringa, PG, Jakhesara, SJ, Rank, DN, Nauriyal, DS, et al. Milk microbiome signatures of subclinical mastitis-affected cattle analysed by shotgun sequencing. J Appl Microbiol. (2012) 112:639–50. doi: 10.1111/j.1365-2672.2012.05244.x
7. Oultram, JWH, Ganda, EK, Boulding, SC, Bicalho, RC, and Oikonomou, G. A metataxonomic approach could be considered for cattle clinical mastitis diagnostics. Front Vet Sci. (2017) 4:36. doi: 10.3389/fvets.2017.00036
8. Falentin, H, Rault, L, Nicolas, A, Bouchard, DS, Lassalas, J, Lamberton, P, et al. Bovine teat microbiome analysis revealed reduced alpha diversity and significant changes in taxonomic profiles in quarters with a history of mastitis. Front Microbiol. (2016) 7:480. doi: 10.3389/fmicb.2016.00480
9. Kaczorowski, Ł, Powierska-Czarny, J, Wolko, Ł, Piotrowska-Cyplik, A, Cyplik, P, and Czarny, J. The influence of Bacteria causing subclinical mastitis on the structure of the cow’s Milk microbiome. Molecules. (2022) 27:1829. doi: 10.3390/molecules27061829
10. Burakova, I, Gryaznova, M, Smirnova, Y, Morozova, P, Mikhalev, V, Zimnikov, V, et al. Association of milk microbiome with bovine mastitis before and after antibiotic therapy. Vet World. (2023) 16:2389–402. doi: 10.14202/vetworld.2023.2389-2402
11. Johnson, JS, Spakowicz, DJ, Hong, B-Y, Petersen, LM, Demkowicz, P, Chen, L, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. (2019) 10:5029. doi: 10.1038/s41467-019-13036-1
12. Abellan-Schneyder, I, Matchado, MS, Reitmeier, S, Sommer, A, Sewald, Z, Baumbach, J, et al. Primer, pipelines, parameters: issues in 16S rRNA gene sequencing. mSphere. (2021) 6:e01202–20. doi: 10.1128/mSphere.01202-20
13. Poretsky, R, Rodriguez-R, LM, Luo, C, Tsementzi, D, and Konstantinidis, KT. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS One. (2014) 9:e93827. doi: 10.1371/journal.pone.0093827
14. da Silva Duarte, V, Franklin, FV, Krysmann, AM, and Porcellato, D. Longitudinal study of the udder microbiome of Norwegian red dairy cows using metataxonomic and shotgun metagenomic approaches: insights into pathogen-driven microbial adaptation and succession. bioRxiv. (2025). doi: 10.1101/2025.04.04.647183
15. Butowski, CF, Dixit, Y, Reis, MM, and Mu, C. Metatranscriptomics for understanding the microbiome in food and nutrition science. Meta. (2025) 15:185. doi: 10.3390/metabo15030185
16. de Almeida, OGG, Pereira, MG, Oxaran, V, De Martinis, ECP, and Alves, VF. In silico metatranscriptomic approach for tracking biofilm-related effectors in dairies and its importance for improving food safety. Front Microbiol. (2022) 13:928480. doi: 10.3389/fmicb.2022.928480
17. Raghavan, V, Kraft, L, Mesny, F, and Rigerte, L. A simple guide to de novo transcriptome assembly and annotation. Brief Bioinform. (2022) 23:bbab563. doi: 10.1093/bib/bbab563
18. Martin, JA, and Wang, Z. Next-generation transcriptome assembly. Nat Rev Genet. (2011) 12:671–82. doi: 10.1038/nrg3068
19. Chen, S, Zhou, Y, Chen, Y, and Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
20. Dobin, A, Davis, CA, Schlesinger, F, Drenkow, J, Zaleski, C, Jha, S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. (2012) 29:15–21. doi: 10.1093/bioinformatics/bts635
21. Kopylova, E, Noé, L, and Touzet, H. Sortmerna: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. (2012) 28:3211–7. doi: 10.1093/bioinformatics/bts611
22. Haas, BJ, Papanicolaou, A, Yassour, M, Grabherr, M, Blood, PD, Bowden, J, et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. (2013) 8:1494–512. doi: 10.1038/nprot.2013.084
23. Liao, Y, Smyth, GK, and Shi, W. Featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2013) 30:923–30. doi: 10.1093/bioinformatics/btt656
24. Yu, G. Microbiomeprofiler: an R/shiny package for microbiome functional enrichment analysis. (2025). Available online at: https://bioconductor.org/packages/MicrobiomeProfiler (Accessed October 06, 2025).
25. Altschul, SF, Gish, W, Miller, W, Myers, EW, and Lipman, DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
26. Cantalapiedra, CP, Hernández-Plaza, A, Letunic, I, Bork, P, and Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. bioRxiv. (2021). doi: 10.1093/molbev/msab293
27. Blum, M, Chang, H-Y, Chuguransky, S, Grego, T, Kandasaamy, S, Mitchell, A, et al. The interpro protein families and domains database: 20 years on. Nucleic Acids Res. (2020) 49:D344–54. doi: 10.1093/nar/gkaa977
28. Garg, A, and Gupta, D. VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. (2008) 9:62. doi: 10.1186/1471-2105-9-62
29. Almagro Armenteros, JJ, Tsirigos, KD, Sønderby, CK, Petersen, TN, Winther, O, Brunak, S, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. (2019) 37:420–3. doi: 10.1038/s41587-019-0036-z
30. McArthur, AG, Waglechner, N, Nizam, F, Yan, A, Azad, MA, Baylay, AJ, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. (2013) 57:3348–57. doi: 10.1128/AAC.00419-13
31. Gentleman RIaR. R is a programming language for statistical computing and data visualization (1993) Available online at: https://www.r-project.org/ (Accessed October 06, 2025).
32. Pinchuk, IV, Bressollier, P, Verneuil, B, Fenet, B, Sorokulova, IB, Mégraud, F, et al. In vitro anti-Helicobacter pylori activity of the probiotic strain Bacillus subtilis 3 is due to secretion of antibiotics. Antimicrob Agents Chemother. (2001) 45:3156–61. doi: 10.1128/AAC.45.11.3156-3161.2001
33. Jain, N, and Chapman, MR. Bacterial functional amyloids: order from disorder. Biochim Biophys Acta. (2019) 1867:954–60. doi: 10.1016/j.bbapap.2019.05.010
34. Chen, Y, and Wei, J. Identification of pathogen signatures in prostate Cancer using RNA-seq. PLoS One. (2015) 10:e0128955. doi: 10.1371/journal.pone.0128955
35. Ai, D, Xing, Y, Zhang, Q, Wang, Y, Liu, X, Liu, G, et al. Joint analysis of microbial and immune cell abundance in liver cancer tissue using a gene expression profile deconvolution algorithm combined with foreign read remapping. Front Immunol. (2022) 13:853213. doi: 10.3389/fimmu.2022.853213
36. Storz, G, Wolf, YI, and Ramamurthi, KS. Small proteins can no longer be ignored. Annu Rev Biochem. (2014) 83:753–77. doi: 10.1146/annurev-biochem-070611-102400
37. Green, ER, and Mecsas, J. Bacterial secretion systems: an overview. Microbiol Spectr. (2016) 4. doi: 10.1128/microbiolspec.VMBF-0012-2015
38. Hall-Stoodley, L, Costerton, JW, and Stoodley, P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. (2004) 2:95–108. doi: 10.1038/nrmicro821
39. Hood, MI, and Skaar, EP. Nutritional immunity: transition metals at the pathogen–host interface. Nat Rev Microbiol. (2012) 10:525–37. doi: 10.1038/nrmicro2836
40. Potempa, J, and Pike, RN. Bacterial Peptidases In: W Russell and H Herwald, editors. Concepts in bacterial virulence, vol. 12. Allschwilerstrasse, Basel, Switzerland: S. Karger AG (2004)
41. Zhang, Y, Liang, S, Zhang, S, Bai, Q, Dai, L, Wang, J, et al. Streptococcal arginine deiminase system defences macrophage bactericidal effect mediated by XRE family protein XtrSs. Virulence. (2024) 15:2306719. doi: 10.1080/21505594.2024.2306719
42. Trivedi, R, and Nagarajaram, HA. Intrinsically disordered proteins: an overview. Int J Mol Sci. (2022) 23:14050. doi: 10.3390/ijms232214050
43. Marín, M, Uversky, VN, and Ott, T. Intrinsic disorder in pathogen effectors: protein flexibility as an evolutionary hallmark in a molecular arms race. Plant Cell. (2013) 25:3153–7. doi: 10.1105/tpc.113.116319
44. Sharifi, F, and Ye, Y. Identification and classification of reverse transcriptases in bacterial genomes and metagenomes. Nucleic Acids Res. (2021) 50:e29-e. doi: 10.1093/nar/gkab1207
45. Hacker, J, Blum-Oehler, G, Mühldorfer, I, and Tschäpe, H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol. (1997) 23:1089–97. doi: 10.1046/j.1365-2958.1997.3101672.x
46. Mallick, TT, Rahman, MM, Siddique, N, Shuvo, KH, Arafat, KY, Homa, SF, et al. Molecular and genomic investigation unveils Pseudomonas putida as an emerging multidrug-resistant pathogen linked to bovine clinical mastitis. Microb Pathog. (2025) 203:107461. doi: 10.1016/j.micpath.2025.107461
47. Daly, M, Power, E, Björkroth, J, Sheehan, P, O'Connell, A, Colgan, M, et al. Molecular analysis of Pseudomonas aeruginosa: epidemiological investigation of mastitis outbreaks in Irish dairy herds. Appl Environ Microbiol. (1999) 65:2723–9. doi: 10.1128/AEM.65.6.2723-2729.1999
48. Schauer, B, Wald, R, Urbantke, V, Loncaric, I, and Baumgartner, M. Tracing mastitis pathogens-epidemiological investigations of a Pseudomonas aeruginosa mastitis outbreak in an Austrian dairy herd. Animals. (2021) 11:279. doi: 10.3390/ani11020279
49. Banerjee, S, Batabyal, K, Joardar, SN, Isore, DP, Dey, S, Samanta, I, et al. Detection and characterization of pathogenic Pseudomonas aeruginosa from bovine subclinical mastitis in West Bengal, India. Vet World. (2017) 10:738–42. doi: 10.14202/vetworld.2017.738-742
50. Brooke, JS. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev. (2012) 25:2–41. doi: 10.1128/CMR.00019-11
51. He, Z, Zhao, C, He, Y, Liu, Z, Fan, G, Zhu, K, et al. Enterogenic Stenotrophomonas maltophilia migrates to the mammary gland to induce mastitis by activating the calcium-ROS-AMPK-mTOR-autophagy pathway. J Anim Sci Biotechnol. (2023) 14:157. doi: 10.1186/s40104-023-00952-y
52. Ismail, ZB, and Abutarbush, SM. Molecular characterization of antimicrobial resistance and virulence genes of Escherichia coli isolates from bovine mastitis. Vet World. (2020) 13:1588–93. doi: 10.14202/vetworld.2020.1588-1593
53. Janus, A, Deepa, PM, Vergis, J, Rajasekhar, R, Habeeb, BP, Bipin, KC, et al. Unravelling the complex mechanisms of multidrug resistance in bovine mastitis pathogens: insights into antimicrobial resistance genes, biofilm dynamics, and efflux systems. Microb Pathog. (2024) 195:106902. doi: 10.1016/j.micpath.2024.106902
54. Kerek, Á, Németh, V, Szabó, Á, Papp, M, Bányai, K, Kardos, G, et al. Monitoring changes in the antimicrobial-resistance gene set (ARG) of raw milk and dairy products in a cattle farm, from production to consumption. Vet Sci. (2024) 11:265. doi: 10.3390/vetsci11060265
55. Park, HR, Hong, MK, Hwang, SY, Park, YK, Kwon, KH, Yoon, JW, et al. Characterisation of Pseudomonas aeruginosa related to bovine mastitis. Acta Vet Hung. (2014) 62:1–12. doi: 10.1556/avet.2013.054
56. Bains, M, Fernández, L, and Hancock, RE. Phosphate starvation promotes swarming motility and cytotoxicity of Pseudomonas aeruginosa. Appl Environ Microbiol. (2012) 78:6762–8. doi: 10.1128/AEM.01015-12
57. Darbandi, A, Asadi, A, Mahdizade Ari, M, Ohadi, E, Talebi, M, Halaj Zadeh, M, et al. Bacteriocins: properties and potential use as antimicrobials. J Clin Lab Anal. (2022) 36:e24093. doi: 10.1002/jcla.24093
58. Ruegg, PL. A 100-year review: mastitis detection, management, and prevention. J Dairy Sci. (2017) 100:10381–97. doi: 10.3168/jds.2017-13023
59. Ripert, G, Racedo, SM, Elie, AM, Jacquot, C, Bressollier, P, and Urdaci, MC. Secreted compounds of the probiotic Bacillus clausii strain O/C inhibit the cytotoxic effects induced by Clostridium difficile and Bacillus cereus toxins. Antimicrob Agents Chemother. (2016) 60:3445–54. doi: 10.1128/AAC.02815-15
60. Nam, HM, Lim, SK, Kang, HM, Kim, JM, Moon, JS, Jang, KC, et al. Prevalence and antimicrobial susceptibility of gram-negative bacteria isolated from bovine mastitis between 2003 and 2008 in Korea. J Dairy Sci. (2009) 92:2020–6. doi: 10.3168/jds.2008-1739
61. Kurt, S, and Eşki, F. Pathogen isolation and antibiogram analysis in dairy cows with clinical mastitis in Adana region, Turkey. Etlik Vet Mikrobiyol Derg. (2021) 32:20–6. doi: 10.35864/evmd.906990
62. Ohnishi, M, Sawada, T, Marumo, K, Harada, K, Hirose, K, Shimizu, A, et al. Antimicrobial susceptibility and genetic relatedness of bovine Stenotrophomonas maltophilia isolates from a mastitis outbreak. Lett Appl Microbiol. (2012) 54:572–6. doi: 10.1111/j.1472-765X.2012.03246.x
63. El-Prince, E, Amin, WF, Thabet, SS, and Hanna, MIL. Stenotrophomonas species in milk and some dairy products. J Adv Vet Res. (2019) 9:11–3. Available online at: https://advetresearch.com/index.php/AVR/article/view/337/293
64. Zeinhom, MMA, Hassan, GM, Salem, HAM, and Corke, H. Prevalence and survival of Stenotrophomonas species in Milk and dairy products in Egypt. Foodborne Pathog Dis. (2021) 18:337–45. doi: 10.1089/fpd.2020.2893
65. Ocak, F, and Turkyilmaz, S. Investigation of genetic relatedness, antimicrobial resistance, biofilm formation, biofilm-related virulence genes and integron-related genes of Stenotrophomonas maltophilia isolates obtained from bovine milk samples with mastitis: characteristics of Stenotrophomonas maltophilia isolates. J Hellenic Vet Med Soc. (2023) 74:5979–92. doi: 10.12681/jhvms.30641
Keywords: bovine mastitis, RNA-Seq, metatranscriptomics, virulent protein, AMR, Pseudomonas , Stenotrophomonas , Comamonas
Citation: Naveenprasath T, Ummat BH, Tarique F, Chawade A and Kushwaha S (2025) Metatranscriptome analysis to unveil the molecular signatures of transcriptionally active pathogens associated with bovine mastitis. Front. Vet. Sci. 12:1642351. doi: 10.3389/fvets.2025.1642351
Edited by:
Camila Hamond, University of Connecticut, United StatesCopyright © 2025 Naveenprasath, Ummat, Tarique, Chawade and Kushwaha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aakash Chawade, YWFrYXNoLmNoYXdhZGVAc2x1LnNl Sandeep Kushwaha, U2FuZGVlcEBuaWFiLm9yZy5pbg==