Commentary: Thiamine Deficiency in Tropical Pediatrics: New Insights into a Neglected but Vital Metabolic Challenge
 Laurent Hiffler1*
Laurent Hiffler1* Daniel Martinez Garcia
Daniel Martinez Garcia- 1Dakar Unit, Medical Department, Médecins Sans Frontières (MSF), Dakar, Senegal
- 2Unit of Nutrition Physiology, University of Antananarivo, Antananarivo, Madagascar
- 3Pediatric Team, Medical Department, Médecins Sans Frontières (MSF), Barcelona, Spain
In humans, thiamine is a micronutrient prone to depletion that may result in severe clinical abnormalities. This narrative review summarizes current knowledge on thiamine deficiency (TD) and bridges the gap between pathophysiology and clinical presentation by integrating thiamine metabolism at subcellular level with its function to vital organs. The broad clinical spectrum of TD is outlined, with emphasis on conditions encountered in tropical pediatric practice. In particular, TD is associated with type B lactic acidosis and classic forms of beriberi in children, but it is often unrecognized. Other severe acute conditions are associated with hypermetabolism, inducing a functional TD. The crucial role of thiamine in infant cognitive development is also highlighted in this review, along with analysis of the potential impact of TD in refeeding syndrome during severe acute malnutrition (SAM). This review aims to increase clinical awareness of TD in tropical settings where access to diagnostic tests is poor, and advocates for an early therapeutic thiamine challenge in resource-limited settings. Moreover, it provides evidence for thiamine as treatment in critical conditions requiring metabolic resuscitation, and gives rationale to the consideration of increased thiamine supplementation in therapeutic foods for malnourished children.
Introduction
Thiamine (Vitamin B1) is an essential micronutrient with dual coenzymatic and non-coenzymatic functions. It is involved in carbohydrate and branched-chain amino acid metabolism, as well as in the production of neurotransmitters, myelin, and nucleic acids. There is also evidence that thiamine plays a role in immune and anti-inflammatory processes and gene regulation (1–4).
As an essential micronutrient, the body’s requirements are exclusively dependent on dietary supply as there is no endogenous synthesis. The combination of limited body storage and a high turnover rate (half-life <10 days) results in potential depletion of thiamine stores within 2 weeks if it is not continuously replaced. In addition, thiamine’s hydrosolubility, coupled with its renal clearance profile, contributes to a propensity to thiamine deficiency (TD) throughout life.
In pediatrics, the overall clinical picture of TD is not easy to recognize, mimicking or being confused with other diseases. Not surprisingly, the likelihood of misdiagnosis of TD is even greater in resource-limited settings (5–7). Despite being easily treatable, TD continues to be seen in all age groups in both high and low resource countries with potentially severe and life-threatening consequences (8–12).
This article focuses initially on thiamine physiology from ingestion in the diet to uptake and metabolism at mitochondrion level and the circumstances leading to TD in tropical pediatrics. By highlighting the essential role of thiamine in various body functions, this article enables us to better understand the spectrum of clinical presentations of TD in children, including the various manifestations of beriberi, refeeding syndrome in malnutrition, severe clinical states requiring resuscitation, and conditions linked to anti-thiamine factors. A brief overview of diagnosis and treatment of TD concludes the paper and gives suggestions for the way forward in the management of TD.
Thiamine Metabolism: A Physiological Snapshot of Thiamine in Humans
Ingested dietary thiamine is essential to meet body requirements in humans who are unable to synthesize it naturally. The daily requirement of thiamine depends on age, body weight, physiological conditions, and individual metabolism with respect to the overall energy content in the diet.
Unlike for adults, recommendations of the optimal thiamine-caloric ratio (mg/1000 kcal/day of energy consumed) are not yet firmly established in pediatrics (9, 13). In children, whose caloric needs are age dependent, the estimated daily recommended dietary allowance (RDA) is 0.2 mg up to 6 months of age, 0.3 mg from 7 months to 3 years of age, and 0.6 mg between 4 and 8 years of age. Over 8 years, daily RDA varies from 0.9 mg up to 1.2 mg.
The content of thiamine and its derivatives in breast milk is around 0.21 mg/l, but varies widely according to diet and countries. Nevertheless, thiamine-deficient breast milk is not uncommon and can have a serious effect on infant thiamine status during exclusive breastfeeding (14, 15).
Body Thiamine Distribution
After ingestion, free thiamine absorption occurs mainly in the proximal small intestine either via a saturable carrier-mediated system for low thiamine concentrations, or by passive diffusion for high concentrations. Inside the polarized enterocyte, the vectorial transport of thiamine is through specific human thiamine transporters-hTHTRs (ThTr1 and 2) (16–18), after which free thiamine disperses throughout the cell. Genetic defects in ThTr1 (SLC19A2) and ThTr2 (SLC19A3) result in thiamine-responsive megaloblastic anemia (TRMA) and biotin-responsive basal ganglia disease (BBGD), respectively (2, 19, 20).
Intracellular-free thiamine is phosphorylated into thiamine monophosphate (ThMP), then thiamine diphosphate (ThDP) [also known as thiamine pyrophosphate (ThPP)], and finally thiamine triphosphate (ThTP). In the enterocyte, thiamine is phosphorylated to ThDP by thiamine pyrophosphokinase (TPK), before being transported from the cell apex toward the opposite pole. Most of this ThDP is then hydrolyzed to cross the basolateral membrane using an adenosine triphosphate (ATP)-dependent transporter. From here, thiamine gets into the portal blood to reach the liver and tissues via different pathways. Besides the classic hTHTRs, other carriers include alkaline/acid phosphatase transport and the organic cation transporter groups (OCT1), acting as hydrosoluble organic cations (1, 16–18, 21). Thiamine subsequently joins the systemic bloodstream either as a free form or incorporated in circulating cells, mainly erythrocytes (Figure 1A). It targets body cells that utilize glucose as their main fuel; however, thiamine tissue tropism depends on the degree of expression of key transporters on cell membranes in the major body systems (splanchnic, nervous, muscular and renal systems, and the placenta) (1, 2, 16–18, 21, 22).
FIGURE 1
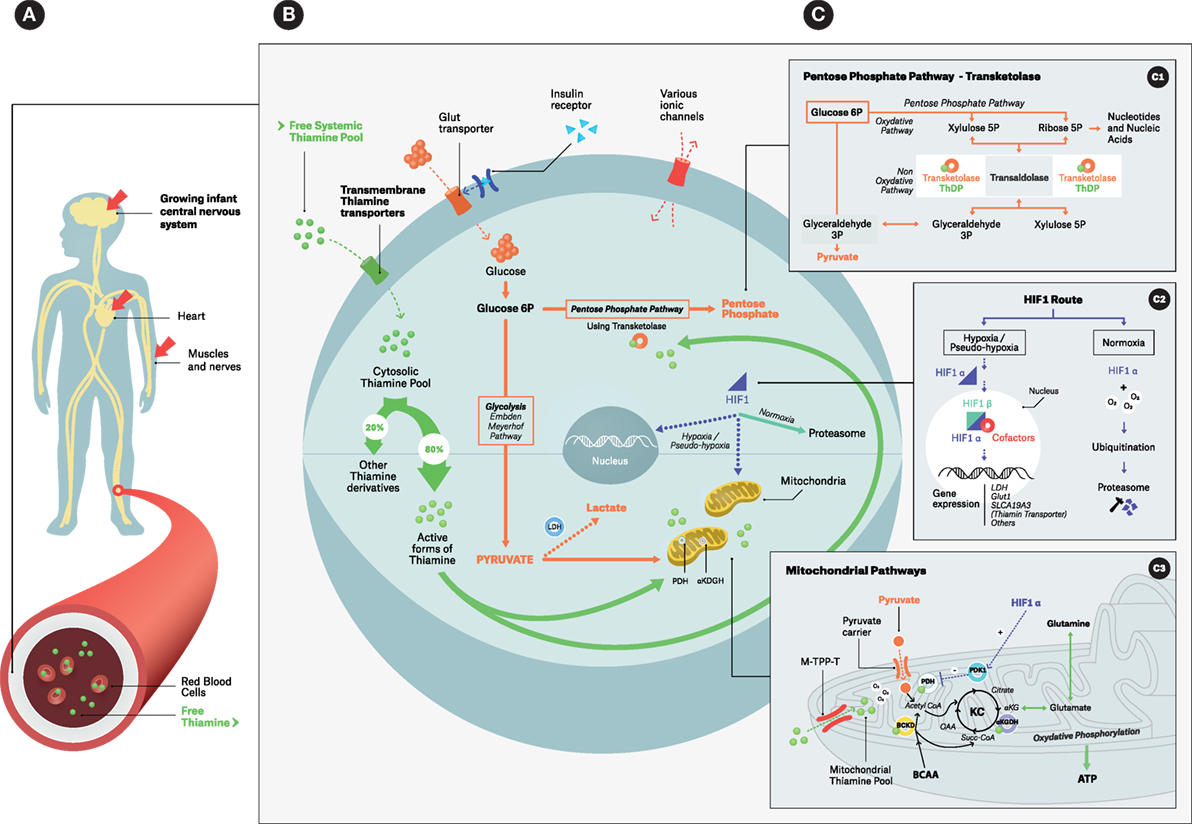
Figure 1. Simplified overview of thiamine intracellular action: focus on coenzymatic functions. (A) Infant thiamine distribution with its three main target organs; (B) thiamine cellular metabolism with its main pools and thiamine-related metabolic pathways; (C) zooming boxes: (C1) – cytosolic pentose phosphate pathways using transketolase, (C2) – HIF-1 alternative routes, (C3) – mitochondrial events; ThDP is a cofactor for PDH, α-KGDH and BCKD. Under hypoxia/pseudo-hypoxia HIF1-α upregulates PDK1 that in turn downregulates PDH. Abbreviations: BCKD, branched-chain ketoacid dehydrogenase complex; LDH, lactic dehydrogenase leading to Type B acidosis; PDH, pyruvate dehydrogenase complex; PDK1, pyruvate dehydrogenase kinase; α-KGDH, alpha-ketoglutarate dehydrogenase. ATP, adenosine triphosphate; BCAA, branched-chain amino acids; Glut transporter, glucose transporter; HIF, hypoxia-inducible factor, as a dimeric (α, β unit) transcriptional factor; KC, Krebs cycle; M-TPP-T, mitochondrial thiamine pyrophosphate transporter; OAA, oxaloacetate; Succ-CoA, succinyl-coenzyme A; ThDP (or TPP), thiamine diphosphate; α-KG: α-ketoglutarate. (B) Intracellular dotted arrows: alternative pathways for pyruvate–lactate axis and for HIF-1 during Hypoxia/pseudo-hypoxia (e.g., TD).
In the splanchnic system, the liver and the pancreas are the two most important targets (21, 23). Recent evidence indicates that human colonic epithelial cells express specific ThDP transporters (SCL44A4) able to uptake thiamine, which is a local by-product of gut microbiota in infancy (Prevotella enterotype 2). It is not yet fully understood whether this microbiome-related thiamine pool is solely used in situ or contributes to whole body thiamine homeostasis (24, 25).
In the nervous system, central neurons – known for their high oxidative metabolism – astrocytes, and peripheral nerves are involved in the biosynthesis of a number of neurotransmitters, including acetylcholine (26, 27). Evidence suggests that the brain appears to be the ultimate target of thiamine (1, 2, 28, 29). Thiamine crosses the blood–brain barrier via carrier proteins to reach select regions of the brain, but the exact pathophysiology of this tropism is poorly understood (30–32). There are key differences in the distribution of thiamine derivatives throughout the brain, providing some insight into thiamine’s apparent preference for certain neuronal tissues. The ThDP form is mainly present in neurons and is involved in cerebral oxidative metabolism. Conversely, the ThTP form, approximately 10% of the cerebral thiamine pool, is involved in membrane excitability and nerve conduction, acting as a modulator of the permeability of sodium chloride channels (29, 30, 33, 34). Similarly, the distribution of thiamine-related enzymes is compartmentalized, especially those with rate-limiting properties, which play a role in cerebral energy utilization. Therefore TD might cause brain tissue injury by inhibiting metabolism in cerebral regions with higher metabolic demands and high thiamine turnover (34–36). The magnitude of the rate of thiamine uptake by the blood–brain barrier highlights the huge cerebral demand for thiamine and the need for its continuous supply for brain activity (22, 30, 32). In vitro and in vivo kinetics reinforce the idea that thiamine turnover is not homogeneous in the brain. It is higher in areas vulnerable to TD, and lower in other structures, such as the cerebral cortex (a relatively less sensitive region) (1, 30, 32, 34, 35). TD neurotropic effects are observed in various histological pathways of the brain, specifically causing disruption to the blood–brain barrier rich in endothelial mitochondria, choroid plexus dysfunction, differential damage in aerobic neurons and anaerobic astrocytes, microglial activation, perturbation in the glutamate system (with its specific transporters), altered dynamics of intracellular calcium and endoplasmic reticulum stress (27, 29, 31, 37–41).
Smooth, skeletal and particularly cardiac myocytes, known for their high aerobic metabolism, are all affected by TD. Their dysfunction might result in the early manifestations of TD, such as gastrointestinal paresis and heart failure (6, 42, 43). In the kidneys, the determinants of thiamine renal clearance are glomerular filtration rate (GFR) and the saturable reabsorptive capacity of proximal tubules through the thiamine/H+ antiport using hTHTRs, similar to polarized enterocytes (2, 18, 20, 44).
Lastly, during pregnancy, thiamine crosses the placental brush-border epithelium through a specific carrier (45, 46). This active transfer is essential for thiamine supply and may have a major impact on fetal growth and development (47).
Intracellular Thiamine Metabolism
As previously outlined, inside the cytosolic compartment, free thiamine is first converted into the phosphorylated metabolites ThMP, ThDP, and ThTP. Adenosine forms have also been identified, e.g., adenosine thiamine triphosphate (AThTP) and diphosphate (AThDP), however their function remains unclear (33).
The synthesis of ThDP from free thiamine requires TPK, ATP, and magnesium, the deficit of which can limit clinical response to thiamine treatment (48, 49). ThDP is the biologically active form in the body (>80%) and serves as the most potent cofactor for thiamine-requiring enzymes (Figure 1B) (34). Most of the ThDP enters the mitochondrial matrix via the mitochondrial thiamine pyrophosphate transporter family (M-ThPP-T) located in its inner membrane (Figure 1C3) (50). Of note, mutation in M-ThPP-T results in Amish lethal microcephaly–neuropathy (50).
Thiamine acts as a crucial cellular cofactor for several metabolic enzymes studied in biochemical models (1, 2), namely the pyruvate dehydrogenase (PDH) multi-enzyme complex responsible for the mitochondrial decarboxylation of pyruvate to acetyl-CoA, the precursor to the Krebs cycle (Figure 1C3) (51), and alpha-ketoglutarate dehydrogenase (α-KGDH), responsible for the mitochondrial oxidative decarboxylation of α-ketoglutarate to succinyl-CoA (Figure 1C3) (36, 52). It also serves as a cofactor of cytosolic transketolase, a rate-limiting enzyme in the pentose monophosphate non-oxidative pathway leading to ribose then to nucleic acid synthesis (Figure 1C1) (53).
Upstream of the Krebs cycle, any functional defect in the PDH complex leads to anaerobic metabolism with an increase in cellular pyruvate and lactate (Figure 1B). This results in a build-up of these substrates in cerebrospinal fluid (CSF) and blood causing an elevation in the plasma anion gap (54–57). A recent report indicates that the consequent detrimental type B lactic acidosis is associated with a pseudo-hypoxia with no actual evidence of poor tissue perfusion or oxygenation. As a consequence, hypoxia-inducible transcription factor 1-alpha (HIF1-α) is increased and upregulates thiamine transporter gene expression [ThTr2 (SLCA19A3)] (Figures 1B,C2,C3) (58). Within the mitochondrial matrix, any earlier failed activities of PDH and α-KGDH can result in reduced ATP synthesis, which can induce an oxidative insult to the cell. Cascade reactions lead to the generation of damaging reactive oxygen species (ROS) and, ultimately, to apoptotic cell death (28, 31, 36, 59, 60). Additionally, in order for the Krebs cycle to function correctly it requires the operative integrity of PDH and α-KGDH enzymes that contribute to the synthesis of myelin and the neurotransmitter acetylcholine. Fully functional α-KGDH also preserves the physiological levels of other substrates (glutamate, gamma-amino butyric acid, and aspartate) (29, 36, 51).
Current research is investigating the role of thiamine in immune system regulation. There is evidence that its non-coenzymatic function is involved in modulation of anti-inflammatory processes and the expression of cytokine mediators (1, 3, 4).
Conditions and Risk Factors Leading to TD in Tropical Pediatrics
Thiamine deficiency global prevalence is poorly documented. It principally affects precarious communities where children are most vulnerable and where dietary habits rely on refined processed cereals or tubers (e.g., rice, wheat, cassava), notably in Southeast Asia (5, 6, 10, 61–64), Africa (65–67) and, the Americas (68). These reports on TD prevalence vary from 13.4% of children admitted to hospital without signs of beriberi in Laos, 30% of Laotian children with uncomplicated malaria, 40% of severely malnourished children from Jamaica and Ghana, and up to 100% of children with respiratory distress and tachycardia in one Indian study.
Thiamine deficiency can result from various mechanisms that are not mutually exclusive. The most frequent mechanisms are insufficiency of dietary thiamine (enteral–parenteral supply) and poor intestinal absorptive capacity during malnutrition, tropical enteropathy, or secondary to surgical resection of large portions of the gastrointestinal tract (11, 69–71). A relative inadequacy of thiamine content to caloric ratio (high carbohydrate diets) is also common. It is this thiamine–calorie imbalance that is responsible for TD in heavy drinkers of sweet drinks (72, 73) and contributes to the onset of TD when dextrose-based fluids are administered without thiamine supplementation in critically ill patients (74).
Poor thiamine intake may be due to losses from food secondary to pre-cooking and food processing (e.g., repetitive rice washing), a restrictive diet due to cultural habits (75) and ingestion of anti-thiamine factors (tea leaves, betel nuts, and coffee) or thiaminases (e.g., fermented raw fish, mycotoxins, stored food, and larvae) that break down and, thus, inactivate thiamine (76, 77). Excessive loss of thiamine from the body, such as renal (loop diuretics, osmotic diabetic diuresis) or digestive losses (chronic diarrhea, hyperemesis), can also precipitate TD. At cellular level, two main mechanisms contribute to TD – impaired uptake or increased demand. Impaired cellular uptake of thiamine can be due to defects in thiamine transporters or specific enzymes (mutation, hypomagnesemia, drug-induced interaction), or reduced levels of ThDP. Increased cellular demand of thiamine occurs in hypermetabolic states during critical illness, e.g., severe infections, shock, burns, fever, hyperthyroidism.
Exposure to toxic substances, such as alcohol can also affect thiamine metabolism. When this occurs during pregnancy, the fetus may be indirectly affected, as observed in fetal alcohol syndrome (45–47, 78). Maternal TD associated with excessive alcohol consumption is usually secondary to inadequate thiamine intake or decreased intestinal absorption due to reduced expression of ThTr-1. In addition, impaired intracellular thiamine utilization affecting the TPK activity results in a low bioavailability of the active cellular ThDP due to ethanol and acetaldehyde exposure (2, 79–81).
In humanitarian fields, TD occurring in epidemic proportions has been described following abrupt food shortage caused by disasters, famines, conflicts, or large population displacement (66, 67, 76, 77, 82). In tropical pediatrics, the main cause of TD is poor thiamine intake, either by the child or by the mother for young infants. Associated co-morbidities are common and increase TD risk, for example, sepsis and shock during complicated severe acute malnutrition (SAM) are frequent and lead to an increase in mortality (83). Poor absorption, increased digestive losses and systemic inflammation can be aggravating cofactors in critically ill children. Additionally, pediatric intensive care practices in many resource-limited settings do not include thiamine as a component of treatment, increasing TD risk even further. In emergency and tropical contexts, awareness of the clinical spectrum of TD and recognition of such cases could have a major impact on the management of sick children and, consequently, on under-five mortality (10, 56, 61, 67, 68, 82–84).
Syndromes of TD in Children
Classic Forms of Beriberi in Infants
The earliest presentation of TD is in breastfeeding infants (1–3 months of age) with non-specific signs, including a “loud piercing cry” and colic (85). Refusal to breastfeed, constipation, vomiting, and agitation may also be present. Eventually edema, cyanosis, and congestive heart failure may appear. Shoshin beriberi, a fulminant form of congestive heart failure (cyanosis without edema) with type B lactic acidosis, has also been documented in infants (6, 54–56, 67, 71, 86). The administration of loop diuretics in such cases may unexpectedly worsen TD-related congestive cardiac failure due to increased renal thiamine clearance (87, 88), and TD should be suspected in any cardiogenic shock not responding to appropriate therapy (6). If undetected at this stage, death can occur within hours but prompt recognition and treatment with injectable thiamine can rapidly reverse the clinical picture and drastically improve the prognosis.
In this acute infantile form, TD results from a low thiamine content in breast milk among deficient, but mostly asymptomatic, mothers. Lactic acidosis may result in generalized symptoms, such as lethargy, irritability, anorexia, tachycardia, and tachypnea (64, 89). These clinical manifestations are probably secondary to dysfunction of mitochondrial energetics in the heart and smooth muscle (particularly the gastrointestinal tract), and an autonomic nervous system insult (6, 42, 43, 90). Due to the non-specific presentation, TD is often overlooked or misdiagnosed as typhoid fever, sepsis, malaria, pneumonia, or decompensated congenital cardiopathy in infants (5, 6, 71). TD may even be a cause of sudden infant death in this young age group (86, 90).
Later, at 4–7 months, the infant is more likely to present with an aphonic form. After increasing cough and dyspnea, the cry changes from hoarse to soundless (“aphonic cry”). Similar to the younger infant, without treatment this condition can evolve into severe acute congestive heart failure, edema, respiratory distress, and eventually death within a few days (86). The clinical appearance at this stage is easily mistaken for severe croup, though TD is usually an afebrile condition. Laryngeal symptoms in TD are due to a Xth cranial nerve palsy caused by local nerve damage and voice changes may be worsened by laryngeal edema and generalized edema secondary to congestive heart failure, which are both observed in TD (6, 67, 89, 91).
Pediatric Neurological Manifestations of TD
Thiamine deficiency can present with a broad range of neurological signs in children, such as anorexia, irritability, agitation, muscle pain, diminished or abolished deep tendon reflexes, ataxia, paralysis, and a progressively altered level of consciousness (67, 86, 92). Given the varied clinical presentations, any unexplained severe neurological signs or symptoms should raise the suspicion of TD. Nevertheless, three main neurological syndromes are described in children: pseudomeningitic beriberi in the first year of life, Enrick’s encephalopathy, and impaired cognitive development associated with mild chronic TD throughout infancy.
The onset of the pseudomeningitic form of infantile beriberi occurs at around 6–12 months of age. Clinical manifestations include muscular fasciculation, nystagmus, ophthalmoplegia, tense fontanel, seizures, and coma. If a lumbar puncture is performed, CSF is found to be clear, with slightly high lactate and low 5-hydroxyindoleacetic acid (5-HIAA) levels (22, 86, 93, 94). This form of TD can be easily confused with encephalitis, meningitis, cerebral-malaria, and vitamin A intoxication and should be included in the differential diagnosis of meningism in children (91, 95).
Nystagmus and ophthalmoplegia are attributed to lesions of the pons vestibular nuclei, and the nuclei of the third and the sixth cranial nerves of the periaqueductal region, respectively. The absence of significant destruction of neurons in these nuclei might explain the rapid reversal of the ocular and vestibular disturbances following thiamine administration. This suggests a continuous evolution from the “biochemical stage” to the irreversible organic lesions that are seen if left untreated (36, 52).
Although classically associated with alcoholism, Wernicke syndrome as a manifestation of TD is not limited to adults. Some children may develop a truncated Wernicke-like syndrome without ataxia, while the classic triad of ophthalmoplegia or nystagmus, ataxia and altered consciousness, is more common in older children (69, 96–100). Typical lesions of TD should be sought in the cerebral targets of thiamine: the cerebellar–pons axis, the midbrain (periaqueductal gray matter and tectum), the mammillary bodies, and the adjacent hypothalamic and thalamic regions (97, 98). Unlike in adults, pediatric brain MRI of Wernicke-like syndrome displays particular lesions in the frontal lobe and basal ganglia, notably the striatum and putamen. However, both adults and children affected by TD show the same symmetrical high-intensity signal on T2 weighting in mammillary bodies and periaqueductal and thalamic areas (97, 98, 100). Cerebral Spectrum MR displays a significant lactate doublet spike with changes in N-acetylaspartate/creatine and choline/creatine ratios, markers of neuronal damage and disruption to myelination, respectively (97, 100). Although rarely accessible in resource-limited settings, early detection of these lesions on neuroimaging could significantly improve clinical outcome (97, 98, 100).
In addition to the acute neurological presentations described, more subtle neurological impairments due to chronic TD in infancy have been identified, particularly affecting cognitive development. Animal studies and clinical observations suggest that an undetected impairment in neuropsychological development occurs during mild chronic thiamine depletion. In the long term, there is neurological damage causing cognitive and psychomotor troubles, alteration in the syntactic and lexical modalities of language acquisition, and seizures causing neurological sequelae (94, 101, 102). This highlights the importance of early diagnosis of TD during this critical period in infancy of accelerated learning and development of essential cognitive skills.
Sepsis and Severe Acute Conditions Associated With, or Exacerbated by Thiamine Deficiency
Severe acute conditions are often associated with functional or true TD in adults (103). Lima et al. (104) found that prevalence of TD on hospital admission was 28% in a Brazilian pediatric intensive care unit,(104) while Khounnorath et al. (5) documented biochemical TD in up to 13.4% of sick infants without evidence of overt beriberi (5). In tropical pediatrics, severe infections often occur on the background of malnutrition, with significant macro and micronutrient deficiencies already present. This may explain the much worse prognosis of septic shock in children with SAM, who potentially have an underlying TD exacerbated by sepsis (83). TD has been linked to an increase in mortality in severe acute conditions and shock, associated with both type A and B lactic acidosis (5, 57, 103). However, although Costa et al. (105) confirmed the high prevalence of TD in septic shock in intensive care patients (up to 71%), they were unable to show a consistent correlation with increased mortality (105).
Complex metabolic conditions may indirectly lead to TD or functional TD, for example, through increased digestive losses, poor thiamine intake during severe illnesses, and high turnover associated with hypermetabolic states, which leads to exhaustion of thiamine stores (74, 106). Critically ill children are often fasting for the first 24 to 48 h of admission and parenteral nutrition in resource-poor settings is lacking. The use of dextrose-based maintenance fluids further potentiates TD risk due to the high consumption of thiamine in the glucose metabolic pathway, as previously outlined.
In sepsis, shock syndromes and severe acute conditions, the current understanding is that hyperlactatemia (a prognostic indicator) is secondary to both type A acidosis, related to hypoxia, and type B acidosis, related caused by dysfunction of PDH in TD. Severe acute conditions lead to endogenous hypermetabolism with mitochondrial respiratory dysfunction. This causes an increase in both oxygen and thiamine consumption (as a PDH cofactor) because of a higher metabolic demand. The resulting functional TD induces a pseudohypoxic state, with the accumulation of pyruvate and lactate, which in turn trigger the release of HIF1-α. HIF1-α, bound to its β subunit, regulates the expression of many genes, notably those involved in metabolic adaptability to hypoxia (gene expression for angiogenesis and erythropoietin synthesis) and innate system modulation (Figure 1C2) (58, 107). HIF1-α also favors anaerobic pathways via LDH gene expression and downregulation of the PDH complex (through PDK activation) in the mitochondria (Figure 1C3). In addition, HIF1-α is known to upregulate both Glut (glucose transporter) to increase uptake of glucose into the cell to meet the immediate demand for ATP via anaerobic pathways, and ThTr2 (thiamine transporter) synthesis at the transcriptional level in order to increase the intracellular thiamine pool pending the recovery of mitochondrial function (Figure 1C2).
This adaptive HIF1-α response may be beneficial in the face of rapid depletion of the thiamine pool caused by excessive utilization in severe acute conditions. In other words, the HIF1-α system creates the optimal cellular conditions to restart aerobic respiration once oxygenation and thiamine supply are restored. This is consistent with Donnino et al.’s (108) recent findings that show the significant positive effect of administration of high-dose thiamine (400 mg) in a subgroup of adults with septic shock and TD (108). Moreover, the Zhu study (109) suggests that sepsis per se might differentially block thiamine-carrier protein synthesis at the mRNA level and impair mitochondrial energetics in rat diaphragms. This would limit cellular thiamine entry, thus, another precipitating factor for functional TD (109). In these circumstances, much higher thiamine dosage than that recommended for beriberi would be required to counteract this phenomenon, as observed in Donnino’s trial (108).
The physician should suspect underlying TD in critically ill children with persistent lactic acidosis or increased plasma anion gap, as well as in any condition resulting in a hypermetabolic state such as shock, sepsis, large burns, poly-trauma, unexplained congestive heart failure, congenital heart disease, diabetic ketoacidosis, and severe malaria (5, 54, 63, 71, 110–114).
Induced-Thiamine Deficiency in Refeeding Syndrome
Thiamine deficiency or subclinical low vitamin B1 levels may be present in children with SAM, as reported in Jamaican and Ghanaian studies (~40–43%) (65, 68). This is most likely the result of a poor and monotonous diet, associated with malabsorption and thiamine loss caused by malnutrition-related diarrhea as well as a degree of intestinal epithelial atrophy (69, 84).
Refeeding syndrome is a potentially fatal complication of SAM management, especially when the introduction of food is too fast. Rapid initiation of nutritional rehabilitation also increases intracellular thiamine turnover which, on a background of pre-existing low whole body thiamine status, can precipitate the onset of true TD and may contribute to the mortality linked with refeeding syndrome (115–117).
Despite overlapping clinical pictures, refeeding syndrome and TD associated with refeeding do not share the same pathogenesis, with TD being a consequence of refeeding rather than a part of the syndrome (117). Refeeding syndrome itself is characterized by electrolyte imbalances caused by intracellular shift (e.g., hypophosphatemia, hypokalemia, hypomagnesemia), and leads to congestive heart failure with edema, neurological, and hematological complications. In induced TD, refeeding stimulates insulin production leading to protein synthesis and increased thiamine demand due to glucose utilization. Pre-existing low thiamine levels in SAM are further reduced by this increased cellular consumption of thiamine. As a result, mitochondrial enzyme complexes (PDH and alpha-KGDH, dependent on their cofactor level) become dysfunctional and the glucose pathway is blocked at the level of pyruvate. The only alternative is for pyruvate to be channeled through the anaerobic pathway leading to type B lactic acidosis, a hallmark of acute TD.
Signs of refeeding syndrome and induced TD are often overlooked or misinterpreted as sepsis, cardiac failure, pneumonia, or sudden death (116). In addition, thiamine assays are usually processed in highly specialized laboratory facilities, therefore, are rarely available in settings where SAM commonly occurs, making confirmed diagnosis rare in resource-limited settings (115). For this reason, diagnosis of TD in this context relies on a high level of clinical awareness and suspicion.
Refeeding syndrome requires careful management, including adjustment of therapeutic milk volumes with consideration of calorie load, gradual correction of electrolyte imbalances and administration of a thiamine loading dose to prevent or correct refeeding-induced TD. Sydney Children’s Hospital Guidelines and Cape Town Pediatric Interest Group suggest 2 mg/kg of thiamine daily during the first week of SAM treatment to prevent refeeding syndrome and its consequences (118, 119). Although current international therapeutic feeding guidelines are designed to provide optimal SAM management and avoid complications, high-dose thiamine at initiation of treatment is not the current practice in humanitarian settings (84). Since we know that a number of children with complicated SAM have borderline thiamine stores, even very cautious introduction of feeds may trigger refeeding syndrome and induced TD without the addition thiamine supplementation.
Acute Clinical Syndromes Associated with Thiaminases
Traditional diets, including fermented fish (containing thiaminase) and betel nut (containing thiamine antagonists) can precipitate symptoms of TD in a nutritional context of high carbohydrate monotonous diet, such as polished rice (66, 77).
However, the most striking clinical manifestation of TD associated with thiaminases is African Seasonal Ataxia. Small case series’ or large outbreaks (described in western Nigeria), related to the consumption of thiaminase-containing Anaphe venata larva, are described during the rainy season (91, 120). Patients develop nausea, vomiting, and dizziness within hours of thiaminase ingestion. Subsequently, fulminant neurological signs develop with nystagmus, tremor, cerebellar ataxia, dysarthria, confusion, and eventually coma. If the diagnosis is suspected early, treatment with thiamine bolus rapidly relieves symptoms and reverses coma within 72 h, suggesting a massive depletion of whole body thiamine caused by thiaminase.
A recent report of infant botulism was found to be associated with clostridium production of thiaminase I, inducing TD. It was speculated that the paralysis of this patient due to clostridium neurotoxins was in fact aggravated by TD. Indeed, the administration of thiamine resulted in faster improvement and a shorter clinical course (121).
Diagnosis and Treatment
Thiamine deficiency can be investigated using plasma, erythrocytes, whole blood ThPP levels (22, 33), and urinary excretion of thiamine before and after exogenous thiamine administration (7). However, serum or whole blood thiamine has poor sensitivity and specificity in severe acute conditions as it decreases during systemic inflammation, and represents only a small part of the whole body thiamine pool. Erythrocyte transketolase activity more accurately evaluates the thiamine status of the body and is, therefore, used as standard (9). MR imaging showing specific lesions can also be very helpful in early detection of neurologic features of TD (97, 100). However, none of these investigations allow immediate diagnosis of TD in life-threatening conditions and they are rarely available in resource-limited settings.
Consequently, the diagnosis of TD in such contexts is a real challenge, especially considering its wide and non-specific clinical spectrum and potentially fatal prognosis. A high level of clinical suspicion should be demonstrated in the following situations (5, 6, 54): suspicion of infantile beriberi; unexplained neurological signs, encephalitis, and cardiac failure; early clinical deterioration after initiation of feeds in malnutrition; sepsis (including in SAM); severe burns; major trauma; hypoxia; and unresponsive lactic acidosis. Rapid point of care lactate testing can be used to confirm persistent lactic acidosis despite correction of shock, and could help to raise the suspicion of TD (54, 57, 58). However, in the absence of specific diagnostic tests, the only way to diagnose TD is to carry out a therapeutic thiamine challenge. Considering its safety profile and wide dosage range, in such cases, thiamine can be administered by slow intravenous injection over 30 min. In severe acute conditions caused by TD, rapid clinical improvement will be seen (within hours or days) following thiamine administration.
Evidence-based pediatric thiamine dosage recommendations for severe acute illness are lacking. Doses found in this literature review vary from 50 to 1500 mg depending on the clinical condition (7, 9, 86, 91, 122), with neurological presentations potentially requiring higher doses and having a longer recovery time (a few days). A recent study showed that thiamine-deficient adults with septic shock in ICU had a significantly lower mortality if they received thiamine 200 mg twice daily for a week compared to placebo (108). Rao and Chandak (6) used 150 mg IV thiamine to treat breast-fed infants under 6 months of age presenting with cardiac failure and/or tachypnea (6), while Qureshi et al. successfully treated lactic acidosis in younger infants (aged 32 days to 4 months) with 100 mg of thiamine daily (89). In both studies, thiamine was well tolerated.
Given this evidence, early thiamine injection in children with severe acute conditions should be considered as a complementary resuscitation tool, especially when there is co-existing malnutrition or where dextrose-based fluid is used to resuscitate (6, 90, 99). Switching to the enteral route should be considered as soon as possible after the initial thiamine bolus (54, 123, 124). In addition to optimal dosage, practical aspects of drug dilution and calculation must be taken into account in resource-poor settings where staff are not always highly qualified. For example, 100 mg is an easy dose to prepare and administer for preparations of 100 mg/ml of thiamine.
According to the literature, prevention of TD during the early refeeding phase in complicated SAM initially requires 10–30 mg of thiamine daily followed by 5–10 mg daily for a month (74, 119, 125). This is not standard practice in humanitarian programs and would provide 10–20 times more than currently recommended in therapeutic protocols, as shown in Table 1 (84, 119). One sachet of ready-to-use therapeutic food (RUTF) or 600 ml of therapeutic milk [either of F-75 (75 kcal/100 ml) or F-100 (100 kcal/100 ml)] contain an average of 0.5 mg of thiamine (84); thus, the average daily intake represents 1–2 mg at most. This is even more significant in infants under 6 months of age with SAM who are at greater risk of developing TD. These babies receive neither RUTF, F-75 nor F-100 but only breast milk or specially diluted F-100, and breastfeeding mothers are rarely supplemented. Infants under 6 months should, therefore, be supplemented with thiamine (2 mg/kg) in order to minimize the risk of precipitating TD.
TABLE 1

Table 1. Thiamine content of therapeutic milk and estimated thiamine needs in children during the acute phase of refeeding in SAM.
Adding weight to the benefits of thiamine supplementation, Luxemburger et al. (82) showed thiamine distribution in a population with high prevalence of TD (Karen refugees’ camps) to be clinically significant. Pregnant women with overt signs of TD received 100 mg of thiamine daily until delivery, followed by 10 mg weekly until 9 months after birth for both mother and infant. Highly relevant results showed overall reductions in infant mortality rate from 183 to 78 per 1000 live births, case fatality of infantile beriberi from 100 to 7%, postnatal deaths by 79% (CI95% 65–87%), and mortality attributable to beriberi from 73 to 5 per 1000 (P < 0.0001) (82).
In summary, based on current evidence, we propose that a therapeutic challenge of 100 mg of thiamine should be given in acute severe illness and in clinically evident TD. For prevention of TD in high-risk situations, such as nutritional and humanitarian crises, refugee, and displaced populations, mass thiamine weekly distribution of 10–30 mg could be considered. The thiamine content of therapeutic foods should also be re-evaluated to minimize TD during the initial phase of refeeding. This must include RUTF, as most SAM patients are treated as out-patients. Finally, pregnant women with a suspicion of TD should receive thiamine treatment during pregnancy, and both mother and baby should continue supplementation while breastfeeding.
Conclusion
This review highlights new perspectives on pathophysiological and clinical correlations in pediatric TD. Thiamine is an intracellular micronutrient with ubiquitous body distribution and plays a fundamental role in mitochondrial energetics within its key target organs. The article outlines the intracellular thiamine transporter traffic and explains the association of TD with type B lactic acidosis and dysfunction of the Krebs cycle, indicating that TD should be considered as a form of acquired functional mitochondrial disease in pediatrics. This review also sheds light on the important role of thiamine in refeeding syndrome and in critical illness. In such circumstances, there is evidence of a mismatch between the increased endogenous metabolic requirements and cellular thiamine availability.
Thiamine deficiency has a large clinical spectrum, and as such it is frequently misdiagnosed, sometimes with fatal consequences or permanent neurological sequelae. Even classic beriberi is still often unrecognized, and increased clinical awareness is essential. Early treatment with thiamine has the potential to rapidly reverse clinical signs and minimize sequelae, before the onset of fixed lesions. The lack of diagnostic capacity in resource-poor settings justifies the use of a therapeutic thiamine challenge in cases with high clinical suspicion. It is an effective, inexpensive, and easy to administer medication.
Many risk factors for TD coexist in tropical pediatrics, making it a highly relevant public health issue. This review provides an opportunity to raise awareness of TD prevalence in malnourished and critically ill children, and to consider early systematic thiamine treatment and clarification of the role of thiamine in the algorithms of metabolic resuscitation. Finally, it calls into question the current thiamine content of RUTF and milk in therapeutic feeding programs, and provides arguments for added thiamine supplementation in therapeutic foods for malnutrition.
Author Contributions
LH is in charge of clinical and therapeutic aspects. BR is in charge of physiological and some clinical aspects. NL is in charge of critical review, relevance, medical, and English editing. DMG is in charge of references and critical medicine aspects. All authors contributed to the coherence of the final draft. All authors collectively agreed with the scientific content of this article.
Conflict of Interest Statement
Authors declare having no conflict of interest and received no specific funding for this study.
Acknowledgments
Prof. Michael Shea for English review and M. Lapuyade for figure design.
Funding
Médecins Sans Frontières contributed to the cost of figure 1 production. No other funding received.
Abbreviations
AThDP, adenosine thiamine diphosphate; AThTP, adenosine thiamine triphosphate; hTHTRs, human thiamine transporters; M-ThPP-T, mitochondrial thiamine pyrophosphate transporter; Spectrum MR, spectrum magnetic resonance; TD, thiamine deficiency; ThDP, thiamine diphosphate; ThTP, thiamine triphosphate; ThTr1, thiamine transporter 1; ThTr2, thiamine transporter 2; TPK, thiamine pyrophosphokinase.
References
1. Manzetti S, Zhang J, van der Spoel D. Thiamine function, metabolism, uptake, and transport. Biochemistry (2014) 53(5):821–35. doi: 10.1021/bi401618y
2. Singleton CK, Martin PR. Molecular mechanisms of thiamine utilization. Curr Mol Med (2001) 1(2):197–207. doi:10.2174/1566524013363870
3. Bettendorff L. Thiamine in excitable tissues: reflections on a non-cofactor role. Metab Brain Dis (1994) 9(3):183–209. doi:10.1007/BF01991194
4. Bettendorff L, Wins P. Biological functions of thiamine derivatives: focus on non-coenzyme roles. OA Biochem (2013) 1(1):10. doi:10.13172/2052-9651
5. Khounnorath S, Chamberlain K, Taylor AM, Soukaloun D, Mayxay M, Lee SJ, et al. Clinically unapparent infantile thiamine deficiency in Vientiane, Laos. PLoS Negl Trop Dis (2011) 5(2):e969. doi:10.1371/journal.pntd.0000969
6. Rao SN, Chandak GR. Cardiac beriberi: often a missed diagnosis. J Trop Pediatr (2010) 56(4):284–5. doi:10.1093/tropej/fmp108
7. Crook MA, Sriram K. Thiamine deficiency: the importance of recognition and prompt management. Nutrition (2014) 30(7–8):953–4. doi:10.1016/j.nut.2014.03.003
8. Fattal-Valevski A, Kesler A, Sela BA, Nitzan-Kaluski D, Rotstein M, Mesterman R, et al. Outbreak of life-threatening thiamine deficiency in infants in Israel caused by a defective soy-based formula. Pediatrics (2005) 115(2):e233–8. doi:10.1542/peds.2004-1255
9. Frank LL. Thiamine in clinical practice. JPEN J Parenter Enteral Nutr (2015) 39(5):503–20. doi:10.1177/0148607114565245
10. Barennes H, Sengkhamyong K, René JP, Phimmasane M. Beriberi (thiamine deficiency) and high infant mortality in northern Laos. PLoS Negl Trop Dis (2015) 9(3):e0003581. doi:10.1371/journal.pntd.0003581
11. Kerns JC, Arundel C, Chawla LS. Thiamine deficiency in people with obesity. Adv Nutr (2015) 6(2):147–53. doi:10.3945/an.114.007526
12. Klein M, Weksler N, Gurman GM. Fatal metabolic acidosis caused by thiamine deficiency. J Emerg Med (2004) 26(3):301–3. doi:10.1016/j.jemermed.2003.11.014
13. The National Academies Press. Nutrition – Dietary Reference Intakes (DRIs): Recommended Dietary Allowances and Adequate Intakes, Vitamins. USA: (2010). Available from: http://iom.edu/Activities/Nutrition/SummaryDRIs/~/media/Files/ActivityFiles/Nutrition/DRIs/RDAandAIs_VitaminandElements.pdf
14. Allen LH. B vitamins in breast milk: relative importance of maternal status and intake, and effects on infant status and function. Adv Nutr (2012) 3(3):362–9. doi:10.3945/an.111.001172
15. Stuetz W, Carrara VI, McGready R, Lee SJ, Biesalski HK, Nosten FH. Thiamine diphosphate in whole blood, thiamine and thiamine monophosphate in breast-milk in a refugee population. PLoS One (2012) 7(6):e36280. doi:10.1371/journal.pone.0036280
16. Dudeja PK, Tyagi S, Kavilaveettil RJ, Gill R, Said HM. Mechanism of thiamine uptake by human jejunal brush-border membrane vesicles. Am J Physiol Cell Physiol (2001) 281(3):C786–92.
17. Said HM, Balamurugan K, Subramanian VS, Marchant JS. Expression and functional contribution of hTHTR-2 in thiamine absorption in human intestine. Am J Physiol Gastrointest Liver Physiol (2004) 286(3):G491–8. doi:10.1152/ajpgi.00361.2003
18. Subramanian VS, Marchant JS, Parker I, Said HM. Cell biology of the human thiamine transporter-1 (hTHTR1). Intracellular trafficking and membrane targeting mechanisms. J Biol Chem (2003) 278(6):3976–84. doi:10.1074/jbc.M210717200
19. Zhao R, Goldman ID. Folate and thiamine transporters mediated by facilitative carriers (SLC19A1-3 and SLC46A1) and folate receptors. Mol Aspects Med (2013) 34(2–3):373–85. doi:10.1016/j.mam.2012.07.006
20. Rajgopal A, Edmondnson A, Goldman ID, Zhao R. SLC19A3 encodes a second thiamine transporter ThTr2. Biochim Biophys Acta (2001) 1537(3):175–8. doi:10.1016/S0925-4439(01)00073-4
21. Chen L, Yee SW, Giacomini KM. OCT1 in hepatic steatosis and thiamine disposition. Cell Cycle (2015) 14(3):283–4. doi:10.1080/15384101.2015.1006532
22. Wyatt DT, Nelson D, Hillman RE. Age-dependent changes in thiamine concentrations in whole blood and cerebrospinal fluid in infants and children. Am J Clin Nutr (1991) 53(2):530–6.
23. Mee L, Nabokina SM, Sekar VT, Subramanian VS, Maedler K, Said HM. Pancreatic beta cells and islets take up thiamine by a regulated carrier-mediated process: studies using mice and human pancreatic preparations. Am J Physiol Gastrointest Liver Physiol (2009) 297(1):G197–206. doi:10.1152/ajpgi.00092.2009
24. Nabokina SM, Inoue K, Subramanian VS, Valle JE, Yuasa H, Said HM. Molecular identification and functional characterization of the human colonic thiamine pyrophosphate transporter. J Biol Chem (2014) 289(7):4405–16. doi:10.1074/jbc.M113.528257
25. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4578–85. doi:10.1073/pnas.1000081107
26. Afadlal S, Labetoulle R, Hazell AS. Role of astrocytes in thiamine deficiency. Metab Brain Dis (2014) 29(4):1061–8. doi:10.1007/s11011-014-9571-y
27. Wang X, Wang B, Fan Z, Shi X, Ke ZJ, Luo J. Thiamine deficiency induces endoplasmic reticulum stress in neurons. Neuroscience (2007) 144(3):1045–56. doi:10.1016/j.neuroscience.2006.10.008
28. Abdou E, Hazell AS. Thiamine deficiency: an update of pathophysiologic mechanisms and future therapeutic considerations. Neurochem Res (2015) 40(2):353–61. doi:10.1007/s11064-014-1430-z
29. Claus D, Eggers R, Warecka K. Neundörfer B. thiamine deficiency and nervous system function disturbances. Eur Arch Psychiatry Neurol Sci (1985) 234(6):390–4. doi:10.1007/BF00386056
30. Rindi G, Comincioli V, Reggiani C, Patrini C. Nervous tissue thiamine metabolism in vivo. II. Thiamine and its phosphoesters dynamics in different brain regions and sciatic nerve of the rat. Brain Res (1984) 293(2):329–42. doi:10.1016/0006-8993(84)91240-X
31. Todd K, Butterworth RF. Mechanisms of selective neuronal cell death due to thiamine deficiency. Ann N Y Acad Sci (1999) 893:404–11. doi:10.1111/j.1749-6632.1999.tb07866.x
32. Greenwood J, Love ER, Pratt OE. Kinetics of thiamine transport across the Blood-brain barrier in the rat. J Physiol (1982) 327:95–103. doi:10.1113/jphysiol.1982.sp014222
33. Gangolf M, Czerniecki J, Radermecker M, Detry O, Nisolle M, Jouan C, et al. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS One (2010) 5(10):e13616. doi:10.1371/journal.pone.0013616
34. Bettendorff L, Mastrogiacomo F, Kish SJ, Grisar T. Thiamine, thiamine phosphates, and their metabolizing enzymes in human brain. J Neurochem (1996) 66(1):250–8. doi:10.1046/j.1471-4159.1996.66010250.x
35. Bettendorff L, Wins P, Lesourd M. Subcellular localization and compartmentation of thiamine derivatives in rat brain. Biochim Biophys Acta (1994) 1222(1):1–6. doi:10.1016/0167-4889(94)90018-3
36. Butterworth RF, Giguère JF, Besnard AM. Activities of thiamine-dependent enzymes in two experimental models of thiamine-deficiency encephalopathy. 2. alpha-Ketoglutarate dehydrogenase. Neurochem Res (1986) 11(4):567–77. doi:10.1007/BF00965326
37. Calingasan NY, Baker H, Sheu KF, Gibson GE. Blood-brain barrier abnormalities in vulnerable brain regions during thiamine deficiency. Exp Neurol (1995) 134(1):64–72. doi:10.1006/exnr.1995.1037
38. Todd KG, Butterworth RF. Early microglial response in experimental thiamine deficiency: an immunohistochemical analysis. Glia (1999) 25(2):190–8. doi:10.1002/(SICI)1098-1136(19990115)25:2<190::AID-GLIA9>3.0.CO;2-B
39. Nixon PF. Glutamate export at the choroid plexus in health, thiamine deficiency, and ethanol intoxication: review and hypothesis. Alcohol Clin Exp Res (2008) 32(8):1339–49. doi:10.1111/j.1530-0277.2008.00727.x
40. Hazell AS, Butterworth RF, Hakim AM. Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. J Neurochem (1993) 61(3):1155–8. doi:10.1111/j.1471-4159.1993.tb03635.x
41. Lee S, Yang G, Yong Y, Liu Y, Zhao L, Xu J, et al. ADAR2-dependent RNA editing of GluR2 is involved in thiamine deficiency-induced alteration of calcium dynamics. Mol Neurodegener (2010) 5:54. doi:10.1186/1750-1326-5-54
42. Romanenko AV, Gnatenko VM, Vladimirova IA. Effect of thiamine on neuromuscular transmission in smooth muscles. Neurophysiology (1994) 26(6):370–7. doi:10.1007/BF01053581
43. Gioda CR, Roman-Campos D, Carneiro-Júnior MA, da Silva KA, de Souza MO, Mendes LJ, et al. Impaired cellular contractile function in thiamine-deficient rat cardiomyocytes. Eur J Heart Fail (2009) 11(12):1126–8. doi:10.1093/eurjhf/hfp146
44. Yui Y, Itokawa Y, Kawai C. Furosemide-induced thiamine deficiency. Cardiovasc Res (1980) 14(9):537–40. doi:10.1093/cvr/14.9.537
45. Grassl SM. Thiamine transport in human placental brush border membrane vesicles. Biochim Biophys Acta (1998) 1371(2):213–22. doi:10.1016/S0005-2736(98)00019-4
46. Schenker S, Johnson RF, Hoyumpa AM, Henderson GI. Thiamine-transfer by human placenta: normal transport and effects of ethanol. J Lab Clin Med (1990) 116(1):106–15.
47. Roecklein B, Levin SW, Comly M, Mukherjee AB. Intrauterine growth retardation induced by thiamine deficiency and pyrithiamine during pregnancy in the rat. Am J Obstet Gynecol (1985) 151(4):455–60. doi:10.1016/0002-9378(85)90269-8
48. Dyckner T, Ek B, Nyhlin H, Wester PO. Aggravation of thiamine deficiency by magnesium depletion. A case report. Acta Med Scand (1985) 218(1):129–31. doi:10.1111/j.0954-6820.1985.tb08836.x
49. Traviesa DC. Magnesium deficiency: a possible cause of thiamine refractoriness in Wernicke-Korsakoff encephalopathy. J Neurol Neurosurg Psychiatry (1974) 37(8):959–62. doi:10.1136/jnnp.37.8.959
50. Subramanian VS, Nabokina SM, Lin-Moshier Y, Marchant JS, Said HM. Mitochondrial uptake of thiamine pyrophosphate: physiological and cell biological aspects. PLoS One (2013) 8(8):e73503. doi:10.1371/journal.pone.0073503
51. Butterworth RF, Giguere JF, Besnard AM. Activities of thiamine-dependent enzymes in two experimental models of thiamine-deficiency encephalopathy: 1. The pyruvate dehydrogenase complex. Neurochem Res (1985) 10(10):1417–28. doi:10.1007/BF00964982
52. Butterworth RF. Effects of thiamine deficiency on brain metabolism: implications for the pathogenesis of the Wernicke-Korsakoff syndrome. Alcohol Alcohol (1989) 24(4):271–9.
53. Giguère JF, Butterworth RF. Activities of thiamine-dependent enzymes in two experimental models of thiamine deficiency encephalopathy: 3. Transketolase. Neurochem Res (1987) 12(3):305–10. doi:10.1007/BF00972141
54. Shah S, Wald E. Type B lactic acidosis secondary to thiamine deficiency in a child with malignancy. Pediatrics (2015) 135(1):e221–4. doi:10.1542/peds.2014-2289
55. Madl C, Kranz A, Liebisch B, Traindl O, Lenz K, Druml W. Lactic acidosis in thiamine deficiency. Clin Nutr (1993) 12(2):108–11. doi:10.1016/0261-5614(93)90060-H
56. Boonsiri P, Tangrassameeprasert R, Panthongviriyakul C, Yongvanit P. A preliminary study of thiamine status in northeastern Thai children with acute diarrhea. Southeast Asian J Trop Med Public Health (2007) 38(6):1120–5.
57. Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc (2013) 88(10):1127–40. doi:10.1016/j.mayocp.2013.06.012
58. Sweet RL, Zastre JA. HIF1-α-mediated gene expression induced by vitamin B1 deficiency. Int J Vitam Nutr Res (2013) 83(3):188–97. doi:10.1024/0300-9831/a000159
59. Desjardins P, Butterworth RF. Role of mitochondrial dysfunction and oxidative stress in the pathogenesis of selective neuronal loss in Wernicke’s encephalopathy. Mol Neurobiol (2005) 31(1–3):17–25. doi:10.1385/MN:31:1-3:017
60. Hazell AS, Butterworth RF. Update of cell damage mechanisms in thiamine deficiency: focus on oxidative stress, excitotoxicity and inflammation. Alcohol Alcohol (2009) 44(2):141–7. doi:10.1093/alcalc/agn120
61. Coats D, Shelton-Dodge K, Ou K, Khun V, Seab S, Sok K, et al. Thiamine deficiency in Cambodian infants with and without beriberi. J Pediatr (2012) 161(5):843–7. doi:10.1016/j.jpeds.2012.05.006
62. Liu S, Zhang KN, Riley M. Thiamine deficiency is associated with ethnicity in a subtropical area of China. Asia Pac J Clin Nutr (1994) 3(4):211–5.
63. Mayxay M, Taylor AM, Khanthavong M, Keola S, Pongvongsa T, Phompida S, et al. Thiamin deficiency and uncomplicated falciparum malaria in Laos. Trop Med Int Health (2007) 12(3):363–9. doi:10.1111/j.1365-3156.2006.01804.x
64. Keating EM, Nget P, Kea S, Kuong S, Daly L, Phearom S, et al. Thiamine deficiency in tachypnoeic Cambodian infants. Paediatr Int Child Health (2015) 35(4):312–8. doi:10.1080/20469047.2015.1109226
65. Neumann CG, Swendseid ME, Jacob M, Stiehm ER, Dirige OV. Biochemical evidence of thiamine deficiency in young Ghanian children. Am J Clin Nutr (1979) 32(1):99–104.
66. Tang CM, Wells JC, Rolfe M, Cham K. Outbreak of beriberi in the Gambia. Lancet (1989) 2(8656):206–7. doi:10.1016/S0140-6736(89)90383-8
67. Duce M, Escriba JM, Masuet C, Farias P, Fernandez E, de la Rosa O. Suspected thiamine deficiency in Angola. Field Exch (2003) (20):26–8. Available from: http://files.ennonline.net/attachments/1639/FEX-20.pdf
68. Hailemariam B, Landman JP, Jackson AA. Thiamine status in normal and malnourished children in Jamaica. Br J Nutr (1985) 53(3):477–83. doi:10.1079/BJN19850057
69. Pfeiffer RF. Neurologic manifestations of malabsorption syndromes. Handb Clin Neurol (2014) 120:621–32. doi:10.1016/B978-0-7020-4087-0.00042-5
70. Nightingale J, Woodward JM. Small Bowel and Nutrition Committee of the British Society of Gastroenterology. Guidelines for management of patients with a short bowel. Gut (2006) 55(Suppl 4):iv1–12. doi:10.1136/gut.2006.091108
71. Greenspon J, Perrone EE, Alaish SM. Shoshin beriberi mimicking central line sepsis in a child with short bowel syndrome. World J Pediatr (2010) 6(4):366–8. doi:10.1007/s12519-010-0022-5
72. Kawai C, Wakabayashi A, Matsumura T, Yui Y. Reappearance of beriberi heart disease in Japan. A study of 23 cases. Am J Med (1980) 69(3):383–6. doi:10.1016/0002-9343(80)90008-X
73. Bell D, Robertson CE, Muir AL. Carbonated drinks, thiamine deficiency and right ventricular failure. Scott Med J (1987) 32(5):137–8.
74. Leite HP, Lima LF. Thiamine (vitamin B1) deficiency in intensive care: physiology, risk factors, diagnosis, and treatment. In: Rajendram, Rajkumar, Preedy, Victor R, Patel, Vinood B, editors. Diet and Nutrition in Critical Care. New York: Springer Science+ Business Media (2014). p. 1–16.
75. Barennes H, Simmala C, Odermatt P, Thaybouavone T, Vallee J, Martinez-Aussel B, et al. Postpartum traditions and nutrition practices among urban Lao women and their infants in Vientiane, Lao PDR. Eur J Clin Nutr (2009) 63(3):323–31. doi:10.1038/sj.ejcn.1602928
76. Macias-Matos C, Rodriguez-Ojea A, Chi N, Jimenez S, Zulueta D, Bates CJ. Biochemical evidence of thiamine depletion during the Cuban neuropathy epidemic, 1992-1993. Am J Clin Nutr (1996) 64(3):347–53.
77. Vimokesant SL, Hilker DM, Nakornchai S, Rungruangsak K, Dhanamitta S. Effects of betel nut and fermented fish on the thiamine status of northeastern Thais. Am J Clin Nutr (1975) 28(12):1458–63.
78. Bâ A. Comparative effects of alcohol and thiamine deficiency on the developing central nervous system. Behav Brain Res (2011) 225(1):235–42. doi:10.1016/j.bbr.2011.07.015
79. Hoyumpa AM Jr. Mechanisms of thiamine deficiency in chronic alcoholism. Am J Clin Nutr (1980) 33(12):2750–61.
80. Subramanya SB, Subramanian VS, Said HM. Chronic alcohol consumption and intestinal thiamine absorption: effects on physiological and molecular parameters of the uptake process. Am J Physiol Gastrointest Liver Physiol (2010) 299(1):G23–31. doi:10.1152/ajpgi.00132.2010
81. Laforenza U, Patrini C, Gastaldi G, Rindi G. Effects of acute and chronic ethanol administration on thiamine metabolizing enzymes in some brain areas and in other organs of the rat. Alcohol Alcohol (1990) 25(6):591–603.
82. Luxemburger C, White NJ, ter Kuile F, Singh HM, Allier-Frachon I, Ohn M, et al. Beri-beri: the major cause of infant mortality in Karen refugees. Trans R Soc Trop Med Hyg (2003) 97(2):251–5. doi:10.1016/S0035-9203(03)90134-9
83. Iqbal H, Jobayer C, Shoji Y, Rehana Y, Tahmeed A. Features in septic children with or without severe acute malnutrition and the risk factors of mortality. Pediatrics (2015) 135:S10. doi:10.1542/peds.2014-3330Q
84. World Health Organisation. Guideline: Updates on the Management of Severe Acute Malnutrition in Infants and Children. Geneva: WHO (2013).
85. Christopher D, Watkins J, Allan WW. Nutrition in pediatrics. 4th ed. Pmph U S A. Halmilton: BC Decker Inc. (2008). p. 106.
86. Rabinowitz SS. Paediatric Beri Beri. [Medscape]. (2014). Available from: emedicine.medscape.com/article/984721-overview.
87. Sica DA. Loop diuretic therapy, thiamine balance, and heart failure. Congest Heart Fail (2007) 13(4):244–7. doi:10.1111/j.1527-5299.2007.06260.x
88. Roman-Campos D, Cruz JS. Current aspects of thiamine deficiency on heart function. Life Sci (2014) 98(1):1–5. doi:10.1016/j.lfs.2013.12.029
89. Qureshi UA, Sami A, Altaf U, Ahmad K, Iqbal J, Wani NA, et al. Thiamine responsive acute life threatening metabolic acidosis in exclusively breast-fed infants. Nutrition (2016) 32(2):213–6. doi:10.1016/j.nut.2015.08.007
90. Jeffrey HE, McCleary BV, Hensley WJ, Read DJ. Thiamine deficiency – a neglected problem of infants and mothers – possible relationships to sudden infant death syndrome. Aust N Z J Obstet Gynaecol (1985) 25(3):198–202. doi:10.1111/j.1479-828X.1985.tb00643.x
91. McCandless DW. Thiamine deficiency and associated clinical disorders. Contemporary Clinical Neuroscience. Humana Press (2009) p. 31–46. doi:10.1007/978-1-60761-311-4_4.
92. Ogunlesi TA. Thiamine deficiency: a cause of childhood ataxia not to be ignored. Ann Trop Paediatr (2004) 24(4):357–60. doi:10.1179/146532804X10835
93. Botez MI, Young SN, Bachevalier J, Gauthier S. Thiamine deficiency and cerebrospinal fluid 5-hydroxyindoleacetic acid: a preliminary study. J Neurol Neurosurg Psychiatry (1982) 45(8):731–3. doi:10.1136/jnnp.45.8.731
94. Vigil FA, Oliveira-Silva Ide F, Ferreira LF, Pereira SR, Ribeiro AM. Spatial memory deficits and thalamic serotonergic metabolite change in thiamine deficient rats. Behav Brain Res (2010) 210(1):140–2. doi:10.1016/j.bbr.2010.02.019
95. Newland JG, Shah SS, Zaoutis TE. The child with aseptic meningitis. Pediatr Case Rev (2003) 3(4):218–21. doi:10.1097/01.PCA.0000083250.03358.9F
96. Kesler A, Stolovitch C, Hoffmann C, Avni I, Morad Y. Acute ophthalmoplegia and nystagmus in infants fed a thiamine-deficient formula: an epidemic of Wernicke encephalopathy. J Neuroophthalmol (2005) 25(3):169–72. doi:10.1097/01.wno.0000177293.06727.d5
97. Kornreich L, Bron-Harlev E, Hoffmann C, Schwarz M, Konen O, Schoenfeld T, et al. Thiamine deficiency in infants: MR findings in the brain. AJNR Am J Neuroradiol (2005) 26(7):1668–74.
98. Xin Y, Wan DH, Chu Q, Li AM, Gao XJ. Severe sepsis as an initial presentation in children with Wernicke’s encephalopathy: report of a case and literature review [English abstract only]. Zhonghua Er Ke Za Zhi (2011) 49(8):612–6.
99. Qureshi UA, Wani NA, Ahmad K, Irshad M, Ali I. Infantile Wernicke’s encephalopathy. Arch Dis Child (2015) 100(7):648. doi:10.1136/archdischild-2014-307949
100. Zuccoli G, Siddiqui N, Bailey A, Bartoletti SC. Neuroimaging findings in pediatric Wernicke encephalopathy: a review. Neuroradiology (2010) 52(6):523–9. doi:10.1007/s00234-009-0604-x
101. Mimouni-Bloch A, Goldberg-Stern H, Strausberg R, Brezner A, Heyman E, Inbar D, et al. Thiamine deficiency in infancy: long-term follow-up. Pediatr Neurol (2014) 51(3):311–6. doi:10.1016/j.pediatrneurol.2014.05.010
102. Friedmann N, Fattal I, Fattal-valevski A. The effect of thiamine deficiency in infancy on the development of syntactic and lexical abilities. Procedia Soc Behav Sci (2010) 6:168–9. doi:10.1016/j.sbspro.2010.08.083
103. Cruickshank AM, Telfer AB, Shenkin A. Thiamine deficiency in the critically ill. Intensive Care Med (1988) 14(4):384–7. doi:10.1007/BF00262893
104. Lima LF, Leite HP, Taddei JA. Low blood thiamine concentrations in children upon admission to the intensive care unit: risk factors and prognostic significance. Am J Clin Nutr (2011) 93(1):57–61. doi:10.3945/ajcn.2009.29078
105. Costa NA, Gut AL, de Souza Dorna M, Pimentel JA, Cozzolino SM, Azevedo PS, et al. Serum thiamine concentration and oxidative stress as predictors of mortality in patients with septic shock. J Crit Care (2014) 29(2):249–52. doi:10.1016/j.jcrc.2013.12.004
106. Manzanares W, Hardy G. Thiamine supplementation in the critically ill. Curr Opin Clin Nutr Metab Care (2011) 14(6):610–7. doi:10.1097/MCO.0b013e32834b8911
107. Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med (Berl) (2007) 85(12):1339–46. doi:10.1007/s00109-007-0282-2
108. Donnino MW, Andersen LW, Chase M, Berg KM, Tidswell M, Giberson T, et al. Randomized, double-blind, placebo-controlled trial of thiamine as a metabolic resuscitator in septic shock: a pilot study. Crit Care Med (2016) 44(2):360–7. doi:10.1097/CCM.0000000000001572
109. Zhu E, Fang L, Subramanian VS, Said HM, Sassoon CS. Expression of thiamine Transporter Mrna in plasmic microvesicles of rats with sepsis. Am J Respir Crit Care Med (2014) (189):A6218.
110. Krishna S, Taylor AM, Supanaranond W, Pukrittayakamee S, ter Kuile F, Tawfiq KM, et al. Thiamine deficiency and malaria in adults from southeast Asia. Lancet (1999) 353(9152):546–9. doi:10.1016/S0140-6736(98)06316-8
111. McConachie I, Haskew A. Thiamine status after major trauma. Intensive Care Med (1988) 14(6):628–31. doi:10.1007/BF00256767
112. Clark JA, Burny I, Sarnaik AP, Audhya TK. Acute thiamine deficiency in diabetic ketoacidosis: diagnosis and management. Pediatr Crit Care Med (2006) 7(6):595–9. doi:10.1097/01.PCC.0000244463.59230.DA
113. Rosner EA, Strezlecki KD, Clark JA, Lieh-Lai M. Low thiamine levels in children with type 1 diabetes and diabetic ketoacidosis: a pilot study. Pediatr Crit Care Med (2015) 16(2):114–8. doi:10.1097/PCC.0000000000000302
114. Shamir R, Dagan O, Abramovitch D, Abramovitch T, Vidne BA, Dinari G. Thiamine deficiency in children with congenital heart disease before and after corrective surgery. JPEN J Parenter Enteral Nutr (2000) 24(3):154–8. doi:10.1177/0148607100024003154
115. Manary M, Trehan I, Weisz A. Systematic Review of Transition Phase Feeding of Children with Severe Acute Malnutrition as in Patients. World Health Organization (WHO) (2012). Available from: www.who.int/nutrition/publications/guidelines/updates_management_SAM_infantandchildren_review5.pdf
116. Fuentebella J, Kerner JA. Refeeding syndrome. Pediatr Clin North Am (2009) 56(5):1201–10. doi:10.1016/j.pcl.2009.06.006
117. Maiorana A, Vergine G, Coletti V, Luciani M, Rizzo C, Emma F, et al. Acute thiamine deficiency and refeeding syndrome: similar findings but different pathogenesis. Nutrition (2014) 30(7–8):948–52. doi:10.1016/j.nut.2014.02.019
118. Refeeding Syndrome: Guidelines. Cape Town Metropole Paediatric Interest Group (2009). Available from: http://www.adsa.org.za/Portals/14/Documents/Clinical20Guidelines20Refeeding20Syndrome20Paeds20Section20Only20_.pdf
119. Refeeding Syndrome: Prevention and Management – SCH Practice Guideline. Australia: Sydney Children’s Hospital Guidelines (2013). Available from: http://www.schn.health.nsw.gov.au/_policies/pdf/2013-7036.pdf
120. Nishimune T, Watanabe Y, Okazaki H, Akai H. Thiamin is decomposed due to Anaphe spp. entomophagy in seasonal ataxia patients in Nigeria. J Nutr (2000) 130(6):1625–8.
121. Ringe H, Schuelke M, Weber S, Dorner BG, Kirchner S, Dorner MB. Infant botulism: is there an association with thiamine deficiency? Pediatrics (2014) 134(5):e1436–40. doi:10.1542/peds.2013-3378
122. Park SW, Yi YY, Han JW, Kim HD, Lee JS, Kang HC. Wernicke’s encephalopathy in a child with high dose thiamine therapy. Korean J Pediatr (2014) 57(11):496–9. doi:10.3345/kjp.2014.57.11.496
123. Smithline HA, Donnino M, Greenblatt DJ. Pharmacokinetics of high-dose oral thiamine hydrochloride in healthy subjects. BMC Clin Pharmacol (2012) 12:4. doi:10.1186/1472-6904-12-4
124. Giacalone M, Martinelli R, Abramo A, Rubino A, Pavoni V, Iacconi P, et al. Rapid reversal of severe lactic acidosis after thiamine administration in critically ill adults: a report of 3 cases. Nutr Clin Pract (2015) 30(1):104–10. doi:10.1177/0884533614561790
Keywords: thiamine, thiamine deficiency, tropical pediatrics, beriberi, refeeding syndrome, pediatric critical care, type B lactic acidosis, thiamine transporters
Citation: Hiffler L, Rakotoambinina B, Lafferty N and Martinez Garcia D (2016) Thiamine Deficiency in Tropical Pediatrics: New Insights into a Neglected but Vital Metabolic Challenge. Front. Nutr. 3:16. doi: 10.3389/fnut.2016.00016
Received: 05 January 2016; Accepted: 25 May 2016;
Published: 14 June 2016
Edited by:
Maurizio Muscaritoli, Sapienza University of Rome, ItalyReviewed by:
Luigi Iuliano, Sapienza University of Rome, ItalyTianan Alan Jiang, McCormick and Compay, Inc., USA
Copyright: © 2016 Hiffler, Rakotoambinina, Lafferty and Martinez Garcia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Hiffler, laurent.hiffler@barcelona.msf.org;
Daniel Martinez Garcia, daniel.martinez@barcelona.msf.org