 Mehul P. Jariwala
Mehul P. Jariwala Ronald M. Laxer
Ronald M. Laxer- 1Saskatoon Health Region, Saskatoon, SK, Canada
- 2Department of Pediatrics, Royal University Hospital, University of Saskatchewan, Saskatoon, SK, Canada
- 3The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
Childhood onset anti-neutrophilic cytoplasmic antibody (ANCA) associated vasculitis (AAV) is a rare group of primary systemic vasculitides affecting medium and small blood vessels. AAV includes granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA), and renal limited ANCA vasculitis. These disorders are associated with severe clinical manifestations, frequent relapses and a high cumulative morbidity, and often present with multisystem involvement. Renal involvement is common in the pediatric age group, characterized by pauci-immune necrotizing and crescentic glomerulonephritis which frequently progresses to chronic kidney disease in adulthood. ANCAs against proteinase 3 (PR3-ANCA) or myeloperoxidase (MPO) (MPO-ANCA) remain the hallmark of AAV and are integral to the disease pathogenesis. Newer understanding of neutrophil extracellular traps and complement activation have provided better insights into disease pathogenesis. A pediatric vasculitis working group has developed and validated childhood vasculitis classification criteria and disease activity and damage scores. No specific pediatric treatment recommendations exist due to rare nature of the illness in pediatric population. Smaller case series have been published on the efficacy of adult treatment regimens in pediatric patients. The prognosis often remains guarded with frequent relapses and a high cumulative morbidity. The aim of this article is to provide a comprehensive review on pediatric AAV with a focus on recent observations regarding epidemiology, disease pathogenesis, treatment, and prognosis.
Introduction
Anti-neutrophilic cytoplasmic antibody (ANCA) associated vasculitides (AAVs) are primary systemic vasculitides characterized by necrotizing arteritis with few or no immune deposits in small to medium-sized arteries in multiple organs. This group comprises granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA), and renal limited ANCA vasculitis (1). These diseases often have the presence of circulating autoantibodies (ANCA) that are usually directed against myeloperoxidase (MPO) or proteinase 3 (PR3) antigens. In the pediatric population, GPA is more common than MPA and EGPA. Renal involvement in AAV is characterized by rapidly progressive pauci-immune necrotizing and crescentic glomerulonephritis contributing significantly to the morbidity and progression to end stage renal disease. Treatment is often extrapolated from adult studies due to rare nature of this illness in the pediatric population. Early recognition and treatment remains pivotal to the better outcome in these patients. This review focuses on recent publications on epidemiology, update on AAV pathogenesis, recently described pediatric cohorts, disease outcome measures and the Canadian Vasculitis research network (CanVasc) endorsed pediatric treatment guidelines.
Epidemiology
AAV is much more common in adults compared to the pediatric population. GPA is the most frequent, followed by MPA and EGPA. The peak incidence of GPA is in fourth-fifth decade of life and is more common in males. Multiple adult studies have been published on the epidemiology of AAV from Europe, Japan, the USA, New Zealand and Australia. Europe reports overall incidence rates of AAV from 13 to 20 per million (2).
Epidemiological data in childhood AAV are scarce. The French registry reported an increasing annual incidence of AAV over the 25-year period from 0.10 in 1986–90 to 0.45 per million children from 2006 to 2010 (3). The reported annual incidence rate per million children from a Swedish study was 1.4 (4). In contrast to this, the reported incidence in Southern Alberta, Canada continues to increase from 2.75 cases/million/year in the last 15 years to 6.39 cases/million/year in the last 5 years (5). Childhood AAV has a higher female preponderance with a peak age at onset in second decade and median age at diagnosis of approximately 12–14 years (3, 5, 6).
Pathogenesis
The precise etiopathogenesis of AAV is not fully elucidated. There appears to be a complex interplay of genetic susceptibility factors, environmental triggers and dysregulation in both the innate and adaptive immune systems contributing to the development of AAV. Multiple theories have been proposed to identify the pathogenic pathways in AAV.
Role of ANCA: ANCA have a central role in pathogenesis of AAV. The presence of ANCA indicates the involvement of the neutrophil as the effector cell. Higher levels of ANCA target antigens myeloperoxidase (MPO) or proteinase 3 (PR3) are noted on the surface of circulating neutrophils in AAV, which could be secondary to disturbed epigenetic modification (7, 8). In vitro studies have also demonstrated IgG-ANCA capable of inducing an oxidative burst releasing toxic oxygen radicals, primary granule release and surface activation in cytokine primed neutrophils with IgG-ANCA (9). This process eventually leads to endothelial damage and activation of the alternate complement pathway (10). The development of ANCA may result from a breakdown of tolerance.
Mechanisms of Tolerance Breakdown
a) Complementary Peptide Model: This theory hypothesizes that the initial immune response is to a peptide with complementary structure relative to the autoantigen. In AAV, these complementary peptides are derived from antisense transcription of the antisense strand of the autoantigen at the PRTN3 (the gene encoding PR3) or MPO loci. Alternatively, the complementary peptide can be a mimic of an antisense peptide that is produced by a symbiotic or pathogenic microbe. These can stimulate a B cell adaptive immune response leading to anti-idiotype antibodies which cross react with the autoantigen epitopes (Figure 1) (11).
b) Molecular mimicry models: An infectious link to autoimmunity is well known in AAV. Chronic nasal carriage of Staphylococcus aureus has been identified as an independent risk factor in relapse of GPA (12). Kain and colleagues proposed a model of molecular mimicry wherein rats injected with gram-negative bacillus adhesion protein FimH developed pauci-immune focal necrotizing glomerulonephritis. Autoantibodies to human LAMP-2 are highly prevalent in pauci-immune FNGN. These antibodies share considerable homology to FimH and could induce antibodies to human LAMP-2 and initiate pauci-immune FNGN (13).
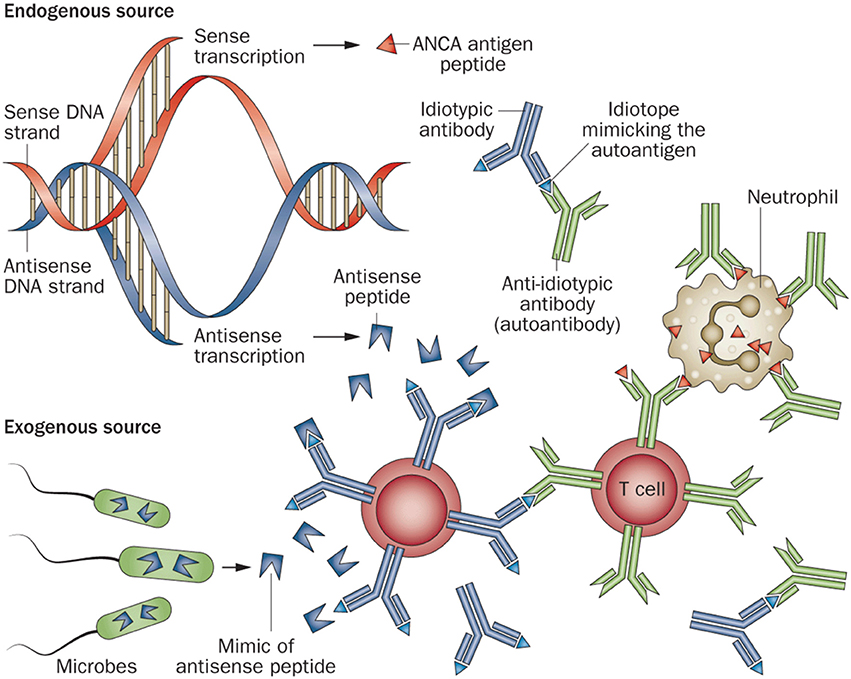
Figure 1. Diagram of the induction of an ANCA-mediated autoimmune response by an initial immune response to a peptide that is complementary to an autoantigen peptide. This complementary peptide immunogen could arise from antisense transcription of the antisense strand of the autoantigen gene, or could be a mimic of an antisense peptide that is produced by a symbiotic or pathogenic microbe. The anti-complementary peptide antibody idiotopes would engender an anti-idiotypic antibody response that cross reacts with the autoantigen epitopes that are complementary to the initial immunogenic peptide. Reprinted by permission from Springer Nature Terms and Conditions for RightsLink Permissions Springer Customer Service Centre GmbH: Nature. Jennette and Falk (11).
NETosis: Traditionally it was hypothesized that neutrophils die in small vessels by necrosis. However neutrophil extracellular traps (NETs) have been identified at the site of the vasculitic lesion (14, 15). NETosis is a type of programed cell death mechanism in which the neutrophils have the ability to extrude their DNA and proinflammatory bactericidal molecules creating NET-like structures. Patients with AAV possess elevated levels of NETs in the circulation (16, 17). NETs can lead to vascular necrosis, endothelial damage, expose immune-stimulatory molecules and can activate alternate complement pathways (18–20). Kessenbrock and colleagues showed that ANCA-stimulated neutrophils are capable of inducing NETs which contain proteinase-3 (PR3) and myeloperoxidase (MPO). This complex promotes the autoimmune response against neutrophil components in individuals with vasculitis (14).
Role of Apoptosis: Apoptosis (programmed cell death) is a vital component of the immune system, promoting resolution of inflammation by clearance of cellular debris by macrophages (21). In patients with GPA, spontaneous apoptosis of neutrophils is significantly less as compared to normal individuals. Neutrophils in these patients express higher membrane bound PR3 without degranulation as compared to healthy controls. The membrane associated PR3 on apoptotic neutrophil delays their clearance by macrophage and also act as a danger signal through the IL-1R1/MyD88 signaling pathway in a NO-dependent manner triggering a proinflammatory response by macrophages recruiting more neutrophils and monocytes (22, 23).
Role of complement pathway: The role of alternate complement pathway (ACP) activation was first described by Xiao et al. (24) who showed that complement component depletion in mice could prevent crescentic glomerulonephritis. Further animal studies by the same group suggested that knock-out mice for C5 and factor B after receiving anti-MPO IgG did not develop disease. These data suggest that ACP and the activation of complement C5 are critical to AAV pathogenesis. The activation of alternate pathway leads to increase anaphylatoxin C3a and C5a resulting in an inflammatory amplification loop which mediates the severe necrotizing inflammation of ANCA disease (24).
Classification
The 1990 American College of Rheumatology (ACR) classification criteria were developed for GPA and EGPA and were derived from adult patient data (25). A new set of classification criteria was proposed in 2006 by European League against Rheumatism/Pediatric Rheumatology European Society (EULAR/PReS) group that included components of the 1990 ACR criteria along with common pediatric manifestations and inclusion of ANCA (26). These criteria were endorsed by EULAR, the Pediatric Rheumatology European Society, and Pediatric Rheumatology International Trial Organization (EULAR/PRINTO/PReS) and were published in 2010 (27). They included CT scan results, ANCA positivity and better descriptive terms for upper and lower respiratory involvement with specific mention of subglottic stenosis which were lacking in ACR criteria. In the ARChiVe cohort, a pediatric cohort of patients with AAV, the EULAR/PRINTO/PRES criteria were found to be more sensitive than the ACR criteria in classifying pediatric GPA (28). The EMA (European Medicine Agency) is a classification algorithm (29) which comprises of ACR criteria, Chapel Hill Consensus Conference (CHCC) definition, Lanham criteria for Churg Strauss syndrome (30) and presence or absence of ANCA. The EMA algorithm was reported as the most sensitive in diagnosing childhood GPA (28).
MPA was defined by Chapel Hill Consensus Conference (CHCC) in the small vessel vasculitis category as: “necrotizing vasculitis, with few or no immune deposits, predominantly affecting small vessels (i.e., capillaries, venules, or arterioles). Necrotizing arteritis involving small and medium arteries may be present. Necrotizing glomerulonephritis is very common. Pulmonary capillaritis often occurs. Granulomatous inflammation is absent” (1). No specific pediatric classification criteria for MPA were endorsed in the EULAR/PReS meeting in 2008. It was proposed more of a clinical syndrome in 2006 by EULAR/PReS group. The only modification to the Chapel Hill report was addition of ANCA to the description of microscopic polyangiitis.
The newly endorsed pediatric vasculitis classification criteria have several limitations such as no pediatric specific MPA criteria were defined, certain degrees of overlap between GPA and MPA have been noted and limited forms of the disease were not described in these criteria.
The criteria for classifying GPA according to the EULAR/PRINTO/PReS criteria are listed in Table 1.

Table 1. Final EULAR/PRINTO/PRES childhood GPA criteria.
Clinical Features of AAV
GPA
GPA is a systemic pauci-immune necrotizing small and medium-size vessel vasculitis associated with granulomatous inflammation (26). The classic form of GPA presents with a triad of upper and lower respiratory tract involvement with renal manifestation presenting as pauci-immune crescentic glomerulonephritis (GN). Disease manifestations in GPA have been described in the larger cohorts listed in Table 2.
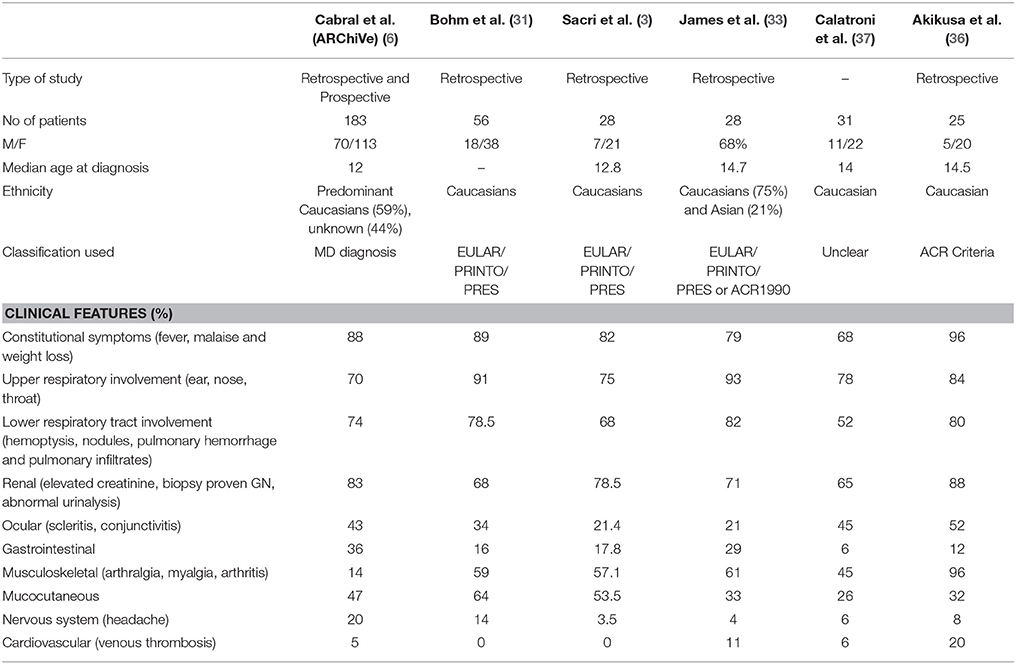
Table 2. Clinical features of GPA at presentation in the largest pediatric cohorts reported.
Involvement of the upper respiratory tract remains the most feature in children with GPA. Childhood GPA presents either as localized granulomatous disease with a chronic course or as an acute small vessel vasculitis characterized by pulmonary hemorrhage and/or rapidly progressive renal involvement. The majority of patients present with symptoms of upper or lower respiratory tract including epistaxis, sinusitis, otitis media, and hearing loss. Subglottic stenosis; one of the severe complication of GPA was reported in at presentation from 10 to 20% (6, 31). It is also more common in pediatric as compared to adult cohorts and hence proposed in pediatric classification criteria (26, 32). Alvelolar, pleural or bronchial tissues can be involved as a part of lower respiratory tract involvement, presenting with cough, wheezing, hemoptysis, bronchial stenosis, or a catastrophic pulmonary hemorrhage. Imaging findings of nodules, fixed pulmonary infiltrates and cavitation were noted in patients ranging from 21 to 80% (6, 33) (Figure 2).
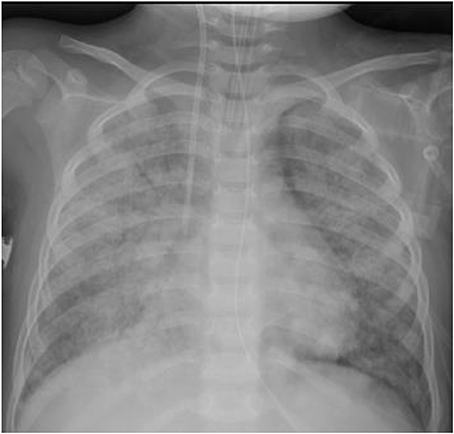
Figure 2. Diffuse pulmonary hemorrhage in 3-year-old presenting with pulmonary-renal syndrome diagnosed as MPA.
MPA
Upper respiratory tract involvement is rarely noted in MPA. In fact, per the case definitions, involvement of upper respiratory tract often precludes the diagnosis of MPA. Lower respiratory tract involvement is common with MPA often presenting as hemoptysis, anemia secondary to chronic, low-grade pulmonary hemorrhage with pulmonary hemosiderosis, or as catastrophic pulmonary hemorrhage noted in up to 42% patients (6). Granulomatous inflammation does not occur in MPA. Pulmonary-renal syndrome is one of the most severe AAV manifestations and can often reveal the disease or occur during its evolution. Renal involvement is very common and noted in 94–100% of the patients (3, 6, 34, 35). Disease manifestations in MPA have been described in the larger cohorts listed in Table 4.
Clinical Features Common to Both GPA and MPA
Pauci-immune necrotizing GN is a severe manifestation of GPA and MPA often leading into renal failure contributing to significant morbidity. Renal involvement is noted in up to 80% of patients with GPA. The clinical features of renal involvement include hypertension, edema, proteinuria, and hematuria. The largest cohort, published by Cabral et al. reported elevated creatinine, requirement of dialysis and end stage renal disease in 60, 25, and 10% respectively (6). Histopathological findings consistent with pauci-immune GN and/or necrotizing glomerulonephritis were noted in 94% of the patients and biopsy findings of vasculitis in 29% (6).
More than 80% of patients across all cohorts reported constitutional symptoms of malaise, weight loss and/or fever. The recently published GPA cohorts report higher incidence of gastrointestinal (GI) manifestation, in 30–36% of patients (6, 33). Common GI manifestations in AAV include chronic nausea, diarrhea, and non-specific abdominal pain. Cabral et al. reported <5% of children with bloody diarrhea or ischemic abdominal pain (6). Mucocutaenous manifestation include oral and genital ulcers, palpable purpura (Figure 3), petechial rash, livedo, and subcutaneous nodules. Eye involvement was reported in up to 20–50% across the different cohorts. Common eye symptoms in order of frequency were conjunctivitis, episcleritis, proptosis secondary to retro-orbital mass, and keratitis. Arthritis, arthralgia, myalgia, and muscle weakness were the common MSK symptoms in both MPA and GPA. Neurologic symptoms were less common across all cohorts and included non-specific findings of headache and dizziness. The highest incidence of venous thrombosis (20%) was reported in GPA by Akikusa et al. but remained less common in other cohorts (36). Cardiovascular involvement in GPA was more common than MPA (Table 3).
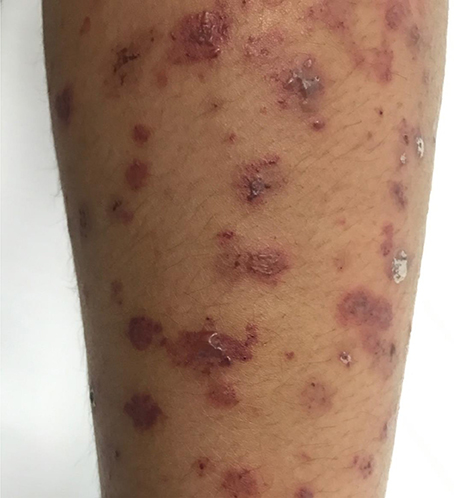
Figure 3. Eight year old diagnosed with GPA with leucocytoclastic vasculitis involving upper and lower limbs.
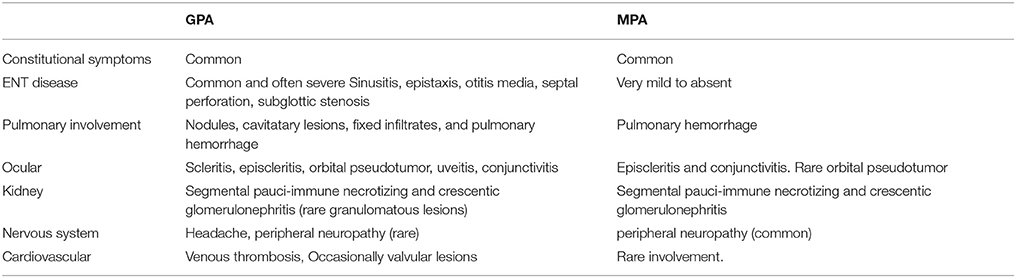
Table 3. Clinical differences between GPA and MPA.
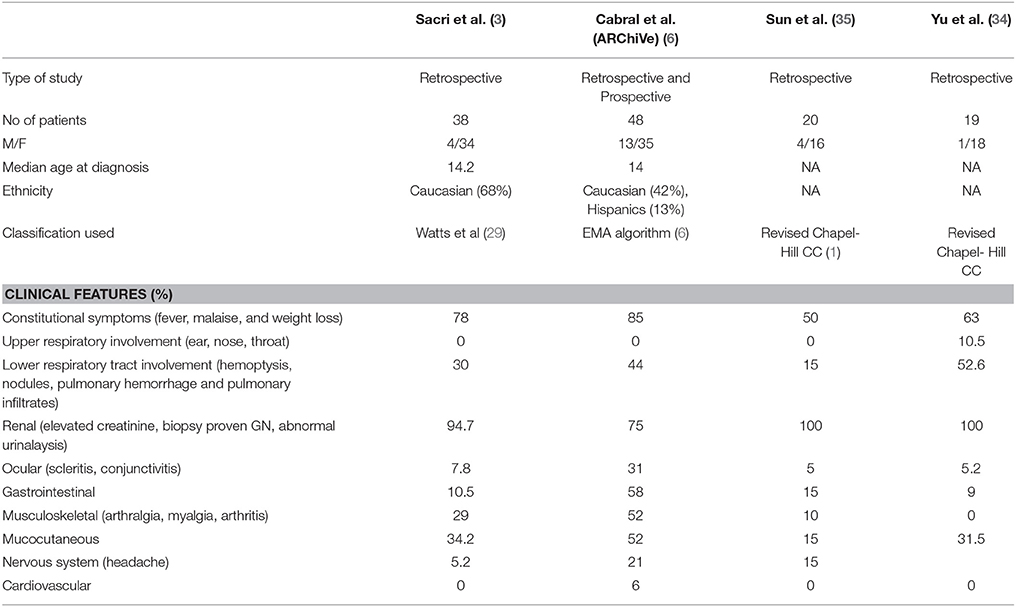
Table 4. Clinical features of MPA in different cohorts at presentation in the largest pediatric cohorts reported.
Disease Activity Scoring
The development of a validated scoring tool to measure disease activity, damage and outcome is crucial for pediatric vasculitis related clinical trials. The Birmingham Vasculits Activity Score (BVAS) is a validated multisystem disease activity tool assessment for primary systemic vasculitis in adults. The latest revision of BVAS is v.3, which was applied to the ARChiVe cohort and showed only weak to moderate correlations with PGA, ESR and treatment decision (38). Dolazelova et al. in 2012 published the Pediatric Vasculitis Activity Score (PVAS) by redefining the BVAS components and adding eight pediatric items in cutaneous, cardiovascular and abdominal sections (39). PVAS has 64 items in nine categories with higher score indicating higher disease activity. PVAS was validated in pediatrics patients with systemic vasculitis and can be used as to define disease activity for clinical trials and future research.
The Vasculitis Damage Index (VDI) is a standardized clinical assessment tool of damage in adult systemic vasculitides (40). There is no validated tool to assess disease damage in children with vasculitis. Pediatric modification of the Vasculitis Damage Index (PVDI) was proposed by Dolezalova et al. in 2014 (41). PVDI contains 72 items in 10 systems. Once validated, PVDI should serve as an important step toward better disease assessment in clinical trials in children with systemic vasculitis.
Treatment
No specific pediatric management guidelines are available to guide the therapeutic approach in pediatric patients with AAV. Therefore, treatment recommendations are adapted from the adult clinical trials and expert consensus (42, 43). Survival rates in AAV has improved secondary to better disease management, the expertise of care teams at academic and referral vasculitis centers and treatment based on intensive remission induction followed by maintenance therapy. The Canadian Vasculitis research network (CanVasc) recommends that children with newly diagnosed AAV should be treated as per adult recommendations for induction of remission and then maintenance (43). Glucocorticoids and cyclophosphamide has been associated with dramatic improvement in patients with AAV, however this combination has not prevented relapses in the majority of patients and is associated with short and long term toxicity risk (44).
Remission Induction for Newly Diagnosed Disease
The EULAR/ERA-EDTA (European League Against Rheumatism/ European Renal Association—European Dialysis and Transplant Association) and CanVasc recommend treatment with a combination of glucocorticoids and either cyclophosphamide (CYC) or rituximab (RTX) (42, 43). RTX is preferred as a first line remission induction therapy for patients in whom CYC is contraindicated or presents a risk of infertility. CYC can be administered either orally or as pulse intravenous dose (3–6 months) but the latter is preferred as it is associated with less cumulative dose and reduced risk of bladder-related complications. However, daily oral low-dose CYC is associated with a slightly lower rate of relapse on long-term follow up (45).
RTX was shown to be non-inferior to CYC at inducing remission in adults with organ or life-threatening disease (46, 47). RTX is usually administered as 4 weekly infusions of 375 mg/m2 as recommended in the RAVE (Rituximab in ANCA- Associated Vasculitis) (46) and RITUXVAS (Rituximab versus Cyclophosphamide in ANCA-Associated Renal Vasculitis) trials (47). RTX is being increasingly used in children with AAV as a first line induction therapy compared to CYC (48) because of reduced toxicity.
Glucocorticoids (GC) remain an important therapy in remission induction and maintenance. No RCTs have been published comparing different GC dosing. In life-threatening disease or those with major organ involvement, pulsed IV methylprednisolone 0.5–1 g/day for 3 consecutive days is recommended. Most adult guidelines recommend initial GC dose of 1 mg/kg/day and tapering to a desirable level of reaching a target GC dose of 10–12.5 mg by 3–5 month. In children (<15 years old), the initial dose of oral prednisone used is 1–2 mg/kg/day with a maximum of 60 mg/day (43). Daily calcium (500–1000 mg) and vitamin D (1000 IU) supplementation is recommended.
Remission-Induction of Limited or Non-Severe Non-Organ-Threatening AAV
Methotrexate (MTX) is recommended in combination with GC in those with limited or non-organ threatening AAV. In the NORAM trial (Non-renal Wegener's Granulomatosis Treated Alternatively with Methotrexate [MTX]), MTX was reported non-inferior to oral CYC in achieving remission induction but subsequent reports indicated less effective disease control than CYC-based induction therapy (49, 50). Mycophenolate mofetil (MMF) was compared to IV CYC in non-severe GPA in MYCYC (mycophenolate mofetil versus cyclophosphamide for remission induction of ANCA-associated vasculitis) trial. MMF was noted to be non-inferior to CYC but was associated with a higher rate of relapse (51).
Remission Maintenance Therapy
The CanVasc as well as EULAR/ERA-EDTA recommends remission maintenance treatment with a combination of low-dose glucocorticoids and either azathioprine (AZA), rituximab, methotrexate or mycophenolate mofetil. This therapy for AAV be continued for at least 24 months following induction of sustained remission.
In the Cyclophosphamide versus Azathioprine for Early Remission Phase of Vasculitis (CYCAZAREM) trial, AZA was shown to be equally efficacious as continuous CYC as maintenance treatment. This regime was also associated with fewer side-effects (52). MTX was shown to be well tolerated and effective in maintaining remission after induction with CYC and was proven to be of comparable efficacy to AZA (53, 54). Leflunomide, though associated with increased frequency of adverse events, was found to be more effective than methotrexate in remission maintenance at a dose of 30 mg/day (55). Leflunomide can be used as an alternative agent in patients with intolerance to MTX and AZA. In the International Mycophenolate Mofetil Protocol to Reduce Outbreaks of Vasculitides (IMPROVE) trial, MMF was shown to be less effective than azathioprine for maintaining disease remission (56).
The role of RTX in maintenance therapy has been investigated in patients with AAV after inducing remission either with CYC or RTX. It is considered a safe and effective alternative to AZA. The MAINRITSAN (Efficacy Study of Two Treatments in the Remission of Vasculitis) was the first randomized trial comparing RTX (500 mg at remission, at 2 weeks and then once every 6 months till 18 months) to daily AZA (which was tapered after 12 months). Patients receiving RTX had sustained remission compared to AZA without significant adverse events (57). MAINRITSAN 2 (https://clinicaltrials.gov/ct2/show/NCT01731561) explores the RTX maintenance treatment based on ANCA estimation and CD19 lymphocytes. The RITAZAREM trial is planned to evaluate two remission-maintenance strategies of repeated doses of RTX compared to daily orally administered AZA for 24 months following induction with RTX (58).
Relapsing Disease
Both the CanVasc and EULAR/ERA-EDTA guidelines recommend switching from RTX to CYC and vice versa for relapsing AAV. In those who continue to have persistent active disease, intravenous immunoglobulin may be used as an adjunctive therapy.
Role of Plasmapheresis (PLEX)
The role of PLEX in AAV is not well defined. It is recommended to be used for rapidly progressive glomerulonephritis in the setting of new or relapsing disease or for the treatment of severe diffuse alveolar hemorrhage (42). The largest trial investigating role of PLEX, the MEPEX trial (High-Dosage Methylprednisolone or Plasma Exchange as Adjunctive Therapy for Severe Renal Vasculitis) (59) showed an increased rate of renal recovery in AAV patients presenting with renal failure when compared with intravenous methylprednisolone. However, this trial enrolled patients who were dialysis dependent or nearing end stage renal disease and it was unable to identify role of PLEX as an adjunctive to conventional therapies. Long-term follow-up of the same cohort failed to identify sustained benefit in PLEX group (60). PEXIVAS is an international study which enrolled patients with AAV with severe renal vasculitis and/or diffuse alveolar hemorrhage. This study aims to determine if the adjunctive plasma exchange with two oral glucocorticoid regimens (standard- and reduced-dose GC) with standard remission induction immunosuppression is effective in reducing death and end-stage renal disease (61).
As mentioned above, clinical trials have not been conducted in pediatric GPA. The data from ARChiVe cohort highlights use of GC and CYC pulses most commonly used as a part of induction therapy (both for GPA and MPA) followed most frequently with methotrexate as a maintenance regime (6). However, most pediatric patients are now treated according to the adult recommendations (42, 43). Many North American centers now prefers RTX as first-line remission induction therapy in children with severe GPA or MPA as it has low toxicity profile. PLEX should be considered in children with severe pulmonary hemorrhage or rapidly progressive renal disease responding inappropriately to GC and CYC or RTX.
Emerging Therapy in ANCA Vasculitis
The better understanding of the pathogenesis of ANCA vasculitis from in vitro and animal studies have helped us to identify the targeted therapies focusing on components of innate and adaptive immune system.
Belimumab (BEL) was investigated in a phase III, multicenter randomized double-blind trial evaluating its role the maintenance of remission in GPA and MPA in combination with AZA (BREVAS: NCT01663623). In BREVAS trial, addition of BEL to maintenance therapy with AZA did not reduce the risk of relapse. Fewer relapses of vasculitis was identified in RTX induced patients compared with placebo (62).
Abatacept, a fusion protein co-stimulatory T cell blocker was evaluated in an open labeled study in non-severe relapsing GPA. Almost 90% patient had disease improvement and >70% patients could discontinue prednisone. The study was limited by a small sample size, continuing background DMARDS and prednisone early during the study (63). Abatacept is currently being investigated in a multicenter, phase III, double-blind, placebo-controlled trial in the treatment of non-severe AAV (ABROGATE: NCT02108860).
The role of blocking complement component/receptor has been explored. Avacopan (CCX168/ selective C5a receptor inhibitor) was investigated in a phase II randomized, placebo-controlled trial (CLEAR: NCT01363388). Results from this study indicate both treatment groups receiving CCX168 were non-inferior to the standard induction and high-dose prednisone. These results highlight the importance of C5a as an important inflammatory mediator in AAV and can be used in future as an alternative to the use of oral glucocorticoids (64). The efficacy of Avacopan is now being evaluated in a larger phase III randomized, double-blind, active-controlled study (ADVOCATE: NCT02994927). Eculizumab is a long-acting humanized monoclonal antibody targeted against complement C5 and inhibits the deployment of the terminal complement system including the formation of membrane attack complex. It has been used successfully in a case report as an add-on treatment with an excellent clinical response with complete recovery of renal function (65).
Gusperimus (15-deoxyspergualin) inhibits mainly T-cell maturation and cytotoxic T-cell proliferation. It was used in a small open labeled trial with high response rates in refractory GPA (66). Clinical trial assessing the efficacy of gusperimus compared to conventional treatment was prematurely terminated (SPARROW: NCT NCT01446211). IL-6 is expressed and produced at sites of active vasculitis and levels are increased in patients with AAV (67). Case reports of successful treatment with Tocilizumab have been published in literature (67, 68). Clinical trial with TCZ and AAV is currently being considered (69).
Outcome/Prognosis
The mortality rates reported in pediatric series are low. The French registry reported a mortality of 10% (70) as compared to none in a single center series reported by Noone et al. (71) and James et al. (33) in pediatric patients with predominant AAV associated glomerulonephritis and GPA respectively. The French vasculitis study group registry reported an increased incidence of ischemic abdominal pain and damage involving the upper respiratory tract. Childhood AAV relapse rates were also reported much higher requiring longer maintenance therapy compared to adult AAV (72). Important morbidity associated with AAV in pediatric patients includes nasal septal perforation with saddle nose deformity, chronic sinusitis, osteoporosis, chronic kidney disease, end stage renal disease, cystitis, infertility, and avascular necrosis.
Recently published early treatment outcome data, reported under 50% of patients achieving remission at 12 months and 61% with inactive disease at 12 months. Improvement of PVAS score of at least 50% from time of diagnosis to post-induction was seen in 92% of patients. Vasculitis associated disease damage with PVDI scores ≥1 was identified in more than 60% of the patient cohort early in their disease course (73).
Conclusion
AAV are rare life threatening severe illnesses of childhood associated with significant organ damage. The pathogenesis of AAV is unclear but both the innate and adaptive arms of the immune system play a role in the disease causation. The collaborative efforts among pediatric rheumatologist have helped in recognizing common clinical features and treatment choices in these rare AAV illness. The treatment of AAV is extrapolated from the adult studies with cyclophosphamide and glucocorticoids continuing to remain the main choices for induction regime, and rituximab gaining increasing popularity recently. Larger collaborative efforts to conduct international multicenter pediatric clinical trials are required to determine the efficacy of the existing treatment, to devise validated disease activity and damage indices and to better define the long-term outcome of pediatric AAV.
Author Contributions
MJ reviewed the literature and prepared the manuscript. RL reviewed the literature and prepared the manuscript.
Funding
This work was supported by funding from College of Medicine, University of Saskatchewan.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of vasculitides. Arthritis Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
2. Watts RA, Mahr A, Mohammad AJ, Gatenby P, Basu N, Flores-Suarez LF. Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol Dial Transplant. (2015) 30(Suppl. 1):i14–22. doi: 10.1093/ndt/gfv022
3. Sacri AS, Chambaraud T, Ranchin B, Florkin B, See H, Decramer S, et al. Clinical characteristics and outcomes of childhood-onset ANCA-associated vasculitis: a French nationwide study. Nephrol Dial Transplant. (2015) 30(Suppl. 1):i104–12. doi: 10.1093/ndt/gfv011
4. Mossberg M, Segelmark M, Kahn R, Englund M, Mohammad AJ. Epidemiology of primary systemic vasculitis in children: a population-based study from southern Sweden. Scand J Rheumatol. (2018) 47:295–302. doi: 10.1080/03009742.2017.1412497
5. Grisaru S, Yuen GW, Miettunen PM, Hamiwka LA. Incidence of Wegener's granulomatosis in children. J Rheumatol. (2010) 37:440–2. doi: 10.3899/jrheum.090688
6. Cabral DA, Canter DL, Muscal E, Nanda K, Wahezi DM, Spalding SJ, et al. Comparing presenting clinical features in 48 children with microscopic polyangiitis to 183 children who have granulomatosis with polyangiitis (Wegener's): an ARChiVe Cohort Study. Arthritis Rheumatol. (2016) 68:2514–26. doi: 10.1002/art.39729
7. Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. (2010) 120:3209–19. doi: 10.1172/JCI40034
8. Schreiber A, Luft FC, Kettritz R. Membrane proteinase 3 expression and ANCA-induced neutrophil activation. Kidney Int. (2004) 65:2172–83. doi: 10.1111/j.1523-1755.2004.00640.x
9. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA (1990) 87:4115–9. doi: 10.1073/pnas.87.11.4115
10. Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. (2013) 83:129–37. doi: 10.1038/ki.2012.313
11. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. (2014) 10:463–73. doi: 10.1038/nrrheum.2014.103
12. Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. (1994) 120:12–7. doi: 10.7326/0003-4819-120-1-199401010-00003
13. Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. (2008) 14:1088–96. doi: 10.1038/nm.1874
14. Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. (2009) 15:623–5. doi: 10.1038/nm.1959
15. Abreu-Velez AM, Smith JG Jr, Howard MS. Presence of neutrophil extracellular traps and antineutrophil cytoplasmic antibodies associated with vasculitides. N Am J Med Sci. (2009) 1:309–13.
17. Soderberg D, Segelmark M. Neutrophil extracellular traps in ANCA-associated vasculitis. Front Immunol. (2016) 7:256. doi: 10.3389/fimmu.2016.00256
18. Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol. (2015) 181:518–27. doi: 10.1111/cei.12654
19. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
20. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
21. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. (1998) 101:890–8. doi: 10.1172/JCI1112
22. Millet A, Martin KR, Bonnefoy F, Saas P, Mocek J, Alkan M, et al. Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J Clin Invest. (2015) 125:4107–21. doi: 10.1172/JCI78182
23. Kantari C, Pederzoli-Ribeil M, Amir-Moazami O, Gausson-Dorey V, Moura IC, Lecomte MC, et al. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood (2007) 110:4086–95. doi: 10.1182/blood-2007-03-080457
24. Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. (2007) 170:52–64. doi: 10.2353/ajpath.2007.060573
25. Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. (1990) 33:1101–7. doi: 10.1002/art.1780330807
26. Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. (2006) 65:936–41. doi: 10.1136/ard.2005.046300
27. Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. Paediatric Rheumatology International Trials, EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. (2010) 69:798–806. doi: 10.1136/ard.2009.116657
28. Uribe AG, Huber AM, Kim S, O'Neil KM, Wahezi DM, Abramson L, et al. Increased sensitivity of the European medicines agency algorithm for classification of childhood granulomatosis with polyangiitis. J Rheumatol. (2012) 39:1687–97. doi: 10.3899/jrheum.111352
29. Watts R, Lane S, Hanslik T, Hauser T, Hellmich B, Koldingsnes W, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. (2007) 66:222–7. doi: 10.1136/ard.2006.054593
30. Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (1984) 63:65–81. doi: 10.1097/00005792-198403000-00001
31. Bohm M, Gonzalez Fernandez MI, Ozen S, Pistorio A, Dolezalova P, Brogan P, et al. Clinical features of childhood granulomatosis with polyangiitis (wegener's granulomatosis). Pediatr Rheumatol Online J. (2014) 12:18. doi: 10.1186/1546-0096-12-18
32. Rottem M, Fauci AS, Hallahan CW, Kerr GS, Lebovics R, Leavitt RY, et al. Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr. (1993) 122:26–31. doi: 10.1016/S0022-3476(05)83482-1
33. James KE, Xiao R, Merkel PA, Weiss PF. Clinical course and outcomes of childhood-onset granulomatosis with polyangiitis. Clin Exp Rheumatol. (2017) 35(Suppl. 103):202–8.
34. Yu F, Huang JP, Zou WZ, Zhao MH. The clinical features of anti-neutrophil cytoplasmic antibody-associated systemic vasculitis in Chinese children. Pediatr Nephrol. (2006) 21:497–502. doi: 10.1007/s00467-006-0028-3
35. Sun L, Wang H, Jiang X, Mo Y, Yue Z, Huang L, et al. Clinical and pathological features of microscopic polyangiitis in 20 children. J Rheumatol. (2014) 41:1712–9. doi: 10.3899/jrheum.131300
36. Akikusa JD, Schneider R, Harvey EA, Hebert D, Thorner PS, Laxer RM, et al. Clinical features and outcome of pediatric Wegener's granulomatosis. Arthritis Rheum. (2007) 57:837–44. doi: 10.1002/art.22774
37. Calatroni M, Oliva E, Gianfreda D, Gregorini G, Allinovi M, Ramirez GA, et al. ANCA-associated vasculitis in childhood: recent advances. Ital J Pediatr. (2017) 43:46. doi: 10.1186/s13052-017-0364-x
38. Morishita K, Li SC, Muscal E, Spalding S, Guzman J, Uribe A, et al. Assessing the performance of the Birmingham vasculitis activity score at diagnosis for children with antineutrophil cytoplasmic antibody-associated vasculitis in A Registry for Childhood Vasculitis (ARChiVe). J Rheumatol. (2012) 39:1088–94. doi: 10.3899/jrheum.111030
39. Dolezalova P, Price-Kuehne FE, Ozen S, Benseler SM, Cabral DA, Anton J, et al. Disease activity assessment in childhood vasculitis: development and preliminary validation of the Paediatric Vasculitis Activity Score (PVAS). Ann Rheum Dis. (2013) 72:1628–33. doi: 10.1136/annrheumdis-2012-202111
40. Exley AR, Bacon PA, Luqmani RA, Kitas GD, Gordon C, Savage CO, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum. (1997) 40:371–80. doi: 10.1002/art.1780400222
41. Dolezalova P, Wilkinson N, Brogan PA, Anton J, Benseler SM, Brunner J, et al. SAT0286 Paediatric vasculitis damage index: a new tool for standardised disease assessment. Ann Rheum Dis. (2014) 73:696. doi: 10.1136/annrheumdis-2014-eular.5893
42. Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis. (2016) 75:1583–94. doi: 10.1136/annrheumdis-2016-209133
43. McGeoch L, Twilt M, Famorca L, Bakowsky V, Barra L, Benseler SM, et al. Canadian vasculitis research, CanVasc recommendations for the management of antineutrophil cytoplasm antibody-associated vasculitides. J Rheumatol. (2016) 43:97–120. doi: 10.3899/jrheum.150376
44. Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. (1992) 116:488–98. doi: 10.7326/0003-4819-116-6-488
45. Harper L, Morgan MD, Walsh M, Hoglund P, Westman K, Flossmann O, et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis. (2012) 71:955–60. doi: 10.1136/annrheumdis-2011-200477
46. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. (2010) 363:221–32. doi: 10.1056/NEJMoa0909905
47. Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. (2010) 363:211–20. doi: 10.1056/NEJMoa0909169
48. James KE, Xiao R, Merkel PA, Weiss PF. Variation in the treatment of children hospitalized with antineutrophil cytoplasmic antibody-associated vasculitis in the US. Arthritis Care Res. (2017) 69:1377–83. doi: 10.1002/acr.23142
49. Faurschou M, Westman K, Rasmussen N, de Groot K, Flossmann O, Hoglund P, et al. Brief Report: long-term outcome of a randomized clinical trial comparing methotrexate to cyclophosphamide for remission induction in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. (2012) 64:3472–7. doi: 10.1002/art.34547
50. De Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. (2005) 52:2461–9. doi: 10.1002/art.21142
51. Jones R, Harper L, Ballarin J, Blockmans D, Brogan P, Bruchfeld A, et al. A randomized trial of mycophenolate mofetil versus cyclophosphamide for remission induction of ANCA-associated vasculitis: “MYCYC”. On behalf of the European vasculitis study group. La Presse Médicale (2013) 42:678–679. doi: 10.1016/j.lpm.2013.02.067
52. Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniene J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. (2003) 349:36–44. doi: 10.1056/NEJMoa020286
53. Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, Kyndt X, Lifermann F, Papo T, Lambert M, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. (2008) 359:2790–803. doi: 10.1056/NEJMoa0802311
54. Langford CA, Talar-Williams C, Barron KS, Sneller MC. Use of a cyclophosphamide-induction methotrexate-maintenance regimen for the treatment of Wegener's granulomatosis: extended follow-up and rate of relapse. Am J Med. (2003) 114:463–9. doi: 10.1016/S0002-9343(03)00077-9
55. Metzler C, Miehle N, Manger K, Iking-Konert C, de Groot K, Hellmich B, Gross WL, et al. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener's granulomatosis. Rheumatology (2007) 46:1087–91. doi: 10.1093/rheumatology/kem029
56. Hiemstra TF, Walsh M, Mahr A, Savage CO, de Groot K, Harper L, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA (2010) 304:2381–8. doi: 10.1001/jama.2010.1658
57. Guillevin L, Pagnoux C, Karras A, Khouatra C, Aumaitre O, Cohen P, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. (2014) 371:1771–80. doi: 10.1056/NEJMoa1404231
58. Gopaluni S, Smith RM, Lewin M, McAlear CA, Mynard K, Jones RB, et al. Rituximab versus azathioprine as therapy for maintenance of remission for anti-neutrophil cytoplasm antibody-associated vasculitis (RITAZAREM): study protocol for a randomized controlled trial. Trials (2017) 18:112. doi: 10.1186/s13063-017-1857-z
59. Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, Mirapeix E, Savage CO, Sinico RA, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. (2007) 18:2180–8. doi: 10.1681/ASN.2007010090
60. Walsh M, Casian A, Flossmann O, Westman K, Hoglund P, Pusey C, Jayne DR, et al. Long-term follow-up of patients with severe ANCA-associated vasculitis comparing plasma exchange to intravenous methylprednisolone treatment is unclear. Kidney Int. (2013) 84:397–402. doi: 10.1038/ki.2013.131
61. Walsh M, Merkel PA, Peh CA, Szpirt W, Guillevin L, Pusey CD, et al. Plasma exchange and glucocorticoid dosing in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis (PEXIVAS): protocol for a randomized controlled trial. Trials (2013) 14:73. doi: 10.1186/1745-6215-14-73
62. Jayne D, Blockmans D, Luqmani R, Ji B, Green Y, Hall L, et al. Efficacy and Safety of Belimumab in Combination with Azathioprine for remission maintenance in granulomatosis with polyangiitis and microscopic polyangiitis: a multicenter randomized, placebo-controlled study. In: 2017 ACR/ARHP Annual Meeting. Hoboken, NJ (2017).
63. Langford CA, Monach PA, Specks U, Seo P, Cuthbertson D, McAlear CA, et al. Vasculitis clinical research, an open-label trial of abatacept (CTLA4-IG) in non-severe relapsing granulomatosis with polyangiitis (Wegener's). Ann Rheum Dis. (2014) 73:1376–9. doi: 10.1136/annrheumdis-2013-204164
64. D.Jayne RW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. (2017) 28:2756–67. doi: 10.1681/ASN.2016111179
65. Manenti L, Urban ML, Maritati F, Galetti M, Vaglio A. Complement blockade in ANCA-associated vasculitis: an index case, current concepts and future perspectives. Intern Emerg Med. (2017) 12:727–31. doi: 10.1007/s11739-017-1636-6
66. Birck R, Warnatz K, Lorenz HM, Choi M, Haubitz M, Grunke M, et al. 15-Deoxyspergualin in patients with refractory ANCA-associated systemic vasculitis: a six-month open-label trial to evaluate safety and efficacy. J Am Soc Nephrol. (2003) 14:440–7. doi: 10.1097/01.ASN.0000048716.42876.14
67. Berti A, Cavalli G, Campochiaro C, Guglielmi B, Baldissera E, Cappio S, et al. Interleukin-6 in ANCA-associated vasculitis: rationale for successful treatment with tocilizumab. Semin Arthritis Rheum. (2015) 45:48–54. doi: 10.1016/j.semarthrit.2015.02.002
68. Sakai R, Kondo T, Kurasawa T, Nishi E, Okuyama A, Chino K, et al. Current clinical evidence of tocilizumab for the treatment of ANCA-associated vasculitis: a prospective case series for microscopic polyangiitis in a combination with corticosteroids and literature review. Clin Rheumatol. (2017) 36:2383–2392. doi: 10.1007/s10067-017-3752-0
69. Harigai M, Arimura Y, Tsutsumino M. A Clinical Trial Of Tocilizumab For Microscopic Polyangiitis And Granulomatosis With Polyangiitis, Rheumatology. Oxford: Oxford Univ Press (2017).
70. Iudici M, Puechal X, Pagnoux C, Quartier P, Agard C, Aouba A, et al. Brief Report: childhood-onset systemic necrotizing vasculitides: long-term data from the french vasculitis study group registry. Arthritis Rheumatol. (2015) 67:1959–65. doi: 10.1002/art.39122
71. Noone DG, Twilt M, Hayes WN, Thorner PS, Benseler S, Laxer RM, et al. The new histopathologic classification of ANCA-associated GN and its association with renal outcomes in childhood. Clin J Am Soc Nephrol. (2014) 9:1684–91. doi: 10.2215/CJN.01210214
72. Iudici M, Pagnoux C, Quartier P, Buchler M, Cevallos R, Cohen P, et al. Vasculitis study, childhood- versus adult-onset ANCA-associated vasculitides: A nested, matched case-control study from the French Vasculitis Study Group Registry. Autoimmun Rev. (2018) 17:108–14. doi: 10.1016/j.autrev.2017.11.014
Keywords: childhood vasculitis, microscopic polyangiitis, granulomatosis with polyangiitis, ANCA—associated vasculitis, small vessel vasculitis
Citation: Jariwala MP and Laxer RM (2018) Primary Vasculitis in Childhood: GPA and MPA in Childhood. Front. Pediatr. 6:226. doi: 10.3389/fped.2018.00226
Received: 18 June 2018; Accepted: 25 July 2018;
Published: 16 August 2018.
Edited by:
Christian Michael Hedrich, University of Liverpool, United KingdomReviewed by:
Klaus Tenbrock, RWTH Aachen Universität, GermanyHermann Girschick, Vivantes Klinikum, Germany
Copyright © 2018 Jariwala and Laxer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mehul P. Jariwala, bWVodWwuamFyaXdhbGFAdXNhc2suY2E=