Abstract
Neurons are post-mitotic cells that allocate huge amounts of energy to the synthesis of new organelles and molecules, neurotransmission and to the maintenance of redox homeostasis. In neurons, autophagy is not only crucial to ensure organelle renewal but it is also essential to balance nutritional needs through the mobilization of internal energy stores. A delicate crosstalk between the pathways that sense nutritional status of the cell and the autophagic processes to recycle organelles and macronutrients is fundamental to guarantee the proper functioning of the neuron in times of energy scarcity. This review provides a detailed overview of the pathways and processes involved in the balance of cellular energy mediated by autophagy, which when defective, precipitate the neurodegenerative cascade of Parkinson’s disease, frontotemporal dementia, amyotrophic lateral sclerosis or Alzheimer’s disease.
Schematic Representation Highlighting the Mechanisms of Nutrient Sensing and Autophagy Under Physiological Conditions.
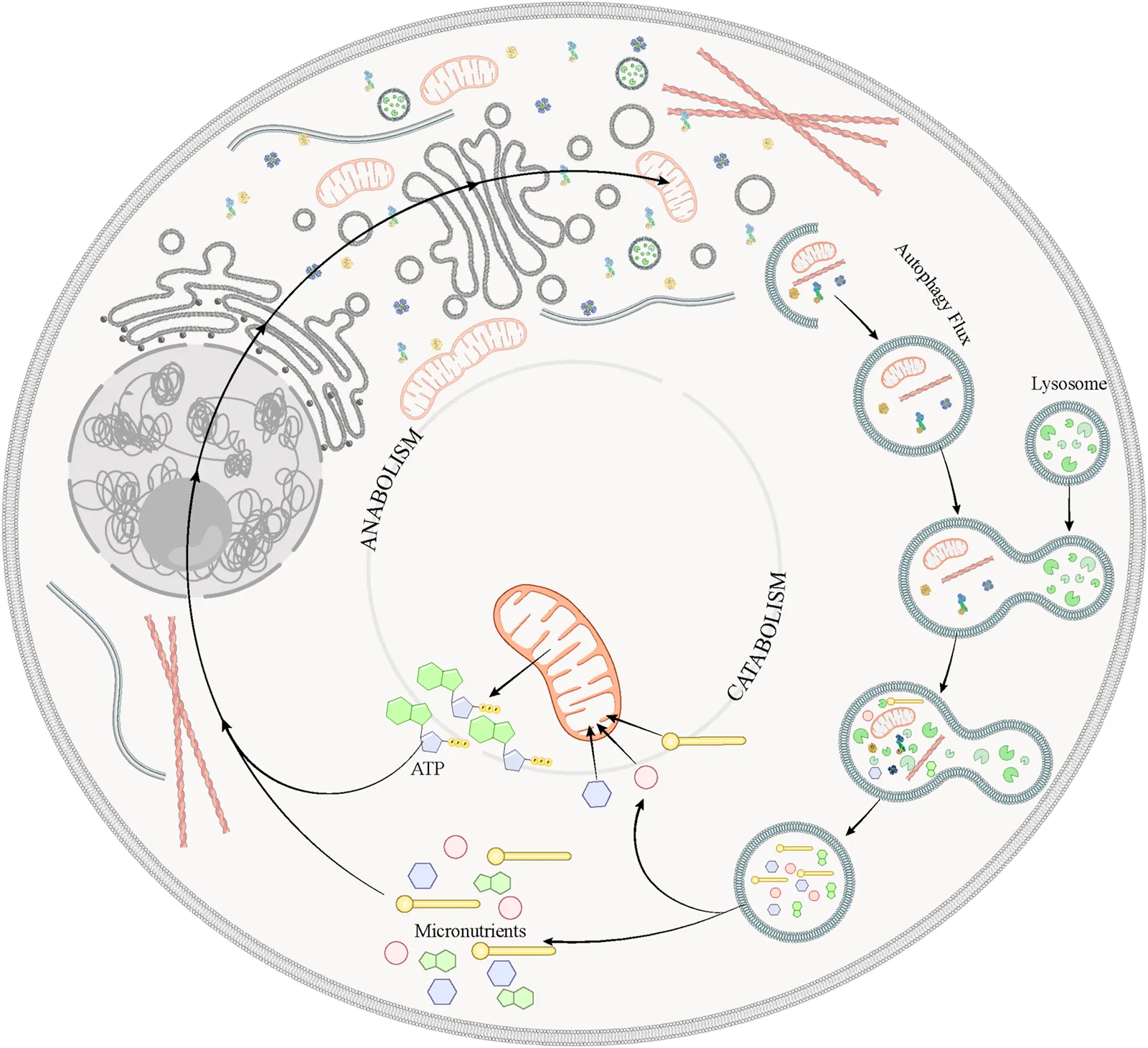
Under physiological conditions, the cell can sense changes in the availability of metabolic fuels and respond to these challenges through the reconfiguration of energy-producing pathways and recycling of accumulated or damaged organelles and proteins. For that purpose, the cell initiates the ALP-in which the damaged or unnecessary organelles and proteins are carried in the autophagosomes and degraded into the lysosomes-, generating micronutrients that will be used to obtain energy. The cell uses the energy produced and the micronutrients as building blocks to synthesize new macromolecules and organelles, which will respond to the cell requirements and maintain cell homeostasis.
1 The Importance of Autophagy to Access the Internal Nutrient Stores in Times of Energy Scarcity
Because the availability of environmental nutrients can be intermittent, cells and organisms have developed efficient ways to store nutrients during periods of abundance, as well as mechanisms to mobilize internal nutrient stores and reconfigure catabolic pathways to withstand periods of scarcity. Autophagy is the main cellular mechanism for the degradation and recycling of intracellular components, and thus of the utmost importance for adapting to and surviving nutrient limitation. This cellular process begins with the encapsulation of a cargo in a double membrane structure formed de novo called autophagosome, and culminates with the fusion of the lysosome and breakdown of the cargo into its basic building blocks, which will be used to produce energy or generate new cellular components. Thanks to pioneering studies in yeasts and later work in mammalian cells, we know that changes in the carbon source, as well as wholesale nutrient restriction, can activate macroautophagy (Mitchener et al., 1976). Multiple signaling mechanisms modulate autophagic activity in response to the nutritional status of the cell (Mehrpour et al., 2010), two of the most important of which are the mammalian target of rapamycin (mTOR) and the AMP activated protein kinase (AMPK).
MTOR comprises the mTOR 1 (mTORC1) and mTOR 2 (mTORC2) complexes. The mTORC1 complex is a nutrient sensing kinase that acts as an integrator of information on cellular energy balance and negatively regulates the initiation of macroautophagy. In the presence of nutrients and growth factors, mTORC1 is activated and supports anabolism and growth, while inhibiting catabolic pathways such as autophagy (Hosokawa et al., 2009) (Bar-Peled and Sabatini, 2014). Once activated, mTORC1 directly inactivates ULK1 (Unc-51 Like Autophagy Activting Kinase) by phoshorylation at Ser757 (Kim et al., 2011), a condition that is sufficient to inhibit autophagy in the presence of sufficient nutrients. Furthermore, mTORC1 can undirectly suppress autophagy by controlling lysosome biogenesis (Roczniak-Ferguson et al., 2012). mTORC1-mediated phosphorylation of the transcription factor EB (TFEB) decreases its transcriptional activity, thus decreasing the overall expression of autophagy-related gene expression (Roczniak-Ferguson et al., 2012). TFEB is the master transcriptional regulator of lysosomal and autophagy-related genes (Settembre et al., 2011).
he lysosome is the main mediator of cellular catabolism, being the key organelle in cellular degradation and recycling processes, which allows this organelle to senses cell’s nutritional status and give the initial response to the environmental changes. Thus, lysosome is at the center of a complex regulatory network for the control of cellular and organism homeostasis. The activation of mTORC1 requires its recruitment to the lysosomal surface, which is mediated by the amino acid-dependent activation of heterodimeric RAG GTPase and their interaction with Ragulator (Sancak et al., 2010; Lawrence et al., 2018; Lawrence and Zoncu, 2019). In this way, the information collected by the lysosome about the nutrient levels is transferred to mTORC1 to further regulate the biosynthesis pathways. The lysosome is also able to initiate the cellular response to a nutrient need through the activation of TFEB (Lysosomal mTORC1 and RAG GTPases modulate the nucleocytoplasmic shuttling of TFEB recruiting it in to the lysosomes) inducing the expression of genes that are related to the biogenesis of autophagosomes and lysosomes (Martina and Puertollano, 2013; Settembre et al., 2013). Recent findings have shown that lysosome is the place where AMPK can sense glucose availability as well in the absence of any changes in cellular energy state (i.e., AMP/ATP) through a mechanism that involves the formation of a complex with v-ATPase, ragulator, axin, liver kinase B1 (LKB1) acting in opposition to mTORC1 (Zhang et al., 2017).
AMPK is activated by nutrient limitation, energy deficiencies and other stress signals, and plays an essential role in the maintenance of metabolic homeostasis by activating key catabolic pathways while inhibiting anabolism. AMPK activation has classically been accepted to be triggerd by increased AMP/ATP ratios, but it was recently shown that it can be activated by glucose withdrawal even before AMP/ATP ratios increase (Zhang et al., 2017). AMPK promotes autophagosome formation through direct phosphorylation of ULK1 and ULK2 at Ser317 and Ser777 (Kim et al., 2011), and it can also indirectly activate autophagy by inhibiting mTOR through phosphorylation of tuberous sclerosis complex 2 (TSC2) and Raptor (Gwinn et al., 2008). Furthermore, AMPK is required during the TFEB-induced lysosomal biogenesis process; therefore, it is also capable of stimulating autophagic flux by increasing the number of lysosomal vesicles (Zhao et al., 2020). These studies have clearly demonstrated that AMPK, being a critical sensor of nutrient availability among other important cellular processes, is capable of coordinating autophagy activity through mTOR-dependent and independent mechanisms. Persistently high AMPK signaling as a consequence of energy shortages or dysregulated nutrient sensing would result in excessive autophagic flux and, consequently, in excessive elimination of certain organelles such as mitochondria, leading to further aggravation of the energy crisis. This is consistent with observations showing that persistent activation of the AMPK signaling pathway results in neuronal death in vivo (Ju et al., 2011; Domise et al., 2019; Belforte et al., 2021).
Although autophagy was originally described 60 years ago, numerous recent studies have reported autophagic alterations in different human disorders such as cancer and neurodegenerative diseases, among others (Mizushima et al., 2008; Mehrpour et al., 2010). Most of the studies that establish a connection between autophagy dysregulation and disease have emphasized the important role of autophagy in the maintenance of prosteostasis, in the quality control of intracellular organelles, its contribution to cell remodeling, or its role in innate and acquired immunity (Mizushima et al., 2008). In recent years, bioenergetic deficiencies combined with impaired autophagic responses to such challenges have emerged as novel potential drivers of disease, since they would ultimately aggravate the energy imbalance and compromise cellular homeostasis (Chakravorty et al., 2019). In highly specialized tissues with high-energy demands, the correct balance between energy production and utilization is essential to guarantee correct physiological functioning. The human brain consumes up to 20% of the body’s energy, predominantly directed towards biosynthesis, neurotransmission and defense against oxidative stress (Tefera et al., 2021). Therefore, during neurodegeneration or aging -both scenarios where energy-producing pathways are challenged and nutrients may be scarce-, the correct functioning of autophagy is not only necessary to recycle dysfunctional organelles and macromolecules, but it is also important for balancing nutrient needs and guaranteeing the availability of energy fuels, mainly in the form of glucose and fatty acids.
Alterations in autophagy and in the pathways and processes of energy production or sensing are a fundamental part of the complex neuropathological cascade in many neurodegenerative diseases, and several of the mechanisms that connect both processes, i.e., AMPK and mTOR, have been widely studied as first-in-class targets to treat neurodegeneration. The following lines are intented to make a comprehensive review on the mechanisms involved in defective nutrient sensing and organelle reclycling that are common or specific to the most prevalent neurodegenerative diseases, and how risk factors, both environmental and genetic, impinge on them. The processes and mechanisms are further summarized in Table 1 and illustrated in Figures 1–3.
TABLE 1
| Risk factors related to nutrient dyshomeostasis | Altered cellular mechanism | mTOR/AMPK dysregulation | References | |
|---|---|---|---|---|
| AD | Gene-related | — | Dysregulated mTOR response to starving | Lee et al. (2010) |
| APOE4 | Poor lipid utilization, Impaired lysosome and endosome sorting | |||
| PICALM | AMPK activation rescues amyloid pathology | Caccamo et al. (2010), Tramutola et al. (2017), Ou et al. (2018), Parcon et al. (2018) | ||
| MAPT | Impaired autophagosome trafficking | |||
| Environment/lifestyle-related | — | Leoni et al. (2013), Vurusaner et al. (2014) | ||
| Insulin resistance | Impaired brain glucose metabolism | |||
| Hypercholesterolemia | Impaired brain cholesterol metabolism | |||
| PD | Gene-related | — | Upregulated mTOR activity | Kitada et al. (1998), Valente et al. (2004), Stefanis et al. (2001) |
| Parkin, PINK1 | Impaired mitochondrial recycling | |||
| SNCA, LRKK2 | Impaired lysosome traffic and Impaired mitochondrial recycling | Ng et al. (2012), Zhu et al. (2019), Alegre-Abarrategui et al., 2009 | ||
| GBA | Defective lysosome activity and Mitochondrial dysfuncion | A et al. (2014), Magalhaes et al. (2016), Osellame et al. (2013); ME and AH. (2016) | ||
| Environment/lifestyle-related | Impaired brain glucose metabolism | Chen et al. (2015), Cai et al. (2019) | ||
| NA | — | |||
| ALS/FTD | Gene-related | — | Chronic overactivation of AMPK signalling likely aggravates neuropathology | Bartolome et al. (2013), Straub et al. (2021), Lim et al. (2012a), Ling et al. (2014), Perera and Turner (2016) |
| TARDBP, VCP, CHCHD10, SOD1 | Impaired energy production in mitochondria | |||
| Impaired glucose uptake in MNs | ||||
| C9ORF72, OPTN, SQSTM1, VCP, TBK1 | Impaired lysosome biogenesis and maturation | Root et al. (2021), Sundaramoorthy et al. (2015), Deng et al. (2020), Ju et al. (2009) | ||
| GRN (FTD) | link to metabolic disease and lysosome storage disorder | Youn et al. (2009), Smith et al. (2012) | ||
| Environment/lifestyle-related (ALS) | — | |||
| Low BMI | — | O’Reilly et al. (2013), Perera and Turner, (2016) | ||
| Type I diabetes mellitus | — | Jawaid et al. (2015) | ||
| Strenuous sport exercising | — | Gallo et al. (2016), Julian et al. (2021) |
Summary of altered mechanisms and related genes across neurodegenerative diseases.
NA, Not applicable.
FIGURE 1
FIGURE 2
FIGURE 3
2 Defects of Nutrient Signaling and Autophagy in Alzheimer’s Disease
Alzheimer’s Disease (AD) is the most prevalent age-related neurological disease worldwide, with its incidence increasing together with the rise of life expectancy. AD is characterized by the deposition of amyloid β protein (Aβ) in the form of extracellular amyloid plaques and by the presence of intracellular Neurofibrilary tangles (NFT) formed by hyperphosporylated Tau protein aggregates (TAU) (De Strooper and Karran, 2016). Aβ is genrated by the cleavage of APP by β-secretases (BACE1) and by Presenilin 1 and 2 (PS1 and PS2). Mutations in APP, PS1, PS2 or BACE1 (Reitz et al., 2020) have been shown to be responsible for the familiar cases of the disease, that show an early onset (EOAD). However, the vast majority of AD cases (95%) are sporadic, present a late onset (LOAD) and the causes remain largely unknown, although its thought to be due to a combination of environmental and genetic risk factors, in addition to lifestyle. Apolipoprotein E4 (ApoE4) allele is the most significant genetic risk factor for LOAD, being present in more than 50% of the cases (Raber et al., 2004). The main role of ApoE is cholesterol transport, but it is also involved in Aβ processing and glucose metabolism (Jiang et al., 2008). Indeed, regional glucose hypometabolism is an eary event in the pathogenesis of the disease, being used even for diagnosis purposes, and ApoE4 carriers show decreased glucose metabolism compared to non-carriers (Ong et al., 2014; Johnson et al., 2017).
Considering that glucose is a major source of energy in the brain, it is possible that in the context of AD, when deficiencies in glucose metabolism start to occur, autophagy plays a key role in order to acces internal sources of nutrients to maintain the bioenergetic homeostasis. However, deregulation of nutrient-sensing pathways and defects in autophagy and in the clearance of dysfunctional proteins have also been linked to AD pathophysiology (Nilsson and Saido, 2014; Hou et al., 2019). Therefore, a better understanding of pathways such as the regulation of mTORC1 and AMPK-induced activation of autophagy may offer novel potential treatments for age-related diseases.
2.1 Evidence of Poor Nutrient Sensing in Alzheimer’s Disease
A seminal work on the role of autophagy in AD identified a higher accumulation of autophagosomes in the frontoparietal cortex of affected patients through immuno-electron microscopy (Nixon et al., 2005). Later, another study showed Beclin-1 to be significantly reduced in AD brains (Pickford et al., 2008). Upregulation of microtubule-associated light chain 3-ll (LC3-ll), which induces autophagy, is seen at early AD stages (Yu et al., 2005). Moreover, genes that regulate autophagy, such as the phosphatidylinositol-binding clathrin assembly protein (PICALM), are correlated with the accumulation of tau in zebrafish and Drosophila models (Moreau et al., 2014) and have emerged as genetic risk factors for AD (Lee et al., 2011; Ortega-Rojas et al., 2016; Santos-Rebouças et al., 2017). Indeed, PICALM cleavage is altered in AD (Carrasquillo et al., 2010; Schnetz-Boutaud et al., 2012), leading to reduced levels that result in a lack of autophagosome maturation of (Lee et al., 2010). Also, autophagy is disrupted by PS1 mutations related to EOAD (Lee et al., 2010). Decreased lysosomal acidification, a cause of autophagy dysfunction, has been observed in fibroblasts from EOAD PS1 A246E mutation carriers and it’s linked to mutations in a vacuolar ATPase (v-ATPase), responsible for pumping protons into the lysosome (Coffey et al., 2014). In PS1 deficient cells, the amino-acid sensing mTORC1 is attached to lysosomal membranes and does not react to starvation, restricting lysosomal function (Reddy et al., 2016). These findings suggest that nutrient-sensing pathways could play a central role in the pathogenesis of AD.
mTORC1 is one of the most studied amino acid nutrient sensing signaling pathwys and plays a key role in both Aβ and TAU pathologies. Aβ accumulation has been associated to the overactivation of mTORC1 signaling, whilst mTOR signaling inhibition leads to reduced Aβ levels (Caccamo et al., 2010; Kioussis et al., 2021). Moreover, accumulating evidence suggests that mTOR activation enhances TAU pathology via autophagy inhibition which, in turn, enhances Aβ accumulation (Tramutola et al., 2017), that further promotes TAU hyperphosphorylation. In both cases, mTORC1 inhibition seems to be beneficial to diminish Aβ and TAU pathology (Wang et al., 2014), although mTORC1 regulation is complex and needs to be better understood in order to design effective treatments with minimal side-effects (Carosi and Sargeant, 2019).
Together with mTORC1, AMPK regulates glucose and lipid metabolism, it is deregulated in AD brains (Jeon, 2016). Numerous reports show that AMPK signalling modulates TAU phosphorylation and pathology in vivo (Domise et al., 2016; Wang et al., 2020b, 2020a). Further, AMPK is an autophagy activator, thus its activation should be beneficial to reduce Aβ pathology. For example, treatment with metformin, a known AMPK activator, induced a decrease of Aβ pathology in an in vivo model of AD (Ou et al., 2018). Also, some studies have shown that metformin is associated with a lower risk of AD (Ng et al., 2014; Chin-Hsiao, 2019). However, this issue remains controversial, as at least one other study found no correlation between metformin and AD risk in diabetic patients (Sluggett et al., 2020), and other studies have found an increased risk of AD with metformin intake (Imfeld et al., 2012; Ha et al., 2021). The discrepancies between these reports might be explained by inter-individual differences in the pathogenesis of AD and highlight the relevance of identifying AD subtypes for personalized treatments (Loera-Valencia et al., 2019a; Caberlotto et al., 2019). Altogether, it becomes evident that nutrient metabolism and autophagy are crucial regulators of pathways with relevant roles in TAU and Aβ aggregation (Tang et al., 2014; Yang et al., 2019).
2.2 Autophagy Dysregulation in the Context of Alzheimer’s Disease: Focus on Disease-Relevant Genes and Metabolic Risk Factors
2.2.1 Amyloid β Protein
The relationship between autophagy and amyloidosis is complex since many features of proteolytic cleavage are involved in both the clearance and production of amyloid-beta. Inhibition of autophagy with chloroquine (CQ) in SH-SY5Y cells and the HEK293T (AβPPsw) model induces intracellular accumulation of Aβ1-42 (Gerenu et al., 2021), while autophagic activation by estrogen receptor beta overexpression, decreased Aβ1-42 levels (Wei et al., 2019). However, Nilsson and co-workers previously reported that a conditional knockout of Atg7 (with impaired autophagy), decreased amyloid-beta secretion by 90%, and normal Aβ secretion was restored when autophagy was stimulated to normal levels (Nilsson et al., 2013; Loera-Valencia et al., 2019b). This is evidence that autophagy is important not only for Aβ degradation but also for its production, this theory is supported also by the presence of the four components of the beta-secretase complex in autophagic vesicles (Yu et al., 2005).
Although the exact mechanism regulating the formation of the isolation membrane that leads to autophagosome formation is not fully understood, it has been reported that mitochondria-endoplasmic reticulum (ER) contact sites (MERCS) are one of the places where this process can occur (Garofalo et al., 2016). MERCS are domains arising from the interaction between specific regions of the ER and the outer mitochondrial membrane. These contact sites have been related to important biological processes such as the ER calcium shuttling into mitochondria and they are altered in AD and in AD-related models (Paillusson et al., 2016). Moroever, all required components for APP processing as well as Aβ peptide can also be found in MERCS (Schreiner et al., 2015). It was recently shown that the connectivity between mitochondria and ER is increased in brains and primary neurons isolated from AD mouse models displaying increased amyloidosis. In addition to MERCS ultrastructural alterations, neurons from AD mouse models also displayed alterations in autophagosome formation, mitochondrial membrane potential and ATP production during starvation (Leal et al., 2020).
2.2.2 MAPT (Tau Protein Aggregates)
Autophagy is also related to the clearance of TAU as an alternative pathway for degradation by the ubiquitin-proteasome system. Swedish amyloid precursor protein gene double-mutation KM670/671NL (APPswe) EOAD patients show hyperphosphorylated tau associated with LC3 and p62 (a ubiquitin-binding protein) in their frontal cortex, which is not present in control subjects (Piras et al., 2016). Again, the conditional knockout model of Atg7 shows impaired autophagy, age-related neurodegeneration, and accumulation of hyperphosphorylated tau in the brain (Inoue et al., 2012). Reciprocally, pathogenic tau mutations such as the tubulin-binding repeats linked to chromosome 17 can induce accumulation of acid lysosomes and interfere with normal autophagic flux (Lim et al., 2001). Surmounting evidence shows the importance of autophagy for AD and other neurodegenerative diseases; nevertheless, the classic view of autophagy as a recycling system of the cell is becoming outdated, since it can actively modulate mechanisms of synaptogenesis and cell death (Nikoletopoulou et al., 2015; Jung et al., 2020). Therefore, we need to study these new roles if a feasible autophagy-based AD therapy is to be developed.
2.2.3 ApoE4
As we mentioned before, ApoE is a lipoprotein that functions as a transporter of cholesterol and other lipids in the body (Holtzman et al., 2012). ApoE has three isoforms (ε2, ε3 and ε4) where ApoE4 confers a higher risk to develop AD and is regarded as the strongest genetic risk factor for LOAD (Kloske and Wilcock, 2020).
Several studies point to ApoE4 involvement in autophagy dysfunction. FoxO3a, a member of a family of transcription factors that play a role in the regulation of several autophagy-related genes (Audesse et al., 2019), is repressed in human postmortem brain samples from AD ApoE4 carriers in comparison to non-carriers (Sohn et al., 2021). This study also showed a decrease in protein expression of autophagy- and mitophagy-related genes, such as ATG12, BECLIN-1, BNIP3 and PINK1 that are regulated by FoxO3a. Another study reported that the coordinated lysosomal expression and regulation (CLEAR) DNA motifs, a region in the DNA where the master autophagy regulator and TFEB induces its downstream genes, can also be targeted by ApoE4, which competitively binds CLEAR motifs, inhibiting the expression of autophagy-related genes (Parcon et al., 2018).
Cell-type-specific studies show that astrocyte metabolism is affected by ApoE4 expression (Figure 1). Upon autophagy-inducing conditions, ApoE4 expressing astrocytes exhibited lower autophagic flux in comparison to ApoE3 astrocytes (Simonovitch et al., 2016). Moreover, APOE4 expression was associated with altered mitochondrial dynamics including, fusion, fission, and mitophagy in comparison to ApoE3-expressing astrocytes (Schmukler et al., 2020). A recent study investigated the role of ApoE4 in regulating fatty-acid metabolism in the brain (Qi et al., 2021) and found that ApoE4 astrocytes had more fragmented mitochondria, lower β-oxidation levels and a higher accumulation of lipid droplets. These effects could in turn contribute to the lipid dysregulation and bioenergetic deficits that are observed in AD (Sato and Morishita, 2015).
2.3 Lipid and Cholesterol Sensing in Alzheimer’s Disease
2.3.1 Alterations in Cholesterol Metabolism Lead to Inflammation and Synaptic Dysfunction in the Brain
Cholesterol nuclear receptors are widespread in neurons and glial cells alike (Olkkonen and Levine, 2004; Ali et al., 2013; Moutinho et al., 2015). Cholesterol levels in the brain are regulated by the interplay between oxidized forms 24-hydroxycholesterol (24-OH) and 27-hydroxycholesterol (27-OH) (Meaney et al., 2007; Båvner et al., 2010; Shafaati et al., 2011; Ali et al., 2013). As cholesterol is metabolized by the enzyme CYP46A1 into 24-OH in neurons, HMG-CoA reductase is activated to promote cholesterol biosynthesis in the brain. Conversely, high levels of 27-OH coming from peripheral cholesterol catabolism decrease HMG-CoA reductase activity and decrease global cholesterol levels in the brain. Neurodegeneration depletes the brain from CYP46A1 and thus decreases cholesterol biosynthesis (Leoni et al., 2013), which is essential for memory function. Similarly, hypercholesterolemia leads to the accumulation of 27-OH in the brain, which disrupts cholesterol metabolism as discussed below.
Cholesterol in the brain is synthesized de novo by glial cells, transporting it to neurons via lipoprotein particles, such as APOE (Figure 1). When cholesterol synthesis disruption causes neuronal function impairment, showing behavioral defects in mice (Ferris et al., 2017). Cholesterol metabolism deregulation can alter astrocytes and microglia physiological states. High levels of 27-OH generated from a high-fat/cholesterol diet led to overproduction of S100A8 alarmin and its receptor RAGE, inducing sterile inflammation that contributes neurodegeneration (Loera-Valencia et al., 2021a).
Besides pro-inflammatory responses, 27-OH is also able to activate cellular pathways that are regulated by reactive oxygen species (ROS) such as autophagy (Vurusaner et al., 2014). By activating the extracellular signal-regulated kinase (ERK) and the phosphoinositide 3-kinase (PI3K)/Akt pathways, 27-OH induces the upregulation of the nuclear factor erythroid 2 p45-related factor 2 (Nrf2) in vitro. Nrf2 is a transcription factor for several antioxidant proteins that plays a role modulating autophagy via p62 in response to oxidative stress (Tang et al., 2019). In vitro treatments of 27-OH induced an increase in Beclin1, LC3II and a decrease in p62 (Vurusaner et al., 2018).
Cholesterol metabolism alterations can also directly impact synaptic function (Figure 1). Evidence shows that APOE4 has low transport affinity and binding capacity for lipids reducing cholesterol transport from astrocytes to neurons and leading to lower synaptic density and cognitive decline (Lee et al., 2021). Cholesterol imbalance in the brain can worse memory performance in mice (Merino-Serrais et al., 2019), lead to synaptic dysfunction by downregulating Psd95 and SNAP-25 (Merino-Serrais et al., 2019).
2.3.2 CD36 and Lipid Sensing in the Brain
While neuroendocrine neurons sense fatty acids to modulate metabolism in the body (Oishi et al., 1990; Tewari et al., 2000; Honen et al., 2003), at the cellular level, the molecular sensor for fatty acids in brain cells is CD36, a transmembrane glycoprotein that is expressed in many cell types, including epithelial cells, adipocytes, dendritic cells and hepatocytes, among others (Mitchell et al., 2011). Although CD36 plays a role in the binding, transport and uptake of fatty acids (Pepino et al., 2014; Ioghen et al., 2021) as well as fatty-acid sensing in oral epithelial cells (Laugerette et al., 2005), it has multiple cellular functions such as angiogenesis or internalization of bacteria depending on the cell type where it is expressed on (Abumrad et al., 2005).
Early studies on Alzheimer’s Disease patients showed that CD36 is highly expressed in cortical samples of AD patients and cognitively normal subjects with the presence of amyloid plaques compared with amyloid-free controls (Ricciarelli et al., 2004). This study pointed at an association between CD36 and Aβ in human brains independently of AD occurrence. However, a genetic study of 859 AD patients revealed that a polymorphism in the CD36 gene significantly associated the risk for developing AD (Šerý et al., 2017). In the early stages of AD, activated microglia and macrophages induce the expression of CD36 in response to Aβ (Ricciarelli et al., 2004). The interaction between Aβ and CD36 has been shown to activate the NLRP3-inflammasome system, inducing the production of pro-inflammatory cytokines (Sheedy et al., 2013) and ROS (Coraci et al., 2002; Bamberger et al., 2003). It is also interesting to consider the possibility that early autophagy induction by CD36 in response to Aβ can also increase the autophagy-mediated secretion of oligomers from multi-vesicular bodies in the brain, thus contributing to seeding and protein aggregation (Figure 1) (Nilsson et al., 2013; Loera-Valencia et al., 2019b).
3 Defects of Nutrient Signaling and Autophagy in Parkinson’s Disease
Parkinson’s disease (PD) is a common neurodegenerative disorder that currently affects 1% of people over 65 years of age (Aarsland et al., 2021), and whose prevalence is expected to double by 2060 in the United States (Savica et al., 2018). As well as bradykinesia, rigidity or rest tremor (Postuma et al., 2015), PD often features non-motor symptoms (NMS), such as cognitive impairment or sensory alterations (Chaudhuri and Schapira, 2009). Pathophysiologically, PD is characterized chiefly by a slow and progressive degeneration of dopaminergic neurons in the Substantia Nigra (Drui et al., 2014), which is the cause of most of the motor symptoms, although non-dopaminergic neurons are also affected in PD (Schneider and Alcalay, 2017; Stoker and Greenland, 2018). While the underlying causes of the neurodegenerative process in PD are not fully understood, it evidently stems from a combination of cell-autonomous and non-cell-autonomous mechanisms. The former include, altered mitochondrial bioenergetics, impaired protein recycling (Al-Bari and Xu, 2020), where the master regulators mTOR and AMPK are crucial nutrient sensors (Hardie et al., 2012; Tan and Miyamoto, 2016), whereas protein aggregation and neuroinflammation are prominent non-cell-autonomous contributors to PD pathology, and it is believed to pathogenesis (Poewe et al., 2017). The development of dopaminergic neurons is an energy demanding process owing to the extensive branching of dendrites. This high-energy demand suggests nutrient and energy sensing, via mTOR and AMPK could be key disease development modifiers. In fact, much of the future work investigating defective organelles and metabolic abnormalities in PD is expected to involve analysis of specific proteins implicated in PD via pathological and genetic studies. Hence, here we focus on the effects of PD causing gene mutations on energy balance and the autophagolysosomal pathway, and their relationship to nutrient-sensing abnormalities.
3.1 Evidence of Poor Nutrient Utilization and Autophagy Dysregulation in Parkinson’s Disease Patients
PD patients are neuropathologically characterized by α-synuclein (α-syn) Lewy Bodies (LBs) neuronal inclusions, not efficiently metabolized by abnormal autophagy observed in dopaminergic neurons in the substantia nigra in PD brains (Anglade et al., 1997). Phosphorylated ubiquitin, as mitophagy tag, is increased in PD patients and correlates with tau tangles and levels of Lewy Bodies (LB) (Fiesel et al., 2015; Hou et al., 2018). Moreover, LC3II has been localized in LBs and its levels were significantly increased in PD samples (Dehay et al., 2010). In contrast, lysosomal enzymes activity is impaired, showing that autophagy is an important cellular process in PD pathophysiology.
Recycling of cell components is most important during starvation and so nutrient sensing and energy producing and regulating systems should always be considered in conjunction with autophagy. Increased expression of mTOR has been detected in the temporal cortex of PD patients with aggregations of α-syn (Crews et al., 2010; Zhu et al., 2019). Moreover, the inhibition of autophagy that accompanies elevated α-syn expression is attributed to mTOR activation (Gao et al., 2015; Zhu et al., 2019). Equally, in cultured cells and neurons with mutant α-syn mTOR signaling is upregulated and autophagy repressed, with the latter being reversed by mTOR inactivation (Jiang et al., 2013; Zhu et al., 2019). Although mTOR inactivation restores autophagy in PD cell models, it is crucial to avoid complete inhibition, because mTOR is essential for neuronal growth and survival and it regulates many important processes, such as synaptic plasticity and memory formation (Bekinschtein et al., 2007). Therefore, a balance between induction of autophagy and a basal mTOR activity has to be achieved for optimal brain function (Zhu et al., 2019).
AMPK plays a major role in regulating energy metabolism, via activation of catabolism and repression of energy-consuming processes (Hang et al., 2015), and AMPK activation can be neuroprotective during glucose starvation (Culmsee et al., 2001). AMPK appears to impact dopaminergic neuronal homeostasis in conjunction with the protein Parkin, is involved in recycling mitochondria (Zong et al., 2002). Concordantly, AMPK activation by metformin ameliorates the locomotion defects of Parkin-null flies, whereas this protective effect disappears when AMPK is silenced. Overexpression of AMPK also partially rescued the mitochondrial abnormalities of Drosophila that express mutated human leucine rich repeat kinase 2 (LRRK2), LRRK2G2019S (Ng et al., 2012). Furthermore, resveratrol, a strong activator of AMPK in neurons, increased mitochondrial biogenesis and autophagic flux in fibroblasts carrying defective Parkin (Ferretta et al., 2014). Conversely, prolonged activation of Parkin (Van Rompuy et al., 2014) or AMPK have been shown to be detrimental for dopaminergic neurons, so both Parkin and AMPK can be neuroprotective or neurotoxic depending on the context, although the reasons behind these diverse effects are not known (Hang et al., 2015). Therefore, further research is needed, to determine the degree and duration of the Parkin-AMPK activation that achieves positive effects on neuron function and survival (Hang et al., 2015).
Autophagolysosomal and mitochondrial function are integrated with lipid metabolism and there are multiple lines of evidence indicating that lipid homeostasis is perturbed in PD. For example, (Zambon et al., 2019), showed a marked reduction in cholesterol levels in dopaminergic neurons of PD patients derived from iPSCs, together with reduced expression of CYP46A1, an ER enzyme crucial for cholesterol metabolism (Liu et al., 2010; Zambon et al., 2019). Moreover, some studies have linked α-syn toxicity with lipid droplet accumulation in human iPSC-derived neurons, (Fanning et al., 2019), whereas an, ultrastructural analysis of LB of PD brain found accumulated lipid vesicles, membrane fragments and cytoskeletal elements (Shahmoradian et al., 2019). Furthermore, lipid accumulation has been implicated in several events associated with PD, such as lysosomal blockade and neuroinflammation (Hallett et al., 2019). Hence, lipid metabolism is a highly promising target for PD.
Although altered cell metabolism is involved in PD pathogenesis, further research is needed to elucidate the fundamental basis of the metabolic alterations, as well as to understand better the network of interactions that encompasses the diverse mechanisms implicated in PD described above. The improved knowledge is expected to clarify and reconcile, or refute, the following contradictory results. While some studies reported a reduced risk of PD in patients with Type 2 diabetes (T2D) (Sääksjärvi et al., 2015), others find the opposite (Hu et al., 2007; Sun et al., 2012), or no relation between them (Palacios et al., 2011). Similar inconsistencies have been described in relation to cholesterol metabolism, indicating that modifying factors could be modulating the association between blood cholesterol and PD risk (de Lau et al., 2006; Mascitelli et al., 2009).
3.2 Evidences of Mitochondrial Dysfunction in Parkinson’s Disease
Mitochondria have been heavily implicated in PD since the landmark finding that PD brains have low respiratory complex I activity (Schapira et al., 1989) and that the complex I inhibitor rotenone induces dopaminergic cell death in rats (Alam and Schmidt, 2002). Although the entire respiratory chain depends on proteins encoded in mitochondrial DNA (mtDNA), defects in mtDNA can predominantly affect complex I (Dunbar et al., 1996). Moreover, mutant mtDNAs accumulate with age, and more so in PD patients (Bender et al., 2006), or alternatively PD patients display low mtDNA numbers (Tzoulis et al., 2013). Mitochondrial DNA is further implicated in the PD developmental cascade by the fact that a variety of defective nuclear genes that adversely impact mtDNA cause parkinsonism, featuring DPN cell death and levodopa responsive motor dysfunction (Miguel et al., 2014; Pedroso et al., 2018).
Further interest in mitochondrial involvement in PD was sparked by the findings that mutations in key factors required for mitochondrial recycling, Parkin and PINK1, cause familial PD (Kitada et al., 1998; Valente et al., 2004). These findings were all the more provocative as other recycling factors were implicated in the disease, and defective autophagy is a recurring theme in other neurodegenerative disorders (Menzies et al., 2015; Guo et al., 2018). Moreover, as impaired autophagy can result from mitochondrial dysfunction, and respiratory complex I is needed for maximal autophagy (Thomas et al., 2018), the activities of mitochondria, autophagosomes and lysosomes are coupled; hence, problems with these organelles are concordant, rather than competing, explanations for the development of PD.
Changes in mitochondrial function in PD could stem from, or be exacerbated by, altered nutrient metabolism given that the brain requires a continuous supply of energy in the form of ATP, much of which is produced from glucose, either via glycolysis or additionally by its further oxidation in mitochondria (Cunnane et al., 2020). In fact, decreased glucose metabolism have been observed in PD patients (Eberling et al., 1994; Ahmed et al., 2009). Interestingly, glucose deprivation promotes α-syn aggregation (Bellucci et al., 2008), and impaired bioenergetics and reduced ATP levels might contribute to the pathogenesis of PD (Cai et al., 2019). Furthermore, Cai and colleagues suggested that glycolysis could be a new therapeutic target for PD, as Terazosin, a drug used to treat prostatic hyperplasia and hypertension, has neuroprotective effects in multiple PD models. Terazosin, in addition to blocking α-adrenergic receptors, enhances glycolysis by stimulating phosphoglycerate kinase 1 (PGK1) activity; and consequently, increases oxidative phosphorylation and thus ATP levels (Chen et al., 2015). This enhancement of PGK1 activity increases dopamine levels, slows or prevents neurodegeneration and improves motor performance in several animal models of PD, such as the MPTP mouse, OHDA rat, and PINK1 and LRRK2 fly models (Cai et al., 2019).
3.3 Parkinson’s Disease Causing Genes as Drivers of Metabolic Alteration
Although a large majority, 85–90%, of PD cases are sporadic there is intense interest in the monogenic causes of PD, as they can potentially reveal the underlying processes that are central to all forms of PD. Among the cellular mechanisms of the mutant proteins that cause familial PD are ones related to protein folding and aggregation, and phosphorylation of α-syn (encoded by the SNCA gene), neuroinflammation, intracellular vesicular trafficking, lysosomal and mitochondrial function (Hong et al., 2011; Hirsch et al., 2013; Ryan et al., 2015; Poewe et al., 2017; Kuhlmann and Milnerwood, 2020). The genetic study of PD started in 1997 with the discovery of a missense variant (A53T) in SNCA in a large Italian family with PD (Polymeropoulos et al., 1996). Currently, more than 20 genes have been identified as PD causing genes (SNCA, PARKIN, PINK1, DJ1, LRRK2, GBA, VSP35…) (Blauwendraat et al., 2020). Here, we focus on three genes with strong links to nutrient-sensing, energy metabolism and recycling pathways (Figure 2).
SNCA. α-syn protein is a natively soluble protein encoded by SNCA gene. Under non-pathological conditions, α-syn is predominantly localized in the presynaptic terminal, where it plays important roles in transmembrane transport, intracellular trafficking and cell recycling pathways, including autophagy, Chaperone-Mediated Autophagy (CMA) or proteasome pathways (Seidel et al., 2010; Hong et al., 2011; Cannon et al., 2013; Eisbach and Outeiro, 2013; Oaks et al., 2013; Blauwendraat et al., 2020). In PD, abnormally aggregated α-syn is the major component of LBs (Iwatsubo, 2003).
Study of the α-syn mutant, A53T, (A53T) has revealed alterations in SIRT1, a NAD+-dependent deacetylase involved in several metabolic functions, including mitochondrial respiration and lipid metabolism suggesting bioenergetic perturbations in dopaminergic neurons (Donmez and Outeiro, 2013; Zambon et al., 2019). Additionally, α-syn interacts directly and indirectly with mitochondria and the mitochondrial recycling pathway. In fact, stable rat neurons expressing A53T, but not wild-type α-syn, showed significant accumulation of autophagic vesicles containing mitochondria (Stefanis et al., 2001). Hence, the inference is that mutant ASYN impairs mitophagy (Stefanis et al., 2001).
Mutations in the LRRK2 gene are the most frequent cause of autosomal dominant monogenic PD. The LRRK2 protein is primarily localized in membrane microdomains, multivesicular bodies, and autophagic vacuoles (Alegre-Abarrategui et al., 2009; Rideout and Stefanis, 2014), and it has been linked to autophagy and phagocytosis (Stoker and Greenland, 2018). Mutations in LRRK2, such as G2019S (kinase domain) or ROC/COR domain (R1441C, R1441H and R1441G), enhance the protein’s kinase activity, and lead to impaired vesicular trafficking, lysosomal activity and mitochondrial function in PD brains (Ryan et al., 2015). Particular mutations alter different steps of macroautophagy (Kuhlmann and Milnerwood, 2020; Madureira et al., 2020), such as autophagosome-lysosomal fusion and cargo degradation (Blauwendraat et al., 2020). Indeed, many LRRK2 mutant cell models have accumulated large autophagic vacuoles and multivesicular bodies containing incompletely degraded material (Alegre-Abarrategui et al., 2009), together with altered mTOR and AMPK activity (Ferree et al., 2012). Collectively, these studies indicate a direct relation between defects in autophagy and LRRK2 gain-of-function. In addition to effects on macroautophagy, LRRK2 mutations were also reported to affect CMA (Orenstein et al., 2013) and mitophagy (Hsieh et al., 2016; Korecka et al., 2019), so most of the clearance routes have been shown to be altered in LRRK2-PD. Furthermore, iPSCs cultures derived from patients carrying G2019S or R1441C mutations have defects in mitochondrial dynamics, as well as alterations in dinucleotide metabolism (Sanders et al., 2014) or abnormal accumulation of autophagic vacuoles (Rosenbusch and Kortholt, 2016). Similarly, animal models overexpressing LRRK2-G2019S show mitochondrial dysfunction (Rosenbusch and Kortholt, 2016). In summary, these data indicate that LRRK2 pathogenic mutations can impair autophagy, and diminish mitochondrial function and vesicular trafficking.
GBA is a glucosylceramide hydroxylase (GCase), which is important for sphingolipid degradation (Alcalay et al., 2015). GBA gene variants are towards the upper limit for risk factors, being the single greatest risk factor for PD. The initial observation of higher incidence of PD in families with Gaucher disease, a lysosomal storage disorder (LSD) caused by deficiency of the GCase, has led to the identification of heterozygous mutations in GBA as important and common risk factors for sporadic PD (Billingsley et al., 2018). Although enzymatic activity of GCase in GBA heterozygous mutants is not well known, an increased production of reactive oxygen species, and affected autophagolysosome function have been shown in fibroblasts from GBA heterozygotes with or without PD (McNeill et al., 2014; Magalhaes et al., 2016). Interestingly, all these findings in GBA mutants suggest a relation between altered lipid metabolism, bioenergetics and autophagy-lysosomal dysfunction in PD pathogenesis. For instance, α-syn accumulation has been described in the brain of patients with lysosomal disorders, as well as in several mouse models of lysosomal storage diseases. Hence, lipid accumulation could contribute to neurotoxicity (Shachar et al., 2011; Nelson et al., 2014, 2018; Smith et al., 2014).
On the other hand, mitochondrial dysfunction and autophagy defects have also been reported in Gaucher disease cellular models, such as iPSC-derived neurons from GBA-PD patients, primary post-mortem brain tissue from GBA heterozygous patients or primary hippocampal neurons from GBA L44P knock-in mouse brains. (Osellame et al., 2013; Gegg and Schapira, 2016; Schöndorf et al., 2018; Li et al., 2019; Hou et al., 2020).
4 Defects of Nutrient Signaling and Autophagy in ALS/FTD
4.1 Evidences of Poor Nutrient Utilization and Autophagy Dysregulation in ALS/FTD Patients
All over these last few decades, ALS has come to be recognised as a complex syndrome, not only due to its heterogeneous clinical display, but also to its newly discovered clinical manifestations affecting fundamental systemic processes such as metabolism and autophagy. ALS is therefore considered a systemic condition, rather than a solely neurological disease (Dupuis et al., 2011; Robberecht and Philips, 2013; Riancho et al., 2019). ALS is often related to another neurodegenerative disorder with non-motor symptoms, the frontotemporal lobar dementia (FTD) (Lau et al., 2018), as they both share common clinical and pathological phenotypes—most notably, the existence of TDP-43 positive aggregates -, and even genetic causality (Robberecht and Philips, 2013; Pang and Hu, 2021); thus, they have been suggested to be part of the same spectrum of disease (Neumann et al., 2006). In this respect, bioenergetic alteration and activation of the unfolded protein response (UPR) are also key events of FTD.
4.1.1 Metabolic Alterations Present in ALS Patients
Neurodegeneration is frequently linked to, and potentially arises from a defective energetic metabolism (Dupuis et al., 2011). In the same fashion as in other neurodegenerative disorders such as Parkinson’s or Alzheimer’s disease (Pradat et al., 2010; Błaszczyk, 2020), metabolic homeostasis of ALS patients is severely altered (Tefera et al., 2021). Of note, most of the occurring abnormalities correlate with life expectancy and have a negative impact on overall pathogenic process (Dupuis et al., 2011).
One of the hallmarks of the metabolic alterations observed in ALS is the loss of the energetic balance, primarily due to a higher energetic expenditure. In other words, patients with ALS frequently exhibit a hypermetabolic state characterised by an increase of energetic expenditure, loss of body mass and depletion of energetic deposits as the disease progresses (Bouteloup et al., 2009; Dupuis et al., 2011; Zufiría et al., 2016; Vandoorne et al., 2018). Despite the uncertainty regarding the importance and causality of such dysregulation, it is worth mentioning that body weight loss is a fundamental prognostic factor for the patients, who are often associated with a lower baseline BMI, and being the disease prevalence 40% lower among obese individuals (O’Reilly et al., 2013; Perera and Turner, 2016; Vandoorne et al., 2018). In this sense, alterations in lipid homeostasis have also been described, reporting an increased LDL/HDL ratio and also higher triglyceride and cholesterol levels in ALS patients compared to controls, exhibiting a rather protective effect (Dupuis et al., 2008). Nonetheless, recently performed case-control studies on peripheral lipid profile of patients with ALS have found little or no discriminatory lipid signature (Fernández-Eulate et al., 2020; Sol et al., 2021).
Remarkably, an obesity-related condition as T2D also seems to act as a protective state for the development of ALS, delaying the disease onset up to 4 years (Mariosa et al., 2015; Tefera et al., 2021). On the other hand, type I diabetes would act as a risk factor (Jawaid et al., 2015). In accordance with this, abnormal glucose tolerance has been observed in ALS patients (Pradat et al., 2010); however, there was controversy as to whether this intolerance was inherently disease-related or due to a lack of glucose utilisation as a result of muscle atrophy, or even to an increased level of free fatty acids (Reyes et al., 1984; Perera and Turner, 2016).
Interestingly, despite the mentioned evidence of the systemic hypermetabolic state of the patients, several performed PET and autoradiography studies using glucose analogues have demonstrated that frontal and occipital cortex in the CNS contrarily exhibit a hypometabolic state, while midbrain areas would remain hypermetabolic (Pagani et al., 2014; Endo et al., 2017; Germeys et al., 2019; Tefera et al., 2021). To understand the controversy between the reported hypermetabolism at a systemic level and hypometabolism at a central level in patients, it is key to acknowledge the different nature of processes to which they refer. The systemic hypermetabolic state indicates increased oxygen consumption and is recognised as an adaptive response to satisfy prolonged increased energy demands or to cope with the inefficiency with which energy is utilized, through the mobilization of energy stores. However, the CNS hypometabolic state is ascribed to an inability of cells to take up glucose due to a state of reduced neuronal cell density, reduced blood flow and reduced glucose transporter expression (Tefera et al., 2021), while the central hypermetabolic state is rather considered a result of glia hyperactivation (Haidet-Phillips et al., 2011; Cistaro et al., 2012). Both states, systemic hypermetabolism and central hypometabolism, appear to have in common one sign: the inability with which glucose is utilized to obtain energy.
The observation of an impaired glucose uptake in motor sensory cortex of ALS patients (Pagani et al., 2014) comes in line with the defective energetic balance of such population. Besides, sporadic ALS patient-derived skin fibroblasts showed markedly lowered levels of glycolytic components (Szelechowski et al., 2018), indicating an alteration of carbohydrate metabolism. Nevertheless, high glycogen concentrations in the spinal cord of autopsied patients have been observed (Vandoorne et al., 2018), both in neurons and glia (Neumann et al., 2006). All these findings together suggest a loss of the natural coupling between blood flow and glucose metabolism in the CNS. Importantly, a widespread prefrontal glucose hypometabolism has been linked to worsened prognosis and shorter survival of ALS patients (Rajagopalan and Pioro, 2019).
It has been suggested that multiple environmental modifiers known to influence energy metabolism, affect the disease onset and/or its course (Zufiría et al., 2016). One of the most controversial activities has been physical exercise, as it is a potent modifier of muscle energy metabolism (Dupuis et al., 2011; Perera and Turner, 2016; Zufiría et al., 2016), but its exact repercussion over the disease is still unknown and many of the performed studies have come up with contradictory results. Case-control studies carried out over a population from various European countries came across the fact that not only strenuous physical exercise was related to the precipitation of the disease, but also that moderate exercise exerts neuroprotective effect (Gallo et al., 2016; Julian et al., 2021). Studies carried out among amateur and professional skiers in Sweden reported that professional athletes—but not amateurs-where related to a higher risk of developing ALS (Fang et al., 2016). Equally, another retrospective cross-sectional observational study carried out on triathletes found a drastically high representation of such athletes as ALS patients (Gotkine et al., 2014). Therefore, these two researches provide evidence suggesting that high-intensity physical exercise might exacerbate the risk of developing ALS.
As essential players in cellular metabolism, several studies have reported mitochondrial abnormalities in ALS patients (Perera and Turner, 2016; Vandoorne et al., 2018; Tefera et al., 2021). Some of the most representative ones are the presence of mitochondrial aggregates in skeletal muscle of patients (Sasaki and Iwata, 2007), and altered dynamics (Tsitkanou et al., 2016) structure, localization, volume and number in motor neurons, muscle and intra-muscular nerves (Perera and Turner, 2016). It is worth noting that mitochondrial function would also be disrupted, particularly regarding alterations in the electron transport chain, displaying reduced activity of the key enzyme complexes of the electron transport chain (Wiedemann et al., 2002; Smith et al., 2019).
Notably, motor neurons appear to be especially vulnerable to these defects in energetic homeostasis (Vandoorne et al., 2018; Ragagnin et al., 2019) due, at least in part, to their need to fulfil unusually high energetic demands to maintain resting membrane potential and propagate action potentials (Perera and Turner, 2016). Importantly, different types of motor neurons have been shown to be differentially sensitive to the disease associated damage, with the neurons innervating type IIb fibres being the most sensitive ones (Nijssen et al., 2017). Interestingly, it is this latter type which most clearly relies on glycolytic metabolism for energy (Frey et al., 2000), again suggesting an alteration in energy metabolism in the mechanism of ALS.
4.1.2 Autophagy-Related Alterations in Patients With ALS
The pathology of ALS/FTD is typically accompanied by marked alterations in a key physiological process that is related to energy homeostasis: autophagy (Sala et al., 2012). Since the deletion of autophagy-related genes is sufficient to cause neuronal death, autophagy is considered an essential homeostatic process for neurons (Evans and Holzbaur, 2019).
In general, most neurodegenerative diseases, including ALS and/or FTD, are characterised by a common pathology comprising misfolded protein inclusions (Lee et al., 2015; Crippa et al., 2016; Torres et al., 2021), which eventually are responsible for degeneration or damage of nerve cells in the form of oxidative and ER stress, mitochondrial dysfunction and neuroinflamation (Ghavami et al., 2014; Corti et al., 2020; Luo et al., 2020). In the specific case of ALS, most of the mutant proteins associated to the familial inheritance of the disease exhibit aggregation-prone domains, and thus must be eliminated to prevent motor neuron toxicity (Zhang et al., 2020). Motor neurons are particularly sensitive to toxicity caused by protein misfolding, although it can also affect other cell types, such as muscle (Crippa et al., 2013). These defective proteins disrupt the ubiquitin-proteasome system, which is one of the central degradation systems for the cell, and thereby initiating a vicious cycle that culminates with further protein deposition leading to the formation of inclusions (Crippa et al., 2010).
The mentioned protein inclusions present in ALS/FTD generally consist of proteins involved in the intracellular degradation systems, various signalling systems, and in particular, TDP-43 (Neumann et al., 2006; De Marco et al., 2011). The neuropathology associated with TDP-43 mutations is characterised by ubiquitin-positive inclusions in neuronal and glial cells in the spinal cord and brain of ALS and FTD patients (Arai et al., 2006; Zhang et al., 2009). Besides, TDP-43 aggregates are characterised by the presence of phosphorylated forms of the protein, both in its full form and in fragments (Crippa et al., 2016). Ubiquitin positive TDP-43 aggregates co-localize with autophagy markers in spinal motor neurons of sporadic ALS patients (Burk and Pasterkamp, 2019).
Several studies suggest that activation of autophagy may be protective in some neurodegenerative diseases, given the potential removal of toxic protein species (Wong and Cuervo, 2010; Chen et al., 2012; Jaronen et al., 2014). Under normal conditions, a HSP protein-driven chaperone-mediated form of autophagy takes place in brain and muscle (Corti et al., 2020). In this very sense, abnormally high concentrations of the autophagy-enhancing chaperone HSPB8 have been detected in the spinal cord (Anagnostou et al., 2010), skeletal muscle (Crippa et al., 2013) and surviving motor neurons (Crippa et al., 2016) of ALS patients, as a possible strategy of the damaged cells trying to get rid of the toxic aggregates. In parallel, a number of post-mortem and experimental neuropathological studies have revealed an increase in the number of autophagosomes in motor neurons of the spinal cord of both sporadic and familial cases of ALS (Chen et al., 2012; Riancho et al., 2019).
In this sense, changes in ER morphology have been reported in patients with ALS (Zufiría et al., 2016). For instance, fragmentation of the rough ER, irregular distention of its cisternae and detachment of ribosomes have been described in degenerating anterior horn cells of post-mortem samples of patients with ALS (Oyanagi et al., 2008). Another study showed deposits of granular or amorphous material in ER lumen of sporadic ALS cases, thereby suggesting that the accumulation of misfolded proteins could be the underlying cause of stress at the ER level (Sasaki, 2010).
The cause behind ER stress in ALS/FTD and subsequent activations of UPR and autophagy is not clear yet. One of the postulated explanations suggests that ER stress could arise from the accumulation of oxidative stress (Jaronen et al., 2014). Supporting this hypothesis, analysis of blood, urine and cerebrospinal fluid samples from ALS patients has revealed an upregulation of oxidative stress markers (Riancho et al., 2019). Another possible explanation is that ER stress arises from an imbalance in calcium signalling (Grosskreutz et al., 2010; Leal and Gomes, 2015) as there is evidence according to which ALS disease has been found to be associated with low calcium content at the ER (Jaiswal and Keller, 2009; Tedeschi et al., 2019). Besides, motor neurons happen to be especially sensitive to calcium signalling dysregulation, as their intracellular Ca2+ levels must be tightly controlled to assure their proper function (Nijssen et al., 2017).
Beyond these postulates, the dysregulation of metabolic pathways and/or the poor utilization of energy resources, either of which are clearly present in ALS/FTD, presumably play key roles in the mechanisms behind the induction of pathological ER stress and autophagic dysregulation in neurons. As further reviewed, the fact that many of the genes that are found mutated in familial cases exert key functions in cellular energy homeostasis and recycling mechanisms provides strong evidence on this link. In this regard, AMPK is plausibly a key mediator of the activation of autophagy in response of energy scarcity in the pathological context of ALS/FTD. AMPK besides its role as metabolic sensor (Hardie et al., 2016), upon activation, AMPK turns on autophagic flux as mentioned above. In this sense, enhanced levels of activated AMPK have been observed in spinal motor neurons from patients with ALS (Lim et al., 2012a; Liu et al., 2015b), providing a compelling explanation for the increased number of autophagosomes in such neurons. However, it is not clear yet whether chronic AMPK activation as an adaptive response of affected neurons against energetic stress may produce beneficial outcomes or further precipitate the pathological crisis as it would interrupt the overall synthesis of proteins and over-activate the degradation of cellular components by the lysosomes (Figure 3).
4.2 Involvement of ALS/FTD Genes in Energetic and Autophagic Homeostasis
In the last decade, multiple studies have identified several disease-causative genes that overlap in both disorders. Such genetic mutations have been linked to different types of specific cytoplasmic inclusions and molecular signatures, which point to convergence, regarding cellular processes and pathomechanisms (Blokhuis et al., 2013). Although the vast majority of ALS-FTD cases have idiopathic or sporadic origin, unrevealing the genetic basis of the disease might be of great interest; and, considering that mutations in disease-causative genes have been also described in sporadic cases (Chen et al., 2013), similar pathogenic mechanisms could be present in different ALS-FTD subtypes (Vandoorne et al., 2018).
4.2.1 ALS/FTD Genes and Regulation of Energy Metabolism
TARDBP, FUS, SQSMT1, VCP, TBK1, CHCHD10, C9ORF72, OPTIN, UBQLN2 and SOD1 are the most significant disease-causative genes that share these two neurodegenerative conditions. Such genes are implicated in several cellular processes, including RNA metabolism, autophagy-lysosome axis and energy metabolism via mitochondrial respiration, pointing out them as the central pathways involved in ALS-FTD disease spectrum.
TDP-43 and FUS-proteinopathies are characterized by the formation of cytoplasmic ubiquitinated and/or hyperphosphorilated inclusions (Neumann et al., 2006; Kwiatkowski et al., 2009; Xu et al., 2010). The translocation of both TDP-43 and FUS onto the cytoplasm implies a loss-of-function in the nucleus disrupting RNA and protein homeostasis, which may provoke a vicious cycle causing further deleterious consequences. It is described that energy homeostasis is impaired in ALS-FTD (Vandoorne et al., 2018). These two ribonucleoprotein aggregates are implicated directly on it by interfering, for example, in mitochondria functionality. Mutations in FUS, specifically P525L, lead mitochondria to fragmentation. Deng et al. (2015), observed in flies carrying mutant-FUS that its translocation occur, mediated by HSP60 (a mitochondrial chaperonin), damaging mitochondrial integrity and disrupting mitochondrial cristae (Deng et al., 2015). Not only in flies, but also in P525L-FUS expressing cells and patients, mutated FUS expression provokes reduced mitochondrial membrane potential and an increment on ROS production. Transgenic mice overexpressing WT-FUS develop a motor phenotype and display early structural abnormalities mitochondrial in the pre-synaptic motor nerve (So et al., 2018).
TDP-43 has been also related with mitochondrial dysfunction, HEK293 cells overexpressing WT or mutant TDP-43 showed reduced mitochondrial membrane potential, oxygen consumption rate, and as a consequence, low ATP levels (Wang et al., 2016). These defects are likely to be mediated by the accumulation of mutant forms of TDP-43 in mitochondria leading to impairments in complex I activity, as shown in neurons from patients with ALS (Tefera et al., 2021).
Besides its involvement in mitochondrial energy production, TDP-43 has also proven to be fundamental for fat and glycolytic metabolism (Stallings et al., 2013). The exact metabolic pathway in which it takes part remains to be clarified, but several studies have suggested that TDP-43 participates in the mechanisms of nutrient sensing and glucose uptake in motor neurons and other cells such as islet β-cells, through regulation of AMPK or adiponectin signalling, or controlling the levels of the Rab GTPase-activating protein Tbc1d1 (Ling et al., 2014; Perera and Turner, 2016; Zufiría et al., 2016). TDP-43 has also emerged as a key regulator of insulin secretion, as it exerts transcriptional control over crucial components of the insulin secretion machinery, such as UNC13A, ci-Ins2 and CaV1.2 RNAs (Araki, 2019; Stoll et al., 2020; Brown et al., 2021).
Coiled-Coil-Helix-Coiled-Coil-Helix Domain 10 (CHCHD10) is a protein with several functions involved in mitochondrial metabolism (e.g., regulation of ETC components’ synthesis) (Zhou Z.-D. et al., 2017). CHCHD10 is located in the intermembrane space, and its mutated forms’ overexpression in HeLa cells leads to fragmentation of mitochondrial network. Furthermore, patients’ muscle as well as fibroblasts showed deficient ETC activity, along with abnormalities in cristae morphology (Bannwarth et al., 2014). A recent study performed with muscle conditional KO-CHCHD10 mice, reported that these mice showed motor defects, aberrant NMJs and thereby disturbed neuromuscular transmission (Xiao et al., 2020). Interestingly, exogenous ATP contributed rescuing the NMJ defects in KO-CHCHD10 muscles. The loss of CHCHD10 function in motor neurons elicits an energy deficit that activates unique responses to nutrient stress in both the mitochondria and ER, including AMPK signaling and UPR (Straub et al., 2021).
Valosin-containing protein (VCP), an ER protein that fundamentally functions as a driver of misfolded proteins to proteosome during ER stress, is also involved in mitochondrial fitness and thereby contribute to the cellular energetic balance. In this regard, patients carrying VCP mutations leads to severe mitochondrial uncoupling, resulting in decreased ATP generation, making neuronal cells more vulnerable to energy-demanding processes (Bartolome et al., 2013).
4.2.2 Role of ALS/FTD Genes in Autophagy
As mentioned above, many of the overlapping genes in ALS-FTD are involved in autophagic pathway. Interestingly, the main hallmark of this neurodegenerative condition are cytoplasmic aggregates that are not being degraded probably due to non-functional autophagic flux. Its impairment results in reduced cell survival, and thereby causes progressive degeneration.
TANK binding kinase 1 (TBK-1) is a protein kinase that belongs to the IKK-kinase family, which is involved in innate immune system. It is also a key player in autophagy, mainly regulating phosphorylation of autophagy and mitophagy adaptors (e.g., SQSTM1 and OPTN) (Pilli et al., 2012). Mutations in TBK1 has been reported as disease-causative in both ALS and FTD, or ALS-FTD continuum (Freischmidt et al., 2015). HEK293T cells transfected with mutant TBK1 compared to WT, showed diminished phosphorylation and reduced homodimerization, which are essential for TBK1 activation. These results were further confirmed using patients’ derived lymphoblastoid cell lines carrying missense mutations in TBK1, where phospho-TBK1 was found reduced, meaning that TBK1 activation is altered because of the reduced phosphorylation by self-interaction or with other kinases (de Majo et al., 2018). Using live-cell imaging to examine mitophagy dynamics after mitochondrial depolarization in HeLa cells, it has been observed that TBK1 and its downstream target OPTIN are required for engulfment of damaged mitochondria. And, specially, it has been seen that loss or chemical inhibition of TBK1, as well as expression of TBK1E696K, OPTINW478G and OPTINQ398X, which are ALS-linked mutations, impair mitophagy, thereby accumulation of damaged mitochondria occurs (Moore and Holzbaur, 2016).
As mentioned above, Optineurin (OPTN) is a downstream target of TBK1, which is involved in several vesicular trafficking pathways. Many ALS-linked mutations in OPTN map within the ubiquitin-binding domain (UBD) (Maruyama et al., 2010), which lead to increased OPTN-immunoreactive cytoplasmic inclusions. Neuron2A cells expressing UBD-mutated OPTIN showed an accumulation of LC3-II-positive inclusion bodies by decreasing autophagy-mediated degradation (Shen et al., 2015). ALS-associated OPTN mutants expression in NSC-34 motor neuron-like cell line, showed disrupted interaction between OPTN and myosin VI, resulting in interrupted protein trafficking, as well as endoplasmic reticulum stress and Golgi fragmentation. Moreover, implication of OPTN in lysosome trafficking during autophagy in association with myosin VI has been reported, since ALS-mutant expression as well as knockdown of OPTN lead to blockage of lysosome-autophagosome fusion, hence accumulating autophagosomes in neuronal cells (Sundaramoorthy et al., 2015).
Sequestosome-1(SQSTM1) encodes p62, a multifunctional protein that has an essential role in selective autophagy, as well as regulating mTOR-signalling pathway. Some of ALS-FTD associated mutations in SQSTM1 map to the LC3-interactin region, whereby lipid-anchored form of LC3 is bound, allowing the phagophore to evolve in autophagosomes (Stamatakou et al., 2020). Goode et al. (2016) demonstrated that the ALS-associated L341V mutation of SQSMT1 affects on the recognition of LC3, reducing binding affinity. These results that were obtained from experiments done in motor neuron-like cells with the L341V mutant SQSMT1, showed more difficulties for incorporation into autophagic vesicles (Goode et al., 2016). In addition to this loss-of-function of the protein, many papers have reported that there is also a gain-of-toxicity which is manifested through p62-positive cytoplasmic inclusion, and not only in neurons, but also in glial cells (Arai et al., 2003). Recent studies have revealed that ALS-FTD linked mutations in SQSTM1 reduce SQSTM1 phosphorylation, which is necessary for activation of selective autophagy, as well as impair KEAP1-SQSMT1 interaction (essential for antioxidant response activation), leading to increased TDP-43 associated aggregates (Deng et al., 2020).
Ubiquilin 2 (UBQLN2) is a chaperon protein that transports ubiquitinated cargoes to be degraded by ubiquitin-proteasome system (Zhang et al., 2014), playing a key role in protein quality control network (Shahheydari et al., 2017). It is also necessary for autophagosome maturation since its reduction leads to decreased autophagosome formation (Rothenberg et al., 2010). Interestingly, UBQLN2 has been related with mTOR, an essential negative regulator of macroautophagy, highlighting its involvement on the autophagic flux (Jung et al., 2010; Şentürk et al., 2019). Mutations in UBQLN2, which are linked to chromosome-X, cause dominantly inherited ALS-FTD (Deng et al., 2011). Recent studies have shown that flies with ALS associated UBQLN2 mutation display defective autophagic flux due to the impaired interaction with the v-ATPase proton pump, which is responsible for lysosome acidification. Therefore, loss of ubqln2 provokes lysosome alkalization, impairing the degradation process; which can be ameliorated in flies by acid nanoparticles for lysosome re-acidification (Şentürk et al., 2019). Consistent with this data, human HeLa cells carrying UBQLN2P497S mutation, show disturbances in proteostasis through interferences in autophagy pathway, as well as in HeLa UBQLN2-KO cell line, in which autophagosome acidification is impeded. Nevertheless, exogenous WT UBQLN2 expression is enough to restore autophagosome acidification almost as normal HeLa cells levels (Wu et al., 2021).
Valosin-containing protein (VCP) in addition to its role in mitochondrial respiration, it is also involved in ubiquitin-containing autophagosomes maturation (Johnson et al., 2010; Tresse et al., 2010). Interestingly, Tresse et al. (2010) demonstrated that VCP is essential for autophagosome maturation at a late stage after acidification; since MEFs expressing tagged-LC3 and mutated-VCP have shown accumulation of immature autophagic vesicles, some of which are acidified and abnormally large and. Other studies have proven the implication of VCP in autophagy by knocking it down, and as a result accumulation of autophagosomes have occur (Ju et al., 2009).
An expansion of GGGGCC hexanucleotide within the C9ORF72 gene was identified as an ALS-FTD causative mutation (DeJesus-Hernandez et al., 2011; Renton et al., 2011). When it was discovered, its biological function persisted unknown for many years. Nowadays, it has been included as an important player in the function and homeostasis of the lysosome (Amick and Ferguson, 2017). In addition, both human and mouse cell models lacking/expressing mutant- C9orf72 have shown abnormal lysosomes, and together with that, defects in autophagy and lysosomal degradation (Root et al., 2021). This may reinforce the aberrant upregulation of mTORC1 seen in c9orf72 and smcr8 double-knockout (dKO) mice, resulting in mTOR signalling overactivation, due to the disruption of autolysosome acidification. (Shao et al., 2020). In Drosophila studies, it has been shown that the repetition of the GGGGCC hexanucleotide impairs the nuclear import of Mitf/TFEB, a transcriptional regulator of autophagolysosomal function, leading to autophagy disruption, accumulation of lysosome-like organelles and proteostasis prior to neurodegeneration (Cunningham et al., 2020).
As discussed above, AMPK is plausibly a key protein mediating a link between the abnormalities of energy producing pathways and autophagy, which are central to the pathological continuum of ALS/FTD; and as such, dysfunction of various causative genes is associated with impaired AMPK signalling. For instance, pathological expression of TDP-43 or mutant SOD1 and CHCHD10 elicits intense activation of AMPK in mice spinal cords and motor neurons (Lim et al., 2012b; Liu et al., 2021; Straub et al., 2021). Importantly, activation of AMPK induces some of the ALS/FTD proteins to mislocalize or change its functional status, as observed for TDP-43 and TBK1 (Zhao et al., 2018; Liu et al., 2021), suggesting that AMPK activation is not a mere epiphenomenon but establishes a bidirectional crosstalk with ALS/FTD pathology. In this sense, suppression of AMPK activity exerted consistent improvements of motor functions in TDP-43 overexpressing mice (Liu et al., 2015a).
4.3 Abnormal Lysosome Function in FTD-GRN as Example of Defective Integration Between Nutrient Sensing and Nutrient Recycling in Neurodegeneration
As explained above, the lysosome is a chief metabolic hub that controls the metabolic status of the cell in response to the environmental changes. Failure of lysosomes has been implicated in the pathogenesis of neurodegeneration, metabolic disease and cancer (Ballabio and Bonifacino, 2020).
The lysosome performs very relevant functions in the central nervous system for the correct maintenance of the cell homeostasis. The central nervous system requires higher rates of macromolecules synthesis and organelles for the proper neuronal plasticity, and autophagy for the degradation of damaged proteins and organelles, which cannot otherwise reduce concentration by cell division (Root et al., 2021). In fact, mutations in genes linked to the lysosome produce complex diseases that are commonly accompanied by neurological syndromes. An extreme example of this is the haploinsufficiency of the lysosomal protein progranulin (PGRN), which causes neurodegeneration of the frontotemporal lobe with TDP-43 pathology (FTD) (Kao et al., 2017).
More than 50 different pathogenic granulin (GRN) mutations have been identified in patients with FTD (Baker et al., 2006; Cruts et al., 2006). Most of the mutations lead to a sequence frameshift and premature stop codons. This results in mutant mRNA transcripts, which undergo nonsense-mediated mRNA decay which leads to a reduction of the precursor protein, PGRN, and its proteolytic products, granulins (Baker et al., 2006; Cruts et al., 2006). While heterozygous mutations of GRN cause an adult age onset FTD with ubiquitinated TDP-43 inclusions and behavioural, agrammatism and motor speech deficits (bvFTD, nfvPPA) (Ferrari et al., 2019), homozygous GRN mutations produce a different and more aggressive juvenile onset of a LSD known as neural ceroid lipofuscinosis (NCL), characterized by abnormal lipopigment deposition in dysfunctional lysosomes (Mole et al., 2019) (Figure 3).
PGRN has been implicated in many physiological processes ranging from cell-cycle progression, cell migration, neurotrophic signalling, wound repair, modulation of inflammation, tumorigenesis, and metabolic fitness (Van Damme et al., 2008; Bateman and Bennett, 2009), acting upstream these processes by keeping lysosomes healthy (Rideout and Stefanis, 2014; Lui et al., 2016; Paushter et al., 2018). PGRN can play a direct or indirect role on the lysosome acidification and enzymatic activity of lysosomal enzymes (Tanaka et al., 2017). Although a portion of PGRN is secreted to the extracellular space, the majority of intracellular PGRN localizes within lysosomes. It has been reported that PGRN and/or granulins can regulate the enzymatic activity of (GBA) (Zhou et al., 2019; Valdez et al., 2020), β-hexosaminidase A (HexA) (Chen et al., 2018) and CTSD, lysosomal enzymes that are important for proper lysosomal activity (Beel et al., 2017; Valdez et al., 2017). On top of that, a relevant evidence linking the role of PGRN with the lysosome is that GRN expression is regulated by the transcription factor TFEB (Belcastro et al., 2011), responsible for the activation of the CLEAR, which regulates the expression of genes involved in autophagy and lysosomal pathway (Settembre et al., 2011).
PGRN was first linked with a lysosomal function when homozygous GRN mutations were discovered to cause NCL (Smith et al., 2012). Importantly, the accumulation of storage material and multillamelar bodies was discovered in post-mortem cortical brain tissue and cells from FTD patients with heterozygous GRN mutations (Ward et al., 2017). Accordingly, GRN-deficient mice models display NCL-like phenotypes (Ahmed et al., 2010; Petkau et al., 2012; Wils et al., 2012; Filiano et al., 2013; Zhou, 2017) together with lysosomal defects in brains (Wils et al., 2012; Zhou, 2017). Collectively, these evidences raise lysosomal dysfunction as a central neurodegenerative process that is caused by PGRN insufficiency and linked to TDP-43 pathology (Ward et al., 2017).
The lysosome is a control hub for cellular growth and survival (Settembre et al., 2013; Mony et al., 2016; Lawrence and Zoncu, 2019). In line with this, PGRN was first related to metabolic disease when it was seen that the serum PGRN levels were increased 1.4-fold times in individuals with visceral obesity and T2D (Youn et al., 2009). Not only increased PGRN levels in blood correlated positively with body mass index (BMI), fat mass, fasting glucose and insulin levels, and insulin resistance (Hossein-Nezhad et al., 2012), but also mediated TNF-a-induced insulin resistance through the regulation of IL-6 expression in 3T3-L1 adipocytes. Matsubara et al. proved that PGRN promotes IL-6 expression preventing the insulin induced phosphorylation of IRS-1 and AKT, suppressing insulin-stimulated glucose uptake and causing TNF-α induced insulin resistance (Matsubara et al., 2012). Chronic inflammation is a well-known consequence of obesity in T2D (Romeo et al., 2012). These patients have increased secretion of cytokines and chemokines as TNF-α and IL-6 and the recruitment and activation of innate immune cells (Taube et al., 2012). The previously mentioned evidence also supports that PGRN acts as a adipokine increasing IL-6 levels and being regulated by TNF-α treatment (Okura et al., 2010). This evidence suggests that PGRN could be regulated by the increased inflammatory cytokines and promote insulin resistance.
Although the role of PGRN is mostly related to the lysosome the primary role that was attributed to the granulin peptides was the modulation of cell growth, so initially it was considered a growth factor (He and Bateman, 2003). The treatment with recombinant PGRN showed stimulation of proliferation of two epithelial cell lines (Daniel et al., 2000). In relation to this PGRN is the only growth factor that is able to induce cell-cycle in IGF-I-receptor negative mouse embryo fibroblasts that are not able to complete the cell cycle (Zanocco-Marani et al., 1999). Moreover, PGRN is able to stimulate endothelial cell migration and vessel growth in vitro and in vivo (He et al., 2003; Toh et al., 2013). PGRN is expressed in proliferating blood vessels and induces the expression of vascular endothelial growth factor (VEGF) a key mediator of angiogenesis (Gonzalez et al., 2003; Tangkeangsirisin and Serrero, 2004). It has been shown that PGRN is able to bind to the receptor Tyrosine Kinase, EPH receptor A2 (EphA2) and activate MAPK and AKT signalling pathways (Zanocco-Marani et al., 1999; Neill et al., 2016). In relation with the mitogenic and angiogenic functions, PGRN has been associated to several cancers, generally with a direct correlation between the PGRN levels and cancer malignancy (Tangkeangsirisin et al., 2004; Abrhale et al., 2011; Serrero et al., 2012; Edelman et al., 2014). In the other hand, reducing PGRN levels by direct antibody treatment against PGRN have a tumour suppressor effect (Ho et al., 2008).
PGRN is a well-established modulator of immune function a very relevant physiological process that takes part in cancer, metabolic disease and neurodegeneration (Toh et al., 2011; Cenik et al., 2012; Jian et al., 2013; Tanaka et al., 2013; Kao et al., 2017). Microglia, the resident immune cells in the brain, are the cells with the highest expression levels of PGRN, being especially elevated in those which have become reactive (Naphade et al., 2010; Zhou X. et al., 2017). It has been shown that PGRN can regulate multiple microglial function, activation, migration, phagocytosis and synapse pruning among others (Van Damme et al., 2008; Yin et al., 2010; Pickford et al., 2011; Martens et al., 2012; Lui et al., 2016). The PGRNs regulation of microglial activation is a complex regulation, where PGRN and GRN peptides have opposite inflammatory functions, with PGRN generally being anti-inflammatory and granulin peptides pro-inflammatory (Zhu et al., 2002; Kojima et al., 2009). Considering the role of PGRN as a growth factor in the periphery and its role in neurodegeneration, it is not surprising that PGRN and GRN E can function as neurotrophic factors. PGRN and GRN E promote neuron survival and neurite outgrowth in vitro and in vivo (Van Damme et al., 2008; Ryan et al., 2009; Gass et al., 2012; De Muynck et al., 2013). Whereas small-interfering RNA (si-RNA) mediated knock down of PGRN in primary rat hippocampal neurons reduces neurite arborisation (Tapia et al., 2011). Since PGRN expression is very low in the developing brain and is a protein that is secreted in an activity-dependent manner in the synapses and regulates microglia activation, its role may be more relevant in the context of neurite plasticity in the adult brain (Tapia et al., 2011; Petkau et al., 2012; Petoukhov et al., 2013).
In summary, PGRN has emerged as an essential protein for the fitness of lysosomes to integrate nutrient sensing with cellular recycling and anabolic processes to respond to energy demands. The function of PGRN in lysosomes provides further compelling explanation for the particular vulnerability of ALS/FTD degenerating neurons to the dismantlement of the fine-tuned crosstalk between energy imbalance and cellular recycling through the ALP.
5 Conclusion and Perspectives
Dysregulation of the energy-producing pathways due to dysfunctional mitochondria or altered glycolysis, as well as defects in the autophagic process are common pathological features of several neurodegenerative diseases, including PD, FTD, ALS and AD. The bulk evidence derived from experimental studies highlights a pathogenic loop in which metabolic perturbations and dysregulated energy-sensing pathways cause a cellular energy crisis. In such scenario of energy scarcity, the main cellular process that responds to this challenge, autophagy, is also altered, further aggravating the energy crisis due to the inability to recycle damaged mitochondria and to provide micronutrients for the biosynthesis of new molecules. Indeed, the fact that many of the genes found mutated in PD, FTD, ALS, and to a lesser extent in AD, share common functions in the regulation of mitochondrial metabolism, autophagic flux and lysosome function provides further compelling evidence for this pathogenic loop.
However, why the different diseases affect distinct brain areas and neuronal populations remains elusive. It is likely that the participation of other pathogenic mechanisms could make some neuron subtypes more vulnerable than others. In any case, the crosstalk between energy metabolism and autophagy regulation appears to be a pillar in the pathological cascade of the different neurodegenerative diseases, and therefore treatments aimed at alleviating such loop might be beneficial for either condition.
Statements
Author contributions
Writing—original draft preparation, JO, HH-E, MG-A, RL-V, FG-B, and GG; writing—review and editing, LR-G, AJ-Z, JG, PR-R, JR-M, FM, IH, AL, FG-B, and GG; coordination of whole manuscript preparation and final edition, FG-B and GG; and funding acquisition: GG. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by Instituto de Salud Carlos III and FEDER, grant number PI19/01637; Basque Country Government, Eusko Jaurlaritzako Osasun Saila grant number 2018111042 and Fundació La Marató, grant number: fmarato20/001. JO, MG-A, and AJ-Z were supported by Basque Country PhD fellowships (PRE_2018_1_0095, PRE_2020_1_0122, PRE_2020_1_0191); JG was supported by the Margaretha af Ugglas (MAU) Foundation; PR-R was supported by MAU foundation and by the Swedish Alzheimer’s Foundation (Alzheimerfonden); FG-B by Roche Stop Fuga de cerebros (BIO19/ROCHE/017/BD); and GG by Juan de la Cierva-Incorporación (Agencia Estatal de Investigación, Ministerio de Ciencia e innovación, Gobierno de España IJC 2019-039965-I).This work was supported by the Biodonostia Health Research Institute, Donostia/San Sebastián, Spain.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors GG.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
AarslandD.BatzuL.HallidayG. M.GeurtsenG. J.BallardC.Ray ChaudhuriK.et al (2021). Parkinson Disease-Associated Cognitive Impairment. Nat. Rev. Dis. Primers7, 1. 10.1038/S41572-021-00280-3
2
AbrhaleT.BrodieA.SabnisG.MacedoL.TianC.YueB.et al (2011). GP88 (PC-Cell Derived Growth Factor, Progranulin) Stimulates Proliferation and Confers Letrozole Resistance to Aromatase Overexpressing Breast Cancer Cells. BMC Cancer11, 231. 10.1186/1471-2407-11-231
3
AbumradN. A.AjmalM.PothakosK.RobinsonJ. K. (2005). CD36 Expression and Brain Function: Does CD36 Deficiency Impact Learning Ability?Prostaglandins & Other Lipid Mediators77, 77–83. 10.1016/j.prostaglandins.2004.09.012
4
AhmedS. S.SantoshW.KumarS.ChristletH. T. T. (2009). Metabolic Profiling of Parkinson's Disease: Evidence of Biomarker from Gene Expression Analysis and Rapid Neural Network Detection. J. Biomed. Sci.16, 63. 10.1186/1423-0127-16-63
5
AhmedZ.ShengH.XuY.-f.LinW.-L.InnesA. E.GassJ.et al (2010). Accelerated Lipofuscinosis and Ubiquitination in Granulin Knockout Mice Suggest a Role for Progranulin in Successful Aging. Am. J. Pathol.177, 311–324. 10.2353/ajpath.2010.090915
6
Al‐BariM. A. A.XuP. (2020). Molecular Regulation of Autophagy Machinery by mTOR‐dependent and ‐independent Pathways. Ann. N.Y. Acad. Sci.1467, 3–20. 10.1111/nyas.14305
7
AlamM.SchmidtW. J. (2002). Rotenone Destroys Dopaminergic Neurons and Induces Parkinsonian Symptoms in Rats. Behav. Brain Res.136, 317–324. 10.1016/S0166-4328(02)00180-8
8
AlcalayR. N.LevyO. A.WatersC. H.FahnS.FordB.KuoS.-H.et al (2015). Glucocerebrosidase Activity in Parkinson's Disease with and withoutGBAmutations. Brain138, 2648–2658. 10.1093/brain/awv179
9
Alegre-AbarrateguiJ.ChristianH.LufinoM. M. P.MutihacR.VendaL. L.AnsorgeO.et al (2009). LRRK2 Regulates Autophagic Activity and Localizes to Specific Membrane Microdomains in a Novel Human Genomic Reporter Cellular Model. Hum. Mol. Genet.18, 4022–4034. 10.1093/hmg/ddp346
10
AliZ.HeverinM.OlinM.AcimovicJ.Lövgren-SandblomA.ShafaatiM.et al (2013). On the Regulatory Role of Side-Chain Hydroxylated Oxysterols in the Brain. Lessons from CYP27A1 Transgenic and Cyp27a1−/− Mice. J. Lipid Res.54, 1033–1043. 10.1194/jlr.M034124
11
AmickJ.FergusonS. M. (2017). C9orf72: At the Intersection of Lysosome Cell Biology and Neurodegenerative Disease. Traffic18, 267–276. 10.1111/tra.12477
12
AnagnostouG.AkbarM. T.PaulP.AngelinettaC.SteinerT. J.de BellerocheJ. (2010). Vesicle Associated Membrane Protein B (VAPB) Is Decreased in ALS Spinal Cord. Neurobiol. Aging31, 969–985. 10.1016/j.neurobiolaging.2008.07.005
13
AngladeP.VyasS.Javoy-AgidF.HerreroM. T.MichelP. P.MarquezJ.et al (1997). Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson's Disease. Histol. Histopathol.12, 25–31.
14
AraiT.HasegawaM.AkiyamaH.IkedaK.NonakaT.MoriH.et al (2006). TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophysical Res. Commun.351, 602–611. 10.1016/j.bbrc.2006.10.093
15
AraiT.NonakaT.HasegawaM.AkiyamaH.YoshidaM.HashizumeY.et al (2003). Neuronal and Glial Inclusions in Frontotemporal Dementia with or without Motor Neuron Disease Are Immunopositive for P62. Neurosci. Lett.342, 41–44. 10.1016/S0304-3940(03)00216-7
16
AudesseA. J.DhakalS.HassellL.-A.GardellZ.NemtsovaY.WebbA. E. (2019). FOXO3 Directly Regulates an Autophagy Network to Functionally Regulate Proteostasis in Adult Neural Stem Cells. Plos Genet.15, e1008097. 10.1371/journal.pgen.1008097
17
BakerM.MackenzieI. R.Pickering-BrownS. M.GassJ.RademakersR.LindholmC.et al (2006). Mutations in Progranulin Cause Tau-Negative Frontotemporal Dementia Linked to Chromosome 17. Nature442, 916–919. 10.1038/nature05016
18
BallabioA.BonifacinoJ. S. (2020). Lysosomes as Dynamic Regulators of Cell and Organismal Homeostasis. Nat. Rev. Mol. Cel Biol.21, 101–118. 10.1038/s41580-019-0185-4
19
BambergerM. E.HarrisM. E.McDonaldD. R.HusemannJ.LandrethG. E. (2003). A Cell Surface Receptor Complex for Fibrillar β-Amyloid Mediates Microglial Activation. J. Neurosci.23, 2665–2674. 10.1523/jneurosci.23-07-02665.2003
20
BannwarthS.Ait-El-MkademS.ChaussenotA.GeninE. C.Lacas-GervaisS.FragakiK.et al (2014). A Mitochondrial Origin for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis through CHCHD10 Involvement. Brain137, 2329–2345. 10.1093/brain/awu138
21
Bar-PeledL.SabatiniD. M. (2014). Regulation of mTORC1 by Amino Acids. Trends Cel Biol.24, 400–406. 10.1016/j.tcb.2014.03.003
22
BartolomeF.WuH.-C.BurchellV. S.PrezaE.WrayS.MahoneyC. J.et al (2013). Pathogenic VCP Mutations Induce Mitochondrial Uncoupling and Reduced ATP Levels. Neuron78, 57–64. 10.1016/j.neuron.2013.02.028
23
BatemanA.BennettH. P. J. (2009). The Granulin Gene Family: from Cancer to Dementia. BioEssays31, 1245–1254. 10.1002/bies.200900086
24
BåvnerA.ShafaatiM.HanssonM.OlinM.ShpitzenS.MeinerV.et al (2010). On the Mechanism of Accumulation of Cholestanol in the Brain of Mice with a Disruption of Sterol 27-hydroxylase. J. Lipid Res.51, 2722–2730. 10.1194/jlr.M008326
25
BeelS.MoisseM.DammeM.De MuynckL.RobberechtW.Van Den BoschL.et al (2017). Progranulin Functions as a Cathepsin D Chaperone to Stimulate Axonal Outgrowth In Vivo. Hum. Mol. Genet.26, 2850–2863. 10.1093/hmg/ddx162
26
BekinschteinP.KatcheC.SlipczukL. N.IgazL. M.CammarotaM.IzquierdoI.et al (2007). mTOR Signaling in the hippocampus Is Necessary for Memory Formation. Neurobiol. Learn. Mem.87, 303–307. 10.1016/j.nlm.2006.08.007
27
BelcastroV.SicilianoV.GregorettiF.MithbaokarP.DharmalingamG.BerlingieriS.et al (2011). Transcriptional Gene Network Inference from a Massive Dataset Elucidates Transcriptome Organization and Gene Function. Nucleic Acids Res.39, 8677–8688. 10.1093/nar/gkr593
28
BelforteN.AgostinoneJ.Alarcon-MartinezL.Villafranca-BaughmanD.DotignyF.Cueva VargasJ. L.et al (2021). AMPK Hyperactivation Promotes Dendrite Retraction, Synaptic Loss, and Neuronal Dysfunction in Glaucoma. Mol. Neurodegeneration16, 43. 10.1186/s13024-021-00466-z
29
BellucciA.ColloG.SarnicoI.BattistinL.MissaleC.SpanoP. (2008). Alpha-synuclein Aggregation and Cell Death Triggered by Energy Deprivation and Dopamine Overload Are Counteracted by D2D3receptor Activation. J. Neurochem.106, 560–577. 10.1111/j.1471-4159.2008.05406.x
30
BenderA.KrishnanK. J.MorrisC. M.TaylorG. A.ReeveA. K.PerryR. H.et al (2006). High Levels of Mitochondrial DNA Deletions in Substantia Nigra Neurons in Aging and Parkinson Disease. Nat. Genet.38, 515–517. 10.1038/NG1769
31
BillingsleyK. J.Bandres-CigaS.Saez-AtienzarS.SingletonA. B. (2018). Genetic Risk Factors in Parkinson's Disease. Cell Tissue Res373, 9–20. 10.1007/s00441-018-2817-y
32
BłaszczykJ. W. (2020). Energy Metabolism Decline in the Aging Brain-Pathogenesis of Neurodegenerative Disorders. Metabolites10, 450. 10.3390/metabo10110450
33
BlauwendraatC.NallsM. A.SingletonA. B. (2020). The Genetic Architecture of Parkinson's Disease. Lancet Neurol.19, 170–178. 10.1016/S1474-4422(19)30287-X
34
BlokhuisA. M.GroenE. J. N.KoppersM.Van Den BergL. H.PasterkampR. J. (2013). Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta Neuropathol.125, 777–794. 10.1007/s00401-013-1125-6
35
BouteloupC.DesportJ.-C.ClavelouP.GuyN.Derumeaux-BurelH.FerrierA.et al (2009). Hypermetabolism in ALS Patients: an Early and Persistent Phenomenon. J. Neurol.256, 1236–1242. 10.1007/s00415-009-5100-z
36
BurkK.PasterkampR. J. (2019). Disrupted Neuronal Trafficking in Amyotrophic Lateral Sclerosis. Acta Neuropathol.137, 859–877. 10.1007/s00401-019-01964-7
37
CaberlottoL.NguyenT.-P.LauriaM.PriamiC.RimondiniR.MaioliS.et al (2019). Cross-disease Analysis of Alzheimer's Disease and Type-2 Diabetes Highlights the Role of Autophagy in the Pathophysiology of Two Highly Comorbid Diseases. Sci. Rep.9, 3965. 10.1038/s41598-019-39828-5
38
CaccamoA.MajumderS.RichardsonA.StrongR.OddoS. (2010). Molecular Interplay between Mammalian Target of Rapamycin (mTOR), Amyloid-β, and Tau. J. Biol. Chem.285, 13107–13120. 10.1074/jbc.M110.100420
39
CaiR.ZhangY.SimmeringJ. E.SchultzJ. L.LiY.Fernandez-CarasaI.et al (2019). Enhancing Glycolysis Attenuates Parkinson's Disease Progression in Models and Clinical Databases. J. Clin. Invest.129, 4539–4549. 10.1172/JCI129987
40
CannonJ. R.GeghmanK. D.TapiasV.SewT.DailM. K.LiC.et al (2013). Expression of Human E46K-Mutated α-synuclein in BAC-Transgenic Rats Replicates Early-Stage Parkinson's Disease Features and Enhances Vulnerability to Mitochondrial Impairment. Exp. Neurol.240, 44–56. 10.1016/J.EXPNEUROL.2012.11.007
41
CarosiJ. M.SargeantT. J. (2019). Rapamycin and Alzheimer Disease: a Double-Edged Sword?Autophagy15, 1460–1462. 10.1080/15548627.2019.1615823
42
CarrasquilloM. M.BelbinO.HunterT. A.MaL.BisceglioG. D.ZouF.et al (2010). Replication of CLU, CR1, and PICALM Associations with Alzheimer Disease. Arch. Neurol.67, 961–964. 10.1001/archneurol.2010.147
43
CenikB.SephtonC. F.Kutluk CenikB.HerzJ.YuG. (2012). Progranulin: A Proteolytically Processed Protein at the Crossroads of Inflammation and Neurodegeneration. J. Biol. Chem.287, 32298–32306. 10.1074/jbc.R112.399170
44
ChakravortyA.JettoC. T.ManjithayaR. (2019). Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer's Disease Pathogenesis. Front. Aging Neurosci.11, 311. 10.3389/fnagi.2019.00311
45
ChaudhuriK. R.SchapiraA. H. (2009). Non-motor Symptoms of Parkinson's Disease: Dopaminergic Pathophysiology and Treatment. Lancet Neurol.8, 464–474. 10.1016/S1474-4422(09)70068-7
46
ChenS.SayanaP.ZhangX.LeW. (2013). Genetics of Amyotrophic Lateral Sclerosis: an Update. Mol. Neurodegeneration8, 28. 10.1186/1750-1326-8-28
47
ChenS.ZhangX.SongL.LeW. (2012). Autophagy Dysregulation in Amyotrophic Lateral Sclerosis. Brain Pathol.22, 110–116. 10.1111/j.1750-3639.2011.00546.x
48
ChenX.ZhaoC.LiX.WangT.LiY.CaoC.et al (2015). Terazosin Activates Pgk1 and Hsp90 to Promote Stress Resistance. Nat. Chem. Biol.11, 19–25. 10.1038/nchembio.1657
49
ChenY.JianJ.HettinghouseA.ZhaoX.SetchellK. D. R.SunY.et al (2018). Progranulin Associates with Hexosaminidase A and Ameliorates GM2 Ganglioside Accumulation and Lysosomal Storage in Tay-Sachs Disease. J. Mol. Med.96, 1359–1373. 10.1007/s00109-018-1703-0
50
Chin-HsiaoT. (2019). Metformin and the Risk of Dementia in Type 2 Diabetes Patients. Aging Dis.10, 37–48. 10.14336/AD.2017.1202
51
CistaroA.ValentiniM. C.ChiòA.NobiliF.CalvoA.MogliaC.et al (2012). Brain Hypermetabolism in Amyotrophic Lateral Sclerosis: a FDG PET Study in ALS of Spinal and Bulbar Onset. Eur. J. Nucl. Med. Mol. Imaging39, 251–259. 10.1007/s00259-011-1979-6
52
CoffeyE. E.BeckelJ. M.LatiesA. M.MitchellC. H. (2014). Lysosomal Alkalization and Dysfunction in Human Fibroblasts with the Alzheimer's Disease-Linked Presenilin 1 A246E Mutation Can Be Reversed with cAMP. Neuroscience263, 111–124. 10.1016/j.neuroscience.2014.01.001
53
CoraciI. S.HusemannJ.BermanJ. W.HuletteC.DufourJ. H.CampanellaG. K.et al (2002). CD36, a Class B Scavenger Receptor, Is Expressed on Microglia in Alzheimer's Disease Brains and Can Mediate Production of Reactive Oxygen Species in Response to β-Amyloid Fibrils. Am. J. Pathol.160, 101–112. 10.1016/s0002-9440(10)64354-4
54
CortiO.BlomgrenK.PolettiA.BeartP. M. (2020). Autophagy in Neurodegeneration: New Insights Underpinning Therapy for Neurological Diseases. J. Neurochem.154, 354–371. 10.1111/jnc.15002
55
CrewsL.SpencerB.DesplatsP.PatrickC.PaulinoA.RockensteinE.et al (2010). Selective Molecular Alterations in the Autophagy Pathway in Patients with Lewy Body Disease and in Models of α-Synucleinopathy. PLoS One5, e9313. 10.1371/JOURNAL.PONE.0009313
56
CrippaV.CarraS.RusminiP.SauD.BolzoniE.BendottiC.et al (2010). A Role of Small Heat Shock Protein B8 (HspB8) in the Autophagic Removal of Misfolded Proteins Responsible for Neurodegenerative Diseases. Autophagy6, 958–960. 10.4161/auto.6.7.13042
57
CrippaV.CicardiM. E.RameshN.SeguinS. J.GanassiM.BigiI.et al (2016). The Chaperone HSPB8 Reduces the Accumulation of Truncated TDP-43 Species in Cells and Protects against TDP-43-Mediated Toxicity. Hum. Mol. Genet.25, 3908–3924. 10.1093/hmg/ddw232
58
CrippaV.GalbiatiM.BoncoraglioA.RusminiP.OnestoE.GiorgettiE.et al (2013). Motoneuronal and Muscle-Selective Removal of ALS-Related Misfolded Proteins. Biochem. Soc. Trans.41, 1598–1604. 10.1042/BST20130118
59
CrutsM.Kumar-SinghS.Van BroeckhovenC. (2006). Progranulin Mutations in Ubiquitin-Positive Frontotemporal Dementia Linked to Chromosome 17q21. Car3, 485–491. 10.2174/156720506779025251
60
CulmseeC.MonnigJ.KempB. E.MattsonM. P. (2001). AMP-activated Protein Kinase Is Highly Expressed in Neurons in the Developing Rat Brain and Promotes Neuronal Survival Following Glucose Deprivation. Jmn17, 45–58. 10.1385/JMN:17:1:45
61
CunnaneS. C.TrushinaE.MorlandC.PrigioneA.CasadesusG.AndrewsZ. B.et al (2020). Brain Energy rescue: an Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov.19, 609–633. 10.1038/s41573-020-0072-x
62
CunninghamK. M.MauldingK.RuanK.SenturkM.GrimaJ. C.SungH.et al (2020). TFEB/Mitf Links Impaired Nuclear Import to Autophagolysosomal Dysfunction in C9-ALS. Elife9, 1. 10.7554/eLife.59419
63
DanielR.HeZ.CarmichaelK. P.HalperJ.BatemanA. (2000). Cellular Localization of Gene Expression for Progranulin. J. Histochem. Cytochem.48, 999–1009. 10.1177/002215540004800713
64
de LauL. M. L.KoudstaalP. J.HofmanA.BretelerM. M. B. (2006). Serum Cholesterol Levels and the Risk of Parkinson's Disease. Am. J. Epidemiol.164, 998–1002. 10.1093/aje/kwj283
65
de MajoM.ToppS. D.SmithB. N.NishimuraA. L.ChenH.-J.GkaziA. S.et al (2018). ALS-associated Missense and Nonsense TBK1 Mutations Can Both Cause Loss of Kinase Function. Neurobiol. Aging71, e1–e1266. e10. 10.1016/j.neurobiolaging.2018.06.015
66
De MarcoG.LupinoE.CalvoA.MogliaC.BuccinnàB.GrifoniS.et al (2011). Cytoplasmic Accumulation of TDP-43 in Circulating Lymphomonocytes of ALS Patients with and without TARDBP Mutations. Acta Neuropathol.121, 611–622. 10.1007/s00401-010-0786-7
67
De MuynckL.HerdewynS.BeelS.ScheveneelsW.Van Den BoschL.RobberechtW.et al (2013). The Neurotrophic Properties of Progranulin Depend on the Granulin E Domain but Do Not Require Sortilin Binding. Neurobiol. Aging34, 2541–2547. 10.1016/j.neurobiolaging.2013.04.022
68
DehayB.BoveJ.Rodriguez-MuelaN.PerierC.RecasensA.BoyaP.et al (2010). Pathogenic Lysosomal Depletion in Parkinson's Disease. J. Neurosci.30, 12535–12544. 10.1523/JNEURISCI.1920-10-201010.1523/jneurosci.1920-10.2010
69
DeJesus-HernandezM.MackenzieI. R.BoeveB. F.BoxerA. L.BakerM.RutherfordN. J.et al (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron72, 245–256. 10.1016/J.NEURON.2011.09.011
70
DengH.-X.ChenW.HongS.-T.BoycottK. M.GorrieG. H.SiddiqueN.et al (2011). Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/dementia. Nature477, 211–215. 10.1038/nature10353
71
DengJ.YangM.ChenY.ChenX.LiuJ.SunS.et al (2015). FUS Interacts with HSP60 to Promote Mitochondrial Damage. Plos Genet.11, e1005357–30. 10.1371/journal.pgen.1005357
72
DengZ.LimJ.WangQ.PurtellK.WuS.PalomoG. M.et al (2020). ALS-FTLD-linked Mutations of SQSTM1/p62 Disrupt Selective Autophagy and NFE2L2/NRF2 Anti-oxidative Stress Pathway. Autophagy16, 917–931. 10.1080/15548627.2019.1644076
73
De StrooperB.KarranE. (2016). The Cellular Phase of Alzheimer's Disease. Cell164, 603–615. 10.1016/j.cell.2015.12.056
74
DomiseM.DidierS.MarinangeliC.ZhaoH.ChandakkarP.BuéeL.et al (2016). AMP-activated Protein Kinase Modulates Tau Phosphorylation and Tau Pathology In Vivo. Sci. Rep.6, 26758. 10.1038/srep26758
75
DomiseM.SauvéF.DidierS.CaillerezR.BégardS.CarrierS.et al (2019). Neuronal AMP-Activated Protein Kinase Hyper-Activation Induces Synaptic Loss by an Autophagy-Mediated Process. Cell Death Dis10, 221. 10.1038/s41419-019-1464-x
76
DonmezG.OuteiroT. F. (2013). SIRT1 and SIRT2: Emerging Targets in Neurodegeneration. EMBO Mol. Med.5, 344–352. 10.1002/emmm.201302451
77
DruiG.CarnicellaS.CarcenacC.FavierM.BertrandA.BouletS.et al (2014). Loss of Dopaminergic Nigrostriatal Neurons Accounts for the Motivational and Affective Deficits in Parkinson's Disease. Mol. Psychiatry19, 358–367. 10.1038/mp.2013.3
78
DunbarD. R.MoonieP. A.ZevianiM.HoltI. J. (1996). Complex I Deficiency Is Associated with 3243G:C Mitochondrial DNA in Osteosarcoma Cell Cybrids. Hum. Mol. Genet.5, 123–129. 10.1093/hmg/5.1.123
79
DupuisL.CorciaP.FerganiA.Gonzalez De AguilarJ.-L.Bonnefont-RousselotD.BittarR.et al (2008). Dyslipidemia Is a Protective Factor in Amyotrophic Lateral Sclerosis. Neurology70, 1004–1009. 10.1212/01.wnl.0000285080.70324.27
80
DupuisL.PradatP.-F.LudolphA. C.LoefflerJ.-P. (2011). Energy Metabolism in Amyotrophic Lateral Sclerosis. Lancet Neurol.10, 75–82. 10.1016/S1474-4422(10)70224-6
81
EberlingJ. L.RichardsonB. C.ReedB. R.WolfeN.JagustW. J. (1994). Cortical Glucose Metabolism in Parkinson's Disease without Dementia. Neurobiol. Aging15, 329–335. 10.1016/0197-4580(94)90028-0
82
EdelmanM. J.FelicianoJ.YueB.BejaranoP.IoffeO.ReismanD.et al (2014). GP88 (Progranulin): a Novel Tissue and Circulating Biomarker for Non-small Cell Lung Carcinoma. Hum. Pathol.45, 1893–1899. 10.1016/j.humpath.2014.05.011
83
EisbachS. E.OuteiroT. F. (2013). Alpha-Synuclein and Intracellular Trafficking: Impact on the Spreading of Parkinson's Disease Pathology. J. Mol. Med.91, 693–703. 10.1007/s00109-013-1038-9
84
EndoH.SekiguchiK.UedaT.KowaH.KandaF.TodaT. (2017). Regional Glucose Hypometabolic Spread within the Primary Motor Cortex Is Associated with Amyotrophic Lateral Sclerosis Disease Progression: A Fluoro-Deoxyglucose Positron Emission Tomography Study. eNeurologicalSci6, 74–79. 10.1016/j.ensci.2017.01.001
85
EvansC. S.HolzbaurE. L. F. (2019). Autophagy and Mitophagy in ALS. Neurobiol. Dis.122, 35–40. 10.1016/j.nbd.2018.07.005
86
FangF.HållmarkerU.JamesS.IngreC.MichaëlssonK.AhlbomA.et al (2016). Amyotrophic Lateral Sclerosis Among Cross-Country Skiers in Sweden. Eur. J. Epidemiol.31, 247–253. 10.1007/s10654-015-0077-7
87
FanningS.HaqueA.ImberdisT.BaruV.BarrasaM. I.NuberS.et al (2019). Lipidomic Analysis of α-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cel73, 1001–1014. e8. 10.1016/j.molcel.2018.11.028
88
FernÁndez-EulateG.Ruiz-SanzJ. I.RianchoJ.ZufirÍaM.GereÑuG.FernÁndez-TorrÓnR.et al (2020). A Comprehensive Serum Lipidome Profiling of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degeneration21, 252–262. 10.1080/21678421.2020.1730904
89
FerrariR.ManzoniC.HardyJ. (2019). Genetics and Molecular Mechanisms of Frontotemporal Lobar Degeneration: an Update and Future Avenues. Neurobiol. Aging78, 98–110. 10.1016/j.neurobiolaging.2019.02.006
90
FerreeA.GuillilyM.LiH.SmithK.TakashimaA.SquillaceR.et al (2012). Regulation of Physiologic Actions of LRRK2: Focus on Autophagy. Neurodegenerative Dis.10, 238–241. 10.1159/000332599
91
FerrettaA.GaballoA.TanzarellaP.PiccoliC.CapitanioN.NicoB.et al (2014). Effect of Resveratrol on Mitochondrial Function: Implications in Parkin-Associated Familiar Parkinson's Disease. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis.1842, 902–915. 10.1016/j.bbadis.2014.02.010
92
FerrisH. A.PerryR. J.MoreiraG. V.ShulmanG. I.HortonJ. D.KahnC. R. (2017). Loss of Astrocyte Cholesterol Synthesis Disrupts Neuronal Function and Alters Whole-Body Metabolism. Proc. Natl. Acad. Sci. USA114, 1189–1194. 10.1073/pnas.1620506114
93
FieselF. C.AndoM.HudecR.HillA. R.Castanedes‐CaseyM.CaulfieldT. R.et al (2015). (Patho‐)physiological Relevance of PINK 1‐dependent Ubiquitin Phosphorylation. EMBO Rep.16, 1114–1130. 10.15252/embr.201540514
94
FilianoA. J.MartensL. H.YoungA. H.WarmusB. A.ZhouP.Diaz-RamirezG.et al (2013). Dissociation of Frontotemporal Dementia-Related Deficits and Neuroinflammation in Progranulin Haploinsufficient Mice. J. Neurosci.33, 5352–5361. 10.1523/JNEUROSCI.6103-11.2013
95
FreischmidtA.WielandT.RichterB.RufW.SchaefferV.MüllerK.et al (2015). Haploinsufficiency of TBK1 Causes Familial ALS and Fronto-Temporal Dementia. Nat. Neurosci.18, 631–636. 10.1038/nn.4000
96
FreyD.SchneiderC.XuL.BorgJ.SpoorenW.CaroniP. (2000). Early and Selective Loss of Neuromuscular Synapse Subtypes with Low Sprouting Competence in Motoneuron Diseases. J. Neurosci.20, 2534–2542. 10.1523/JNEUROSCI.20-07-02534.2000
97
GalloV.VanacoreN.Bueno-de-MesquitaH. B.VermeulenR.BrayneC.PearceN.et al (2016). Physical Activity and Risk of Amyotrophic Lateral Sclerosis in a Prospective Cohort Study. Eur. J. Epidemiol.31, 255–266. 10.1007/s10654-016-0119-9
98
GaoS.DuanC.GaoG.WangX.YangH. (2015). Alpha-synuclein Overexpression Negatively Regulates Insulin Receptor Substrate 1 by Activating mTORC1/S6K1 Signaling. Int. J. Biochem. Cel Biol.64, 25–33. 10.1016/j.biocel.2015.03.006
99
GarofaloT.MatarreseP.ManganelliV.MarconiM.TinariA.GambardellaL.et al (2016). Evidence for the Involvement of Lipid Rafts Localized at the ER-Mitochondria Associated Membranes in Autophagosome Formation. Autophagy12, 917–935. 10.1080/15548627.2016.1160971
100
GassJ.LeeW. C.CookC.FinchN.StetlerC.Jansen-WestK.et al (2012). Progranulin Regulates Neuronal Outgrowth Independent of Sortilin. Mol. Neurodegeneration7, 33. 10.1186/1750-1326-7-33
101
GeggM. E.SchapiraA. H. V. (2016). Mitochondrial Dysfunction Associated with Glucocerebrosidase Deficiency. Neurobiol. Dis.90, 43–50. 10.1016/j.nbd.2015.09.006
102
GerenuG.PerssonT.GoikoleaJ.Calvo-GarridoJ.Loera-ValenciaR.PottmeierP.et al (2021). Thioredoxin-80 Protects against Amyloid-Beta Pathology through Autophagic-Lysosomal Pathway Regulation. Mol. Psychiatry26, 1410–1423. 10.1038/s41380-019-0521-2
103
GermeysC.VandoorneT.BercierV.Van Den BoschL. (2019). Existing and Emerging Metabolomic Tools for ALS Research. Genes10, 1011. 10.3390/genes10121011
104
GhavamiS.ShojaeiS.YeganehB.AndeS. R.JangamreddyJ. R.MehrpourM.et al (2014). Autophagy and Apoptosis Dysfunction in Neurodegenerative Disorders. Prog. Neurobiol.112, 24–49. 10.1016/j.pneurobio.2013.10.004
105
GonzalezE. M.MongiatM.SlaterS. J.BaffaR.IozzoR. V. (2003). A Novel Interaction between Perlecan Protein Core and Progranulin. J. Biol. Chem.278, 38113–38116. 10.1074/jbc.C300310200
106
GoodeA.ButlerK.LongJ.CaveyJ.ScottD.ShawB.et al (2016). Defective Recognition of LC3B by Mutant SQSTM1/p62 Implicates Impairment of Autophagy as a Pathogenic Mechanism in ALS-FTLD. Autophagy12, 1094–1104. 10.1080/15548627.2016.1170257
107
GotkineM.FriedlanderY.HochnerH. (2014). Triathletes Are Over-represented in a Population of Patients with ALS. Amyotroph. Lateral Scler. Frontotemporal Degeneration15, 534–536. 10.3109/21678421.2014.932383
108
GrosskreutzJ.Van Den BoschL.KellerB. U. (2010). Calcium Dysregulation in Amyotrophic Lateral Sclerosis. Cell Calcium47, 165–174. 10.1016/j.ceca.2009.12.002
109
GuoF.LiuX.CaiH.LeW. (2018). Autophagy in Neurodegenerative Diseases: Pathogenesis and Therapy. Brain Pathol.28, 3–13. 10.1111/bpa.12545
110
GwinnD. M.ShackelfordD. B.EganD. F.MihaylovaM. M.MeryA.VasquezD. S.et al (2008). AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cel30, 214–226. 10.1016/j.molcel.2008.03.003
111
HaJ.ChoiD.-W.KimK. J.ChoS. Y.KimH.KimK. Y.et al (2021). Association of Metformin Use with Alzheimer's Disease in Patients with Newly Diagnosed Type 2 Diabetes: a Population-Based Nested Case-Control Study. Sci. Rep.11, 24069. 10.1038/s41598-021-03406-5
112
Haidet-PhillipsA. M.HesterM. E.MirandaC. J.MeyerK.BraunL.FrakesA.et al (2011). Astrocytes from Familial and Sporadic ALS Patients Are Toxic to Motor Neurons. Nat. Biotechnol.29, 824–828. 10.1038/nbt.1957
113
HallettP. J.EngelenderS.IsacsonO. (2019). Lipid and Immune Abnormalities Causing Age-dependent Neurodegeneration and Parkinson's Disease. J. Neuroinflammation16, 153. 10.1186/s12974-019-1532-2
114
HangL.ThundyilJ.LimK.-L. (2015). Mitochondrial Dysfunction and Parkinson Disease: a Parkin-AMPK alliance in Neuroprotection. Ann. N.Y. Acad. Sci.1350, 37–47. 10.1111/nyas.12820
115
HardieD. G.RossF. A.HawleyS. A. (2012). AMPK: a Nutrient and Energy Sensor that Maintains Energy Homeostasis. Nat. Rev. Mol. Cel Biol.13, 251–262. 10.1038/nrm3311
116
HardieD. G.SchafferB. E.BrunetA. (2016). AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cel Biol.26, 190–201. 10.1016/j.tcb.2015.10.013
117
HeZ.BatemanA. (2003). Progranulin (Granulin-epithelin Precursor, PC-Cell-Derived Growth Factor, Acrogranin) Mediates Tissue Repair and Tumorigenesis. J. Mol. Med.81, 600–612. 10.1007/s00109-003-0474-3
118
HeZ.OngC. H. P.HalperJ.BatemanA. (2003). Progranulin Is a Mediator of the Wound Response. Nat. Med.9, 225–229. 10.1038/nm816
119
HirschE. C.JennerP.PrzedborskiS. (2013). Pathogenesis of Parkinson's Disease. Mov. Disord.28, 24–30. 10.1002/mds.25032
120
HoJ. C.IpY. C.CheungS. T.LeeY. T.ChanK. F.WongS. Y.et al (2008). Granulin-epithelin Precursor as a Therapeutic Target for Hepatocellular Carcinoma. Hepatology47, 1524–1532. 10.1002/hep.22191
121
HoltzmanD. M.HerzJ.BuG. (2012). Apolipoprotein E and Apolipoprotein E Receptors: normal Biology and Roles in Alzheimer Disease. Cold Spring Harbor Perspect. Med.2, a006312. 10.1101/cshperspect.a006312
122
HonenB. N.SaintD. A.LaverD. R. (2003). Suppression of Calcium sparks in Rat Ventricular Myocytes and Direct Inhibition of Sheep Cardiac RyR Channels by EPA, DHA and Oleic Acid. J. Membr. Biol.196, 95–103. 10.1007/s00232-003-0628-9
123
HongD.-P.XiongW.ChangJ.-Y.JiangC. (2011). The Role of the C-Terminus of Human α-synuclein: Intra-disulfide Bonds between the C-Terminus and Other Regions Stabilize Non-fibrillar Monomeric Isomers. FEBS Lett.585, 561–566. 10.1016/J.FEBSLET.2011.01.009
124
HosokawaN.HaraT.KaizukaT.KishiC.TakamuraA.MiuraY.et al (2009). Nutrient-dependent mTORC1 Association with the ULK1-Atg13-Fip200 Complex Required for Autophagy. MBoC20, 1981–1991. 10.1091/mbc.e08-12-1248
125
Hossein-NezhadA.MirzaeiK.AnsarH.Emam-GholipourS.TooteeA.KeshavarzS. A. (2012). Obesity, Inflammation and Resting Energy Expenditure: Possible Mechanism of Progranulin in This Pathway. Minerva Endocrinol.37, 255–266.
126
HouX.FieselF. C.TrubanD.Castanedes CaseyM.LinW.-l.SotoA. I.et al (2018). Age- and Disease-dependent Increase of the Mitophagy Marker Phospho-Ubiquitin in normal Aging and Lewy Body Disease. Autophagy14, 1404–1418. 10.1080/15548627.2018.1461294
127
HouX.WatzlawikJ. O.FieselF. C.SpringerW. (2020). Autophagy in Parkinson's Disease. J. Mol. Biol.432, 2651–2672. 10.1016/j.jmb.2020.01.037
128
HouY.DanX.BabbarM.WeiY.HasselbalchS. G.CroteauD. L.et al (2019). Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol.15, 565–581. 10.1038/s41582-019-0244-7
129
HsiehC.-H.ShaltoukiA.GonzalezA. E.Bettencourt da CruzA.BurbullaL. F.St. LawrenceE. E.et al (2016). Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson's Disease. Cell Stem Cell19, 709–724. 10.1016/j.stem.2016.08.002
130
HuG.JousilahtiP.BidelS.AntikainenR.TuomilehtoJ. (2007). Type 2 Diabetes and the Risk of Parkinson's Disease. Diabetes Care30, 842–847. 10.2337/dc06-2011
131
ImfeldP.BodmerM.JickS. S.MeierC. R. (2012). Metformin, Other Antidiabetic Drugs, and Risk of Alzheimer's Disease: A Population-Based Case-Control Study. J. Am. Geriatr. Soc.60, 916–921. 10.1111/j.1532-5415.2012.03916.x
132
InoueK.RispoliJ.KaphzanH.KlannE.ChenE. I.KimJ.et al (2012). Macroautophagy Deficiency Mediates Age-dependent Neurodegeneration through a Phospho-Tau Pathway. Mol. Neurodegeneration7, 48. 10.1186/1750-1326-7-48
133
IoghenO.ChițoiuL.GherghiceanuM.CeafalanL. C.HinescuM. E. (2021). CD36 - A Novel Molecular Target in the Neurovascular Unit. Eur. J. Neurosci.53, 2500–2510. 10.1111/ejn.15147
134
IwatsuboT. (2003). Aggregation of ?-synuclein in the Pathogenesis of Parkinson?s Disease. J. Neurol.250, 1. 10.1007/s00415-003-1303-x
135
JaiswalM. K.KellerB. U. (2009). Cu/Zn Superoxide Dismutase Typical for Familial Amyotrophic Lateral Sclerosis Increases the Vulnerability of Mitochondria and Perturbs Ca2+Homeostasis in SOD1G93AMice. Mol. Pharmacol.75, 478–489. 10.1124/mol.108.050831
136
JaronenM.GoldsteinsG.KoistinahoJ. (2014). ER Stress and Unfolded Protein Response in Amyotrophic Lateral Sclerosisâ€"a Controversial Role of Protein Disulphide Isomerase. Front. Cel. Neurosci.8. 10.3389/fncel.2014.00402
137
JawaidA.BrownJ. A.SchulzP. E. (2015). Diabetes Mellitus in Amyotrophic Lateral Sclerosis: Dr Jekyll or Mr Hyde?Eur. J. Neurol.22, 1419–1420. 10.1111/ene.12660
138
JeonS.-M. (2016). Regulation and Function of AMPK in Physiology and Diseases. Exp. Mol. Med.48, e245. 10.1038/emm.2016.81
139
JianJ.KonopkaJ.LiuC. (2013). Insights into the Role of Progranulin in Immunity, Infection, and Inflammation. J. Leukoc. Biol.93, 199–208. 10.1189/jlb.0812429
140
JiangQ.LeeC. Y. D.MandrekarS.WilkinsonB.CramerP.ZelcerN.et al (2008). ApoE Promotes the Proteolytic Degradation of Aβ. Neuron58, 681–693. 10.1016/j.neuron.2008.04.010
141
JiangT.-F.ZhangY.-J.ZhouH.-Y.WangH.-M.TianL.-P.LiuJ.et al (2013). Curcumin Ameliorates the Neurodegenerative Pathology in A53T α-synuclein Cell Model of Parkinson's Disease through the Downregulation of mTOR/p70S6K Signaling and the Recovery of Macroautophagy. J. Neuroimmune Pharmacol.8, 356–369. 10.1007/s11481-012-9431-7
142
JohnsonJ. O.MandrioliJ.BenatarM.AbramzonY.Van DeerlinV. M.TrojanowskiJ. Q.et al (2010). Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron68, 857–864. 10.1016/j.neuron.2010.11.036
143
JohnsonL. A.TorresE. R. S.ImpeyS.StevensJ. F.RaberJ. (2017). Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome. Sci. Rep.7, 43701. 10.1038/srep43701
144
JuJ.-S.FuentealbaR. A.MillerS. E.JacksonE.Piwnica-WormsD.BalohR. H.et al (2009). Valosin-containing Protein (VCP) Is Required for Autophagy and Is Disrupted in VCP Disease. J. Cel Biol.187, 875–888. 10.1083/jcb.200908115
145
JuT.-C.ChenH.-M.LinJ.-T.ChangC.-P.ChangW.-C.KangJ.-J.et al (2011). Nuclear Translocation of AMPK-Α1 Potentiates Striatal Neurodegeneration in Huntington's Disease. J. Cel Biol.194, 209–227. 10.1083/JCB.201105010
146
JulianT. H.GlascowN.BarryA. D. F.MollT.HarveyC.KlimentidisY. C.et al (2021). Physical Exercise Is a Risk Factor for Amyotrophic Lateral Sclerosis: Convergent Evidence from Mendelian Randomisation, Transcriptomics and Risk Genotypes. EBioMedicine68, 103397. 10.1016/j.ebiom.2021.103397
147
JungC. H.RoS.-H.CaoJ.OttoN. M.KimD.-H. (2010). mTOR Regulation of Autophagy. FEBS Lett.584, 1287–1295. 10.1016/j.febslet.2010.01.017
148
JungS.ChoeS.WooH.JeongH.AnH.-K.MoonH.et al (2020). Autophagic Death of Neural Stem Cells Mediates Chronic Stress-Induced Decline of Adult Hippocampal Neurogenesis and Cognitive Deficits. Autophagy16, 512–530. 10.1080/15548627.2019.1630222
149
KaoA. W.McKayA.SinghP. P.BrunetA.HuangE. J. (2017). Progranulin, Lysosomal Regulation and Neurodegenerative Disease. Nat. Rev. Neurosci.18, 325–333. 10.1038/nrn.2017.36
150
KimJ.KunduM.ViolletB.GuanK.-L. (2011). AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cel Biol.13, 132–141. 10.1038/ncb2152
151
KioussisB.TuttleC. S. L.HeardD. S.KennedyB. K.LautenschlagerN. T.MaierA. B. (2021). Targeting Impaired Nutrient Sensing with Repurposed Therapeutics to Prevent or Treat Age-Related Cognitive Decline and Dementia: A Systematic Review. Ageing Res. Rev.67, 101302. 10.1016/j.arr.2021.101302
152
KitadaT.AsakawaS.HattoriN.MatsumineH.YamamuraY.MinoshimaS.et al (1998). Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature392, 605–608. 10.1038/33416
153
KloskeC. M.WilcockD. M. (2020). The Important Interface between Apolipoprotein E and Neuroinflammation in Alzheimer's Disease. Front. Immunol.11, 754. 10.3389/fimmu.2020.00754
154
KojimaY.OnoK.InoueK.TakagiY.KikutaK.-i.NishimuraM.et al (2009). Progranulin Expression in Advanced Human Atherosclerotic Plaque. Atherosclerosis206, 102–108. 10.1016/j.atherosclerosis.2009.02.017
155
KoreckaJ. A.ThomasR.ChristensenD. P.HinrichA. J.FerrariE. J.LevyS. A.et al (2019). Mitochondrial Clearance and Maturation of Autophagosomes Are Compromised in LRRK2 G2019S Familial Parkinson's Disease Patient Fibroblasts. Hum. Mol. Genet.28, 3232–3243. 10.1093/hmg/ddz126
156
KuhlmannN.MilnerwoodA. J. (2020). A Critical LRRK at the Synapse? the Neurobiological Function and Pathophysiological Dysfunction of LRRK2. Front. Mol. Neurosci.13, 153. 10.3389/fnmol.2020.00153
157
KwiatkowskiT. J.BoscoD. A.LeClercA. L.TamrazianE.VanderburgC. R.RussC.et al (2009). Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science323, 1205–1208. 10.1126/science.1166066
158
LauD. H. W.HartoppN.WelshN. J.MuellerS.GlennonE. B.MórotzG. M.et al (2018). Disruption of ER−mitochondria Signalling in Fronto-Temporal Dementia and Related Amyotrophic Lateral Sclerosis. Cel Death Dis9, 327. 10.1038/s41419-017-0022-7
159
LaugeretteF.Passilly-DegraceP.PatrisB.NiotI.FebbraioM.MontmayeurJ.-P.et al (2005). CD36 Involvement in Orosensory Detection of Dietary Lipids, Spontaneous Fat Preference, and Digestive Secretions. J. Clin. Invest.115, 3177–3184. 10.1172/JCI25299
160
LawrenceR. E.ChoK. F.RappoldR.ThrunA.TofauteM.KimD. J.et al (2018). A Nutrient-Induced Affinity Switch Controls mTORC1 Activation by its Rag GTPase-Ragulator Lysosomal Scaffold. Nat. Cel Biol.20, 1052–1063. 10.1038/s41556-018-0148-6
161
LawrenceR. E.ZoncuR. (2019). The Lysosome as a Cellular centre for Signalling, Metabolism and Quality Control. Nat. Cel Biol.21, 133–142. 10.1038/s41556-018-0244-7
162
LealN. S.DentoniG.SchreinerB.NaiaL.PirasA.GraffC.et al (2020). Amyloid β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer's Disease-Related Models. Cells9, 2552. 10.3390/cells9122552
163
LealS. S.GomesC. u. M. (2015). Calcium Dysregulation Links ALS Defective Proteins and Motor Neuron Selective Vulnerability. Front. Cel. Neurosci.9. 10.3389/fncel.2015.00225
164
LeeJ.-H.YuW. H.KumarA.LeeS.MohanP. S.PeterhoffC. M.et al (2010). Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell141, 1146–1158. 10.1016/j.cell.2010.05.008
165
LeeJ. H.ChengR.BarralS.ReitzC.MedranoM.LantiguaR.et al (2011). Identification of Novel Loci for Alzheimer Disease and Replication of CLU, PICALM, and BIN1 in Caribbean Hispanic Individuals. Arch. Neurol.68, 320–328. 10.1001/archneurol.2010.292
166
LeeJ. K.ShinJ. H.LeeJ. E.ChoiE.-J. (2015). Role of Autophagy in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis.1852, 2517–2524. 10.1016/j.bbadis.2015.08.005
167
LeeS.-I.JeongW.LimH.ChoS.LeeH.JangY.et al (2021). APOE4-carrying Human Astrocytes Oversupply Cholesterol to Promote Neuronal Lipid Raft Expansion and Aβ Generation. Stem Cel. Rep.16, 2128–2137. 10.1016/j.stemcr.2021.07.017
168
LeoniV.SolomonA.Lövgren-SandblomA.MinthonL.BlennowK.HanssonO.et al (2013). Diagnostic Power of 24S-Hydroxycholesterol in Cerebrospinal Fluid: Candidate Marker of Brain Health. Jad36, 739–747. 10.3233/JAD-130035
169
LiH.HamA.MaT. C.KuoS.-H.KanterE.KimD.et al (2019). Mitochondrial Dysfunction and Mitophagy Defect Triggered by Heterozygous GBA Mutations. Autophagy15, 113–130. 10.1080/15548627.2018.1509818
170
LimF.HernándezF.LucasJ. J.Gómez-RamosP.MoránM. A.ÁvilaJ. (2001). FTDP-17 Mutations in Tau Transgenic Mice Provoke Lysosomal Abnormalities and Tau Filaments in Forebrain. Mol. Cell Neurosci.18, 702–714. 10.1006/mcne.2001.1051
171
LimM. A.SelakM. A.XiangZ.KraincD.NeveR. L.KraemerB. C.et al (2012a). Reduced Activity of AMP-Activated Protein Kinase Protects against Genetic Models of Motor Neuron Disease. J. Neurosci.32, 1123–1141. 10.1523/JNEUROSCI.6554-10.2012
172
LimM. A.SelakM. A.XiangZ.KraincD.NeveR. L.KraemerB. C.et al (2012b). Reduced Activity of AMP-Activated Protein Kinase Protects against Genetic Models of Motor Neuron Disease. J. Neurosci.32, 1123–1141. 10.1523/JNEUROSCI.6554-10.2012
173
LingD.MagallanesM.SalvaterraP. M. (2014). Accumulation of Amyloid-like Aβ1-42 in AEL (Autophagy-Endosomal-Lysosomal) Vesicles: Potential Implications for Plaque Biogenesis. ASN Neuro6, AN20130044. 10.1042/AN20130044
174
LiuJ.-P.TangY.ZhouS.TohB. H.McLeanC.LiH. (2010). Cholesterol Involvement in the Pathogenesis of Neurodegenerative Diseases. Mol. Cell Neurosci.43, 33–42. 10.1016/j.mcn.2009.07.013
175
LiuY.-J.JuT.-C.ChenH.-M.JangY.-S.LeeL.-M.LaiH.-L.et al (2015a). Activation of AMP-Activated Protein Kinase α1 Mediates Mislocalization of TDP-43 in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet.24, 787–801. 10.1093/hmg/ddu497
176
LiuY.-J.KuoH.-C.ChernY. (2021). A System-wide Mislocalization of RNA-Binding Proteins in Motor Neurons Is a New Feature of ALS. Neurobiol. Dis.160, 105531. 10.1016/j.nbd.2021.105531
177
LiuY.-J.LeeL.-M.LaiH.-L.ChernY. (2015b). Aberrant Activation of AMP-Activated Protein Kinase Contributes to the Abnormal Distribution of HuR in Amyotrophic Lateral Sclerosis. FEBS Lett.589, 432–439. 10.1016/j.febslet.2014.12.029
178
Loera‐ValenciaR.Cedazo‐MinguezA.KenigsbergP. A.PageG.DuarteA. I.GiustiP.et al (2019a). Current and Emerging Avenues for Alzheimer's Disease Drug Targets. J. Intern. Med.286, 398–437. 10.1111/joim.12959
179
Loera-ValenciaR.IsmailM.-A. -M.GoikoleaJ.LodeiroM.MateosL.BjörkhemI.et al (2021a). Hypercholesterolemia and 27-Hydroxycholesterol Increase S100A8 and RAGE Expression in the Brain: a Link between Cholesterol, Alarmins, and Neurodegeneration. Mol. Neurobiol.58, 6063–6076. 10.1007/s12035-021-02521-8
180
Loera-ValenciaR.IsmailM.-A. -M.NilssonP.WinbladB. (2019b). Restoring Synaptic Function through Multimodal Therapeutics. Prog. Mol. Biol. Transl. Sci.168, 257–275. 10.1016/bs.pmbts.2019.07.003
181
LuiH.ZhangJ.MakinsonS. R.CahillM. K.KelleyK. W.HuangH.-Y.et al (2016). Progranulin Deficiency Promotes Circuit-specific Synaptic Pruning by Microglia via Complement Activation. Cell165, 921–935. 10.1016/j.cell.2016.04.001
182
LuoF.SandhuA. F.RungratanawanichW.WilliamsG. E.AkbarM.ZhouS.et al (2020). Melatonin and Autophagy in Aging-Related Neurodegenerative Diseases. Ijms21, 7174. 10.3390/ijms21197174
183
MadureiraM.Connor-RobsonN.Wade-MartinsR. (2020). "LRRK2: Autophagy and Lysosomal Activity". Front. Neurosci.14, 498. 10.3389/fnins.2020.00498
184
MagalhaesJ.GeggM. E.Migdalska-RichardsA.DohertyM. K.WhitfieldP. D.SchapiraA. H. V. (2016). Autophagic Lysosome Reformation Dysfunction in Glucocerebrosidase Deficient Cells: Relevance to Parkinson Disease. Hum. Mol. Genet.25, 3432–3445. 10.1093/hmg/ddw185
185
MariosaD.KamelF.BelloccoR.YeW.FangF. (2015). Association between Diabetes and Amyotrophic Lateral Sclerosis in Sweden. Eur. J. Neurol.22, 1436–1442. 10.1111/ene.12632
186
MartensL. H.ZhangJ.BarmadaS. J.ZhouP.KamiyaS.SunB.et al (2012). Progranulin Deficiency Promotes Neuroinflammation and Neuron Loss Following Toxin-Induced Injury. J. Clin. Invest.122, 3955–3959. 10.1172/JCI63113
187
MartinaJ. A.PuertollanoR. (2013). Rag GTPases Mediate Amino Acid-dependent Recruitment of TFEB and MITF to Lysosomes. J. Cel Biol.200, 475–491. 10.1083/jcb.201209135
188
MaruyamaH.MorinoH.ItoH.IzumiY.KatoH.WatanabeY.et al (2010). Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature465, 223–226. 10.1038/nature08971
189
MascitelliL.PezzettaF.GoldsteinM. R.TuomilehtoJ. (2009). Total Cholesterol and the Risk of Parkinson Disease. Neurology72, 860–861. 10.1212/01.wnl.0000339395.41343.b3
190
MatsubaraT.MitaA.MinamiK.HosookaT.KitazawaS.TakahashiK.et al (2012). PGRN Is a Key Adipokine Mediating High Fat Diet-Induced Insulin Resistance and Obesity through IL-6 in Adipose Tissue. Cel Metab.15, 38–50. 10.1016/j.cmet.2011.12.002
191
McNeillA.MagalhaesJ.ShenC.ChauK.-Y.HughesD.MehtaA.et al (2014). Ambroxol Improves Lysosomal Biochemistry in Glucocerebrosidase Mutation-Linked Parkinson Disease Cells. Brain137, 1481–1495. 10.1093/brain/awu020
192
MeaneyS.HeverinM.PanzenboeckU.EkströmL.AxelssonM.AnderssonU.et al (2007). Novel Route for Elimination of Brain Oxysterols across the Blood-Brain Barrier: Conversion into 7α-Hydroxy-3-Oxo-4-Cholestenoic Acid. J. Lipid Res.48, 944–951. 10.1194/jlr.M600529-JLR200
193
MehrpourM.EsclatineA.BeauI.CodognoP. (2010). Overview of Macroautophagy Regulation in Mammalian Cells. Cell Res20, 748–762. 10.1038/cr.2010.82
194
MenziesF. M.FlemingA.RubinszteinD. C. (2015). Compromised Autophagy and Neurodegenerative Diseases. Nat. Rev. Neurosci.16, 345–357. 10.1038/nrn3961
195
Merino-SerraisP.Loera-ValenciaR.Rodriguez-RodriguezP.Parrado-FernandezC.IsmailM. A.MaioliS.et al (2019). 27-Hydroxycholesterol Induces Aberrant Morphology and Synaptic Dysfunction in Hippocampal Neurons. Cereb. Cortex29, 429–446. 10.1093/cercor/bhy274
196
MiguelR.GagoM. F.MartinsJ.BarrosP.ValeJ.RosasM. J. (2014). POLG1-related Levodopa-Responsive Parkinsonism. Clin. Neurol. Neurosurg.126, 47–54. 10.1016/j.clineuro.2014.08.020
197
MitchellR. W.OnN. H.Del BigioM. R.MillerD. W.HatchG. M. (2011). Fatty Acid Transport Protein Expression in Human Brain and Potential Role in Fatty Acid Transport across Human Brain Microvessel Endothelial Cells. J. Neurochem.117, no. 10.1111/j.1471-4159.2011.07245.x
198
MitchenerJ. S.ShelburneJ. D.BradfordW. D.HawkinsH. K. (1976). Cellular Autophagocytosis Induced by Deprivation of Serum and Amino Acids in HeLa Cells. Am. J. Pathol.83, 485–492.
199
MizushimaN.LevineB.CuervoA. M.KlionskyD. J. (2008). Autophagy Fights Disease through Cellular Self-Digestion. Nature451, 1069–1075. 10.1038/nature06639
200
MoleS. E.AndersonG.BandH. A.BerkovicS. F.CooperJ. D.Kleine HolthausS.-M.et al (2019). Clinical Challenges and Future Therapeutic Approaches for Neuronal Ceroid Lipofuscinosis. Lancet Neurol.18, 107–116. 10.1016/S1474-4422(18)30368-5
201
MonyV. K.BenjaminS.O'RourkeE. J. (2016). A Lysosome-Centered View of Nutrient Homeostasis. Autophagy12, 619–631. 10.1080/15548627.2016.1147671
202
MooreA. S.HolzbaurE. L. F. (2016). Dynamic Recruitment and Activation of ALS-Associated TBK1 with its Target Optineurin Are Required for Efficient Mitophagy. Proc. Natl. Acad. Sci. USA113, E3349–E3358. 10.1073/pnas.1523810113
203
MoreauK.FlemingA.ImarisioS.Lopez RamirezA.MercerJ. L.Jimenez-SanchezM.et al (2014). PICALM Modulates Autophagy Activity and Tau Accumulation. Nat. Commun.5, 4998. 10.1038/ncomms5998
204
MoutinhoM.NunesM. J.GomesA. Q.GamaM. J.Cedazo-MinguezA.RodriguesC. M. P.et al (2015). Cholesterol 24S-Hydroxylase Overexpression Inhibits the Liver X Receptor (LXR) Pathway by Activating Small Guanosine Triphosphate-Binding Proteins (sGTPases) in Neuronal Cells. Mol. Neurobiol.51, 1489–1503. 10.1007/s12035-014-8828-0
205
NaphadeS. B.KigerlK. A.JakemanL. B.KostykS. K.PopovichP. G.KuretJ. (2010). Progranulin Expression Is Upregulated after Spinal Contusion in Mice. Acta Neuropathol.119, 123–133. 10.1007/s00401-009-0616-y
206
NeillT.BuraschiS.GoyalA.SharpeC.NatkanskiE.SchaeferL.et al (2016). EphA2 Is a Functional Receptor for the Growth Factor Progranulin. J. Cel Biol.215, 687–703. 10.1083/jcb.201603079
207
NelsonM. P.BoutinM.TseT. E.LuH.HaleyE. D.OuyangX.et al (2018). The Lysosomal Enzyme Alpha-Galactosidase A Is Deficient in Parkinson's Disease Brain in Association with the Pathologic Accumulation of Alpha-Synuclein. Neurobiol. Dis.110, 68–81. 10.1016/j.nbd.2017.11.006
208
NelsonM. P.TseT. E.O’QuinnD. B.PercivalS. M.JaimesE. A.WarnockD. G.et al (2014). Autophagy-lysosome Pathway Associated Neuropathology and Axonal Degeneration in the Brains of Alpha-Galactosidase A-Deficient Mice. Acta Neuropathol. Commun.2, 20. 10.1186/2051-5960-2-20
209
NeumannM.SampathuD. M.KwongL. K.TruaxA. C.MicsenyiM. C.ChouT. T.et al (2006). Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science314, 130–133. 10.1126/science.1134108
210
NgC.-H.GuanM. S. H.KohC.OuyangX.YuF.TanE.-K.et al (2012). AMP Kinase Activation Mitigates Dopaminergic Dysfunction and Mitochondrial Abnormalities in Drosophila Models of Parkinson's Disease. J. Neurosci.32, 14311–14317. 10.1523/JNEUROSCI.0499-12.2012
211
NgT. P.FengL.YapK. B.LeeT. S.TanC. H.WinbladB. (2014). Long-term Metformin Usage and Cognitive Function Among Older Adults with Diabetes. Jad41, 61–68. 10.3233/JAD-131901
212
NijssenJ.ComleyL. H.HedlundE. (2017). Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol.133, 863–885. 10.1007/s00401-017-1708-8
213
NikoletopoulouV.PapandreouM.-E.TavernarakisN. (2015). Autophagy in the Physiology and Pathology of the central Nervous System. Cell Death Differ22, 398–407. 10.1038/cdd.2014.204
214
NilssonP.LoganathanK.SekiguchiM.MatsubaY.HuiK.TsubukiS.et al (2013). Aβ Secretion and Plaque Formation Depend on Autophagy. Cel Rep.5, 61–69. 10.1016/j.celrep.2013.08.042
215
NilssonP.SaidoT. C. (2014). Dual Roles for Autophagy: Degradation and Secretion of Alzheimer's Disease Aβ Peptide. Bioessays36, 570–578. 10.1002/bies.201400002
216
NixonR. A.WegielJ.KumarA.YuW. H.PeterhoffC.CataldoA.et al (2005). Extensive Involvement of Autophagy in Alzheimer Disease: an Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol.64, 113–122. 10.1093/jnen/64.2.113
217
O'ReillyÉ. J.WangH.WeisskopfM. G.FitzgeraldK. C.FalconeG.McCulloughM. L.et al (2013). Premorbid Body Mass index and Risk of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degeneration14, 205–211. 10.3109/21678421.2012.735240
218
OaksA. W.FrankfurtM.FinkelsteinD. I.SidhuA. (2013). Age-Dependent Effects of A53T Alpha-Synuclein on Behavior and Dopaminergic Function. PLoS One8, e60378. 10.1371/journal.pone.0060378
219
OishiK.ZhengB.KuoJ. F. (1990). Inhibition of Na,K-ATPase and Sodium Pump by Protein Kinase C Regulators Sphingosine, Lysophosphatidylcholine, and Oleic Acid. J. Biol. Chem.265, 70–75. 10.1016/s0021-9258(19)40196-8
220
OkuraH.YamashitaS.OhamaT.SagaA.Yamamoto-KakutaA.HamadaY.et al (2010). HDL/Apolipoprotein A-I Binds to Macrophage-Derived Progranulin and Suppresses its Conversion into Proinflammatory Granulins. Jat17, 568–577. 10.5551/jat.3921
221
OlkkonenV. M.LevineT. P. (2004). Oxysterol Binding Proteins: in More Than One Place at One Time?Biochem. Cel Biol.82, 87–98. 10.1139/o03-088
222
OngQ.-R.ChanE. S.LimM.-L.ColeG. M.WongB.-S. (2014). Reduced Phosphorylation of Brain Insulin Receptor Substrate and Akt Proteins in Apolipoprotein-E4 Targeted Replacement Mice. Sci. Rep.4, 3754. 10.1038/srep03754
223
OrensteinS. J.KuoS.-H.TassetI.AriasE.KogaH.Fernandez-CarasaI.et al (2013). Interplay of LRRK2 with Chaperone-Mediated Autophagy. Nat. Neurosci.16, 394–406. 10.1038/nn.3350
224
Ortega-RojasJ.MoralesL.GuerreroE.Arboleda-BustosC. E.MejiaA.ForeroD.et al (2016). Association Analysis of Polymorphisms in TOMM40, CR1, PVRL2, SORL1, PICALM, and 14q32.13 Regions in Colombian Alzheimer Disease Patients. Alzheimer Dis. Assoc. Disord.30, 305–309. 10.1097/WAD.0000000000000142
225
OsellameL. D.RahimA. A.HargreavesI. P.GeggM. E.Richard-LondtA.BrandnerS.et al (2013). Mitochondria and Quality Control Defects in a Mouse Model of Gaucher Disease-Links to Parkinson's Disease. Cel Metab.17, 941–953. 10.1016/j.cmet.2013.04.014
226
OuZ.KongX.SunX.HeX.ZhangL.GongZ.et al (2018). Metformin Treatment Prevents Amyloid Plaque Deposition and Memory Impairment in APP/PS1 Mice. Brain Behav. Immun.69, 351–363. 10.1016/j.bbi.2017.12.009
227
OyanagiK.YamazakiM.TakahashiH.WatabeK.WadaM.KomoriT.et al (2008). Spinal Anterior Horn Cells in Sporadic Amyotrophic Lateral Sclerosis Show Ribosomal Detachment from, and Cisternal Distention of the Rough Endoplasmic Reticulum. Neuropathol. Appl. Neurobiol.34, 650–658. 10.1111/j.1365-2990.2008.00941.x
228
PaganiM.ChioA.ValentiniM. C.ObergJ.NobiliF.CalvoA.et al (2014). Functional Pattern of Brain FDG-PET in Amyotrophic Lateral Sclerosis. Neurology83, 1067–1074. 10.1212/WNL.0000000000000792
229
PaillussonS.StoicaR.Gomez-SuagaP.LauD. H. W.MuellerS.MillerT.et al (2016). There's Something Wrong with My MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosciences39, 146–157. 10.1016/j.tins.2016.01.008
230
PalaciosN.GaoX.McCulloughM. L.JacobsE. J.PatelA. V.MayoT.et al (2011). Obesity, Diabetes, and Risk of Parkinson's Disease. Mov. Disord.26, 2253–2259. 10.1002/mds.23855
231
PangW.HuF. (2021). Cellular and Physiological Functions of C9ORF72 and Implications for ALS/FTD. J. Neurochem.157, 334–350. 10.1111/jnc.15255
232
ParconP. A.BalasubramaniamM.AyyadevaraS.JonesR. A.LiuL.Shmookler ReisR. J.et al (2018). Apolipoprotein E4 Inhibits Autophagy Gene Products through Direct, Specific Binding to CLEAR Motifs. Alzheimer's Demen.14, 230–242. 10.1016/j.jalz.2017.07.754
233
PaushterD. H.DuH.FengT.HuF. (2018). The Lysosomal Function of Progranulin, a Guardian against Neurodegeneration. Acta Neuropathol.136, 1–17. 10.1007/s00401-018-1861-8
234
PedrosoJ. L.ValeT. C.BuenoF. L.MarussiV. H. R.AmaralL. L. F. d.FrançaM. C.et al (2018). SPG7 with Parkinsonism Responsive to Levodopa and Dopaminergic Deficit. Parkinsonism Relat. Disord.47, 88–90. 10.1016/j.parkreldis.2017.12.004
235
PepinoM. Y.KudaO.SamovskiD.AbumradN. A. (2014). Structure-function of CD36 and Importance of Fatty Acid Signal Transduction in Fat Metabolism. Annu. Rev. Nutr.34, 281–303. 10.1146/annurev-nutr-071812-161220
236
PereraN. D.TurnerB. J. (2016). AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res.41, 544–553. 10.1007/s11064-015-1665-3
237
PetkauT. L.NealS. J.MilnerwoodA.MewA.HillA. M.OrbanP.et al (2012). Synaptic Dysfunction in Progranulin-Deficient Mice. Neurobiol. Dis.45, 711–722. 10.1016/j.nbd.2011.10.016
238
PetoukhovE.FernandoS.MillsF.ShivjiF.HunterD.KriegerC.et al (2013). Activity-dependent Secretion of Progranulin from Synapses. J. Cel Sci.126, 5412–5421. 10.1242/jcs.132076
239
PickfordF.MarcusJ.CamargoL. M.XiaoQ.GrahamD.MoJ.-R.et al (2011). Progranulin Is a Chemoattractant for Microglia and Stimulates Their Endocytic Activity. Am. J. Pathol.178, 284–295. 10.1016/j.ajpath.2010.11.002
240
PickfordF.MasliahE.BritschgiM.LucinK.NarasimhanR.JaegerP. A.et al (2008). The Autophagy-Related Protein Beclin 1 Shows Reduced Expression in Early Alzheimer Disease and Regulates Amyloid β Accumulation in Mice. J. Clin. Invest.118, 2190–2199. 10.1172/JCI33585
241
PilliM.Arko-MensahJ.PonpuakM.RobertsE.MasterS.MandellM. A.et al (2012). TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity37, 223–234. 10.1016/j.immuni.2012.04.015
242
PirasA.CollinL.GrüningerF.GraffC.RönnbäckA. (2016). Autophagic and Lysosomal Defects in Human Tauopathies: Analysis of post-mortem Brain from Patients with Familial Alzheimer Disease, Corticobasal Degeneration and Progressive Supranuclear Palsy. Acta Neuropathol. Commun.4, 22. 10.1186/s40478-016-0292-9
243
PoeweW.SeppiK.TannerC. M.HallidayG. M.BrundinP.VolkmannJ.et al (2017). Parkinson Disease. Nat. Rev. Dis. Primers3, 17013. 10.1038/nrdp.2017.13
244
PolymeropoulosM. H.HigginsJ. J.GolbeL. I.JohnsonW. G.IdeS. E.Di IorioG.et al (1996). Mapping of a Gene for Parkinson's Disease to Chromosome 4q21-Q23. Science274, 1197–1199. 10.1126/SCIENCE.274.5290.1197
245
PostumaR. B.BergD.SternM.PoeweW.OlanowC. W.OertelW.et al (2015). MDS Clinical Diagnostic Criteria for Parkinson's Disease. Mov Disord.30, 1591–1601. 10.1002/mds.26424
246
PradatP.-F.BruneteauG.GordonP. H.DupuisL.Bonnefont-RousselotD.SimonD.et al (2010). Impaired Glucose Tolerance in Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler.11, 166–171. 10.3109/17482960902822960
247
QiG.MiY.ShiX.GuH.BrintonR. D.YinF. (2021). ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cel Rep.34, 108572. 10.1016/j.celrep.2020.108572
248
RaberJ.HuangY.AshfordJ. W. (2004). ApoE Genotype Accounts for the Vast Majority of AD Risk and AD Pathology. Neurobiol. Aging25, 641–650. 10.1016/j.neurobiolaging.2003.12.023
249
RagagninA. M. G.ShadfarS.VidalM.JamaliM. S.AtkinJ. D. (2019). Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci.13, 1. 10.3389/fnins.2019.00532
250
RajagopalanV.PioroE. P. (2019). Longitudinal 18F-FDG PET and MRI Reveal Evolving Imaging Pathology that Corresponds to Disease Progression in a Patient with ALS-FTD. Front. Neurol.10, 1. 10.3389/fneur.2019.00234
251
ReddyK.CusackC. L.NnahI. C.KhayatiK.SaqcenaC.HuynhT. B.et al (2016). Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cel Rep.14, 2166–2179. 10.1016/j.celrep.2016.02.006
252
ReitzC.RogaevaE.BeechamG. W. (2020). Late-onset vs Nonmendelian Early-Onset Alzheimer Disease. Neurol. Genet.6, e512. 10.1212/NXG.0000000000000512
253
RentonA. E.MajounieE.WaiteA.Simón-SánchezJ.RollinsonS.GibbsJ. R.et al (2011). A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron72, 257–268. 10.1016/J.NEURON.2011.09.010
254
ReyesE. T.PerurenaO. H.FestoffB. W.JorgensenR.MooreW. V. (1984). Insulin Resistance in Amyotrophic Lateral Sclerosis. J. Neurol. Sci.63, 317–324. 10.1016/0022-510X(84)90154-0
255
RianchoJ.Gil-BeaF.SanturtunA.López de MunaínA. (2019). Amyotrophic Lateral Sclerosis: a Complex Syndrome that Needs an Integrated Research Approach. Neural Regen. Res.14, 193. 10.4103/1673-5374.244783
256
RicciarelliR.d'AbramoC.ZinggJ.-M.GilibertoL.MarkesberyW.AzziA.et al (2004). CD36 Overexpression in Human Brain Correlates with β-amyloid Deposition but Not with Alzheimer's Disease. Free Radic. Biol. Med.36, 1018–1024. 10.1016/j.freeradbiomed.2004.01.007
257
RideoutH. J.StefanisL. (2014). The Neurobiology of LRRK2 and its Role in the Pathogenesis of Parkinson's Disease. Neurochem. Res.39, 576–592. 10.1007/s11064-013-1073-5
258
RobberechtW.PhilipsT. (2013). The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci.14, 248–264. 10.1038/nrn3430
259
Roczniak-FergusonA.PetitC. S.FroehlichF.QianS.KyJ.AngarolaB.et al (2012). The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Signal.5. 10.1126/scisignal.2002790
260
RomeoG. R.LeeJ.ShoelsonS. E. (2012). Metabolic Syndrome, Insulin Resistance, and Roles of Inflammation - Mechanisms and Therapeutic Targets. Atvb32, 1771–1776. 10.1161/ATVBAHA.111.241869
261
RootJ.MerinoP.NuckolsA.JohnsonM.KukarT. (2021). Lysosome Dysfunction as a Cause of Neurodegenerative Diseases: Lessons from Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neurobiol. Dis.154, 105360. 10.1016/j.nbd.2021.105360
262
RosenbuschK. E.KortholtA. (20162016). Activation Mechanism of LRRK2 and its Cellular Functions in Parkinson's Disease. Parkinson's Dis.2016, 1–8. 10.1155/2016/7351985
263
RothenbergC.SrinivasanD.MahL.KaushikS.PeterhoffC. M.UgolinoJ.et al (2010). Ubiquilin Functions in Autophagy and Is Degraded by Chaperone-Mediated Autophagy. Hum. Mol. Genet.19, 3219–3232. 10.1093/hmg/ddq231
264
RyanB. J.HoekS.FonE. A.Wade-MartinsR. (2015). Mitochondrial Dysfunction and Mitophagy in Parkinson's: from Familial to Sporadic Disease. Trends Biochem. Sci.40, 200–210. 10.1016/j.tibs.2015.02.003
265
RyanC. L.BaranowskiD. C.ChitramuthuB. P.MalikS.LiZ.CaoM.et al (2009). Progranulin Is Expressed within Motor Neurons and Promotes Neuronal Cell Survival. BMC Neurosci.10, 130. 10.1186/1471-2202-10-130
266
SääksjärviK.KnektP.MännistöS.LyytinenJ.HeliövaaraM. (2015). Prospective Study on the Components of Metabolic Syndrome and the Incidence of Parkinson's Disease. Parkinsonism Relat. Disord.21, 1148–1155. 10.1016/j.parkreldis.2015.07.017
267
SalaG.TremolizzoL.MelchiondaL.StefanoniG.DerosaM.SusaniE.et al (2012). A Panel of Macroautophagy Markers in Lymphomonocytes of Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler.13, 119–124. 10.3109/17482968.2011.611139
268
SancakY.Bar-PeledL.ZoncuR.MarkhardA. L.NadaS.SabatiniD. M. (2010). Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for its Activation by Amino Acids. Cell141, 290–303. 10.1016/j.cell.2010.02.024
269
SandersL. H.LaganièreJ.CooperO.MakS. K.VuB. J.HuangY. A.et al (2014). LRRK2 Mutations Cause Mitochondrial DNA Damage in iPSC-Derived Neural Cells from Parkinson's Disease Patients: Reversal by Gene Correction. Neurobiol. Dis.62, 381–386. 10.1016/j.nbd.2013.10.013
270
Santos-RebouçasC. B.GonçalvesA. P.Dos SantosJ. M.AbdalaB. B.MottaL. B.LaksJ.et al (2017). rs3851179 Polymorphism at 5′ to the PICALM Gene Is Associated with Alzheimer and Parkinson Diseases in Brazilian Population. Neuromol Med.19, 293–299. 10.1007/s12017-017-8444-z
271
SasakiS. (2010). Endoplasmic Reticulum Stress in Motor Neurons of the Spinal Cord in Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol.69, 346–355. 10.1097/NEN.0b013e3181d44992
272
SasakiS.IwataM. (2007). Mitochondrial Alterations in the Spinal Cord of Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol.66, 10–16. 10.1097/nen.0b013e31802c396b
273
SatoN.MorishitaR. (2015). The Roles of Lipid and Glucose Metabolism in Modulation of β-amyloid, Tau, and Neurodegeneration in the Pathogenesis of Alzheimer Disease. Front. Aging Neurosci.7, 199. 10.3389/fnagi.2015.00199
274
SavicaR.GrossardtB. R.RoccaW. A.BowerJ. H. (2018). Parkinson Disease with and without Dementia: A Prevalence Study and Future Projections. Mov Disord.33, 537–543. 10.1002/mds.27277
275
SchapiraA. H. V.CooperJ. M.DexterD.JennerP.ClarkJ. B.MarsdenC. D. (1989). Mitochondrial Complex I Deficiency in Parkinson's Disease. The Lancet333, 1269. 10.1016/S0140-6736(89)92366-0
276
SchmuklerE.SolomonS.SimonovitchS.GoldshmitY.WolfsonE.MichaelsonD. M.et al (2020). Altered Mitochondrial Dynamics and Function in APOE4-Expressing Astrocytes. Cel Death Dis11, 578. 10.1038/s41419-020-02776-4
277
SchneiderS. A.AlcalayR. N. (2017). Neuropathology of Genetic Synucleinopathies with Parkinsonism: Review of the Literature. Mov Disord.32, 1504–1523. 10.1002/mds.27193
278
Schnetz-BoutaudN. C.HoffmanJ.CoeJ. E.MurdockD. G.Pericak-VanceM. A.HainesJ. L. (2012). Identification and Confirmation of an Exonic Splicing Enhancer Variation in Exon 5 of the Alzheimer Disease AssociatedPICALMGene. Ann. Hum. Genet.76, 448–453. 10.1111/j.1469-1809.2012.00727.x
279
SchöndorfD. C.IvanyukD.BadenP.Sanchez-MartinezA.De CiccoS.YuC.et al (2018). The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson's Disease. Cel Rep.23, 2976–2988. 10.1016/j.celrep.2018.05.009
280
SchreinerB.HedskogL.WiehagerB.AnkarcronaM. (2014). Amyloid-β Peptides Are Generated in Mitochondria-Associated Endoplasmic Reticulum Membranes. Jad43, 369–374. 10.3233/JAD-132543
281
SeidelK.SchölsL.NuberS.Petrasch-ParwezE.GiergaK.WszolekZ.et al (2010). First Appraisal of Brain Pathology Owing to A30P Mutant Alpha-Synuclein. Ann. Neurol.67, NA. 10.1002/ana.21966
282
ŞentürkM.LinG.ZuoZ.MaoD.WatsonE.MikosA. G.et al (2019). Ubiquilins Regulate Autophagic Flux through mTOR Signalling and Lysosomal Acidification. Nat. Cel Biol.21, 384–396. 10.1038/s41556-019-0281-x
283
SerreroG.HawkinsD. M.YueB.IoffeO.BejaranoP.PhillipsJ. T.et al (2012). Progranulin (GP88) Tumor Tissue Expression Is Associated with Increased Risk of Recurrence in Breast Cancer Patients Diagnosed with Estrogen Receptor Positive Invasive Ductal Carcinoma. Breast Cancer Res.14, R26. 10.1186/bcr3111
284
ŠerýO.JanoutováJ.EwerlingováL.HálováA.LochmanJ.JanoutV.et al (2017). CD36 Gene Polymorphism Is Associated with Alzheimer's Disease. Biochimie135, 46–53. 10.1016/j.biochi.2017.01.009
285
SettembreC.Di MaltaC.PolitoV. A.ArencibiaM. G.VetriniF.ErdinS.et al (2011). TFEB Links Autophagy to Lysosomal Biogenesis. Science332, 1429–1433. 10.1126/science.1204592
286
SettembreC.FraldiA.MedinaD. L.BallabioA. (2013). Signals from the Lysosome: a Control centre for Cellular Clearance and Energy Metabolism. Nat. Rev. Mol. Cel Biol.14, 283–296. 10.1038/nrm3565
287
ShacharT.BiancoC. L.LoA.RecchiaC.Raas-RothschildA.FutermanA. H. (2011). Lysosomal Storage Disorders and Parkinson's Disease: Gaucher Disease and beyond. Mov. Disord.26, 1593–1604. 10.1002/mds.23774
288
ShafaatiM.OlinM.BåvnerA.PetterssonH.RozellB.MeaneyS.et al (2011). Enhanced Production of 24S-Hydroxycholesterol Is Not Sufficient to Drive Liver X Receptor Target Genes In Vivo. J. Intern. Med.270, 377–387. 10.1111/j.1365-2796.2011.02389.x
289
ShahheydariH.RagagninA.WalkerA. K.TothR. P.VidalM.JagarajC. J.et al (2017). Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci.10, 1. 10.3389/fnmol.2017.00119
290
ShahmoradianS. H.LewisA. J.GenoudC.HenchJ.MoorsT. E.NavarroP. P.et al (2019). Lewy Pathology in Parkinson's Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci.22, 1099–1109. 10.1038/s41593-019-0423-2
291
ShaoQ.YangM.LiangC.MaL.ZhangW.JiangZ.et al (2020). C9orf72 and Smcr8 Mutant Mice Reveal MTORC1 Activation Due to Impaired Lysosomal Degradation and Exocytosis. Autophagy16, 1635–1650. 10.1080/15548627.2019.1703353
292
SheedyF. J.GrebeA.RaynerK. J.KalantariP.RamkhelawonB.CarpenterS. B.et al (2013). CD36 Coordinates NLRP3 Inflammasome Activation by Facilitating Intracellular Nucleation of Soluble Ligands into Particulate Ligands in Sterile Inflammation. Nat. Immunol.14, 812–820. 10.1038/ni.2639
293
ShenW.-C.LiH.-Y.ChenG.-C.ChernY.TuP.-h. (2015). Mutations in the Ubiquitin-Binding Domain of OPTN/optineurin Interfere with Autophagy-Mediated Degradation of Misfolded Proteins by a Dominant-Negative Mechanism. Autophagy11, 685–700. 10.4161/auto.36098
294
SimonovitchS.SchmuklerE.BespalkoA.IramT.FrenkelD.HoltzmanD. M.et al (2016). Impaired Autophagy in APOE4 Astrocytes. Jad51, 915–927. 10.3233/JAD-151101
295
SluggettJ. K.KoponenM.BellJ. S.TaipaleH.TanskanenA.TiihonenJ.et al (2020). Metformin and Risk of Alzheimer's Disease Among Community-Dwelling People with Diabetes: A National Case-Control Study. J. Clin. Endocrinol. Metab.105, e963–e972. 10.1210/clinem/dgz234
296
SmithB. R.SantosM. B.MarshallM. S.Cantuti-CastelvetriL.Lopez-RosasA.LiG.et al (2014). Neuronal Inclusions of α-synuclein Contribute to the Pathogenesis of Krabbe Disease. J. Pathol.232, 509–521. 10.1002/path.4328
297
SmithE. F.ShawP. J.De VosK. J. (2019). The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett.710, 132933. 10.1016/j.neulet.2017.06.052
298
SmithK. R.DamianoJ.FranceschettiS.CarpenterS.CanafogliaL.MorbinM.et al (2012). Strikingly Different Clinicopathological Phenotypes Determined by Progranulin-Mutation Dosage. Am. J. Hum. Genet.90, 1102–1107. 10.1016/j.ajhg.2012.04.021
299
SoE.MitchellJ. C.MemmiC.ChennellG.Vizcay-BarrenaG.AllisonL.et al (2018). Mitochondrial Abnormalities and Disruption of the Neuromuscular junction Precede the Clinical Phenotype and Motor Neuron Loss in hFUSWT Transgenic Mice. Hum. Mol. Genet.27, 463–474. 10.1093/hmg/ddx415
300
SohnH.-Y.KimS.-I.ParkJ.-Y.ParkS.-H.KohY. H.KimJ.et al (2021). ApoE4 Attenuates Autophagy via FoxO3a Repression in the Brain. Sci. Rep.11, 17604. 10.1038/s41598-021-97117-6
301
SolJ.JovéM.PovedanoM.SprovieroW.DomínguezR.Piñol-RipollG.et al (2021). Lipidomic Traits of Plasma and Cerebrospinal Fluid in Amyotrophic Lateral Sclerosis Correlate with Disease Progression. Brain Commun.3. 10.1093/braincomms/fcab143
302
StallingsN. R.PuttaparthiK.DowlingK. J.LutherC. M.BurnsD. K.DavisK.et al (2013). TDP-43, an ALS Linked Protein, Regulates Fat Deposition and Glucose Homeostasis. PLoS One8, e71793. 10.1371/journal.pone.0071793
303
StamatakouE.WróbelL.HillS. M.PuriC.SonS. M.FujimakiM.et al (2020). Mendelian Neurodegenerative Disease Genes Involved in Autophagy. Cell Discov6, 24. 10.1038/s41421-020-0158-y
304
StefanisL.LarsenK. E.RideoutH. J.SulzerD.GreeneL. A. (2001). Expression of A53T Mutant but Not Wild-type α-Synuclein in PC12 Cells Induces Alterations of the Ubiquitin-dependent Degradation System, Loss of Dopamine Release, and Autophagic Cell Death. J. Neurosci.21, 9549–9560. 10.1523/JNEUROSCI.21-24-09549.2001
305
StokerT. B.GreenlandJ. C. (2018). Parkinson's Disease: Pathogenesis and Clinical Aspects. Brisbane, Australia: Codon Publications. 10.15586/codonpublications.parkinsonsdisease.2018
306
StraubI. R.WeraarpachaiW.ShoubridgeE. A. (2021). Multi-OMICS Study of a CHCHD10 Variant Causing ALS Demonstrates Metabolic Rewiring and Activation of Endoplasmic Reticulum and Mitochondrial Unfolded Protein Responses. Hum. Mol. Genet.30, 687–705. 10.1093/hmg/ddab078
307
SunY.ChangY.-H.ChenH.-F.SuY.-H.SuH.-F.LiC.-Y. (2012). Risk of Parkinson Disease Onset in Patients with Diabetes: a 9-year Population-Based Cohort Study with Age and Sex Stratifications. Diabetes Care35, 1047–1049. 10.2337/dc11-1511
308
SundaramoorthyV.WalkerA. K.TanV.FifitaJ. A.MccannE. P.WilliamsK. L.et al (2015). Defects in Optineurin- and Myosin VI-mediated Cellular Trafficking in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet.24, 3830–3846. 10.1093/hmg/ddv126
309
SzelechowskiM.AmoedoN.ObreE.LégerC.AllardL.BonneuM.et al (2018). Metabolic Reprogramming in Amyotrophic Lateral Sclerosis. Sci. Rep.8, 3953. 10.1038/s41598-018-22318-5
310
TanV. P.MiyamotoS. (2016). Nutrient-sensing mTORC1: Integration of Metabolic and Autophagic Signals. J. Mol. Cell Cardiol.95, 31–41. 10.1016/j.yjmcc.2016.01.005
311
TanakaY.MatsuwakiT.YamanouchiK.NishiharaM. (2013). Exacerbated Inflammatory Responses Related to Activated Microglia after Traumatic Brain Injury in Progranulin-Deficient Mice. Neuroscience231, 49–60. 10.1016/j.neuroscience.2012.11.032
312
TanakaY.SuzukiG.MatsuwakiT.HosokawaM.SerranoG.BeachT. G.et al (2017). Progranulin Regulates Lysosomal Function and Biogenesis through Acidification of Lysosomes. Hum. Mol. Genet.26 (5), 969–988. 10.1093/hmg/ddx011
313
TangG.GudsnukK.KuoS.-H.CotrinaM. L.RosoklijaG.SosunovA.et al (2014). Loss of mTOR-dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron83, 1131–1143. 10.1016/j.neuron.2014.07.040
314
TangZ.HuB.ZangF.WangJ.ZhangX.ChenH. (2019). Nrf2 Drives Oxidative Stress-Induced Autophagy in Nucleus Pulposus Cells via a Keap1/Nrf2/p62 Feedback Loop to Protect Intervertebral Disc from Degeneration. Cel Death Dis10, 510. 10.1038/s41419-019-1701-3
315
TangkeangsirisinW.HayashiJ.SerreroG. (2004). PC Cell-Derived Growth Factor Mediates Tamoxifen Resistance and Promotes Tumor Growth of Human Breast Cancer Cells. Cancer Res.64, 1737–1743. 10.1158/0008-5472.CAN-03-2364
316
TangkeangsirisinW.SerreroG. (2004). PC Cell-Derived Growth Factor (PCDGF/GP88, Progranulin) Stimulates Migration, Invasiveness and VEGF Expression in Breast Cancer Cells. Carcinogenesis25, 1587–1592. 10.1093/carcin/bgh171
317
TapiaL.MilnerwoodA.GuoA.MillsF.YoshidaE.VasutaC.et al (2011). Progranulin Deficiency Decreases Gross Neural Connectivity but Enhances Transmission at Individual Synapses. J. Neurosci.31, 11126–11132. 10.1523/JNEUROSCI.6244-10.2011
318
TaubeA.SchlichR.SellH.EckardtK.EckelJ. (2012). Inflammation and Metabolic Dysfunction: Links to Cardiovascular Diseases. Am. J. Physiology-Heart Circulatory Physiol.302, H2148–H2165. 10.1152/ajpheart.00907.2011
319
TedeschiV.PetrozzielloT.SecondoA. (2019). Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis. Cells8, 1216. 10.3390/cells8101216
320
TeferaT. W.SteynF. J.NgoS. T.BorgesK. (2021). CNS Glucose Metabolism in Amyotrophic Lateral Sclerosis: a Therapeutic Target?Cell Biosci11, 14. 10.1186/s13578-020-00511-2
321
TewariK. P.MalinowskaD. H.SherryA. M.CuppolettiJ. (2000). PKA and Arachidonic Acid Activation of Human Recombinant ClC-2 Chloride Channels. Am. J. Physiology-Cell Physiol.279, C40–C50. 10.1152/ajpcell.2000.279.1.C40
322
ThomasH. E.ZhangY.StefelyJ. A.VeigaS. R.ThomasG.KozmaS. C.et al (2018). Mitochondrial Complex I Activity Is Required for Maximal Autophagy. Cel Rep.24, 2404–2417. e8. 10.1016/j.celrep.2018.07.101
323
TohH.CaoM.DanielsE.BatemanA. (2013). Expression of the Growth Factor Progranulin in Endothelial Cells Influences Growth and Development of Blood Vessels: A Novel Mouse Model. PLoS One8, e64989. 10.1371/journal.pone.0064989
324
TohH.ChitramuthuB. P.BennettH. P. J.BatemanA. (2011). Structure, Function, and Mechanism of Progranulin; the Brain and beyond. J. Mol. Neurosci.45, 538–548. 10.1007/s12031-011-9569-4
325
TorresP.Cabral‐MirandaF.Gonzalez‐TeuberV.HetzC. (2021). Proteostasis Deregulation as a Driver of C9ORF72 Pathogenesis. J. Neurochem.159, 941–957. 10.1111/jnc.15529
326
TramutolaA.LanzillottaC.Di DomenicoF. (2017). Targeting mTOR to Reduce Alzheimer-Related Cognitive Decline: from Current Hits to Future Therapies. Expert Rev. Neurotherapeutics17, 33–45. 10.1080/14737175.2017.1244482
327
TresseE.SalomonsF. A.VesaJ.BottL. C.KimonisV.YaoT.-P.et al (2010). VCP/p97 Is Essential for Maturation of Ubiquitin-Containing Autophagosomes and This Function Is Impaired by Mutations that Cause IBMPFD. Autophagy6, 217–227. 10.4161/auto.6.2.11014
328
TsitkanouS.Della GattaP. A.RussellA. P. (2016). Skeletal Muscle Satellite Cells, Mitochondria, and MicroRNAs: Their Involvement in the Pathogenesis of ALS. Front. Physiol.7, 1. 10.3389/fphys.2016.00403
329
TzoulisC.TranG. T.SchwarzlmüllerT.SpechtK.HaugarvollK.BalafkanN.et al (2013). Severe Nigrostriatal Degeneration without Clinical Parkinsonism in Patients with Polymerase Gamma Mutations. Brain136, 2393–2404. 10.1093/brain/awt103
330
ValdezC.WongY. C.SchwakeM.BuG.WszolekZ. K.KraincD. (2017). Progranulin-mediated Deficiency of Cathepsin D Results in FTD and NCL-like Phenotypes in Neurons Derived from FTD Patients. Hum. Mol. Genet.26, 4861–4872. 10.1093/hmg/ddx364
331
ValdezC.YsselsteinD.YoungT. J.ZhengJ.KraincD. (2020). Progranulin Mutations Result in Impaired Processing of Prosaposin and Reduced Glucocerebrosidase Activity. Hum. Mol. Genet.29, 716–726. 10.1093/hmg/ddz229
332
ValenteE. M.SalviS.IalongoT.MarongiuR.EliaA. E.CaputoV.et al (2004). PINK1 Mutations Are Associated with Sporadic Early-Onset Parkinsonism. Ann. Neurol.56, 336–341. 10.1002/ANA.20256
333
Van DammeP.Van HoeckeA.LambrechtsD.VanackerP.BogaertE.van SwietenJ.et al (2008). Progranulin Functions as a Neurotrophic Factor to Regulate Neurite Outgrowth and Enhance Neuronal Survival. J. Cel Biol.181, 37–41. 10.1083/jcb.200712039
334
Van RompuyA.-S.LobbestaelE.Van der PerrenA.Van den HauteC.BaekelandtV. (2014). Long-term Overexpression of Human Wild-type and T240R Mutant Parkin in Rat Substantia Nigra Induces Progressive Dopaminergic Neurodegeneration. J. Neuropathol. Exp. Neurol.73, 159–174. 10.1097/NEN.0000000000000039
335
VandoorneT.De BockK.Van Den BoschL. (2018). Energy Metabolism in ALS: an Underappreciated Opportunity?Acta Neuropathol.135, 489–509. 10.1007/s00401-018-1835-x
336
VurusanerB.GambaP.TestaG.GargiuloS.BiasiF.ZerbinatiC.et al (2014). Survival Signaling Elicited by 27-hydroxycholesterol through the Combined Modulation of Cellular Redox State and ERK/Akt Phosphorylation. Free Radic. Biol. Med.77, 376–385. 10.1016/j.freeradbiomed.2014.07.026
337
VurusanerB.GargiuloS.TestaG.GambaP.LeonarduzziG.PoliG.et al (2018). The Role of Autophagy in Survival Response Induced by 27-hydroxycholesterol in Human Promonocytic Cells. Redox Biol.17, 400–410. 10.1016/j.redox.2018.05.010
338
WangC.YuJ.-T.MiaoD.WuZ.-C.TanM.-S.TanL. (2014). Targeting the mTOR Signaling Network for Alzheimer's Disease Therapy. Mol. Neurobiol.49, 120–135. 10.1007/s12035-013-8505-8
339
WangL.LiN.ShiF.-X.XuW.-Q.CaoY.LeiY.et al (2020a). Upregulation of AMPK Ameliorates Alzheimer's Disease-like Tau Pathology and Memory Impairment. Mol. Neurobiol.57, 3349–3361. 10.1007/s12035-020-01955-w
340
WangL.ShiF.-X.LiN.CaoY.LeiY.WangJ.-Z.et al (2020b). AMPK Ameliorates Tau Acetylation and Memory Impairment through Sirt1. Mol. Neurobiol.57, 5011–5025. 10.1007/s12035-020-02079-x
341
WangW.WangL.LuJ.SiedlakS. L.FujiokaH.LiangJ.et al (2016). The Inhibition of TDP-43 Mitochondrial Localization Blocks its Neuronal Toxicity. Nat. Med.22, 869–878. 10.1038/nm.4130
342
WardM. E.ChenR.HuangH.-Y.LudwigC.TelpoukhovskaiaM.TaubesA.et al (2017). Individuals with Progranulin Haploinsufficiency Exhibit Features of Neuronal Ceroid Lipofuscinosis. Sci. Transl. Med.9, eaah5642. 10.1126/scitranslmed.aah5642
343
WeiY.ZhouJ.WuJ.HuangJ. (2019). ERβ Promotes Aβ Degradation via the Modulation of Autophagy. Cel Death Dis10, 565. 10.1038/s41419-019-1786-8
344
WiedemannF. R.ManfrediG.MawrinC.BealM. F.SchonE. A. (2002). Mitochondrial DNA and Respiratory Chain Function in Spinal Cords of ALS Patients. J. Neurochem.80, 616–625. 10.1046/j.0022-3042.2001.00731.x
345
WilsH.KleinbergerG.PeresonS.JanssensJ.CapellA.Van DamD.et al (2012). Cellular Ageing, Increased Mortality and FTLD-TDP-Associated Neuropathology in Progranulin Knockout Mice. J. Pathol.228 (1), 67–76. 10.1002/path.4043
346
WongE.CuervoA. M. (2010). Autophagy Gone Awry in Neurodegenerative Diseases. Nat. Neurosci.13, 805–811. 10.1038/nn.2575
347
WuJ. J.CaiA.GreensladeJ. E.HigginsR.FanC.LeN. T. T.et al (2021). Correction for Wu et al., ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc. Natl. Acad. Sci. USA118, e2114051118. 10.1073/pnas.2114051118
348
XiaoY.ZhangJ.ShuX.BaiL.XuW.WangA.et al (2020). Loss of Mitochondrial Protein CHCHD10 in Skeletal Muscle Causes Neuromuscular junction Impairment. Hum. Mol. Genet.29, 1784–1796. 10.1093/hmg/ddz154
349
XuY.-F.GendronT. F.ZhangY.-J.LinW.-L.D'AltonS.ShengH.et al (2010). Wild-Type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J. Neurosci.30, 10851–10859. 10.1523/JNEUROSCI.1630-10.2010
350
YangW.TianZ.-K.YangH.-X.FengZ.-J.SunJ.-M.JiangH.et al (2019). Fisetin Improves lead-induced Neuroinflammation, Apoptosis and Synaptic Dysfunction in Mice Associated with the AMPK/SIRT1 and Autophagy Pathway. Food Chem. Toxicol.134, 110824. 10.1016/j.fct.2019.110824
351
YinF.BanerjeeR.ThomasB.ZhouP.QianL.JiaT.et al (2010). Exaggerated Inflammation, Impaired Host Defense, and Neuropathology in Progranulin-Deficient Mice. J. Exp. Med.207, 117–128. 10.1084/jem.20091568
352
YounB.-S.BangS.-I.KlötingN.ParkJ. W.LeeN.OhJ.-E.et al (2009). Serum Progranulin Concentrations May Be Associated with Macrophage Infiltration into Omental Adipose Tissue. Diabetes58, 627–636. 10.2337/db08-1147
353
YuW. H.CuervoA. M.KumarA.PeterhoffC. M.SchmidtS. D.LeeJ.-H.et al (2005). Macroautophagy-a Novel β-amyloid Peptide-Generating Pathway Activated in Alzheimer's Disease. J. Cel Biol.171, 87–98. 10.1083/jcb.200505082
354
ZambonF.CherubiniM.FernandesH. J. R.LangC.RyanB. J.VolpatoV.et al (2019). Cellular α-synuclein Pathology Is Associated with Bioenergetic Dysfunction in Parkinson's iPSC-Derived Dopamine Neurons. Hum. Mol. Genet.28, 2001–2013. 10.1093/hmg/ddz038
355
Zanocco-MaraniT.BatemanA.RomanoG.ValentinisB.HeZ. H.BasergaR. (1999). Biological Activities and Signaling Pathways of the Granulin/epithelin Precursor. Cancer Res.59, 5331–5340.
356
ZhangC.-S.HawleyS. A.ZongY.LiM.WangZ.GrayA.et al (2017). Fructose-1,6-bisphosphate and Aldolase Mediate Glucose Sensing by AMPK. Nature548, 112–116. 10.1038/nature23275
357
ZhangK. Y.YangS.WarraichS. T.BlairI. P. (2014). Ubiquilin 2: A Component of the Ubiquitin-Proteasome System with an Emerging Role in Neurodegeneration. Int. J. Biochem. Cel Biol.50, 123–126. 10.1016/j.biocel.2014.02.018
358
ZhangX.YangK.LeW. (2020). Autophagy and Motor Neuron Diseases. Prog. Mol. Biol. Transl Sci.172, 53–74. 10.1007/978-981-15-4272-5_3
359
ZhangY.-J.XuY.-F.CookC.GendronT. F.RoettgesP.LinkC. D.et al (2009). Aberrant Cleavage of TDP-43 Enhances Aggregation and Cellular Toxicity. Proc. Natl. Acad. Sci.106, 7607–7612. 10.1073/pnas.0900688106
360
ZhaoB.DierichsL.GuJ.-N.Trajkovic-ArsicM.Axel HilgerR.SavvatakisK.et al (2020). TFEB-mediated Lysosomal Biogenesis and Lysosomal Drug Sequestration Confer Resistance to MEK Inhibition in Pancreatic Cancer. Cell Death Discov.6, 12. 10.1038/s41420-020-0246-7
361
ZhaoP.WongK. i.SunX.ReillyS. M.UhmM.LiaoZ.et al (2018). TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell172, 731–743. e12. 10.1016/j.cell.2018.01.007
362
ZhouX.PaushterD. H.PaganM. D.KimD.Nunez SantosM.LiebermanR. L.et al (2019). Progranulin Deficiency Leads to Reduced Glucocerebrosidase Activity. PLoS One14, e0212382. 10.1371/journal.pone.0212382
363
ZhouX. (2017). “Research on Optimization of Queuing System Based on Computer Simulation,” in 2017 International Conference on Computer Technology, Electronics and Communication (ICCTEC) (IEEE), 9–12. 10.1109/ICCTEC.2017.00011
364
ZhouX.SunL.BrackoO.ChoiJ. W.JiaY.NanaA. L.et al (2017a). Impaired Prosaposin Lysosomal Trafficking in Frontotemporal Lobar Degeneration Due to Progranulin Mutations. Nat. Commun.8, 15277. 10.1038/ncomms15277
365
ZhouZ.-D.SawW.-T.TanE.-K. (2017b). Mitochondrial CHCHD-Containing Proteins: Physiologic Functions and Link with Neurodegenerative Diseases. Mol. Neurobiol.54, 5534–5546. 10.1007/s12035-016-0099-5
366
ZhuJ.NathanC.JinW.SimD.AshcroftG. S.WahlS. M.et al (2002). Conversion of Proepithelin to Epithelins. Cell111, 867–878. 10.1016/S0092-8674(02)01141-8
367
ZhuZ.YangC.IyaswamyA.KrishnamoorthiS.SreenivasmurthyS. G.LiuJ.et al (2019). Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson's Disease. Ijms20, 728. 10.3390/ijms20030728
368
ZongH.RenJ. M.YoungL. H.PypaertM.MuJ.BirnbaumM. J.et al (2002). AMP Kinase Is Required for Mitochondrial Biogenesis in Skeletal Muscle in Response to Chronic Energy Deprivation. Proc. Natl. Acad. Sci.99, 15983–15987. 10.1073/PNAS.252625599
369
ZufiríaM.Gil-BeaF. J.Fernández-TorrónR.PozaJ. J.Muñoz-BlancoJ. L.Rojas-GarcíaR.et al (2016). ALS: A Bucket of Genes, Environment, Metabolism and Unknown Ingredients. Prog. Neurobiol.142, 104–129. 10.1016/j.pneurobio.2016.05.004
Summary
Keywords
neurodegenerative diseases, autophagy, nutrient sensing pathways, mitochondrial metabolism, lipid metabolism, glucose metabolism, mTORC1 (mechanistic target of rapamycin complex 1), AMPK
Citation
Ondaro J, Hernandez-Eguiazu H, Garciandia-Arcelus M, Loera-Valencia R, Rodriguez-Gómez L, Jiménez-Zúñiga A, Goikolea J, Rodriguez-Rodriguez P, Ruiz-Martinez J, Moreno F, Lopez de Munain A, Holt IJ, Gil-Bea FJ and Gereñu G (2022) Defects of Nutrient Signaling and Autophagy in Neurodegeneration. Front. Cell Dev. Biol. 10:836196. doi: 10.3389/fcell.2022.836196
Received
15 December 2021
Accepted
21 February 2022
Published
28 March 2022
Volume
10 - 2022
Edited by
Javier Calvo Garrido, Karolinska Institutet (KI), Sweden
Reviewed by
Kunikazu Tanji, Hirosaki University, Japan
Danielle Sliter, Regenxbio Inc., United States
Zvulun Elazar, Weizmann Institute of Science, Israel
Updates
Copyright
© 2022 Ondaro, Hernandez-Eguiazu, Garciandia-Arcelus, Loera-Valencia, Rodriguez-Gómez, Jiménez-Zúñiga, Goikolea, Rodriguez-Rodriguez, Ruiz-Martinez, Moreno, Lopez de Munain, Holt, Gil-Bea and Gereñu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gorka Gereñu, gorka.gerenu@biodonostia.org; Francisco Javier Gil-Bea, francisco.gilbea@biodonostia.org
†These authors have contributed equally to this work
This article was submitted to Molecular and Cellular Pathology, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.