 Charalampia Papadopoulou
Charalampia Papadopoulou Liza J. McCann
Liza J. McCann- 1Infection, Inflammation, and Rheumatology Section, UCL Great Ormond Street Institute of Child Health, London, United Kingdom
- 2Great Ormond Street Hospital NHS Foundation Trust, London, United Kingdom
- 3Department of Pediatric Rheumatology, Alder Hey Children's NHS Foundation Trust, Liverpool, United Kingdom
Juvenile dermatomyositis (JDM) is a rare autoimmune disease mainly characterized by muscle and skin involvement. Vasculopathy is considered central to the pathogenesis of the disease. The exact nature of vasculopathy is not yet understood but it is a complex process with both an inflammatory and a non-inflammatory, occlusive component. Impaired function of JDM vasculature includes immune complex deposition, altered expression of cell adhesion molecules predominantly inducing Th17 cell infiltration, and endothelial cell dysfunction. Development of vasculopathy is associated with the severe extra-muscular manifestations of JDM, such as gastrointestinal and cardiac manifestations, interstitial lung disease, ulcerative skin disease or development of calcinosis, and portends a poor prognosis. Correlation of histopathological findings, autoantibodies, and extensive diagnostic workup represent key elements to the early detection of vasculopathic features and early aggressive treatment. Monitoring of vasculopathy remains challenging due to the lack of non-invasive biomarkers. Current treatment approaches provide variable benefit, but better understanding of the essential pathogenic mechanisms should help lead to improved outcomes. Whilst acknowledging that evidence is limited, this review aims to describe the vasculopathy of JDM in the context of pathophysiology, clinical features, and treatment of disease.
Introduction
Idiopathic Inflammatory Myopathies (IIM) consist of a group of highly heterogenous diseases characterized by a systemic inflammatory process. Muscle weakness and skin involvement are common characteristics but other organs can also be affected (1). Juvenile Dermatomyositis (JDM) is the commonest childhood IIM seen in ~85% of cases, while polymyositis consists of <5% of the pediatric IIM cases (2). The reported annual incidence of JDM ranges between two to four cases per one million children per year (2). It is more common in females compared to males, with reported female: male ratios ranging between 2:1 and 5:1 in different cohorts (2).
The exact etiology of JDM is not fully understood and is thought to be multifactorial. The prevailing hypothesis is that JDM is the result of genetic susceptibility and environmental triggers, which subsequently cause dysregulation and dysfunction of the immune system resulting in tissue inflammation (3). Vasculopathy seems to play a central role in the pathogenesis of myositis and cutaneous manifestations (4), but is also central to other severe systemic features that contribute significantly to the burden of disease in children. The vasculopathy associated with JDM is thought to underlie the development of intestinal ischaemia and perforation, cutaneous ulceration, interstitial lung disease, and calcinosis (5, 6).
The identification of vascular involvement in JDM is challenging, and depends on extensive immune, histopathologic and imaging diagnostic approaches together with clinical experience and expertise. It may influence choice of therapeutic strategy and prediction of individual patient prognosis. Therefore, to better understand the pathophysiology, histopathological findings, clinical manifestations, and associated complications is of great clinical significance.
Pathophysiology
The exact nature of vasculopathy remains unclear and is likely to change at different stages of the disease. Early on there is evidence of a true inflammatory small vessel vasculitis driven by interferons and other cytokines (4, 7); but also a later, non-inflammatory occlusive vasculopathy with capillary drop out (5, 8). Necrotising capillaritis is seen with complement deposition in lesional muscle biopsies suggestive of a small vessel vasculitis mediated by immune complexes (4, 9) (Figures 1A,B). Immunoglobulin and complement have been identified in the vessel walls of skeletal muscle in children with JDM (4). Endothelial cells in the affected muscle produce IL-1 along with other cytokines (10) and promote inflammation through paracrine upregulation of endothelial intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (11). Moreover, activated endothelium expresses binding sites for various chemokines, resulting in attraction of other inflammatory cells, and has an important effect on angiogenesis. Angiostatic chemokines are expressed at high levels in biopsy specimens of untreated JDM patients, correlating with the degree of capillary loss and mononuclear cell infiltration (12). Neovascularization may then occur later in the disease process (13).
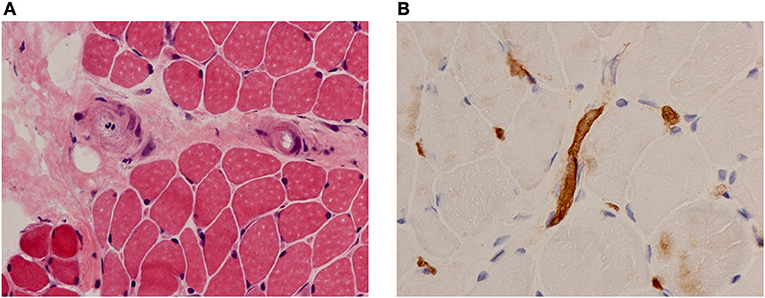
Figure 1. Muscle biopsy findings in a patient with JDM. (A) Haematoxylin and eosin (H&E) stain showing vessel endothelial swelling. (B) Immunohistochemical staining for membrane attack complex (MAC) showing deposition on muscle capillaries. Magnification 40x.
Activated dendritic cells present within lesional muscle release interferons which have a range of biological effects on the endothelium, such as increased expression of major histocompatibility complex (MHC) class I and class II and adhesion molecules promoting T-cell migration (7, 12, 14, 15), such as macrophage inflammatory protein (MIP), monocyte chemoattractant protein (MCP)-1, and MCP-2. Involvement of T cells occurs via lesional T helper type 17 (Th17) cells. The participation of Th17 cells also seems to lead to induction of IL-6 and IL-17, which correlate with the interferon response and with active disease. Indeed, interferons are considered central to the pathogenesis of JDM and important drivers of the associated vasculopathy (7).
Later in the course of JDM, the vasculopathy is characterized by endothelial cell swelling, necrosis, and luminal occlusion of capillaries and arterioles (5, 8). Another common and well documented observation in JDM is capillary drop-out, which is the predominant vasculopathic feature observed in muscle biopsies and in the skin (8, 16, 17). Occlusive vasculopathy also contributes significantly to many of the severe late sequelae of JDM including cutaneous ulceration, intestinal ischaemia, and ultimately gut infarction (18). It is possible that occlusive vasculopathy contributes to chronic subcutaneous calcinosis (19).
The vasculopathy of JDM may present with different clinical phenotypes. The extent of symptoms includes mild forms limited to cutaneous vessels, and severe, life-threatening manifestations with organ involvement. The following section addresses the pathophysiology and clinical manifestations of disease relating them where possible to vasculopathy.
Cutaneous Manifestations
JDM is associated with a wide range of skin rashes, illustrated and described in detail by Dugan et al. (20), but most pathognomonic is the heliotrope rash involving the upper eye-lids and Gottron's papules over the small joints of the hands and large joints (Figures 2A,B). Periungual erythema and nailfold capillary loop changes are often present and palmar erythema can be seen (20) (Figure 2C). Skin ulceration is a serious manifestation of JDM that can be life threatening. Ulcers presumably mirror significant vasculopathy in the skin, caused by hypoxia and ischaemia of the affected tissues and may signal vasculopathy in other organs (i.e., intestinal ischaemia and perforation, pulmonary fibrosis and interstitial lung disease) (21, 22). Patients with ulcerative skin disease are considered to have a more severe disease course with a worse long-term prognosis (23). Vasculitic ulcers are present in 23–30% patients (24) and can be seen in particular at the corner of the eyes (usually heal leaving a chicken-pox-like scar), and over elbows or other pressure points (5). Periorbital or generalized edema has been reported in 32% of JDM cases in the UK cohort (25); usually present at diagnosis or during significant disease flare. It can relate to a more severe disease course and indicate resistance to treatment (26). It is considered the result of a generalized capillary leak due to damage of the vascular endothelium (26).
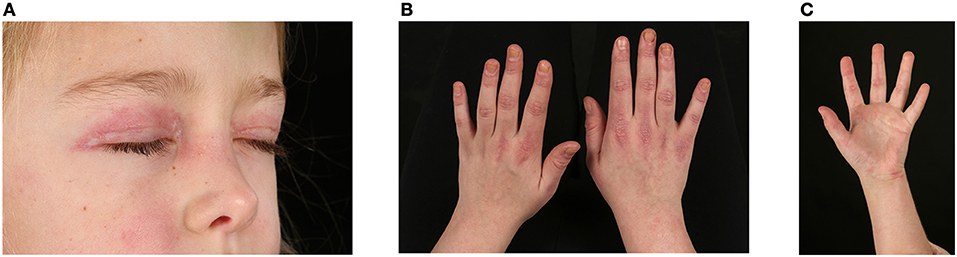
Figure 2. Skin changes seen in juvenile dermatomyositis. (A) Heliotrope rash – erythema involving both upper eyelids in a patient with JDM. Written informed parent consent was obtained for the publication of this image. (B) Gottron's papules over metacarpal and interphalangeal joints with linear extensor erythema. (C) Palmar vasculopathy- palmar erythema, most prominent over the joint creases.
Dystrophic calcification of tissues occurs in 20–40% of patients with JDM, although seen much less frequently in adults with DM (19). Calcinosis can be complicated by ulceration of the overlying skin, contractures of the joints when crossing joint margins (27), pain due to entrapment neuropathy, or topical inflammation with redness, tenderness, and swelling (24, 28). Nailfold capillary changes seen in JDM include dilatation, occlusion, bushy loops, hemorrhages, and capillary drop-out (29), and are present in 80–91% of children at the time of diagnosis. Nailfold capillaroscopy is a non-invasive technique that provides quantitative information about the loss of capillary end-row loops (ERL), areas of decreased or absent vascularity, and formation of arboreal loops (30). Visualization of the nailfolds can be easily performed at the bedside with the use of a light and magnification from a dermatoscope, ophthalmoscope, or auroscope with or without water soluble gel (24). The skin around the nail beds can be erythematous (peri-ungual erythema) and sensitive. Cuticular overgrowth can be seen. Nailfold changes correlate with prolonged disease course, overall disease activity, skin disease activity, and poor response to treatment (30, 31). A similar vessel pattern has been observed in the marginal gingiva that could be part of the vasculopathy associated with JDM (32).
Cardiac Involvement
Recent case-control echocardiographic studies in children with JDM have suggested that up to 25% of patients have evidence of subclinical left ventricular diastolic dysfunction, and a high prevalence of pathological electrocardiographic (ECG) abnormalities (33). Children with JDM have also been found to have reduced heart rate variability which was associated with impaired myocardial function suggestive of reduced cardiac vagal control (34). Whilst studies in children are limited, Rosenbohm et al. demonstrated signs of subclinical myocardial inflammation on cardiac MRI scanning in ~40% of patients with adult onset inflammatory myositis (35), while pericarditis has also been reported (36). Hypertension is seen in 25 to 50% patients probably attributed to both the microvasculopathy of the disease but also corticosteroid treatment (31). In a recent study of adults with juvenile onset DM, subclinical cardiovascular disease and increased carotid intima-media thickness was demonstrated suggestive of early onset atherosclerosis despite their young age and absence of other traditional cardiovascular risk factors (37). Persistence of skin disease activity was shown to correlate with persistence of cardiac dysfunction in a small follow-up study, suggesting that a common vasculopathic process affects the skin and myocardium (30). The significance of this subclinical cardiac involvement and potential contribution to cardiovascular morbidity is currently unknown.
Accelerated atherosclerosis has been demonstrated as an important factor of mortality and morbidity in patients with autoimmune diseases (38). A recent study comparing patients with rheumatoid arthritis and patients with diabetes found equal frequency of atherosclerosis between the two diseases (39). Regardless of the exact nature of small vessel vasculopathy in JDM, in the longer term there may be a generalized secondary systemic vasculopathy affecting larger arteries, ultimately leading to accelerated atherosclerosis and premature cardiovascular morbidity later in adulthood. This could be the result of the primary chronic vasculopathic nature of the disease causing “polyangiitis overlap,” and/or uncontrolled chronic systemic inflammation, as in other autoimmune diseases (40). In support of this, studies of adults with dermatomyositis demonstrate an almost twice-higher risk of acute myocardial infarction and stroke when compared to the general population (41, 42). Interestingly, a small pilot study compared carotid intima-media thickness (CIMT) and flow-mediated dilatation (FMD) as surrogate markers of atherosclerosis in 8 adults with a history of JDM and revealed increased CIMT in JDM patients compared to 8 healthy controls despite the young age (37). Moreover, recently a large retrospective study demonstrated that JDM was associated with higher odds of cardiovascular and cerebrovascular disease in adolescents, including atherosclerosis, transient ischaemic attacks and cerebral infarction (43). It is thus possible that the combination of chronic endothelial injury caused by persistent small vessel vasculitis, chronic systemic inflammation, long-term corticosteroid use, sedentary lifestyle, and conventional cardiovascular risk factors predispose patients with JDM to early atherosclerosis.
Gastrointestinal Tract Involvement
Gastrointestinal tract involvement occurs in 5–37% of JDM cases (24). This includes dysphagia, bowel dysmotility, vasculitis with associated malabsorption, and other more severe features of gastro-intestinal vasculopathy that can be life threatening. Vasculopathy in the gastrointestinal tract may present with abdominal pain, rectal bleeding, intestinal ischaemia, pneumatosis, and finally perforation (44). Acute inflammatory vasculitis and chronic gastrointestinal occlusive arteriopathy have been described in JDM patients, indicating that the underlying pathology is complex and multifactorial (21). Initially inflammatory cells seem to infiltrate the gastric mucosa and play a significant role in pathogenesis. Persistent severe abdominal pain is a worrying sign that warrants prompt investigation and radiographic imaging. The main radiographic finding is thickening of the bowel mucosal folds demonstrated by barium swallow and follow-through studies (45).
Pulmonary Vasculitis
Pulmonary involvement is much less common in children with JDM than adult-onset IIM, but interstitial lung disease (ILD) may still complicate childhood cases (46). Connective tissue disease associated-ILD is thought to be initiated by microvascular injury leading to endothelial cell damage and alveolar epithelial injury (47) leading to the release of numerous cytokines and growth factors which play a key role in the development of lung disease. The exact mechanisms are still not fully understood. Abnormal pulmonary function tests are recorded in more than half of JDM cases (48). In particular, diffusion capacity may be decreased at early stages of ILD. Progression can be rapid and may be life-threatening unless treated aggressively (49). High serum anti-melanoma differentiation-associated gene 5 (MDA5) antibodies and anti-synthetase antibodies are associated with rapidly progressive interstitial lung disease. Typically, ILD presents with increasing cough and progressive dyspnoea, although it may be asymptomatic. Diffuse alveolar hemorrhage (DAH) and pneumomediastinum are rare but life-threatening manifestations of JDM (50–52). Interstitial fibrosis, vasculitis and infarction have been suggested as possible mechanisms in adults with DM (53).
Other Organ Involvement
Although rare, vasculopathy of the central nervous system has been reported in children with JDM (54, 55). It can be very difficult to diagnose this life-threatening complication. It most commonly presents with hallucinations and seizures, and MRI/MRA can be useful diagnostic tools (55). Most reported cases had active retinal vasculitis (54, 55) suggesting that the eyes represent another organ that can be affected by JDM vasculopathy. However, a retrospective review of 82 patients at a single center demonstrated that retinopathy was rare and concluded that routine assessment was not warranted for patients without visual symptoms (56).
Myositis Specific Antibodies
Myositis specific antibodies (MSA) and myositis associated antibodies (MAA) are present in at least 60% of JDM cases (57). Different types of antibodies are associated with specific clinical manifestations but phenotype may differ according to the age of the patient at time of disease onset (57). The commonest MSAs in JDM patients include anti-TIF1γ, anti-NXP2 and anti-MDA5. Anti-TIF1γ (p155/140) is reported in 23–29% of patients with JDM and is associated with more severe skin disease, development of skin ulceration and a long disease course (58), while anti-NXP2 (p140) is mainly connected with the development of calcinosis in pediatric cases (59). Anti-MDA5 antibodies are associated with mild myositis, arthritis mainly of the small joints, ulceration of the skin and increased risk of the disease to be complicated with interstitial lung disease (60). Recently, a number of anti-endothelial cell antibodies (AECA) have been identified in the plasma of JDM cases (61), which are typical of vasculitis and vascular thrombosis in lupus patients (62). Their clinical and diagnostic significance remains unclear and needs to be further elucidated. It is not known if specific antibodies pre-exist the development of the specific disease features with which they are associated; if so, they could be useful as prognostic biomarkers and guide further therapeutic management (57). In support of this, Deakin et al. demonstrated that MSA in combination with muscle biopsies severity scores can be predictive of long-term treatment status and prognosis in children with JDM (63).
Treatment
The intensity of initial therapy is determined by the severity of the presenting symptoms (64) including the presence of life-threatening weakness, major organ involvement, ulcerative skin lesions or extensive calcinosis. To date, management approaches for JDM cases have been mainly based on small case series and anecdotal experience as only a few randomized clinical trials exist to guide decisions. In an attempt to standardize treatment strategies, several therapeutic algorithms for the treatment of JDM have been published based on expert consensus in North America and Europe (65–69). The Childhood Arthritis and Rheumatology Research Alliance (CARRA) have developed Consensus Treatment Plans (CTPs) for the initial treatment of JDM (69), treatment after the first 3 months (68), treatment of skin predominant disease (65), and persistent skin rash (66). It is important to highlight that these CTPs do not consist of therapeutic guidelines per se, but they represent arms for proposed comparative effectiveness studies, developed via consensus methodologies. Moreover, evidence-driven consensus-based recommendations for diagnosis and treatment of JDM have also been published as part of a European initiative called Single Hub and Access point for pediatric Rheumatology in Europe (SHARE) (67).
Delayed or inadequate treatment has been associated with poorer prognosis and early aggressive treatment has been demonstrated to improve the long-term outcome (70). Pharmacological interventions for JDM include corticosteroids, disease-modifying drugs (DMARDs), and biologics agents. Different steroid regimes have been proposed for the initial treatment of JDM with the most commonly used being oral prednisolone or intravenous (IV) pulses with methylprednisolone. IV methylprednisolone is preferred for patients with evidence of active vasculopathy as it has been demonstrated that the absorption of oral prednisolone may be decreased in children with evidence of loss of nail-fold capillary ERL by capillaroscopy (71). The exact role of the two different regimes in the treatment of JDM needs to be further elucidated (72). Regardless of the regime used, corticosteroid dose is then tapered over a period of months depending on the response. Methotrexate (MTX) has become the first line treatment of JDM, recommended in combination with corticosteroids at time of diagnosis (66). Ciclosporin has also been used in many centers as a steroid-sparing agent. In a recent randomized controlled trial (RCT) where monotherapy with corticosteroids was compared with the combination of prednisone with either MTX or ciclosporin, combination therapy had greater efficacy than corticosteroid alone. More adverse effects were seen with ciclosporin than MTX, supporting the usual practice of corticosteroid and MTX as first line treatment (73). Intravenous immunoglobulin (IVIG) is recommended for patients with JDM refractory to steroids and methotrexate (67, 68). An alternative DMARD suggested for severe/refractory cases of JDM or where methotrexate is not tolerated, is mycophenolate mofetil (67), beneficial for both muscle and skin disease. MMF has also been shown to be effective in adult patients with myositis associated interstitial lung disease (ILD) not responding well to steroid treatment (74). Azathioprine is less commonly used in JDM (75).
Several therapeutic agents have been proposed for the treatment of severe/refractory JDM cases. A randomized trial that included 152 adults with myositis and 48 children with JDM demonstrated that 83% of patients met the definition of improvement on rituximab (76), even though the study failed to reach its primary or secondary endpoints possibly due to limitations concerning the trial design (77). The overall response rate, steroid-sparing effect and re-treatment response suggests that rituximab has a beneficial effect, particularly in children and those with antibody positive disease (76). In 2008, a case series reported the efficacy of infliximab in children with refractory JDM and development of calcinosis (78). Evaluation of 66 JDM patients, recruited from the UK JDM Cohort and Biomarker Study and actively treated with anti-TNF agents, both infliximab and adalimumab showed significant improvements in overall disease activity, including both muscle and skin involvement (79). In contrast, etanercept failed to demonstrate efficacy in two prospective case studies in adult and juvenile dermatomyositis, respectively (80, 81). There have been reports of TNF inhibitor induction or exacerbation of DM (82, 83) with the exact pathophysiological mechanism not yet understood. Abatacept, a soluble fusion protein comprising the extracellular domain of human cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) and a fragment of the Fc domain of human immunoglobulin G1, has been reported to be effective in a recalcitrant JDM case with ulcerations and calcinosis (84) and a clinical trial (NCT number: NCT02594735) is currently underway.
Cyclophosphamide (CyC) is another third line therapeutic agent mainly reserved for JDM cases refractory to most other therapies and for cases with significant organ involvement such as cardiopulmonary involvement, skin ulceration or gastrointestinal vasculopathy. Several case studies demonstrate the efficacy of treatment with CyC in both pediatric and adult patients with IIMs (74, 85). A recent analysis of a UK cohort of JDM patients treated with CyC indicated good efficacy and low rates of adverse events (86).
As JDM is now increasingly recognized as a condition that falls into the category of diseases driven by interferons (87), JAK inhibition has been suggested as a potential new targeted therapeutic regime in refractory cases of adult dermatomyositis (88).
Conclusions
JDM represents the commonest idiopathic inflammatory myopathy of childhood. When looking at affected tissues, vascular and perivascular inflammation is a predominant feature of the disease, thus early descriptions recognize JDM as a systemic angiopathy of the childhood. The presence and persistence of vascular involvement is associated with more severe and refractory disease and the development of life-threatening complications. With recent therapeutic strategies, survival, and outcomes of JDM have improved significantly with a reported mortality of <2% (89), but the long-term outcome differs substantially between patients. The exact pathophysiology of JDM vasculopathy remains unclear and may change over the course of disease. Complement deposition and endothelial cells activation with pronounced expression and activation of adhesive molecules are considered key factors in the pathogenesis of the disease, while the role of autoantibodies needs to be further elucidated. Early recognition of vasculopathic features and early aggressive treatment are key components to a better outcome. Predicting and monitoring the development of vasculopathy throughout the disease course remains challenging. Current therapeutic regimes still lack specificity and in severe cases fail to control disease activity. Targeting candidate immune pathways that may be contributing to disease pathogenesis is an active area of research and an unmet need.
Author Contributions
CP and LM have contributed to the draft and made significant intellectual contribution to the generation of the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors would like to thank the patients and their families for permission to use the images. We would also wish to acknowledge Dr Thomas Jacques and Dr Shireena Yasin, Developmental Biology and Cancer Programme, UCL Great Ormond Street Institute of Child Health and Department of Histopathology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, UK, for providing the muscle biopsy images. All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. CP is funded in part by ReMission.
References
1. Wu Q, Wedderburn LR, McCann LJ. Juvenile dermatomyositis: latest advances. Best Prac Res Clin Rheumatol. (2017) 31:535–57. doi: 10.1016/j.berh.2017.12.003
2. Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology. (2015) 54:50–63. doi: 10.1093/rheumatology/keu289
3. Wedderburn LR, Rider LG. Juvenile dermatomyositis: new developments in pathogenesis, assessment and treatment. Best Pract Res Clin Rheumatol. (2009) 23:665–78. doi: 10.1016/j.berh.2009.07.007
4. Whitaker JN, Engel WK. Vascular deposits of immunoglobulin and complement in idiopathic inflammatory myopathy. N Engl J Med. (1972) 286:333–8. doi: 10.1056/NEJM197202172860701
5. Crowe WE, Bove KE, Levinson JE, Hilton PK. Clinical and pathogenetic implications of histopathology in childhood polydermatomyositis. Arthr Rheum. (1982) 25:126–39. doi: 10.1002/art.1780250203
6. McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, et al. The Juvenile Dermatomyositis national registry and repository (UK and Ireland)–clinical characteristics of children recruited within the first 5 yr. Rheumatology. (2006) 45:1255–60. doi: 10.1093/rheumatology/kel099
7. Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. (2007) 13:59–68. doi: 10.2119/2006-00085.Baechler
8. Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol. (1990) 27:343–56. doi: 10.1002/ana.410270402
9. Scott JP, Arroyave C. Activation of complement and coagulation in juvenile dermatomyositis. Arthr Rheum. (1987) 30:572–6. doi: 10.1002/art.1780300513
10. Lundberg I, Ulfgren AK, Nyberg P, Andersson U, Klareskog L. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthr Rheum. (1997) 40:865–74. doi: 10.1002/art.1780400514
11. Sallum AM, Marie SK, Wakamatsu A, Sachetti S, Vianna MA, Silva CA, et al. Immunohistochemical analysis of adhesion molecule expression on muscle biopsy specimens from patients with juvenile dermatomyositis. J Rheumatol. (2004) 31:801–7.
12. Fall N, Bove KE, Stringer K, Lovell DJ, Brunner HI, Weiss J, et al. Association between lack of angiogenic response in muscle tissue and high expression of angiostatic ELR-negative CXC chemokines in patients with juvenile dermatomyositis: possible link to vasculopathy. Arthr Rheum. (2005) 52:3175–80. doi: 10.1002/art.21303
13. Nagaraju K, Rider LG, Fan C, Chen YW, Mitsak M, Rawat R, et al. Endothelial cell activation and neovascularization are prominent in dermatomyositis. J Autoimmune Dis. (2006) 3:2. doi: 10.1186/1740-2557-3-2
14. Lopez de Padilla CM, Vallejo AN, McNallan KT, Vehe R, Smith SA, Dietz AB, et al. Plasmacytoid dendritic cells in inflamed muscle of patients with juvenile dermatomyositis. Arthr Rheum. (2007) 56:1658–68. doi: 10.1002/art.22558
15. Eloranta ML, Barbasso Helmers S, Ulfgren AK, Ronnblom L, Alm GV, Lundberg IE. A possible mechanism for endogenous activation of the type I interferon system in myositis patients with anti-Jo-1 or anti-Ro 52/anti-Ro 60 autoantibodies. Arthr Rheum. (2007) 56:3112–24. doi: 10.1002/art.22860
16. Miles L, Bove KE, Lovell D, Wargula JC, Bukulmez H, Shao M, et al. Predictability of the clinical course of juvenile dermatomyositis based on initial muscle biopsy: a retrospective study of 72 patients. Arthr Rheum. (2007) 57:1183–91. doi: 10.1002/art.22993
17. Wedderburn LR, Varsani H, Li CK, Newton KR, Amato AA, Banwell B, et al. International consensus on a proposed score system for muscle biopsy evaluation in patients with juvenile dermatomyositis: a tool for potential use in clinical trials. Arthr Rheum. (2007) 57:1192–201. doi: 10.1002/art.23012
18. Pachman LM, Hayford JR, Chung A, Daugherty CA, Pallansch MA, Fink CW, et al. Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol. (1998) 25:1198–204.
19. Hoeltzel MF, Oberle EJ, Robinson AB, Agarwal A, Rider LG. The presentation, assessment, pathogenesis, and treatment of calcinosis in juvenile dermatomyositis. Curr Rheumatol Rep. (2014) 16:467. doi: 10.1007/s11926-014-0467-y
20. Dugan EM, Huber AM, Miller FW, Rider LG, International Myositis A, Clinical Studies G. Review of the classification and assessment of the cutaneous manifestations of the idiopathic inflammatory myopathies. Dermatol Online J. (2009) 15:2. Available online at: https://escholarship.org/uc/item/0f26q4rm
21. Mamyrova G, Kleiner DE, James-Newton L, Shaham B, Miller FW, Rider LG. Late-onset gastrointestinal pain in juvenile dermatomyositis as a manifestation of ischemic ulceration from chronic endarteropathy. Arthr Rheum. (2007) 57:881–4. doi: 10.1002/art.22782
22. Pachman LM, Cooke N. Juvenile dermatomyositis: a clinical and immunologic study. J Pediatr. (1980) 96:226–34. doi: 10.1016/S0022-3476(80)80807-9
23. Bowyer SL, Blane CE, Sullivan DB, Cassidy JT. Childhood dermatomyositis: factors predicting functional outcome and development of dystrophic calcification. J Pediatr. (1983) 103:882–8. doi: 10.1016/S0022-3476(83)80706-9
24. Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. (2008) 371:2201–12. doi: 10.1016/S0140-6736(08)60955-1
25. Lowry CA, Pilkington CA. Juvenile dermatomyositis: extramuscular manifestations and their management. Curr Opin Rheumatol. (2009) 21:575–80. doi: 10.1097/BOR.0b013e328331927e
26. Mitchell JP, Dennis GJ, Rider LG. Juvenile dermatomyositis presenting with anasarca: a possible indicator of severe disease activity. J Pediatr. (2001) 138:942–5. doi: 10.1067/mpd.2001.113363
27. Huber AM, Lang B, LeBlanc CM, Birdi N, Bolaria RK, Malleson P, et al. Medium- and long-term functional outcomes in a multicenter cohort of children with juvenile dermatomyositis. Arthr Rheum. (2000) 43:541–9. doi: 10.1002/1529-0131(200003)43:3<541::AID-ANR9>3.0.CO;2-T
28. Moore EC, Cohen F, Douglas SD, Gutta V. Staphylococcal infections in childhood dermatomyositis–association with the development of calcinosis, raised IgE concentrations and granulocyte chemotactic defect. Ann Rheum Dis. (1992) 51:378–83. doi: 10.1136/ard.51.3.378
29. Spencer-Green G, Crowe WE, Levinson JE. Nailfold capillary abnormalities and clinical outcome in childhood dermatomyositis. Arthr Rheum. (1982) 25:954–8. doi: 10.1002/art.1780250807
30. Christen-Zaech S, Seshadri R, Sundberg J, Paller AS, Pachman LM. Persistent association of nailfold capillaroscopy changes and skin involvement over thirty-six months with duration of untreated disease in patients with juvenile dermatomyositis. Arthr Rheum. (2008) 58:571–6. doi: 10.1002/art.23299
31. Gitiaux C, De Antonio M, Aouizerate J, Gherardi RK, Guilbert T, Barnerias C, et al. Vasculopathy-related clinical and pathological features are associated with severe juvenile dermatomyositis. Rheumatology. (2016) 55:470–9. doi: 10.1093/rheumatology/kev359
32. Rider LG, Atkinson JC. Images in clinical medicine. Gingival and periungual vasculopathy of juvenile dermatomyositis. N Engl J Med. (2009) 360:e21. doi: 10.1056/NEJMicm062850
33. Schwartz T, Sanner H, Gjesdal O, Flato B, Sjaastad I. In juvenile dermatomyositis, cardiac systolic dysfunction is present after long-term follow-up and is predicted by sustained early skin activity. Ann Rheum Dis. (2014) 73:1805–10. doi: 10.1136/annrheumdis-2013-203279
34. Barth Z, Nomeland Witczak B, Schwartz T, Gjesdal K, Flato B, Koller A, et al. In juvenile dermatomyositis, heart rate variability is reduced, and associated with both cardiac dysfunction and markers of inflammation: a cross-sectional study median 13.5 years after symptom onset. Rheumatology. (2016) 55:535–43. doi: 10.1093/rheumatology/kev376
35. Rosenbohm A, Buckert D, Gerischer N, Walcher T, Kassubek J, Rottbauer W, et al. Early diagnosis of cardiac involvement in idiopathic inflammatory myopathy by cardiac magnetic resonance tomography. J Neurol. (2015) 262:949–56. doi: 10.1007/s00415-014-7623-1
36. Schwartz T, Diederichsen LP, Lundberg IE, Sjaastad I, Sanner H. Cardiac involvement in adult and juvenile idiopathic inflammatory myopathies. RMD Open (2016) 2:e000291. doi: 10.1136/rmdopen-2016-000291
37. Eimer MJ, Brickman WJ, Seshadri R, Ramsey-Goldman R, McPherson DD, Smulevitz B, et al. Clinical status and cardiovascular risk profile of adults with a history of juvenile dermatomyositis. J Pediatr. (2011) 159:795–801. doi: 10.1016/j.jpeds.2011.05.015
38. Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, du Berger R, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthr Rheum. (2001) 44:2331–7. doi: 10.1002/1529-0131(200110)44:10<2331::AID-ART395>3.0.CO;2-I
39. Stamatelopoulos KS, Kitas GD, Papamichael CM, Chryssohoou E, Kyrkou K, Georgiopoulos G, et al. Atherosclerosis in rheumatoid arthritis versus diabetes: a comparative study. Arterioscler Thromb Vasc Biol. (2009) 29:1702–8. doi: 10.1161/ATVBAHA.109.190108
40. Svenungsson E, Jensen-Urstad K, Heimburger M, Silveira A, Hamsten A, de Faire U, et al. Risk factors for cardiovascular disease in systemic lupus erythematosus. Circulation (2001) 104:1887–93. doi: 10.1161/hc4101.097518
41. Tisseverasinghe A, Bernatsky S, Pineau CA. Arterial events in persons with dermatomyositis and polymyositis. J Rheumatol. (2009) 36:1943–6. doi: 10.3899/jrheum.090061
42. Zaller B, Li X, Sundquist J, Sundquist K. Risk of subsequent coronary heart disease in patients hospitalized for immune-mediated diseases: a nationwide follow-up study from Sweden. PLoS ONE (2012) 7:e33442. doi: 10.1371/journal.pone.0033442
43. Silverberg JI, Kwa L, Kwa MC, Laumann AE, Ardalan K. Cardiovascular and cerebrovascular comorbidities of juvenile dermatomyositis in US children: an analysis of the National Inpatient Sample. Rheumatology (2018) 57:694–702. doi: 10.1093/rheumatology/kex465
44. Downey EC Jr., Woolley MM, Hanson V. Required surgical therapy in the pediatric patient with dermatomyositis. Arch Surg. (1988) 123:1117–20. doi: 10.1001/archsurg.1988.01400330093014
45. Laskin BL, Choyke P, Keenan GF, Miller FW, Rider LG. Novel gastrointestinal tract manifestations in juvenile dermatomyositis. J Pediatr. (1999) 135:371–4. doi: 10.1016/S0022-3476(99)70137-X
46. Kobayashi I, Yamada M, Takahashi Y, Kawamura N, Okano M, Sakiyama Y, et al. Interstitial lung disease associated with juvenile dermatomyositis: clinical features and efficacy of cyclosporin A. Rheumatology. (2003) 42:371–4. doi: 10.1093/rheumatology/keg040
47. Castelino FV, Varga J. Interstitial lung disease in connective tissue diseases: evolving concepts of pathogenesis and management. Arthritis Res Ther. (2010) 12:213. doi: 10.1186/ar3097
48. Trapani S, Camiciottoli G, Vierucci A, Pistolesi M, Falcini F. Pulmonary involvement in juvenile dermatomyositis: a two-year longitudinal study. Rheumatology. (2001) 40:216–20. doi: 10.1093/rheumatology/40.2.216
49. Park S, Nyhan WL. Fatal pulmonary involvement in dermatomyositis. Am J Dis Child. (1975) 129:723–6.
50. Carneiro da SilvaI E, Monteiro BugniI V, TerreriII M, CastroIII M, IshigaiIV M, HilárioV MOE. Juvenile dermatomyositis (JDM) and severe pulmonary involvement: case report. Rev Bras Reumatol. (2009) 49:462–7. doi: 10.1590/S0482-50042009000400012
51. Dogra S, Suri D, Shah R, Rawat A, Sodhi K, Singh S. Spontaneous pneumomediastinum: a rare complication of juvenile dermatomyositis. Int J Rheum Dis. (2012) 15:e131–e3. doi: 10.1111/j.1756-185X.2011.01603.x
52. Kono H, Inokuma S, Nakayama H, Suzuki M. Pneumomediastinum in dermatomyositis: association with cutaneous vasculopathy. Ann Rheum Dis. (2000) 59:372–6. doi: 10.1136/ard.59.5.372
53. Lim JU, Kang HS, Kim YH, Kim TJ. Amyopathic Dermatomyositis associated with histopathological findings of organizing pneumonia and pulmonary vasculitis. Balkan Med J. (2017) 34:374–7. doi: 10.4274/balkanmedj.2016.1061
54. Ramanan AV, Sawhney S, Murray KJ. Central nervous system complications in two cases of juvenile onset dermatomyositis. Rheumatology. (2001) 40:1293–8. doi: 10.1093/rheumatology/40.11.1293
55. Elst EF, Kamphuis SS, Prakken BJ, Wulffraat NM, van der Net J, Peters AC, et al. Case report: severe central nervous system involvement in juvenile dermatomyositis. J Rheumatol. (2003) 30:2059–63.
56. Akikusa JD, Tennankore DK, Levin AV, Feldman BM. Eye findings in patients with juvenile dermatomyositis. J Rheumatol. (2005) 32:1986–91.
57. Tansley S, Wedderburn LR. Comparing and contrasting clinical and serological features of juvenile and adult-onset myositis: implications for pathogenesis and outcomes. Curr Opin Rheumatol. (2015) 27:601–7. doi: 10.1097/BOR.0000000000000224
58. Gunawardena H, Wedderburn LR, North J, Betteridge Z, Dunphy J, Chinoy H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology. (2008) 47:324–8. doi: 10.1093/rheumatology/kem359
59. Gunawardena H, Wedderburn LR, Chinoy H, Betteridge ZE, North J, Ollier WE, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthr Rheum. (2009) 60:1807–14. doi: 10.1002/art.24547
60. Tansley SL, Betteridge ZE, Gunawardena H, Jacques TS, Owens CM, Pilkington C, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther. (2014) 16:R138. doi: 10.1186/ar4600
61. Karasawa R, Tamaki M, Sato T, Tanaka M, Nawa M, Yudoh K, et al. Multiple target autoantigens on endothelial cells identified in juvenile dermatomyositis using proteomics. Rheumatology. (2018) 57:671–6. doi: 10.1093/rheumatology/kex468
62. Cieslik P, Hrycek A, Klucinski P. Vasculopathy and vasculitis in systemic lupus erythematosus. Pol Arch Med Wewn. (2008) 118:57–63. doi: 10.20452/pamw.306
63. Deakin CT, Yasin SA, Simou S, Arnold KA, Tansley SL, Betteridge ZE, et al. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in Juvenile Dermatomyositis. Arthritis Rheumatol. (2016) 68:2806–16. doi: 10.1002/art.39753
64. Stringer E, Bohnsack J, Bowyer SL, Griffin TA, Huber AM, Lang B, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the childhood arthritis and rheumatology research alliance (CARRA) JDM treatment survey. J Rheumatol. (2010) 37:1953–61. doi: 10.3899/jrheum.090953
65. Kim S, Kahn P, Robinson AB, Lang B, Shulman A, Oberle EJ, et al. Childhood arthritis and rheumatology research alliance consensus clinical treatment plans for juvenile dermatomyositis with skin predominant disease. Pediatr Rheum. (2017) 15:1. doi: 10.1186/s12969-016-0134-0
66. Huber AM, Kim S, Reed AM, Carrasco R, Feldman BM, Hong SD, et al. Childhood arthritis and rheumatology research alliance consensus clinical treatment plans for juvenile dermatomyositis with persistent skin rash. J Rheumatol. (2017) 44:110–6. doi: 10.3899/jrheum.160688
67. Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. (2017) 76:329–40. doi: 10.1136/annrheumdis-2016-209247
68. Huber AM, Robinson AB, Reed AM, Abramson L, Bout-Tabaku S, Carrasco R, et al. Consensus treatments for moderate juvenile dermatomyositis: beyond the first two months. results of the second Childhood Arthritis and Rheumatology Research Alliance consensus conference. Arthritis Care Res. (2012) 64:546–53. doi: 10.1002/acr.20695
69. Huber AM, Giannini EH, Bowyer SL, Kim S, Lang B, Lindsley CB, et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Children's Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res. (2010) 62:219–25. doi: 10.1002/acr.20071
70. Kim S, El-Hallak M, Dedeoglu F, Zurakowski D, Fuhlbrigge RC, Sundel RP. Complete and sustained remission of juvenile dermatomyositis resulting from aggressive treatment. Arthr Rheum. (2009) 60:1825–30. doi: 10.1002/art.24571
71. Rouster-Stevens KA, Gursahaney A, Ngai KL, Daru JA, Pachman LM. Pharmacokinetic study of oral prednisolone compared with intravenous methylprednisolone in patients with juvenile dermatomyositis. Arthr Rheum. (2008) 59:222–6. doi: 10.1002/art.23341
72. Seshadri R, Feldman BM, Ilowite N, Cawkwell G, Pachman LM. The role of aggressive corticosteroid therapy in patients with juvenile dermatomyositis: a propensity score analysis. Arthr Rheum. (2008) 59:989–95. doi: 10.1002/art.23829
73. Ruperto N, Pistorio A, Oliveira S, Zulian F, Cuttica R, Ravelli A, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet (2016) 387:671–8. doi: 10.1016/S0140-6736(15)01021-1
74. Kawasumi H, Gono T, Kawaguchi Y, Yamanaka H. Recent treatment of interstitial lung disease with idiopathic inflammatory myopathies. Clin Med. (2015) 9(Suppl 1):9–17. doi: 10.4137/CCRPM.S23313
75. Jacobs JC. Methotrexate and azathioprine treatment of childhood dermatomyositis. Pediatrics (1977) 59:212–8.
76. Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthr Rheum. (2013) 65:314–24. doi: 10.1002/art.37754
77. de Visser M. The efficacy of rituximab in refractory myositis: the jury is still out. Arthr Rheum. (2013) 65:303–6. doi: 10.1002/art.37758
78. Riley P, McCann LJ, Maillard SM, Woo P, Murray KJ, Pilkington CA. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology. (2008) 47:877–80. doi: 10.1093/rheumatology/ken074
79. Campanilho-Marques R, Deakin C, Simou S, Wedderburn L, Pilkington C. Efficacy and safety of tumour necrosis factor antagonists in a large cohort of juvenile dermatomyositis patients [abstract]. Arthritis Rheumatol. (2016) 68 (suppl 10):1708–9.
80. Iannone F, Scioscia C, Falappone PC, Covelli M, Lapadula G. Use of etanercept in the treatment of dermatomyositis: a case series. J Rheumatol. (2006) 33:1802–4. doi: 10.1016/s0093-3619(08)70497-0
81. Rouster-Stevens KA, Ferguson L, Morgan G, Huang CC, Pachman LM. Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res. (2014) 66:783–7. doi: 10.1002/acr.22198
82. Klein R, Rosenbach M, Kim EJ, Kim B, Werth VP, Dunham J. Tumor necrosis factor inhibitor-associated dermatomyositis. Arch Dermatol. (2010) 146:780–4. doi: 10.1001/archdermatol.2010.142
83. Liu SW, Velez NF, Lam C, Femia A, Granter SR, Townsend HB, et al. Dermatomyositis induced by anti-tumor necrosis factor in a patient with juvenile idiopathic arthritis. JAMA Dermatol. (2013) 149:1204–8. doi: 10.1001/jamadermatol.2013.5220
84. Arabshahi B, Silverman RA, Jones OY, Rider LG. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatr. (2012) 160:520–2. doi: 10.1016/j.jpeds.2011.11.057
85. Riley P, Maillard SM, Wedderburn LR, Woo P, Murray KJ, Pilkington CA. Intravenous cyclophosphamide pulse therapy in juvenile dermatomyositis. A review of efficacy and safety. Rheumatology. (2004) 43:491–6. doi: 10.1093/rheumatology/keh082
86. Deakin CT, Campanilho-Marques R, Simou S, Moraitis E, Wedderburn LR, Pullenayegum E, et al. Efficacy and safety of cyclophosphamide treatment in severe juvenile dermatomyositis shown by marginal structural modeling. Arthritis Rheumatol. (2018) 70:785–93. doi: 10.1002/art.40418
87. Reed AM, Peterson E, Bilgic H, Ytterberg SR, Amin S, Hein MS, et al. Changes in novel biomarkers of disease activity in juvenile and adult dermatomyositis are sensitive biomarkers of disease course. Arthr Rheum. (2012) 64:4078–86. doi: 10.1002/art.34659
88. Kurtzman DJ, Wright NA, Lin J, Femia AN, Merola JF, Patel M, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: an alternative treatment. JAMA Dermatol. (2016) 152:944–5. doi: 10.1001/jamadermatol.2016.0866
Keywords: Juvenile, dermatomyositis, vasculopathy, antibodies, pathophysiology
Citation: Papadopoulou C and McCann LJ (2018) The Vasculopathy of Juvenile Dermatomyositis. Front. Pediatr. 6:284. doi: 10.3389/fped.2018.00284
Received: 01 July 2018; Accepted: 17 September 2018;
Published: 09 October 2018.
Edited by:
Mindy S. Lo, Boston Children's Hospital, Harvard University, United StatesReviewed by:
Kaveh Ardalan, Northwestern Medicine, United StatesTiphanie Phillips Vogel, Baylor College of Medicine, United States
Copyright © 2018 Papadopoulou and McCann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charalampia Papadopoulou, c2VqamNwNkB1Y2wuYWMudWs=