 Mariana Mateos1*
Mariana Mateos1* Du Kang2
Du Kang2 Christophe Klopp3
Christophe Klopp3 Hugues Parrinello4
Hugues Parrinello4 Mateo García-Olazábal2,5
Mateo García-Olazábal2,5 Molly Schumer6
Molly Schumer6 Nathaniel K. Jue7
Nathaniel K. Jue7 Yann Guiguen8
Yann Guiguen8 Manfred Schartl2,5,9,10*
Manfred Schartl2,5,9,10*- 1Department of Wildlife and Fisheries Sciences, Texas A&M University, College Station, TX, United States
- 2Physiological Chemistry, Biocenter, University of Wuerzburg, Würzburg, Germany
- 3Mathématiques et Informatique Appliquées de Toulouse (MIAT) and Système d'information du projet d'analyse des génomes des animaux d'élevage (SIGENAE), INRA, Toulouse, France
- 4MGX, Bio Montpellier, CNRS, INSERM, University of Montpellier, Montpellier, France
- 5Department of Biology, Texas A&M University, College Station, TX, United States
- 6Howard Hughes Medical Institute (HHMI) and Department of Biology, Stanford University, Stanford, CA, United States
- 7Department of Biology and Chemistry, California State University Monterey Bay, Seaside, CA, United States
- 8INRA, UR1037 Fish Physiology and Genomics, Rennes, France
- 9Developmental Biochemistry, Biocenter, University of Wuerzburg, Würzburg, Germany
- 10Hagler Institute for Advanced Studies, Texas A&M University, College Station, TX, United States
Introduction
The freshwater livebearing fish genus Poeciliopsis (Poeciliidae) constitutes a valuable research system for questions within the field of evolutionary ecology, including life history evolution (e.g., multiple origins of placentas), intergenomic conflict, evolution of sex (with the existence of several asexual hybrid biotypes), and biogeography (reviewed in Mateos et al., 2019). Despite its importance, a robust phylogenetic framework, and genomic resources are lacking for this taxon. Herein, we report the first whole genome draft sequence of a member of this genus: Poeciliopsis occidentalis Baird and Girard (1853), the Gila topminnow. Poeciliopsis occidentalis, along with its sister lineage P. sonoriensis Girard (1859) (the Yaqui topminnow), are currently considered separate species (Miller et al., 2005). They are distributed in Mexico and the United States, where they are listed (as subspecies of P. occidentalis sensu lato; Sonoran topminnow, “guatopote de Sonora” in Spanish) as threatened and endangered, respectively. P. occidentalis s.l. has several interesting biological features, whose study would benefit from annotated genomes. First, it has an intermediate level of placentation (matrotrophy index) within a clade (i.e., Leptorhaphis group) that contains members with higher (i.e., P. prolifica) and lower (i.e., P. infans) matrotrophy indices (Reznick et al., 2002). Secondly, it is the sexual host of the oldest known asexual hybrid biotype of the genus Poeciliopsis (i.e., the hybridogen Poeciliopsis monacha-occidentalis; Quattro et al., 1992). Moreover, P. occidentalis s.l. has an unresolved phylogenetic position, possibly due to incomplete lineage sorting, and/or reticulation (Mateos et al., 2019). In addition, the taxonomy and status of evolutionary significant units (ESUs) within P. occidentalis s.l. are controversial, as additional ESUs have been proposed (Vrijenhoek et al., 1985; Hedrick and Hurt, 2012). The genome sequence of P. occidentalis will thus be a valuable resource for macroevolutionary and molecular evolution studies of the genus, as well as for phylogeographic and conservation genetics research. In the work presented herein, we used the “linked-reads” Chromium System (10x Genomics, Pleasanton, CA, USA) to sequence, assemble, and annotate a draft genome of P. occidentalis. The resulting assembly had a contig and scaffold N50 of 0.103 and 1.540 Mbp, respectively.
Source of Specimens
The specimen used for the genome assembly was a snap-frozen (sampled in the early 2000's, stored at −80°C) lab-bred male of P. occidentalis s.s. (sample ID MS-8/9/10 AV76-7; strain originally collected at Oquitoa, Rio Altar, Concepcion drainage, Sonora, Mexico, 1976; permits 13 and 4,962 from Departamento de Pesca). Genomic DNA for 10X Genomics Chromium libraries (average fragment size range 50–120 kb) was extracted from the whole body (excluding gut) using a conventional phenol/chloroform method (Sambrook et al., 1989). The specimens used for RNA-sequencing (used as transcriptomic evidence for genome annotation) included: (1) a snap-frozen (gut removed) field-collected male Poeciliopsis sonoriensis ID MVH99-3, lower Yaqui, Sonora, Mexico, 1999; permit 020299-213-03 from SEMARNAP; and (2) snap-frozen individual tissue samples provided from the captive stock populations for P. occidentalis s.s. from Cienega Creek housed at Arizona State University. Total RNA (RIN > 7) was extracted from the P. sonoriensis individual with TRIzol Reagent (Thermo Fisher Scientific, Waltham, USA) according to the supplier's recommendation; and from the P. occidentalis s.s. tissues using the Qiagen RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA).
Libraries Construction and Sequencing
High molecular weight (HMW) genomic DNA was quantified using microfluorimetry (Qubit High sensitivity dsDNA kit, Invitrogen, Carlsbad, CA) and diluted to 0.8 ng/μl. After denaturation, diluted single stranded DNA was processed using the Chromium Genome Library Kit & Gel Bead Kit v2 and Chromium Controller and Next GEM Accessory Kit (10x Genomics, Pleasanton, California) following the manufacturer's instructions. Briefly, high molecular weight DNA in a master mix was combined with a library of Genome Gel Beads and partitioning oil to create Gel Bead-In-EMulsions (GEMs) in a microfluidic Genome Chip on the Chromium Controller. Emulsion was then recovered from the microfluidic chip and underwent an isothermal incubation. This isothermal incubation leads to the dissolution of the Genome Gel Bead, releasing primers containing an Illumina R1 sequence (Read 1 sequencing primer), a 16 bp 10x Barcode (specific to each Genome Gel Bead), and a 6 bp random primer sequence. Those 6 bp random primer sequences hybridize on the HMW DNA and the isothermal incubation produces barcoded fragments ranging from a few to several hundred base pairs. After incubation, the GEMs were broken and the pooled fractions were recovered. The pool of barcoded DNA fragments was repaired and adenylated on their 3′ ends. 10x Genomics adapters were ligated to the ends of each fragment. The ligated fragments underwent an 8-cycle PCR, which enabled the indexing of the library. The final library was verified on a fragment analyzer and quantified by qPCR (Light Cycler 480, Roche Applied Science, Penzberg, Germany). The library was sequenced on a full paired end 2 × 150 nt lane on a Hiseq2500 (Illumina, San Diego, CA, USA) for a total of 235 million sequences.
The RNA library for P. sonoriensis was prepared with the KAPA mRNA HyperPrep Kit (KAPABiosystems, Wilmington, MA, USA) following the manufacturer's instructions at the Bauer core at Harvard University. One microgram of total RNA, quantified with the 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), was used as input. The library was amplified for 10 cycles and purified using KAPA Pure Beads (KAPABiosystems). Library size distribution and quality were examined with Agilent High Sensitivity D1000 ScreenTape assay. Libraries were sequenced at the Harvard Bauer Core on a NextSeq 500 machine (Illumina) to collect paired-end 75 bp reads. The RNA libraries for all P. occidentalis s.s. samples were prepared by removing rRNA with the Epicenter Ribo-Zero rRNA removal kit (Illumina), and processed into a sequencing library using standard library prep methods for ABI SOLiD sequencing using the Total RNA-Seq Kit (ThermoFisher Scientific, Waltham, MA, USA). Libraries were then sequenced at the University of Connecticut Center for Genome Innovation on a SOLiD 5500xl (ThermoFisher Scientific) to collect paired-end 60-bp reads.
Assembly
The linked reads were assembled with Supernova (Weisenfeld et al., 2017) version 1.0 (“supernova1_complete”) and twice with version 2.0.0, the first with all reads (“supernova2_complete”) and the second with the –maxreads parameter set to use only reads corresponding to a 56X genome coverage (“supernova2_reduced”), following the software best practices. The assembly metrics were calculated using the assemblathon_stats.pl script (Bradnam, 2012). To obtain k-mer spectrum graphs (shown in Supporting Figure S1), k-mers were counted with Jellyfish mer counter v.2.1.1 (Marcais and Kingsford, 2011; parameters: count -C -m 21 -s 100 M). The K-mer Analysis Toolkit v.2.4.1 (KAT; Mapleson et al., 2017) was used to compare k-mers between raw reads and assembly (kat comp; parameters: -m 21) and to draw plots (kat plot spectra-cn; parameters: -w 30 -l 20 -× 100 –dpi 300). The assembly gene content was assessed with BUSCO version 3.0.2 (Waterhouse et al., 2017) using actinopterygii_odb9 as reference data set.
The three P. occidentalis genome assemblies gave different statistics (Supporting Table S1). Depending on the software version and method the P. occidentalis genome assembly was comprised of 7,753, 7,444, and 15,410 scaffolds corresponding to 19,800, 15,621, and 23,570 contigs having a scaffold N50 of 2.77, 0.99, and 1.54 Mb, and scaffold L50 of 60, 178, and 103, respectively. The total assembly lengths are 613, 608, and 725 Mb; i.e., within the range of the Feulgen densitometry estimated size of 680 Mb for this species (Cimino, 1974). The unknown base assembly fraction is low, corresponding to 3.26, 1.52, and 1.62% of the scaffold lengths, respectively. Because of the closest length to genome size estimation, the supernova2_reduced assembly (i.e., 725 Mb) was chosen for annotation.
The three assemblies harbor 94, 91.5, and 92% of complete BUSCO genes, respectively (Supporting Table S1). The fragmented genes correspond to 5.6, 4.0, and 5.2% and the missing genes to 2.9, 2.0, and 2.8%, respectively. This validation was performed using a large set that included 4,584 Actinopterygii genes.
The assembly was compared to the chromosomal-scale assembly of the Southern platyfish, Xiphophorus maculatus (NCBI GCF_002775205.1), a phylogenetically related species, using D-GENIES (Cabanettes and Klopp, 2018). The dot plot between the P. occidentalis and Xiphophorus maculatus assemblies (Supporting Figure S2) shows a good correspondence between long scaffolds and chromosomes. Only 12.1% of the Gila topminnow assembly did not align to the Southern platyfish chromosomes. The alignment identities are distributed as follows: 17.14% above zero and lower than 25%; 70.39% between 25 and 50%; 0.36% between 50 and 75%; and 0.01% over 75%.
Annotation
Gene prediction was achieved with a pipeline that collects and synthesizes evidence from genes and intergenic regions, repeat identification, homology annotation, RNA-seq read mapping, and de novo gene prediction. For each homology annotation, transcript evidence and ab-initio gene model annotation, two independent threads were run. Before starting the annotation process, we used the Actinopterygii odb9 database of BUSCO (Simao et al., 2015) to train AUGUSTUS 3.2.3(Stanke et al., 2006).
Repeat elements were first identified de novo using RepeatModeler (Smit et al., 2013-2015) (http://www.repeatmasker.org/). The result, along with a fish-specific repeat database (unpublished) and the database from Shao et al. (2018), was used as a custom library for RepeatMasker to identify repeats in a comprehensive way. The repeats from known families were masked (replaced with N) from the genome assembly, and their location was collected as intergenic evidence.
Next we collected gene evidence from homology alignments. Proteins from Swiss-Prot (www.uniprot.org) and 13 Ensembl genomes (version 95, http://www.ensembl.org): human (Homo sapiens), mouse (Mus musculus), coelacanth (Latimeria chalumnae), spotted gar (Lepisosteus oculatus), zebrafish (Danio rerio), cod (Gadus morhua), tilapia (Oreochromis niloticus), medaka (Oryzias latipes), platyfish (Xiphophorus maculatus), fugu (Takifugu rubripes), tetraodon (Tetraodon nigrovirdis), stickleback (Gasterosteus aculeatus), and sea lamprey (P. marinus) were collected and processed using CD-HIT to form 544,476 non-redundant proteins. They were aligned to the genome assembly with exonerate2.2.0 (Slater and Birney, 2005) https://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate and Genewise2-2-0 (Birney et al., 2004) independently.
To collect gene evidence from RNA-seq data, we used two independent threads: one with Tophat to map reads and Cufflinks 2.1.1 (Trapnell et al., 2012) to form gene models; the other one with HISAT2 2.1.0 (Kim et al., 2015) and Trinity 2.4.0 to assemble transcripts guided by the genome, and PASA 2.2.0 to align transcripts and form gene models (Haas et al., 2003, 2013).
For de novo annotation, we used SNAP 2006-07-28 (Korf, 2004) (http://korflab.ucdavis.edu) and GeneMark-ES (Ter-Hovhannisyan et al., 2008) independently. Confirmed by all lines of evidence described above, a group of high-quality gene models were selected using EVidenceModeler1.1.1 (Haas et al., 2008). They were used to train AUGUSTUS again. Then the species-specifically trained AUGUSTUS was run to predict genes in the assembly, taking as hint all those intergenic and gene evidences collected above. Finally, low-quality genes, which had low confidence score and no BLAST hit to Swiss-Prot (www.uniprot.org), were removed.
The repeat elements, identified using RepeatModeler and RepeatMasker, account for 19.98% (144 Mbp) of the assembly. DNA elements account for 44.69%; SINE, 0.95%; LINE, 11.36%; and LTR elements for 4.10% of the repetitive fraction of the genome (Figure 1). The majority of the represented TE landscape is composed of DNA elements. This makes it distinct from more ancient fishes. In the elephant shark (Callorhinchus milii), TEs are mostly composed of SINE and LINE elements. The Gila topminnow genome shares with other poeciliid genomes (i.e., southern platyfish, guppy, and Amazon molly) an ancient wave of transposable element (TE) expansion. Nonetheless, the more recent TE expansion that is typically seen in poeciliids appears to have started relatively more recently in the Gila topminnow, where its peak in the most recent elements implies that this expansion is probably ongoing. Detailed studies on TE expression and activity in the Gila topminnow will yield insights into how TEs shape the evolution of their host genomes.
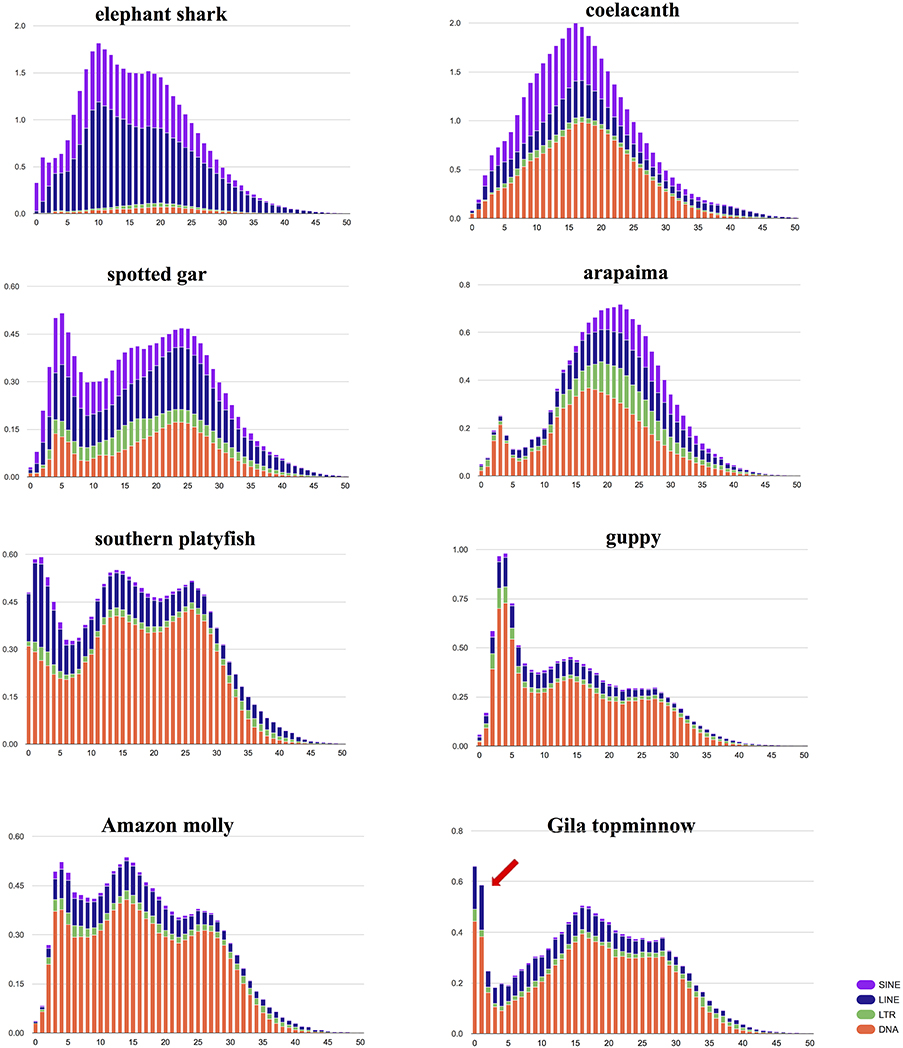
Figure 1. Interspersed repeat landscape. The interspersed repeat landscape, revealing the copy-divergence analysis of Transposable Elements (TE) classes, based on Kimura distances. Percentages of the in genome represented by TEs (Y-axis) are clustered based on their Kimura values (X-axis; K-values from 0 to 50; arbitrary values). Older copies are located on the right side of the graphs while rather recent copies are located on the left side. Arrow indicates the very recent (possibly ongoing) expansion of TE in the Gila topminnow.
In total, 41,501 genes were predicted of which 10,625 were removed because they were deemed of low-quality [i.e., no hit in BUSCO, Swissprot, and Pfam database, failing to present an intact structure (both start and stop codon predicted) and with Augustus score <80). Among the 30,976 retained genes, 27,947 (90.22%) were annotated with start and stop codons, 28,141 (90.84%) have BLAST hit to database Swiss-Prot (www.uniprot.org), 24,912 (80.42%) were suggested by InterProScan (http://www.ebi.ac.uk/interpro/interproscan.html) to contain functional protein domains, and 28,722 (92.72%) were supported by RNA-seq reads. The mean coding sequence length in the retained genes is 1,463 bp and the longest is 54,050 bp. A quality assessment by BUSCO analysis revealed 95.4% complete conserved Actinopterygii genes, 3.6% fragmented and only 1.0% missing genes (Supporting Table S1). The annotation process thus increased the quality of all BUSCO parameters evaluated in this assembly.
Orthology Assignment
Orthology relationships between genes of P. occidentalis and other fish were inferred based on sequence similarity and species phylogeny. First, the annotated genomes of Poecilia formosa (Amazon molly), Xiphophorus maculatus (Platyfish), Gambusia affinis (Western mosquitofish), Fundulus heteroclitus (Mummichog), Kryptolebias marmoratus (Mangrove rivulus), Oryzias latipes (Japanese medaka), Takifugu rubripes (Fugu), Gasterosteus aculeatus (Stickleback), and Lepisosteus oculatus (Spotted gar) were downloaded from Ensemble (http://www.ensembl.org/info/data/ftp/index.html), and of Poecilia reticulata (Guppy) from NCBI (https://www.ncbi.nlm.nih.gov/genome/?term=Poecilia%20reticulata). Second, an all-vs.-all blast was performed among the protein sequences of these fish and P. occidentalis (Camacho et al., 2009). Based on the BLAST raw score, H-score, defined as score (Gene1Gene2)/max [score (Gene1Gene1), score (Gene2Gene2)] (Cho et al., 2013), was calculated to evaluate the sequence similarity between any of two genes. Then with H-scores and L. oculatus set as the outgroup, genes were clustered into groups using Hcluster_sg (Ruan et al., 2007). Third, for each group, a gene tree was reconstructed in the guild of species tree using TreeBeST (Ponting, 2007). Finally, according to the tree, genes were assigned as “XtoX” orthologs to each other (X refers to a positive integer).
We collected 251,212 genes from the genomes listed above and clustered them into 20,823 groups based on sequence similarity. Among them 11,639 were shared by P. occidentalis, T. rubripes, X. maculatus, G. aculeatus, and O. latipes (Supporting Figure S3). Five thousand two hundred seventy-six groups contained only one gene from each of the fish. These genes were identified as one-to-one orthologs and were used to construct the phylogenomic tree using RaxML (Figure 2), as follows. One-to-one orthologs were identified during the orthology assignment after genes were clustered into groups. Protein sequences of each ortholog were then aligned among species using MAFFT (Nakamura et al., 2018). Alignment regions with bad quality were trimmed using trimAI (Capella-Gutiérrez et al., 2009). After trimming, the alignments of orthologs were concatenated into a massive alignment. Based on the massive alignment and with L. oculatus set as the outgroup, RaxML was used to reconstruct the phylogeny (Stamatakis, 2014). Clade support was assessed by means of a bootstrap analysis (100 replicates). Inferred relationships among all taxa were as expected (e.g., Betancur et al., 2013; Reznick et al., 2017; Bragança et al., 2018). As observed in Figure 2, five genes are absent from all Poeciliid branches and thus likely lost in their common ancestor (Solute carrier family 27 member 6; zgc:55888; Leucine-rich repeat neuronal protein 1-like; TGF-beta receptor type-2-like; the other one not characterized). Two genes (both not characterized; “InPoecil” Figure 2) are present in all poeciliid branches and absent in all others; thus, likely gained in the common ancestor of poeciliids.
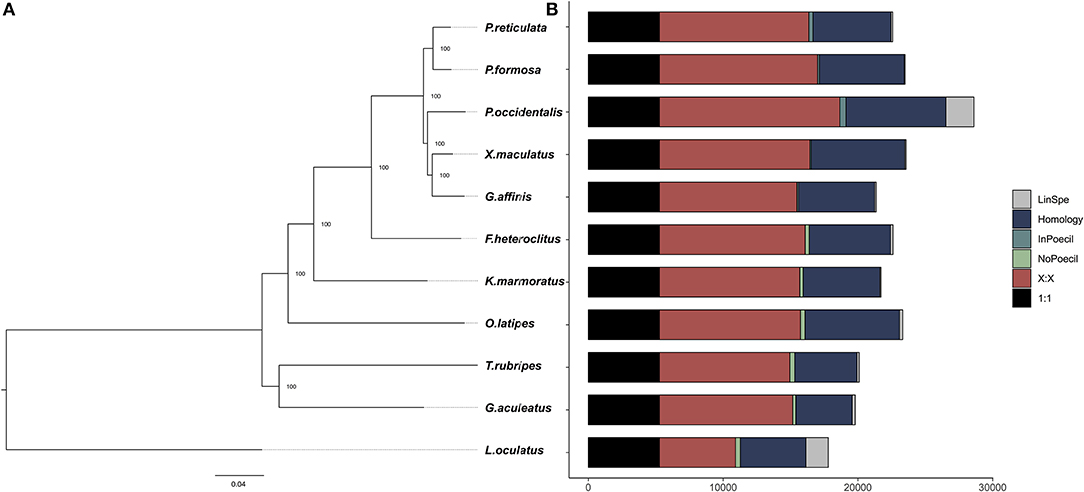
Figure 2. Phylogenetic relationships and gene repertoire of P. occidentalis with other fish species. (A) A phylogenetic tree reconstructed using RaxML based on 5,276 one-to-one orthologs, the numbers on the nodes refer to the support value calculated from 100 bootstraps. (B) A bar chart revealing the percentage of orthologous genes of different types. “1:1” refers to universal single-copy genes; “X:X” orthologs exist in all species but not always as single copy; “NoPoecil,” orthologs exist in none of the Poeciliid branches (Poecilia reticulata, Poecilia formosa, P. occidentalis, Xiphophorus maculatus, and Gambusia affinis) but in at least two non-Poeciliid branches; “InTeleo,” orthologs exist in at least two Poeciliids but none of non-Poeciliid branches; “Homology,” orthologs exist in both Poeciliids and non-Poeciliids but are missing from at least one branch; “LinSpe,” lineage-specific genes where no ortholog was found in other branches.
Assembly and Annotation of Mitochondrial Genomes From P. occidentalis and P. sonoriensis
A BLAST search using the cytochrome b sequence of P. occidentalis was used to search among the assembled contigs to identify the mitochondrial genome. A contig 16,912 bp long was recovered and annotated using the MitoFish webserver (Iwasaki et al., 2013; Sato et al., 2018). The ND2 gene, which spanned the ends of the contig, was corrected with Sanger sequences obtained from PCR products of the same specimen, leading to a shorter contig of 16,772 bp, which was circularized and annotated (NCBI Acc. No. MK860198) based on the P. occidentalis mitochondrial genome available at NCBI (Acc. No. KP013108). We also assembled the entire mitochondrial genome of P. sonoriensis (Acc. No. MK860197) by mapping the RNAseq reads from this species to the mitochondrial genome of P. occidentalis. Comparison of the three mitochondrial genomes revealed 0.23% divergence (uncorrected p) between the two samples of P. occidentalis s.s., and 0.94–0.99% divergence between P. occidentalis s.s. and P. sonoriensis.
Conclusion
We confirm the utility of 10X Genomics technology and the Supernova assembler to generate an assembly with high contiguity and high quality (e.g., Ozerov et al., 2018 and references therein). Our results demonstrate the utility of long-term frozen material for this purpose. The scaffold N50 above 1 Mb that we obtained is in the range of the best assemblies based exclusively on Illumina (i.e., short reads), but only those that have employed jumping libraries (also known as long-insert paired-end reads or mate pair libraries) (e.g., Liu et al., 2017; Schartl et al., 2019). As the first published genome assembly for the genus Poeciliopsis, we expect it will serve as a valuable resource for research in phylogenomics, enabling the generation of a robust framework for macroevolutionary questions, including speciation, hybridization, and adaptation. Furthermore, this resource is expected to facilitate research aimed at taxonomic delimitation and conservation genetics of this endangered taxon.
Data Availability Statement
The genomic and transcriptomic raw reads have been deposited at NCBI under BioProject PRJNA532900, BioSamples SAMN11418710, SAMN11418711. The genome assembly has been deposited at NCBI under Accession No. SZYC00000000. The annotation files have been deposited at GitHub (https://www.github.com/marianamateos/Poeciliopsis-occidentalis-draft-genome-version1). The assembled and annotated mitochondrial genomes are available under NCBI Acc. Nos. MK860197 and MK860198.
Ethics Statement
The animal study was reviewed and approved by the University of Connecticut's Institutional Animal Care and Use Committee for a subset of the specimens. The other specimens have been frozen for 18–20 years at institutions that did not have an animal ethics committee at the time.
Author Contributions
MM and MScha designed the study, supervised all steps of the project, analyzed the data, and drafted the manuscript. MM provided the biological material. HP performed the genome sequencing. CK assembled the genome. YG and MScha analyzed the genome data. DK did the genome annotation. MG-O and MScha isolated and processed the HMW DNA and the RNA. MSchu performed the RNA-seq from the whole fish. NJ provided RNA-seq read data from ovary and embryo. All authors were involved in the manuscript writing and editing.
Funding
This work was supported by NSF DEB 9902224 to R. C. Vrijenhoek and MM. MScha was supported by the Hagler Institute of Advanced Study of Texas A&M University and funds provided by the Department of Biology of Texas A&M University and the Biocenter, Chair Physiologische Chemie, University of Würzburg. MG-O was supported by a Heep fellowship. HP acknowledges financial support from the France Génomique National infrastructure, funded as part of Investissement d'Avenir program managed by Agence Nationale de la Recherche (contract ANR-10-INBS-09). NSF Project Grant IOS-0920088 to M.J. O'Neill and R.J. O'Neill, NSF-MRI 0821466 to Linda Strausbaugh and R.J. O'Neill, and NSF-MRI-R2 0959365 to M.J. O'Neill and R.J. O'Neill. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Robert C. Vrijenhoek and Paul Marsh provided biological material. Michael O'Neill, Rachel O'Neill, and University of Connecticut's Center for Genome Innovation (formerly the Center for Applied Genetics) within the Institute for Systems Genomics for supporting ABI SOLiD RNA-seq sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00404/full#supplementary-material
References
Betancur, R. R., Broughton, R. E., Wiley, E. O., Carpenter, K., Lopez, J. A., Li, C., et al. (2013). The tree of life and a new classification of bony fishes. PLoS Curr 5:ecurrents.tol.53ba26640df0ccaee75bb165c8c26288. doi: 10.1371/currents.tol.53ba26640df0ccaee75bb165c8c26288
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Bradnam, K. (2012). Assemblathon_stats.pl. Available online at: https://github.com/lexnederbragt/sequencetools/blob/master/assemblathon_stats.pl
Bragança, P. H., Amorim, P. F., and Costa, W. J. (2018). Pantanodontidae (Teleostei, Cyprinodontiformes), the sister group to all other cyprinodontoid killifishes as inferred by molecular data. Zoosystem. Evol. 94:137. doi: 10.3897/zse.94.22173
Cabanettes, F., and Klopp, C. (2018). D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6:e4958. doi: 10.7717/peerj.4958
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Cho, Y. S., Hu, L., Hou, H., Lee, H., Xu, J., Kwon, S., et al. (2013). The tiger genome and comparative analysis with lion and snow leopard genomes. Nat. Commun. 4:2433. doi: 10.1038/ncomms3433
Cimino, M. C. (1974). The nuclear DNA content of diploid and triploid P and other poeciliid fishes with reference to the evolution of unisexual forms. Chromosoma 47, 297–307. doi: 10.1007/BF00328863
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K. Jr., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Hedrick, P. W., and Hurt, C. R. (2012). Conservation genetics and evolution in an endangered species: research in Sonoran topminnows. Evol. Appl. 5, 806–819. doi: 10.1111/j.1752-4571.2012.00259.x
Iwasaki, W., Fukunaga, T., Isagozawa, R., Yamada, K., Maeda, Y., Satoh, T. P., et al. (2013). MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 30, 2531–2540. doi: 10.1093/molbev/mst141
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Liu, H., Chen, C., Gao, Z., Min, J., Gu, Y., Jian, J., et al. (2017). The draft genome of blunt snout bream (Megalobrama amblycephala) reveals the development of intermuscular bone and adaptation to herbivorous diet. Gigascience 6, 1–13. doi: 10.1093/gigascience/gix039
Mapleson, D., Garcia Accinelli, G., Kettleborough, G., Wright, J., and Clavijo, B. J. (2017). KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576. doi: 10.1101/064733
Marcais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Mateos, M., Domínguez-Domínguez, O., and Varela-Romero, A. (2019). A multilocus phylogeny of the fish genus Poeciliopsis: solving taxonomic uncertainties and preliminary evidence of reticulation. Ecol. Evol. 9, 1845–1857. doi: 10.1002/ece3.4874
Miller, R. R., Minckley, W. L., and Norris, S. M. (2005). Freshwater Fishes of México. Chicago, IL: The University of Chicago Press.
Nakamura, T., Yamada, K. D., Tomii, K., and Katoh, K. (2018). Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492. doi: 10.1093/bioinformatics/bty121
Ozerov, M. Y., Ahmad, F., Gross, R., Pukk, L., Kahar, S., Kisand, V., et al. (2018). Highly continuous genome assembly of Eurasian Perch (Perca fluviatilis) using linked-read sequencing. G3 (Bethesda) 8, 3737–3743. doi: 10.1534/g3.118.200768
Ponting, C. (2007). TreeBeST: Tree building guided by Species Tree. Available online at: https://sourceforge.net/projects/treesoft/files/treebest/
Quattro, J. M., Avise, J. C., and Vrijenhoek, R. C. (1992). An ancient clonal lineage in the fish genus Poeciliopsis (Atheriniformes: Poeciliidae). Proc. Natl. Acad. Sci. U.S.A. 89, 348–352. doi: 10.1073/pnas.89.1.348
Reznick, D. N., Furness, A. I., Meredith, R. W., and Springer, M. S. (2017). The origin and biogeographic diversification of fishes in the family Poeciliidae. PLoS ONE 12:e0172546. doi: 10.1371/journal.pone.0172546
Reznick, D. N., Mateos, M., and Springer, M. S. (2002). Independent origins and rapid evolution of the placenta in the fish genus Poecil. Sci. 298, 1018–1020. doi: 10.1126/science.1076018
Ruan, J., Li, H., Chen, Z., Coghlan, A., Coin, L. J. M., Guo, Y., et al. (2007). TreeFam: 2008 update. Nucleic Acids Res. 36, D735–D740. doi: 10.1093/nar/gkm1005
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning, A laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Sato, Y., Miya, M., Fukunaga, T., Sado, T., and Iwasaki, W. (2018). MitoFish and MiFish pipeline: a mitochondrial genome database of fish with an analysis pipeline for environmental DNA metabarcoding. Mol. Biol. Evol. 35, 1553–1555. doi: 10.1093/molbev/msy074
Schartl, M., Kneitz, S., Volkoff, H., Adolfi, M., Schmidt, C., Fischer, P., et al. (2019). The piranha draft genome provides molecular insight associated to its unique feeding behavior. Genome Biol. Evol. 11, 2099–2106. doi: 10.1093/gbe/evz139
Shao, F., Wang, J., Xu, H., and Peng, Z. (2018). FishTEDB: a collective database of transposable elements identified in the complete genomes of fish. Database 2018:bax106. doi: 10.1093/database/bax106
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Slater, G. S., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. doi: 10.1186/1471-2105-6-31
Smit, A. F. A., Hubley, R., and Green, P. (2013-2015). RepeatMasker Open-4.0. Available online at: http://www.repeatmasker.org/
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–439. doi: 10.1093/nar/gkl200
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Vrijenhoek, R. C., Douglas, M. E., and Meffe, G. K. (1985). Conservation genetics of endangered fish populations in Arizona. Science 229, 400–402. doi: 10.1126/science.229.4711.400
Waterhouse, R. M., Seppey, M., Simao, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2017). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1101/177485
Keywords: genome assembly, genome annotation, transposable elements, topminnow, mitochondrial genome
Citation: Mateos M, Kang D, Klopp C, Parrinello H, García-Olazábal M, Schumer M, Jue NK, Guiguen Y and Schartl M (2019) Draft Genome Assembly and Annotation of the Gila Topminnow Poeciliopsis occidentalis. Front. Ecol. Evol. 7:404. doi: 10.3389/fevo.2019.00404
Received: 22 June 2019; Accepted: 09 October 2019;
Published: 24 October 2019.
Edited by:
Guillermo Orti, George Washington University, United StatesReviewed by:
Federico Guillermo Hoffmann, Mississippi State University, United StatesKlaus-Peter Koepfli, Smithsonian Conservation Biology Institute (SI), United States
Copyright © 2019 Mateos, Kang, Klopp, Parrinello, García-Olazábal, Schumer, Jue, Guiguen and Schartl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana Mateos, bW1hdGVvc0B0YW11LmVkdQ==; Manfred Schartl, cGhjaDFAYmlvemVudHJ1bS51bmktd3VlcnpidXJnLmRl