 Baoheng Gui1,2,3†
Baoheng Gui1,2,3† Chenxi Yu4,5†
Chenxi Yu4,5† Xiaoxin Li6†
Xiaoxin Li6† Sen Zhao4,5†
Sen Zhao4,5† Hengqiang Zhao4,5
Hengqiang Zhao4,5 Zihui Yan4,5Xi Cheng4,5Jiachen Lin4,5Haiyang Zheng1,2,3Jiashen Shao4,5Zhengye Zhao4,5Lina Zhao6Yuchen Niu6Zhi Zhao6Huizi Wang6
Zihui Yan4,5Xi Cheng4,5Jiachen Lin4,5Haiyang Zheng1,2,3Jiashen Shao4,5Zhengye Zhao4,5Lina Zhao6Yuchen Niu6Zhi Zhao6Huizi Wang6 Bobo Xie1,2,3Xianda Wei1,2Chunrong Gui1,2Chuan Li2,3,7
Bobo Xie1,2,3Xianda Wei1,2Chunrong Gui1,2Chuan Li2,3,7 Shaoke Chen2,3,7Yi Wang8
Shaoke Chen2,3,7Yi Wang8 Yanning Song8Chunxiu Gong8Terry Jianguo Zhang4,5,9,10, on behalf of the Deciphering Disorders Involving Scoliosis, COmorbidities (DISCO) Study,
Yanning Song8Chunxiu Gong8Terry Jianguo Zhang4,5,9,10, on behalf of the Deciphering Disorders Involving Scoliosis, COmorbidities (DISCO) Study,  Xin Fan2,3,7, Zhihong Wu5,6,9, Yujun Chen2,7* and
Xin Fan2,3,7, Zhihong Wu5,6,9, Yujun Chen2,7* and  Nan Wu4,5,9,10*
Nan Wu4,5,9,10*- 1Center for Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
- 2The Guangxi Health Commission Key Laboratory of Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
- 3Department of Pediatric Endocrine and Metabolism, Maternal and Child Health Hospital of Guangxi, Nanning, China
- 4Department of Orthopedic Surgery, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
- 5Beijing Key Laboratory for Genetic Research of Skeletal Deformity, Beijing, China
- 6Medical Research Center, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
- 7Department of Pediatrics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
- 8Department of Endocrinology, Genetics and Metabolism, Beijing Children’s Hospital, National Center for Children’s Health, Capital Medical University, Beijing, China
- 9State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 10Key Laboratory of Big Data for Spinal Deformities, Chinese Academy of Medical Sciences, Beijing, China
Purpose: ROR2, a member of the ROR family, is essential for skeletal development as a receptor of Wnt5a. The present study aims to investigate the mutational spectrum of ROR2 in children with short stature and to identify the underlying molecular mechanisms.
Methods: We retrospectively analyzed clinical phenotype and whole-exome sequencing (WES) data of 426 patients with short stature through mutation screening of ROR2. We subsequently examined the changes in protein expression and subcellular location in ROR2 caused by the mutations. The mRNA expression of downstream signaling molecules of the Wnt5a–ROR2 pathway was also examined.
Results: We identified 12 mutations in ROR2 in 21 patients, including 10 missense, one nonsense, and one frameshift. Among all missense variants, four recurrent missense variants [c.1675G > A(p.Gly559Ser), c.2212C > T(p.Arg738Cys), c.1930G > A(p.Asp644Asn), c.2117G > A(p.Arg706Gln)] were analyzed by experiments in vitro. The c.1675G > A mutation significantly altered the expression and the cellular localization of the ROR2 protein. The c.1675G > A mutation also caused a significantly decreased expression of c-Jun. In contrast, other missense variants did not confer any disruptive effect on the biological functions of ROR2.
Conclusion: We expanded the mutational spectrum of ROR2 in patients with short stature. Functional experiments potentially revealed a novel molecular mechanism that the c.1675G > A mutation in ROR2 might affect the expression of downstream Wnt5a–ROR2 pathway gene by disturbing the subcellular localization and expression of the protein.
Introduction
As a member of ROR family receptor tyrosine kinase, receptor tyrosine kinase like orphan receptor 2 (ROR2) is a 943-amino acid transmembrane protein tyrosine kinase encoded by the ROR2 gene (van Bokhoven et al., 2000). The extracellular domains of ROR2 mainly include an immunoglobulin-like functional domain, a cysteine enrichment domain (CRD), and a kringle domain, while the intracellular domains include a tyrosine kinase domain (TKD), a proline-rich domain, two serine/threonine-rich domains, and a short C-terminal tail (Endo and Minami, 2018; Kamizaki et al., 2020). ROR2 is widely expressed in a variety of tissues, including the heart, brain, and lung and is also involved in the development of the nervous system and the skeletal system (Huang et al., 2015). Recently, it has been reported that ROR2 and ROR1 interact with Wnt9a to regulate the growth of the humerus in vivo (Weissenböck et al., 2019).
Pathogenic mutations in ROR2 are involved in two diseases: autosomal recessive Robinow syndrome (RRS, MIM:268310) and autosomal dominant brachydactyly type B1 (BDB1, MIM:113000) (Afzal et al., 2000; Yang et al., 2020; Zhang et al., 2020). Variants of ROR2 that cause RRS are generally nonsense, missense, and frameshift and are located in all of the domains (Kirat et al., 2020). BDB1-related variants are often nonsense or frameshift variants that reside in the N-terminal region of the protein (Stricker et al., 2017). Truncating variants associated with BDB1 cause a gain-of-function effect, whereas RRS-related variants result in the loss of function of ROR2 (Bacino, 1993). Accumulating evidence from Ror2-knockout mice and RRS patients suggests a significant role of ROR2 in the early formation of chondrocytes as well as the development and formation of bone (DeChiara et al., 2000; Takeuchi et al., 2000; Patton and Afzal, 2002; Schwabe et al., 2004; Weissenböck et al., 2019).
Recent findings indicate that mutations in a single gene, such as SHOX, NPR2, ACAN, or FGFR3, could cause either severe skeletal dysplasia or isolated short stature. Hauer et al. (2018) performed whole-exome sequencing (WES) on 200 patients with idiopathic short stature and found a proportion of patients with pathogenic mutation in genes known to be associated with skeletal dysplasia. We previously revealed distinct genetic architecture and pathophysiological processes in 561 patients with isolated and syndromic short stature using WES and yielded a diagnostic rate of 24.1% (Fan et al., 2021). The milder phenotype (that is, isolated short stature) tends to occur when the mutation only partially disrupts protein function and/or when the mutation occurs in the heterozygous state (Baron et al., 2015).
Although mutations in ROR2 have been implicated in certain congenital skeletal defects, including BDB1 and RRS, the molecular mechanisms underlying isolated short stature still remain elusive. This study aimed to explore the contributions of heterozygous ROR2 variants in short stature patients that remained undiagnosed after WES analysis. We subsequently performed in vitro functional analyses for variants that were recurrent in our cohort.
Materials and Methods
Cohort Recruitment and Whole-Exome Sequencing
From July 2014 to August 2018, we screened 426 WES-undiagnosed patients from three centers in China [Maternal and Child Health Hospital of Guangxi, The Second Affiliated Hospital of Guangxi Medical University, and Beijing Children’s Hospital, as parts of the Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO)] study1.
DNA was extracted from peripheral blood collected from all of the probands and available familial members. In total, 374 patients underwent proband-only WES, while 50 underwent trio-based WES, and two underwent quad-based WES (altogether 532 subjects). The sequencing data were analyzed and annotated using an in-house developed analytical pipeline, Peking Union Medical College Hospital Pipeline (PUMP), as previously described (Wang et al., 2018; Chen et al., 2021; Zhao et al., 2021; Supplementary Methods).
Mutation Analysis
Mutation screening of ROR2 was performed in 426 undiagnosed patients. All mutations in coding exons of ROR2 from our cohort were manually reviewed using Integrative Genomics Viewer (IGV) (Thorvaldsdóttir et al., 2013). Current study use followed criteria to ensure that mutations in ROR2 were highly credible: (1) read depth of mutation > 10; (2) variant allele frequency ≥ 0.2; (3) Combined Annotation Dependent Depletion (CADD) predicted score > 15; (4) frequency in Gnomad database ≤ 0.01 (Figure 1). Sanger sequencing was performed on available subjects and parental samples to validate the variants by an orthogonal sequencing method and to investigate segregation according to Mendelian expectations for the identified variant allele(s).
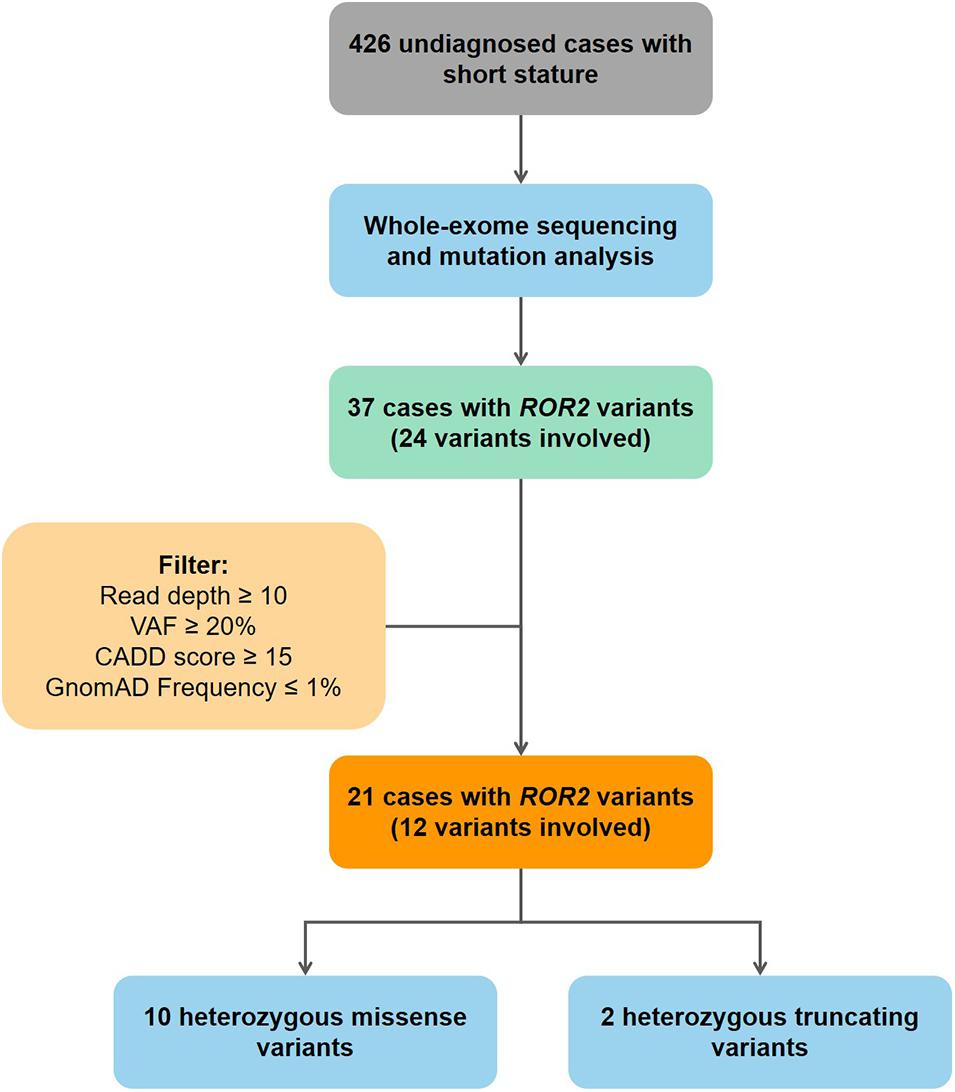
Figure 1. Flowchart of patient enrollment and mutation screening. VAF, variant allele frequency; CADD, combined annotation dependent depletion database.
Plasmid Construction
The wild-type (WT) ROR2 plasmid was constructed through Seamless Cloning reaction. The resulting PCR amplimers were digested with XhoI and BamHI and ligated into the XhoI and BamHI sites of plasmid pEGFP-N1 (Beijing Hitrobio Biotechnology Co., Ltd., Sigma-Aldrich, St. Louis). The resulting plasmid was identified by Sanger sequencing (Forward primer: 5′-CGCTTTGTCCTTCAGCGTTT-3′; Reverse primer: 5′-AATGCCCCTCATTAACCAGC-3′). All ROR2 mutated plasmids were constructed using the strategy of homologous recombination (Primers used for mutated plasmid construction were listed in Supplementary Materials). The resulting plasmid was transformed into Escherichia coli and validated by Sanger sequencing.
Cell Culture and Transfection
The HeLa cells were cultured in Dulbecco’s modified Eagle media/High Glucose (Hyclone), supplemented with 5% fetal bovine serum (FBS; 10099141C, Gibco), 100 U/ml penicillin-streptomycin (15140122, Gibco) at 37°C with 5% CO2. Cells were seeded in six-well plates or confocal dishes and transfected with Lipofectamine 3000 Transfection Reagent (2264840, Invitrogen) according to the manufacturers’ instructions. Six hours after transfection for Western blotting (WB) analysis, the HeLa cells were treated with 100 ng/ml Wnt5a (645-WN-010, R&D Systems) for 48 h.
Immunofluorescence
The HeLa cells were implanted on confocal dishes and transfected with plasmids using Lipofectamine 3000 Transfection Reagent (2264840, Invitrogen) for 48 h. The confocal dishes were rinsed three times with phosphate-buffered saline (PBS; SH0021, Beijing Haicheng Yuanhong Technology Co., Ltd.). The cells were fixed in 4% fixative solution (P1110, Solarbio, China) and blocked with bovine serum albumin buffer (ZLI-9022, ZSGB-bio, China). The first antibody (rabbit polyclonal calnexin antibody, ab22595, Abcam) was incubated with PBS containing 0.1% Triton X-100 (T8200, Solarbio, China) at room temperature (RT; 25°C) for 1 h. Goat anti-rabbit IgG H&L (Alexa Fluor 647) was adopted as secondary antibody (ab150079, Abcam). The dyed dishes were mounted in fluorescent mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; ZLI-9557, ZSGB-BIO, China) and observed using confocal microscopy. All experiments were replicated three times.
Western Blotting Analysis
Cells were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (C1053, Beijing Applygen Technologies Inc.), and protein concentrations were determined using the bicinchoninic acid (BCA) Protein Assay Kit (PC0020, Solarbio, China). Total protein of 20 μg was separated on a 10% NuPAGETM Bis-Tris Welcome Pack (NP030B, Invitrogen), and the electrophoresed products were transferred to iBlotTM 2 NC Regular Stacks (IB23001, Life Technologies). Membranes were blocked for 30 min at RT using 5% powdered milk, and primary antibodies mouse anti-eGFP mAb (TA-06, ZSGB-BIO, China) and mouse anti-GAPDH mAb (TA-08, ZSGB-BIO, China) were incubated overnight at 4°C. After washing the membranes, the secondary antibody Goat anti-Mouse IgG (H + L) (ZB-2305, ZSGB-BIO, China) was incubated for 2 h at RT. Bands were visualized with Pro-light HRP substrate chemiluminescent system (PA112, Tiangen Biotech Co., Ltd.). Chemiluminescent signals were quantified using ImageJ software.
Quantitative Real-Time Polymerase Chain Reaction
The levels of c-Jun and Axin2 mRNA in HeLa cells after Wnt5a treatment were analyzed via qRT-PCR. Total RNA was extracted using FastPure cell/tissue total RNA isolation kit V2 (RC112-01, Nanjing Vazyme Biotech Co., Ltd.), according to the manufacturer’s instructions. The RNA was subsequently reverse transcribed to yield single-stranded cDNAs using PrimeScriptTM RT reagent kit with gDNA eraser (perfect real time, RR047A, TaKaRa) based on the manufacturer’s instructions. The qRT-PCR reaction was performed using TB Green Premix Ex TaqTM II (Tli RNaseH Plus, RR820A, TaKaRa) and the 7500 Fast Dx Real-Time PCR Instrument (Applied Biosystems, United States) based on the manufacturer’s instructions. The primer sequences used for PCR amplification in our study were designed based on the sequences of the cDNA clones as follows: the primers of c-Jun were as previously described (Davoulou et al., 2020) and the primers of Axin2 (137 bp: NM_011359): Forward primer: 5′-CGATGAGTTTGCCTGTGGAG-3′; Reverse primer: 5′-TCAATCGATCCGCTCCACTT-3′.
Statistical Analysis
Comparison among groups was performed by the analysis of Student’s T-test. P-value less than 0.05 was statistically significant as calculated by SPSS 21.0.
Results
Cohort Demographic Characteristics
We found that 21 patients carried variants in ROR2 from 426 patients with short stature. Among these patients, there were 12 males (57%) and 9 females (43%). The mean age, bone age, and height SDs of all cases with ROR2 variants were 6.38 ± 3.99 years, 4.67 ± 2.19 years, and −2.92 ± 1.31, respectively. Ten cases presented other systemic symptoms besides short stature. Six cases presented symptoms of developmental delay, and only one case had a symptom of gonadal dysplasia (Table 1).
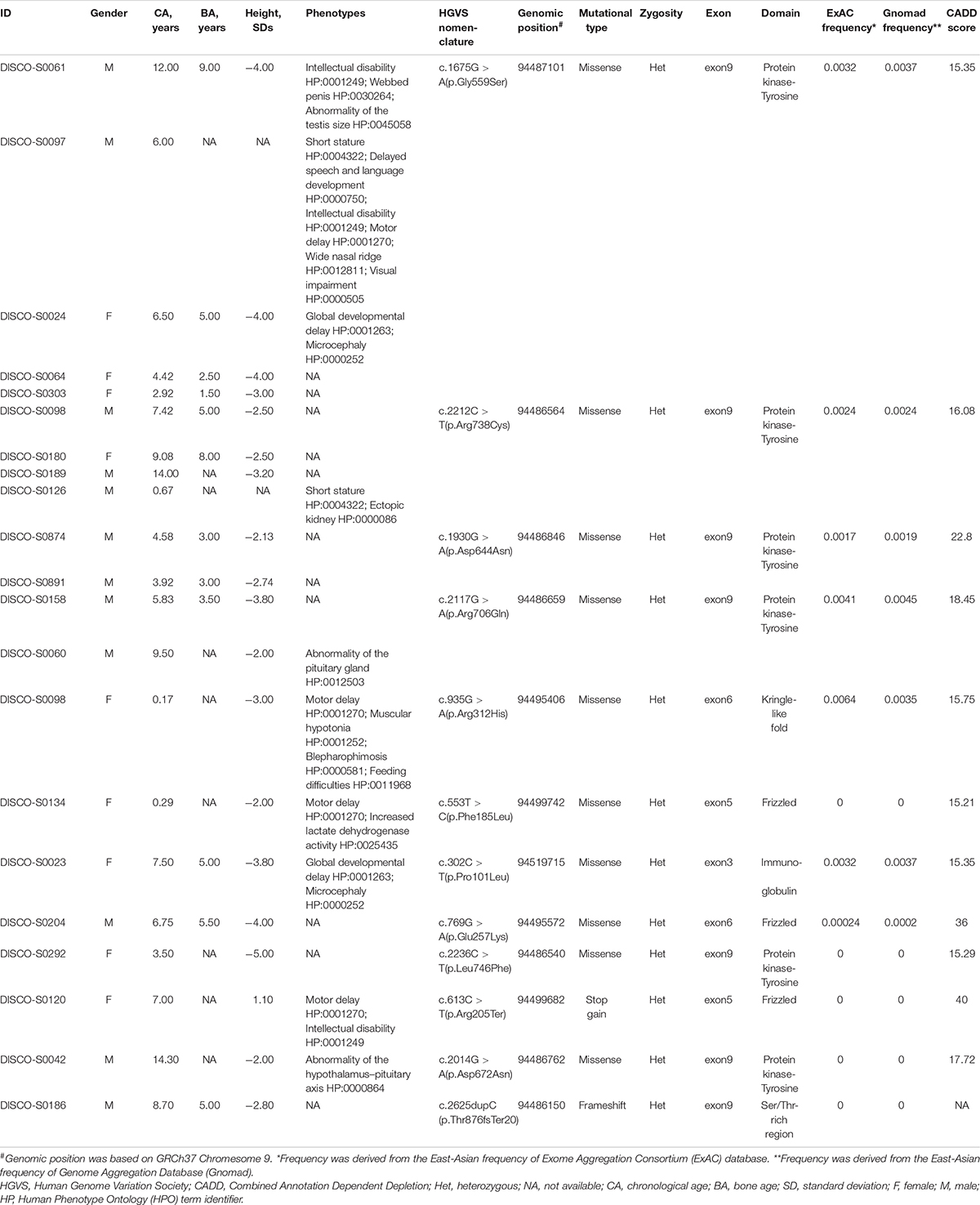
Table 1. Phenotype and genotype of patients carrying mutations in ROR2.
Mutational Screening of ROR2 in the Cohort
Through mutation screening analysis, we identified 12 mutations in ROR2, including two truncating mutations and 10 missense mutations (Table 1). The heterozygous stop-gain mutation [c.613C > T (p.Arg205Ter)] was found in the patient DISCO-S0120 who manifested dwarfism with mild motor retardation and mental retardation, which were partly consistent with the phenotype of RRS. The other patient DISCO-S0186 carried a heterozygous ROR2 frameshift mutation [c.2625dupC (p.Thr876fsTer20)], which was absent from pubic databases and 942 in-house control from the DISCO study. However, no variant in ROR2 was identified in trans in either patient. Furthermore, four recurrent missense mutations in ROR2 were identified, including c.1675G > A (p.Gly559Ser) in five patients, c.2212C > T (p.Arg738Cys) in four patients, c.1930G > A (p.Asp644Asn) in two patients, and c.2117G > A (p.Arg706Gln) in two patients (Table 1). All recurrent missense mutations were located at the TKD of the intracellular region of the ROR2 protein (Figure 2).
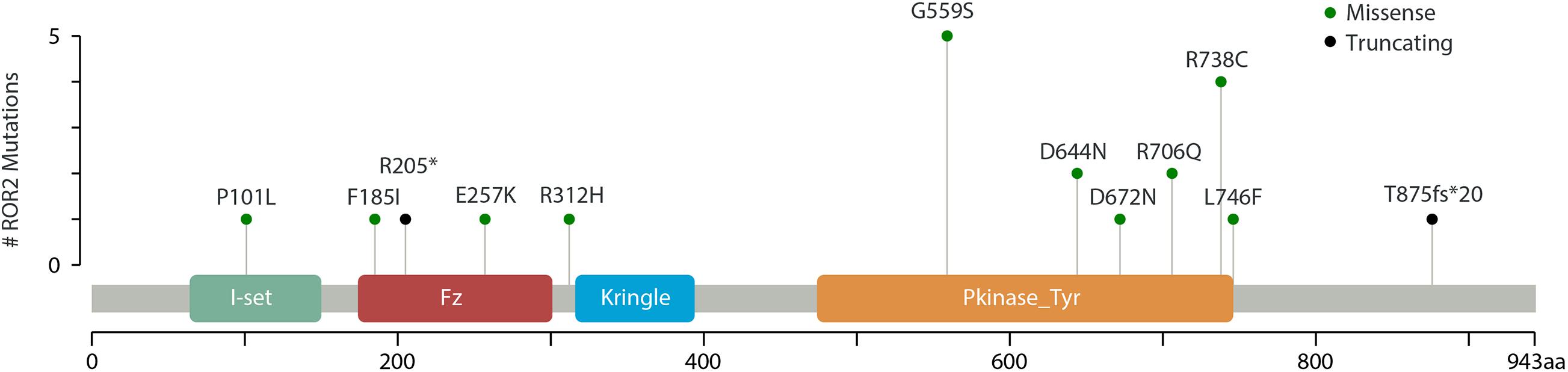
Figure 2. Schematic representation of ROR2 with its domain structure. Mutations identified in this study (green and black) are marked.
Expression of the ROR2 Protein and Wnt5a–ROR2 Pathway-Related Protein
We further conducted in vitro protein expression experiments to examine functional defects caused by recurrent missense mutations in ROR2. WB showed that c.1675G > A mutation could lead to a significant decrease in the expression of ROR2 protein with Wnt5a treatment (P < 0.05; Figures 3A,B). In contrast, the remaining three missense variants did not impact the expression of ROR2 (data not shown). The influence of mutated and WT ROR2 protein on β-catenin and P-Jnk/JNK pathways showed no significant difference with or without Wnt5a treatment (Figures 3A,C,D). To further explore the effect on the Wnt5a pathway conferred by the c.1675G > A variant, we tested the expression of c-Jun, a molecule downstream of the Wnt5a pathway in precursors of osteoclast (Maeda et al., 2012), and Axin2, a regulator of the Wnt pathway, with Wnt5a stimulation (Lammi et al., 2004). As a result, mRNA expression of c-Jun in the cells transfected with mutated ROR2 c.1675G > A was significantly lower than that transfected with WT ROR2 (P < 0.05; Figure 4A). In contrast, there was no significant difference in the expression of Axin2 mRNA after treatment with Wnt5a (Figure 4B).
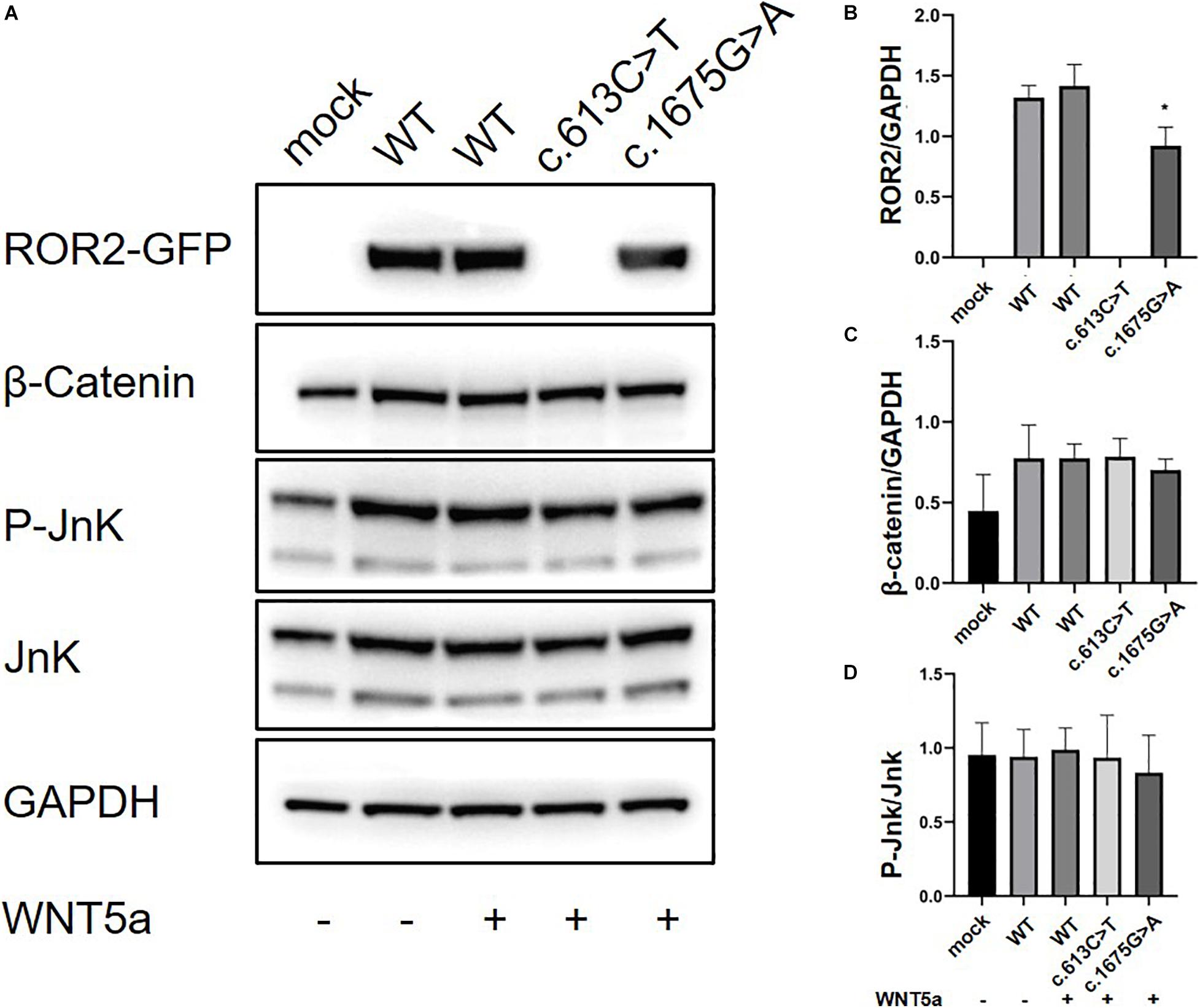
Figure 3. Modulation of non-canonical Wnt signaling in HeLa cells with the supplementation of Wnt5a in general culture conditions. The ROR2-GFP, β-Catenin, P-JnK, JnK levels in HeLa cells with 100 ng/ml concentration of Wnt5a (A) Western blot for Ror2 [wild type (WT) and mutants] in HeLa cells. (B–D) Quantification of protein expression of different ROR2 constructs by ImageJ (n = 3), *P < 0.05 vs. WT + Wnt5a 100 ng/ml.
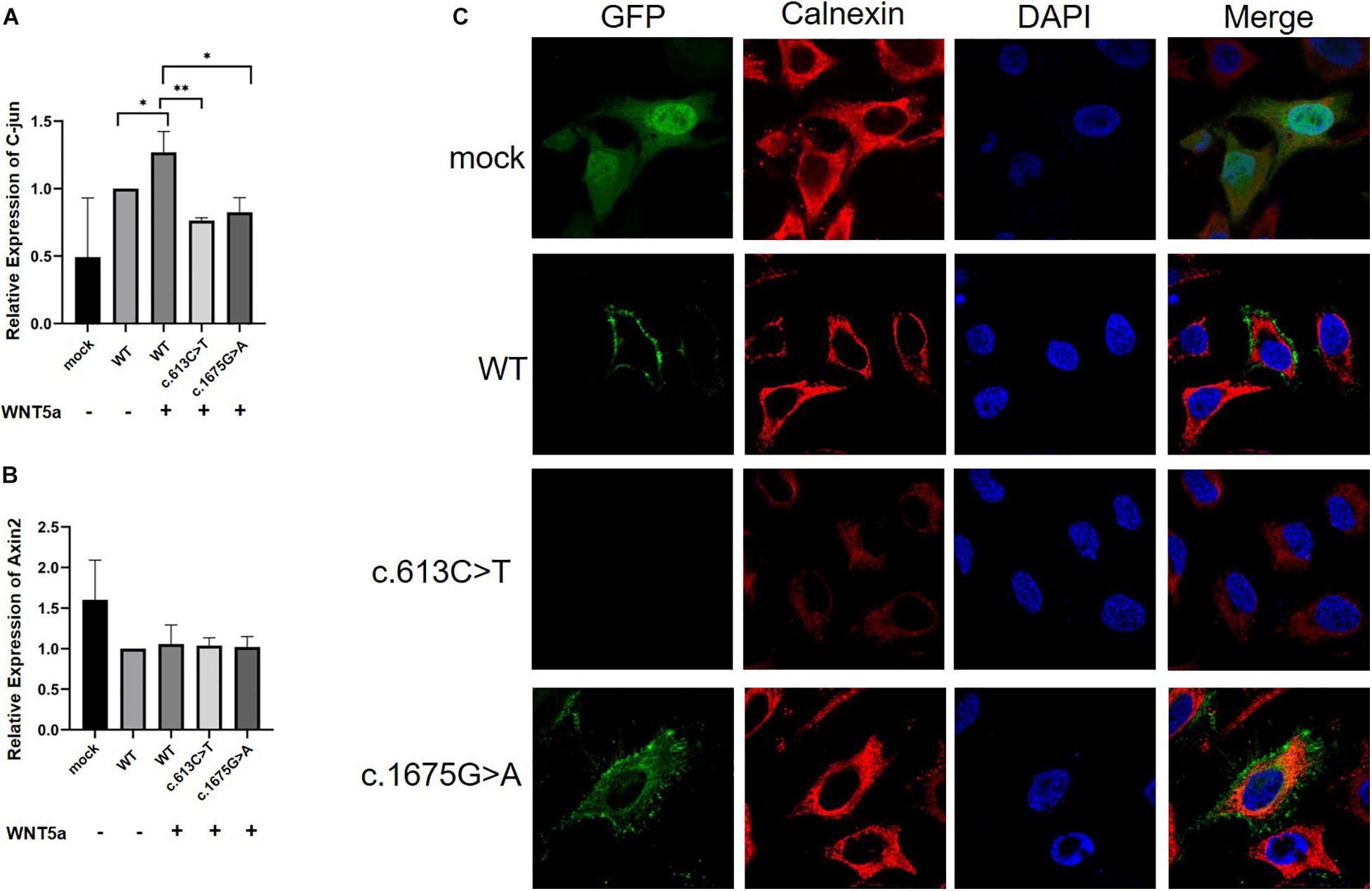
Figure 4. Real-time PCR analysis of mRNA expression of Wnt ligands (C-Jun and Axin2) in HeLa cells (n = 3) and subcellular location of the ROR2 protein. All P-values were calculated by Student’s t-test. ∗P < 0.05 and ∗P < 0.01 were considered statistically significant. (A) qPCR analysis of C-jun mRNA expression under Wnt5a treatment. (B) qPCR analysis of Axin2 mRNA expression under Wnt5a treatment. (C) Subcellular location distribution of ROR2 WT protein, and distribution of mutated ROR2 protein.
Immunofluorescence Colocalization of ROR2
We further performed immunofluorescence experiments to explore the effects of these missense mutations on the subcellular localization of the ROR2 protein. The results showed that the mutated ROR2 c.1675G > A was enriched in the endoplasmic reticulum, and the mutated ROR2 c.613C > T was not detected (Figure 4C). The subcellular localization of other missense mutations in ROR2 was similar to that of WT (data not shown). These in vitro results suggested that the c.1675G > A mutation might affect the expression of downstream genes in the Wnt5a–ROR2 pathway and alter the subcellular localization of the ROR2 protein.
Discussion
With the application of the next-generation sequencing (NGS) in exploring the genetic etiology of disease, complex genetic patterns may explain the genetic causes of many common diseases and relatively rare diseases (Posey et al., 2017). Through mutation screening of ROR2, we found two patients with heterozygous truncating mutations in ROR2 mainly presenting short stature. Genotype–phenotype evaluation confirmed that these two patients were lacking other characteristic features of the RRS, indicating that heterozygous loss-of-function mutation in ROR2 might be associated with isolated short stature.
In addition, we identified six missense mutations in ROR2 including four recurrent missense mutations located within the TKD (Figure 2 and Table 1). Results from functional experiments in vitro demonstrated that the missense mutation in ROR2 [c.1675G > A (p.Gly559Ser)] could perturb the subcellular localization of the ROR2 protein and lead to a decreased expression of downstream molecule of the Wnt5a pathway. Based on these results, we inferred that this heterozygous missense mutation in ROR2 might have a hypomorphic effect, which meant a variant caused partial loss of the gene function (Hrabé de Angelis and Balling, 1998).
In vertebrates, ROR2 and ROR1 bind to non-classical Wnt5a protein through their CRD and act as receptors or co-receptors for Wnt5a to activate non-classical Wnt pathways (β-catenin-independent pathways), including planar cellular polarization pathways and Wnt–Ca2+ pathways (Oishi et al., 2003; Yamamoto et al., 2007; Gao et al., 2011; Kamizaki et al., 2020). Recently, studies showed that the Wnt5a–ROR2 pathway was not only involved in the development of cartilage but also associated with the function of osteoblasts and osteoclasts (Chen et al., 2019; Uehara et al., 2019). Weissenböck et al. (2019) found that knockout of Ror2 also influenced the formation of the trunk bone in vivo. Maeda et al. (2012) found that the Wnt5a–ROR2 pathway could further regulate the differentiation of osteoblast progenitor cells by affecting the dimer of Jun and Sp-1 binding to the transcriptional initiation region of Tnfrsf11a, which is consistent with our findings. According to our in vitro assays, c.1675G > A mutation in ROR2 leads to a decrease in the expression of c-Jun under Wnt5a treatment, which indicated that this missense mutation in ROR2 might disrupt the normal function of the Wnt5a–ROR2 pathway.
The TKD is the key domain in the cytoplasmic region of the ROR2 protein. The biological function of the TKD was controversial in previous studies (Alfaro et al., 2015; Debebe and Rathmell, 2015). In terms of evolutionary biology, mammalian ROR2 exhibits alterations within the highly conserved amino acids in the kinase domains that possibly indicates that the kinase activity may have been evolutionarily degenerated (Forrester, 2002). Also, several studies suggested that both ROR1 and ROR2 might be pseudokinases (Gentile et al., 2011; Debebe and Rathmell, 2015). However, recent studies by Nevenzal et al. (2019) and Sheetz et al. (2020) had found the autophosphorylation activity and autoinhibitory interactions of the TKD in the ROR2 protein from the perspective of high-throughput phosphorylation detection and protein structure. Consistent with results from experiments in vitro, five of our patients carrying the c.1675G > A heterozygous mutation mainly displayed short stature. As shown by both in vitro assays and data from patient cohorts, we confirmed that missense mutation c.1675G > A in the TKD region might be associated with linear growth attenuation among children. However, the function of the TKD needs to be confirmed by further molecular biological experiments. Also, our findings from cohort and in vitro results both highlighted a possiblely novel mechanism through which hypomorphic mutation in ROR2 leads to short stature.
Conclusion
We expanded the mutational spectrum of ROR2 in patients with short stature. The c.1675G > A in ROR2 was recurrently seen in five patients and was revealed to confer a hypomorphic effect on the function and expression of the protein and the normal activity of the Wnt5a pathway.
Data Availability Statement
The data presented in the study are deposited in the ClinVar repository, and accession number are SCV001499871– SCV001499881.
Ethics Statement
Approval for the study was obtained from the Ethics Committee at the Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region (G-1-1), The Second Affiliated Hospital of Guangxi Medical University [2020-KY(0112)] and Beijing Children’s Hospital (Y-028-A-01). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
CY, SZ, BG, XL, and NW contributed to the conceptualization. CY, BG, ZY, SZ, XC, XL, and NW contributed to data curation. CY, BG, XL, SZ, HeZ, and JL contributed to the formal analysis. SC, TZ, ZW, CGo, BG, and NW contributed to funding acquisition. XL, JL, HaZ, JS, ZheZhao, ZhiZhao, HW, BX, XW, CGu, and CL contributed to the investigation. SC, CL, CGo, XF, YC, and NW contributed resources. ZY, HeZ, and YN contributed the software. CGo, XF, YC, and NW contributed to supervision, writing, review, and editing. HeZ and CY contributed to visualization. CY, BG, XL, and SZ contributed to writing the original draft. All authors provided crucial input on several iterations of the manuscript, and approved the final version.
Funding
This research was funded in part by the Capital’s Funds for Health Improvement and Research (2020-4-40114 to NW), National Natural Science Foundation of China (81822030 and 82072391 to NW, 81930068 and 81772299 to ZW, 81972037 to TZ, and 81860272 and 82001531 to BG), Beijing Natural Science Foundation (JQ20032 to NW and 7191007 to ZW), Tsinghua University-Peking Union Medical College Hospital Initiative Scientific Research Program, and Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT320025 to NW), Guangxi Natural Science Foundation (2018GXNSFAA281067 to BG), The Initial Scientific Research Fund for Advanced Talents from The Second Affiliated Hospital of Guangxi Medical University to BG, and The Special Scientific Research Fund of Guangxi Ten-Hundred-Thousand Talents Project to BG.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients and their families who participated in this research. We also thank the Nanjing Geneseeq Technology Inc. for the technical help in sequencing and Ekitech Technology Inc. for the technical help in database.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.661747/full#supplementary-material
Footnotes
References
Afzal, A. R., Rajab, A., Fenske, C. D., Oldridge, M., Elanko, N., Ternes-Pereira, E., et al. (2000). Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 25, 419–422. doi: 10.1038/78107
Alfaro, I. E., Varela-Nallar, L., Varas-Godoy, M., and Inestrosa, N. C. (2015). The ROR2 tyrosine kinase receptor regulates dendritic spine morphogenesis in hippocampal neurons. Mol. Cell. Neurosci. 67, 22–30. doi: 10.1016/j.mcn.2015.05.002
Bacino, C. A. (1993). “ROR2-related robinow syndrome,” in GeneReviews® eds MP Adam, HH Ardinger, RA Pagon, SE Wallace, and LJH Bean (Seattle (WA): University of Washington, Seattle).
Baron, J., Savendahl, L., De Luca, F., Dauber, A., Phillip, M., Wit, J. M., et al. (2015). Short and tall stature: a new paradigm emerges. Nat. Rev. Endocrinol. 11, 735–746. doi: 10.1038/nrendo.2015.165
Chen, K., Ng, P. Y., Chen, R., Hu, D., Berry, S., Baron, R., et al. (2019). Sfrp4 repression of the Ror2/Jnk cascade in osteoclasts protects cortical bone from excessive endosteal resorption. Proc. Natl. Acad. Sci. U.S.A. 116, 14138–14143. doi: 10.1073/pnas.1900881116
Chen, N., Zhao, S., Jolly, A., Wang, L., Pan, H., Yuan, J., et al. (2021). Perturbations of genes essential for Mullerian duct and Wolffian duct development in Mayer-Rokitansky-Kuster-Hauser syndrome. Am. J. Hum. Genet. 108, 337–345.
Davoulou, P., Aggeletopoulou, I., Panagoulias, I., Georgakopoulos, T., and Mouzaki, A. (2020). Transcription factor Ets-2 regulates the expression of key lymphotropic factors. Mol. Biol. Rep. 47, 7871–7881. doi: 10.1007/s11033-020-05865-x
Debebe, Z., and Rathmell, W. K. (2015). Ror2 as a therapeutic target in cancer. Pharmacol. Ther. 150, 143–148. doi: 10.1016/j.pharmthera.2015.01.010
DeChiara, T. M., Kimble, R. B., Poueymirou, W. T., Rojas, J., Masiakowski, P., Valenzuela, D. M., et al. (2000). Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nat. Genet. 24, 271–274. doi: 10.1038/73488
Endo, M., and Minami, Y. (2018). Diverse roles for the ror-family receptor tyrosine kinases in neurons and glial cells during development and repair of the nervous system. Dev. Dyn. 247, 24–32. doi: 10.1002/dvdy.24515
Fan, X., Zhao, S., Yu, C., Wu, D., Yan, Z., Fan, L., et al. (2021). Exome sequencing reveals genetic architecture in patients with isolated or syndromic short stature. J. Genet. Genom. (in press). doi: 10.1016/j.jgg.2021.02.008
Forrester, W. C. (2002). The Ror receptor tyrosine kinase family. Cell. Mol. Life Sci. 59, 83–96. doi: 10.1007/s00018-002-8407-9
Gao, B., Song, H., Bishop, K., Elliot, G., Garrett, L., English, M. A., et al. (2011). Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev. Cell 20, 163–176. doi: 10.1016/j.devcel.2011.01.001
Gentile, A., Lazzari, L., Benvenuti, S., Trusolino, L., and Comoglio, P. M. (2011). Ror1 is a pseudokinase that is crucial for Met-driven tumorigenesis. Cancer Res. 71, 3132–3141. doi: 10.1158/0008-5472.can-10-2662
Hauer, N. N., Popp, B., Schoeller, E., Schuhmann, S., Heath, K. E., Hisado-Oliva, A., et al. (2018). Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet. Med. 20, 630–638. doi: 10.1038/gim.2017.159
Hrabé de Angelis, M., and Balling, R. (1998). Large scale ENU screens in the mouse: genetics meets genomics. Mutat. Res. 400, 25–32. doi: 10.1016/s0027-5107(98)00061-x
Huang, J., Shi, Y., Li, H., Tan, D., Yang, M., and Wu, X. (2015). Knockdown of receptor tyrosine kinase-like orphan receptor 2 inhibits cell proliferation and colony formation in osteosarcoma cells by inducing arrest in cell cycle progression. Oncol. Lett. 10, 3705–3711. doi: 10.3892/ol.2015.3797
Kamizaki, K., Endo, M., Minami, Y., and Kobayashi, Y. (2020). Role of noncanonical Wnt ligands and Ror-family receptor tyrosine kinases in the development, regeneration, and diseases of the musculoskeletal system. Dev. Dyn. 250, 27–38. doi: 10.1002/dvdy.151
Kirat, E., Mutlu Albayrak, H., Sahinoglu, B., Gurler, A. I., and Karaer, K. (2020). Autosomal recessive Robinow syndrome with novel ROR2 variants: distinct cases exhibiting the clinical variability. Clin. Dysmorphol. 29, 137–140. doi: 10.1097/mcd.0000000000000319
Lammi, L., Arte, S., Somer, M., Jarvinen, H., Lahermo, P., Thesleff, I., et al. (2004). Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet. 74, 1043–1050. doi: 10.1086/386293
Maeda, K., Kobayashi, Y., Udagawa, N., Uehara, S., Ishihara, A., Mizoguchi, T., et al. (2012). Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 18, 405–412. doi: 10.1038/nm.2653
Nevenzal, H., Noach-Hirsh, M., Skornik-Bustan, O., Brio, L., Barbiro-Michaely, E., Glick, Y., et al. (2019). A high-throughput integrated microfluidics method enables tyrosine autophosphorylation discovery. Commun. Biol. 2:42.
Oishi, I., Suzuki, H., Onishi, N., Takada, R., Kani, S., Ohkawara, B., et al. (2003). The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 8, 645–654. doi: 10.1046/j.1365-2443.2003.00662.x
Posey, J. E., Harel, T., Liu, P., Rosenfeld, J. A., James, R. A., Coban Akdemir, Z. H., et al. (2017). Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 376, 21–31. doi: 10.1056/nejmoa1516767
Schwabe, G. C., Trepczik, B., Süring, K., Brieske, N., Tucker, A. S., Sharpe, P. T., et al. (2004). Ror2 knockout mouse as a model for the developmental pathology of autosomal recessive Robinow syndrome. Dev. Dyn. 229, 400–410. doi: 10.1002/dvdy.10466
Sheetz, J. B., Mathea, S., Karvonen, H., Malhotra, K., Chatterjee, D., Niininen, W., et al. (2020). Structural insights into pseudokinase domains of receptor tyrosine kinases. Mol. Cell 79, 390–405.e7.
Stricker, S., Rauschenberger, V., and Schambony, A. (2017). ROR-family receptor tyrosine kinases. Curr. Top. Dev. Biol. 123, 105–142.
Takeuchi, S., Takeda, K., Oishi, I., Nomi, M., Ikeya, M., Itoh, K., et al. (2000). Mouse Ror2 receptor tyrosine kinase is required for the heart development and limb formation. Genes Cells 5, 71–78. doi: 10.1046/j.1365-2443.2000.00300.x
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Uehara, S., Udagawa, N., and Kobayashi, Y. (2019). Regulation of osteoclast function via Rho-Pkn3-c-Src pathways. J. Oral Biosci. 61, 135–140. doi: 10.1016/j.job.2019.07.002
van Bokhoven, H., Celli, J., Kayserili, H., van Beusekom, E., Balci, S., Brussel, W., et al. (2000). Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive robinow syndrome. Nat. Genet. 25, 423–426. doi: 10.1038/78113
Wang, K., Zhao, S., Liu, B., Zhang, Q., Li, Y., Liu, J., et al. (2018). Perturbations of BMP/TGF-beta and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J. Med. Genet. 55, 675–684. doi: 10.1136/jmedgenet-2017-105224
Weissenböck, M., Latham, R., Nishita, M., Wolff, L. I., Ho, H. H., Minami, Y., et al. (2019). Genetic interactions between Ror2 and Wnt9a, Ror1 and Wnt9a and Ror2 and Ror1: phenotypic analysis of the limb skeleton and palate in compound mutants. Genes Cells 24, 307–317. doi: 10.1111/gtc.12676
Yamamoto, H., Yoo, S. K., Nishita, M., Kikuchi, A., and Minami, Y. (2007). Wnt5a modulates glycogen synthase kinase 3 to induce phosphorylation of receptor tyrosine kinase Ror2. Genes Cells 12, 1215–1223. doi: 10.1111/j.1365-2443.2007.01128.x
Yang, K., Zhu, J., Tan, Y., Sun, X., Zhao, H., Tang, G., et al. (2020). Whole-exome sequencing identified compound heterozygous variants in ROR2 gene in a fetus with Robinow syndrome. J. Clin. Lab. Anal. 34:e23074.
Zhang, C., Mazzeu, J. F., Eisfeldt, J., Grochowski, C. M., White, J., Akdemir, Z. C., et al. (2020). Novel pathogenic genomic variants leading to autosomal dominant and recessive Robinow syndrome. Am. J. Med. Genet. A doi: 10.1002/ajmg.a.61908 Online ahead of print.
Keywords: short stature, ROR2 gene, recurrent mutations, dysfunction, skeletal development
Citation: Gui B, Yu C, Li X, Zhao S, Zhao H, Yan Z, Cheng X, Lin J, Zheng H, Shao J, Zhao Z, Zhao L, Niu Y, Zhao Z, Wang H, Xie B, Wei X, Gui C, Li C, Chen S, Wang Y, Song Y, Gong C, Zhang TJ, Fan X, Wu Z, Chen Y and Wu N (2021) Heterozygous Recurrent Mutations Inducing Dysfunction of ROR2 Gene in Patients With Short Stature. Front. Cell Dev. Biol. 9:661747. doi: 10.3389/fcell.2021.661747
Received: 31 January 2021; Accepted: 12 March 2021;
Published: 14 April 2021.
Edited by:
Desheng Liang, Central South University, ChinaReviewed by:
Jianqiu Xiao, Washington University in St. Louis, United StatesWeiyu Li, University of California, San Francisco, United States
Copyright © 2021 Gui, Yu, Li, Zhao, Zhao, Yan, Cheng, Lin, Zheng, Shao, Zhao, Zhao, Niu, Zhao, Wang, Xie, Wei, Gui, Li, Chen, Wang, Song, Gong, Zhang, Fan, Wu, Chen and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nan Wu, ZHIud3VuYW5AcHVtY2guY24=; Yujun Chen, Y2hlbnl1anVuMTAwNkAxNjMuY29t
†These authors have contributed equally to this work