 Anette B. Ellingsen1
Anette B. Ellingsen1 Narjol González-Escalona
Narjol González-Escalona- 1Department of Food Safety and Infection Biology, Norwegian School of Veterinary Science, Oslo, Norway
- 2Norwegian Defence Research Establishment, Kjeller, Norway
- 3Food and Drug Administration, Center for Food Safety and Applied Nutrition, College Park, MD, USA
The thermostable direct hemolysin (TDH) and/or TDH-related hemolysin (TRH) genes are carried by most virulent Vibrio parahaemolyticus serovars. In Norway, trh+ V. parahaemolyticus constitute 4.4 and 4.5% of the total number of V. parahaemolyticus isolated from blue mussel (Mytilus edulis) and water, respectively. The trh gene is located in a region close to the gene cluster for urease production (ure). This region was characterized in V. parahaemolyticus strain TH3996 and it was found that a nickel transport operon (nik) was located between the first gene (ureR) and the rest of the ure cluster genes. The organization of the trh-ureR-nik-ure gene cluster in the Norwegian trh+ isolates was unknown. In this study, we explore the gene organization within the trh-ureR-nik-ure cluster for these isolates. PCR analyses revealed that the genes within the trh-ureR-nik-ure gene cluster of Norwegian trh+ isolates were organized in a similar fashion as reported previously for TH33996. Additionally, the phylogenetic relationship among these trh+ isolates was investigated using Multilocus Sequence Typing (MLST). Analysis by MLST or ureR-trh sequences generated two different phylogenetic trees for the same strains analyzed, suggesting that ureR-trh genes have been acquired at different times in Norwegian V. parahaemolyticus isolates. MLST results revealed that some pathogenic and non-pathogenic V. parahaemolyticus isolates in Norway appear to be highly genetically related.
Introduction
Vibrio parahaemolyticus is a halophilic, Gram-negative proteobacterium ubiquitous in the marine and estuarine environment worldwide (Iida and Honda, 2006). It is one of the main bacterial pathogens associated with raw and undercooked seafood in Asia and the U.S. (Su and Liu, 2007). During the last decade, a V. parahaemolyticus clonal complex (mainly composed of O3:K6 serotype strains) has emerged, encompassing at least 14 serotypes (Okura et al., 2008), and described by the term “pandemic group” (Matsumoto et al., 2000; Okura et al., 2003; Nair et al., 2007).
Although the mechanism by which V. parahaemolyticus causes enteric disease is not fully understood, two virulence factors are usually associated with clinical isolates: the genes encoding for thermostable direct hemolysin (TDH), (tdh) and TDH-related hemolysin (trh) (Gonzalez-Escalona et al., 2006). Of the two, trh is less frequently observed than tdh (Nishibuchi and Kaper, 1995). Several variants of tdh have been identified, all of which are about 98% identical (Nishibuchi and Kaper, 1995). In the case of trh, only two gene variants have been described, denoted trh1 and trh2; these are about 84% identical in sequence (Kishishita et al., 1992). The differences between trh and tdh are greater, showing only about 68% similarity (Nishibuchi et al., 1989). While V. parahaemolyticus can carry both genes and simultaneously express them, it appears that such isolates produces less TDH than trh negative isolates (Xu et al., 1994).
The frequency of tdh and/or trh expression among environmental V. parahaemolyticus isolates is typically <1%, but this may depend on location, sample source and detection method (Kaysner et al., 1990; Alam et al., 2002; Cook et al., 2002; Hervio-Heath et al., 2002; Martinez-Urtaza et al., 2008b). For example, between 49 and 78% of the sediment, water, or oyster samples from Willapa Bay (WA, U.S.) contained trh+ V. parahaemolyticus (Kaysner et al., 1990). In Norway, trh+ V. parahaemolyticus was isolated from only 4.4% of the V. parahaemolyticus positive blue mussel (Mytilus edulis) samples (Bauer et al., 2006) and 4.5% of water samples, and the tdh gene was not detected at all (Ellingsen et al., 2008).
Little is known about how these virulence factors are acquired by V. parahaemolyticus, although it has been suggested that the horizontal gene transfer of pathogenicity island(s) might be the acquisition mechanism (Nishibuchi and Kaper, 1995; Hurley et al., 2006; Izutsu et al., 2008). The trh gene is located in a region of ~16 kb that containing the nik (nickel acquisition system) and ure genes. This characteristic was observed in all trh+ strains analyzed (Park et al., 2000). A transposase gene has been identified next to the trh gene, suggesting the possibility that the entire region might have been transmitted into V. parahaemolyticus strains by an insertion sequence (IS)-mediated mechanism (Park et al., 2000). Isolates exhibiting trh+ are almost exclusively urease positive (ure+), which is not a typical characteristic for V. parahaemolyticus (Kaysner et al., 1994). This association was also observed in all Norwegian trh+ isolates (Bauer et al., 2006). Using long and accurate PCR (LA-PCR), Iida et al. (1998) found that the region between the trh and ureC genes was less than 8.5 kb. The gene organization of the trh-ureR-nik-ure gene cluster was determined in V. parahaemolyticus strain (TH3996) (Park et al., 2000), but whether other trh+ isolates have the same organization remains unknown.
Various typing methods are used to distinguish bacterial strains for epidemiological purposes (Foxman et al., 2005). However, conventional serotyping against O and K antigens appears to be of limited epidemiological value for V. parahaemolyticus, particularly given the recent emergence of O3:K6 and the other serotypes encompassed by the term “pandemic group” (Chowdhury et al., 2004a,b; Gonzalez-Escalona et al., 2008). Pulsed Field Gel Electrophoresis (PFGE) has been a favored method for genotyping V. parahaemolyticus isolates (Marshall et al., 1999) because of its high discrimination index, and has successfully been used in outbreak investigations worldwide. It has also been used to type V. parahaemolyticus isolates from seafood (Wong et al., 1999, 2000; Lu et al., 2000; McLaughlin et al., 2005). As PFGE is based on restriction enzyme digestion of total DNA, one potential drawback is that it may not be sensitive to recent genetic events such as horizontal gene transfer, which may be an important source of variability among isolates. An alternative approach, Multilocus Sequence Typing (MLST), is based on direct sequence analysis of housekeeping genes and a public database has recently been established to archive V. parahaemolyticus sequences (http://pubmlst.org/vparahaemolyticus) (Gonzalez-Escalona et al., 2008). MLST is comparable in cost to PFGE, and provides a different, if not better, details of the genetic relationships among isolates (e.g., evolutionary relationships) (Foxman et al., 2005). However, MLST analyses must be interpreted with caution since it is less sensitive for detecting recent genetic changes in populations that can be detected by PFGE, such as genome inversions, transposons, and plasmid, which in some cases will cause changes in their PFGE profiles.
The main objective of this study was to examine the genetic relationship among trh+ V. parahaemolyticus isolates from Norway by using a combination of MLST and ureR-trh region sequence analysis. Furthermore, the organization of the trh-ureR-nik-ure cluster was was explored in all of the trh+ isolates.
Materials and Methods
Bacterial isolates
A total of 31 V. parahaemolyticus and two V. alginolyticus isolates were included in the present study (Table 1). Twenty-two of these strains were isolated from different regions in Norway (Figure 1) and nine comparator strains were from other regions of the world, including 4 from the United States which have been recently been characterized using MLST (Gonzalez-Escalona et al., 2008). Of the 22 Norwegian strains, 16 were trh+ (13 from environmental sources, 3 from clinical sources). The trh+ environmental strains were from water and blue mussel (Mytilus edulis) samples, collected from four different areas in Norway (Figure 1). The three trh+ clinical isolates were kindly donated by Jørgen Lassen from the Norwegian Institute of Public Health. The 6 trh– strains consisted of 4 environmental strains (3 from blue mussels, 1 from water) and two clinical strains. Norwegian trh– environmental isolates 551 and 580 were also included because they represent a larger PFGE cluster, consisting of trh–V. parahaemolyticus collected from several different locations along the Norwegian coast over several years; the PFGE pattern of these isolates appears very similar to the V. parahaemolyticus trh+ clinical isolate 224 (Ellingsen et al., 2008). Of the nine strains isolated in other countries, 5 were trh– and the other 4 were trh+. Both V. alginolyticus strains, 647va and 751va, were environmental isolates carrying a trh-like gene.
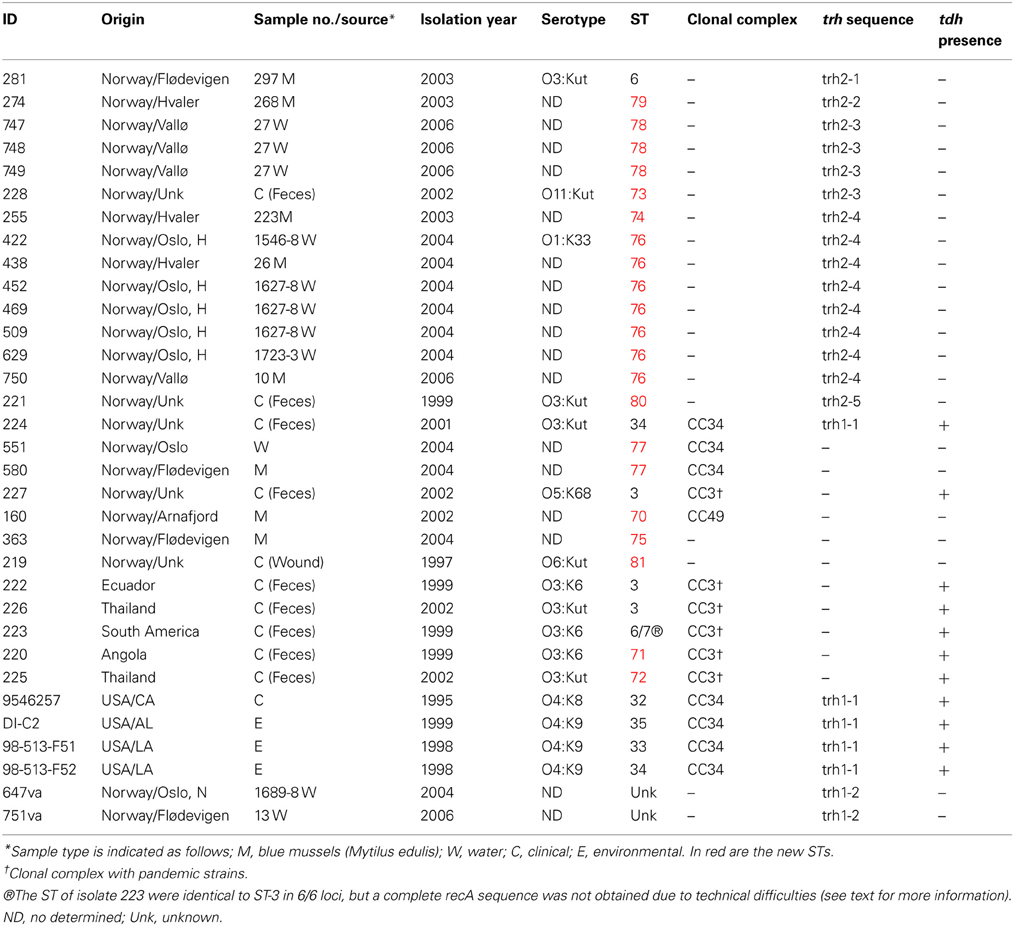
Table 1. Characteristics of the Vibrio spp. strains studied.
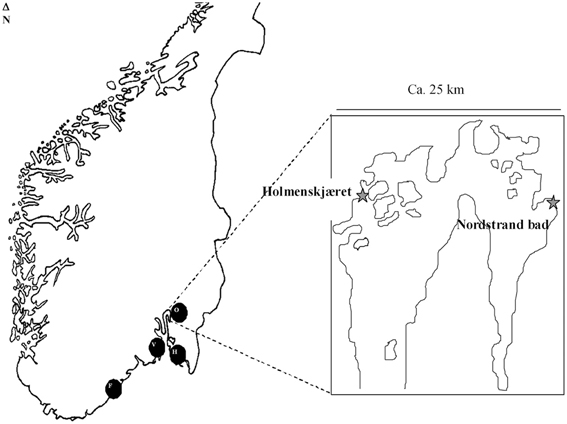
Figure 1. Locations from which the Norwegian trh+ isolates originated (F, Flødevigen; V, Vallø; O, Oslofjord; H, Hvaler). Locations within the Oslo fjord are indicated in the enlargement.
All presumptiveV. parahaemolyticus isolates were analyzed for the presence of oxidase, halophilic characteristics, (growth in presence of: 0, 3, 6, and 10% NaCl) and by the API-20E® Enteric Identification System (BioMerieux, France). The colony morphology was assessed on TCBS agar (Oxoid, Cambridge, UK). In addition, the identity of V. parahaemolyticus isolates were confirmed by V. parahaemolyticus-specific PCR targeting the toxR gene (Bauer and Rorvik, 2007).
DNA preparation
DNA was extracted from one colony of cells suspended in 200 μ l sterile distilled water. The suspension was boiled for 10 min followed by centrifugation at 12,000 × g for 5 min. The supernatant (crude DNA) stored at −20°C until used as template in PCR.
MLST
MLST was performed as described by Gonzalez-Escalona et al. (2008), using a set of PCR primers targeting recA (RecA protein), dnaE (DNA polymerase III, alpha subunit), gyrB (DNA gyrase, subunit B), dtdS (Threonine dehyrogenase), pntA (Transhydrogenase alpha subunit), pyrC (Dihydroorotase), tnaA (Tryptophanase). The PCR amplification and sequencing were performed as described at the V. parahaemolyticus MLST website (http://pubmlst.org/vparahaemolyticus).
Amplification of the trh-ureR-nik-ure region
A set of 12 primer pairs (Table 2) were designed using the Primer2 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and the trh-ureR-nik-ure sequence AB038238.1 (Park et al., 2000) (antisense) as target. The target sequences of these primers were located in regions that spanned contiguous open reading frames (ORFs), as illustrated in Figure 2. Two additional sets of primers were used in order to amplify any partial trh (Honda et al., 1991; Honda and Iida, 1993)and ureR sequences from all trh+ isolates (Table 2). PCR was performed on all trh+ isolates, using the following conditions in a 50 μl reaction solution: 1× buffer (10 mM Tris-Cl pH 8.8, 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton® X-100), 0.24 mM of each nucleotide (dNTP mix), 1.5 U μl−1 Dynazyme II (all from Thermo Scientific, Vantoa, Finland), 20 pmol of the UtoxF primer, 20 pmol of the reverse primer, and 2 μl of sample DNA. The PCR running conditions were: an initial denaturation of 95°C for 4 min, 35 cycles of 94°C for 1 min, 50°C for 1 min and 72°C for 2 min, and a final elongation step of 72°C for 1.5 min in a Bio-Rad Ismart cycler (BioRad, Hercules, CA). Reference V. parahaemolyticus isolate AQ4037 (trh1+) was used as positive control. Reference V. parahaemolyticus isolate 222 (trh–) was used as negative control. All primers were synthesized by Medprobe (Oslo, Norway). All PCR amplicons resulting from V. parahaemolyticus isolate 438 were sequenced by Medprobe in order to confirm specific PCR amplification.
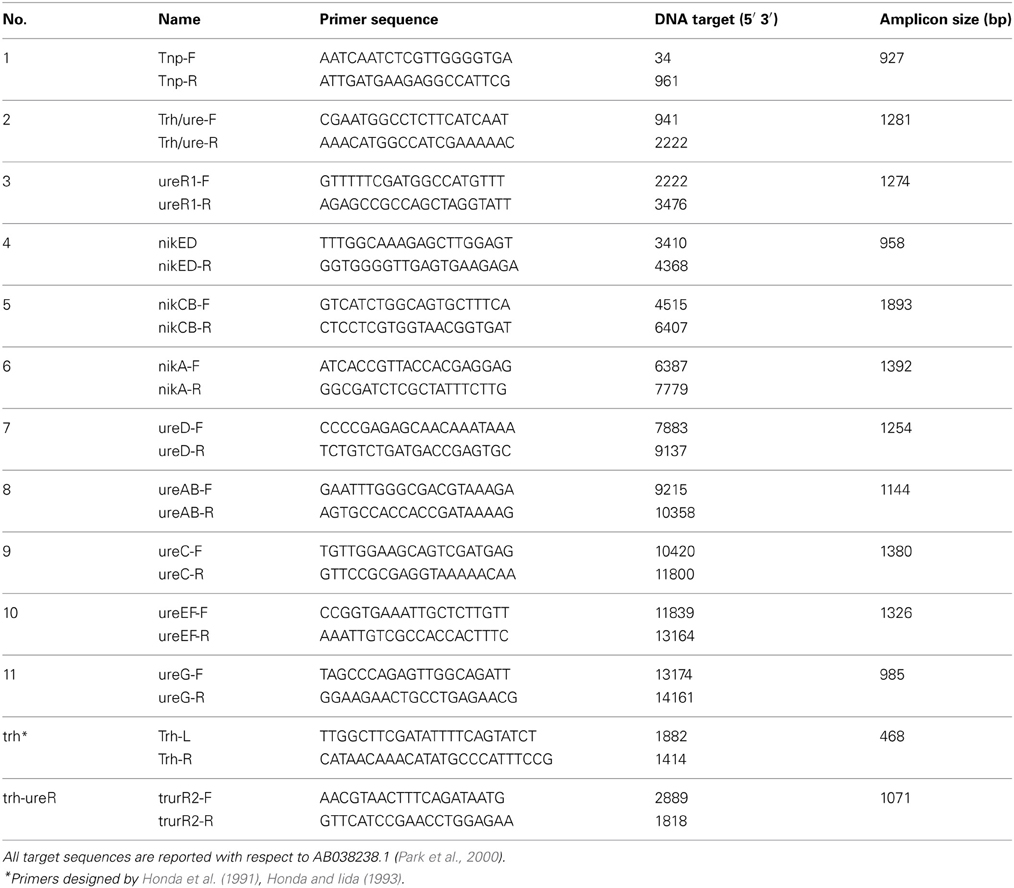
Table 2. Primers employed to explore the organization of the trh-nik-ure region and to sequence the ureR-trh region in the trh+ isolates.
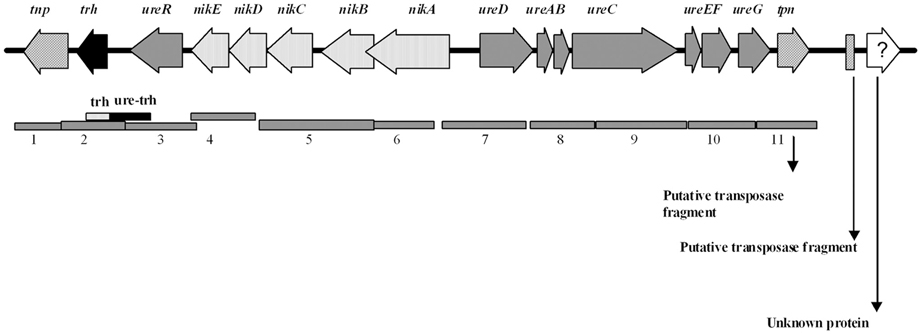
Figure 2. Schematic representation of the trh-ureR-nik-ure gene cluster [adapted from Park et al. (2000)] together with an illustration of the gene products amplified by primer pairs 1–11 designed to assess the genetic organization (gray bars). The fragment of the trh and ureR-trh primers also indicated.
trh and ureR sequence analysis
All partial ureR-trh and trh sequences genes were aligned and manually trimmed to be 1417 and 497 bp in length, respectively, using the Mega 3.1 software (Kumar et al., 2004), starting at position 1436 relative to AB038238.1 (GenBank, http://www.ncbi.nlm.nih.gov). Phylogenetic analysis of those sequences was performed using Mega 3.1 software (Kumar et al., 2004). Minimum evolution (ME) trees for both the ureR-trh sequences and partial trh sequences were constructed using the kimura-2 parameter model to estimate the genetic distances. Additional trh gene sequences available at GenBank database were included in the analysis (Figure 5). The statistical support of the nodes in the ME tree was assessed by 1000 bootstrap re-sampling. All amplified trh and ureR PCR products were sequenced by MedProbe (Oslo, Norway). The new sequences were deposited in GenBank under accession numbers FJ409538–FJ409547.
tdh analysis
The presence of tdh was assessed by colony hybridization (DePaola et al., 2003) and/or by PCR amplification (Bej et al., 1999). The forward and reverse primers were 5′-gttctgatgagatattgtttgttg-′3 and 5′-gttggatatacacattaccaat-′3, respectively. The PCR reactions were performed as described above; except 0.2 mM of each nucleotide (dNTP mix) and 1.0 U μl−1 Dynazyme II were used, with 30 cycles and an annealing temperature of 55°C.
Results and Discussion
The strains of V. parahaemolyticus and V. alginolyticus in this study presented the typical phenotypic characteristics of their type strains, ATCC 17802 and ATCC 17749, respectively. All V. parahaemolyticus strains resulted in positive PCR amplification of the toxR gene, but the two V. alginolyticus isolates (647va and 751va) did not produce any amplicons using the same PCR primers.
MLST
The main objective of the present study was to establish the genetic relationships among trh+ V. parahaemolyticus isolated from Norwegian environmental (mussels and seawater) or clinical sources.
The 22 Norwegian V. parahaemolyticus isolates were differentiated into 15 different sequence types (STs), of which 12 had not previously been reported in the V. parahaemolyticus MLST database (Table 1, Figure 3), indicating a high degree of genotypic diversity among these isolates. This confirms earlier reports of diversity among V. parahaemolyticus found in other geographic locations (Gonzalez-Escalona et al., 2008; Johnson et al., 2009; Rodriguez-Castro et al., 2009). Three of the STs we identified in the Norwegian samples had already been described in the MLST database: ST3, ST6, and ST34. Interestingly, ST3 is believed to be the ancestral type of pandemic CC (Gonzalez-Escalona et al., 2008), ST6 is a singleton that does not appear related to any known CC, and ST34 is believed to be the ancestral type of CC34 (Gonzalez-Escalona et al., 2008). Our analysis also revealed the existence of a new V. parahaemolyticus CC, CC49 (Figure 3, Table 1).
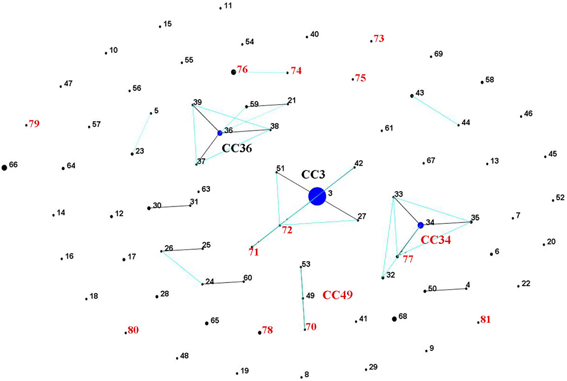
Figure 3. V. parahaemolyticus population “snap-shot” based on the novel STs described in the present study and previously described STs submitted to the MLST database (Gonzalez-Escalona et al., 2008), using eBURST v3 (http://eburst.mlst.net). Four different clusters were observed using stringent criteria (5/7 shared alleles). Isolates from the present study are indicated by red numbers. The clonal complexes CC are indicated (the new one in red CC49), and the predicted ancestral clones are indicated by blue dots. Single locus variants (SLV) are indicated by dark lines, while double locus variants (DLV) are turquoise. The number of isolates in each ST is indicated by different dot sizes.
Of the nine non-Norway isolatesV. parahaemolyticus used as reference strains, four (9546257;DI-C2, 98-513-F51 and 98-513-F52) had been previously analyzed by MLST and belonged to a clonal complex (CC34) (Gonzalez-Escalona et al., 2008) and the other five were ST3, except strain 223. It was difficult to obtain good quality recA sequences for most isolates studied, specifically at both ends of the sequences. In the case of clinical V. parahaemolyticus isolate 223; a complete recA sequence was not obtained. However, the six remaining loci were identical to those of isolates belonging to CC3 (ST3). This suggests that isolate 223 belongs to CC3 and is a single sequence variant (SLV) on recA locus of its ancestral type (ST3). The majority of these CC3 isolates were acquired in Asian or South American countries (Table 1), further emphasizing the worldwide distribution of the pandemic clonal group.
Only 16 of the 22 Norwegian V. parahaemolyticus isolates were carrying the trh gene. These isolates were subdivided into eight different STs. Interestingly, seven of these trh+ V. parahaemolyticus environmental isolates were shown to be identical by MLST (ST-76), even though they originated from different sources, locations and collection years (2004 and 2006). This indicates their persistence in the coastal environs of Norway. One trh+ environmental isolate (281) displayed the same ST (ST6) as an environmental V. parahaemolyticus isolate from Chile (Gonzalez-Escalona et al., 2008). However, the Chilean isolate (PMA 112) was negative for both virulence factors. Further analyses using whole genome sequencing could provide additional evidence about how similar or related these two isolates are.
In contrast to the similarity seen among some environmental strains, each of the three Norwegian trh+ clinical isolates belonged to different STs, two of which were novel (ST-73 and ST-80). The third trh+ clinical isolate (224) belonged to ST-34. This ST was previously observed for an O4:K9 isolate collected from environmental sources in Louisiana in 1998. Intriguingly, ST-34 had been suggested to be the ancestral clone of CC34, which consists of non-pandemic isolates (Gonzalez-Escalona et al., 2008). Isolate 224 may further indicate an association of this cluster with human disease (Gonzalez-Escalona et al., 2008), as well as strengthen the possible role of ST-34 as the ancestral clone of this clonal complex. Clinical isolate 224 (tdh+/trh+) is a single locus variant from two environmental isolates 551 and 580 (tdh–/trh–) belonging to ST-77. The exact origin of 224 is unknown: the patient did not report any stay abroad prior to the infection; however, it is possible the patient ate food that had been re-contaminated during preparation or the patient had consumed imported seafood.
Finding two identical STs in different regions of the world provides evidence that a clonal complex, different from the pandemic clonal complex, is spreading outside its original geographic location. There could be numerous routes for such rapid worldwide dissemination, for transport of contaminated ballast water (McCarthy et al., 1992; McCarthy and Khambaty, 1994), imported food, the motion of oceanic currents, (Martinez-Urtaza et al., 2008a) or human travel activity (Lantagne et al., 2013).
One isolate (novel ST-81) was identified to be trh–/tdh–, and recovered from a wound infection (Table 1). One isolate 227 was ST-3 (“pandemic” clone) and isolated from a clinical case in Norway (Table 1). However, it is unclear whether the pandemic clone has reached the Norwegian environment since there were no epidemiological links to a Norwegian source. Pandemic V. parahaemolyticus had been previously reported in the European environment (Martinez-Urtaza et al., 2005; Quilici et al., 2005; Caburlotto et al., 2008).
MLST was also performed on the two V. alginolyticus sequences using the same conditions as for V. parahaemolyticus. However, the PCR did not result in amplification for five of seven MLST loci. Sequences obtained from gyrB and dtdS genes were not identified in the MLST database (http://pubmlst.org/vparahaemolyticus).
Amplification of the trh-ureR-nik-ure region
The organization of the trh-ureR-nik-ure region previously described in most trh+ isolates (Park et al., 2000) was also explored in Norwegian trh+ V. parahaemolyticus and V. alginolyticus isolates. Amplification of the trh-nik-ure region of these isolates resulted in PCR products of correct length, but with exceptions for six isolates (Figure 4, Table 2). Isolate V. parahaemolyticus 647 Va did not produce any amplicon with primer pair 1 (tnp). Isolates 221, 228, 747, 748, and 749 did not produce any amplicon with primer pair 2 (trh) (Figures 2, 4). Additionally, weak amplicons of the correct size were observed with primer pair 2 and 3 (trh and ureR) for all isolates, except for isolates 224, 647Va, and 751Va for which no amplicons were obtained. Comparing these sequences to those previously submitted to NCBI revealed that the targeted regions of the forward and/or reverse primer of these primer sets (2 and 3) were variable between trh1 and trh2, thus explaining the observation of weak and secondary products of different sizes for the trh2 sequences (data not shown). In conclusion, these primer sets were inadequate for isolates harboring the trh2 gene. We avoided the challenge presented by these two primer pairs by using two additional primer sets (trh and trh-ureR (Honda et al., 1991; Honda and Iida, 1993).
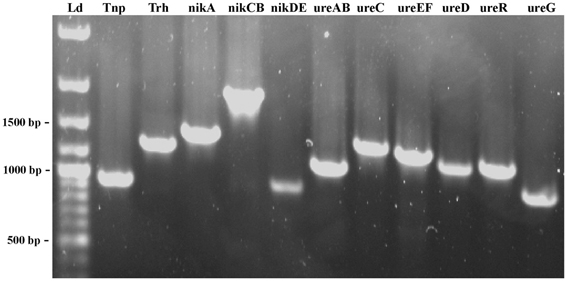
Figure 4. PCR products amplified by each of the primers set designed to determine the organization of the trh-ureR-nik-ure region in each trh+ isolate. The fragments of 500, 1000 and 1500 bp are indicated. The name of each gene target is indicated at the top of each lane. Ld—GeneRuler™ 100 bp Plus DNA Ladder (Fermentas).
Sequences obtained from isolate 438 confirmed that the PCR products were the intended targets (data not shown). The gene organization of the trh-ureR-nik-ure region of all trh+ isolates was shown to be identical to the cluster described by Park et al. (2000). One of the V. alginolyticus isolates (647) was negative for primer pair 1 (tnp). Whether this result is due to the absence of this gene, or if there are mutations in the target sequence(s) is currently unknown. Sequencing the genome of these strains will reveal in which genomic region the trh gene is present, identify the surrounding genes, and also provide a possible explanation for the presence of trh in these isolates.
Partial trh and ureR sequences
Sequence analysis performed on ureR-trh region sequences (1417 bp) obtained from the 22 trh+ isolates (Table 1) showed a total of eight different sequences (Figure 5A). In contrast, an analysis of only the partial trh sequences resulted in seven different sequences (Table 1). This discrepancy was a result of a single nucleotide difference in the ureR-trh intergenic region (position 698) between isolates 747 (ST-78) and 228 (ST-73). Alignment of the partial trh gene sequences of all trh+ isolates with representative trh1 and trh2 sequences retrieved from GenBank revealed that V. parahaemolyticus isolates 224 (ST-34), 9546257 (ST-32), DI-C2 (ST-35), 98-513-F51 (ST-33), and 98-513-F52 (ST-34) and both V. alginolyticus isolates harbored the trh1 sequence type, while the remaining isolates carried trh2 sequence type (Figure 5A). The partial trh sequences of the two V. alginolyticus isolates (647Va and 751Va) were identical. The three trh1 sequences had a four nucleotide insert (GATA) 160 bp upstream of the trh start codon (position 2090, relative to AB038238.1), and a deletion 101 bp upstream. These mutations were in the non-coding region. The trh1 gene is occasionally associated with the presence of tdh (as observed in isolate 224), although the two V. alginolyticus isolates in this study were negative for this gene by PCR. The trh2 type had previously been described in a V. alginolyticus isolated from Alaskan oysters (Gonzalez-Escalona et al., 2006). This is the first report of trh1 in V. alginolyticus. The presence of trh in other Vibrio spp. than V. parahaemolyticus has previously been described, and suggests that environmental Vibrio spp. may be the reservoir for the trh gene (Gonzalez-Escalona et al., 2006; Masini et al., 2007). This is not unique, since another V. parahaemolyticus pathogenicity determinant (tdh gene) was previously identified in a V. alginolyticus isolate (Cai et al., 2007). This re-enforced the reported highly mobile nature of these genes in marine environment among Vibrio.
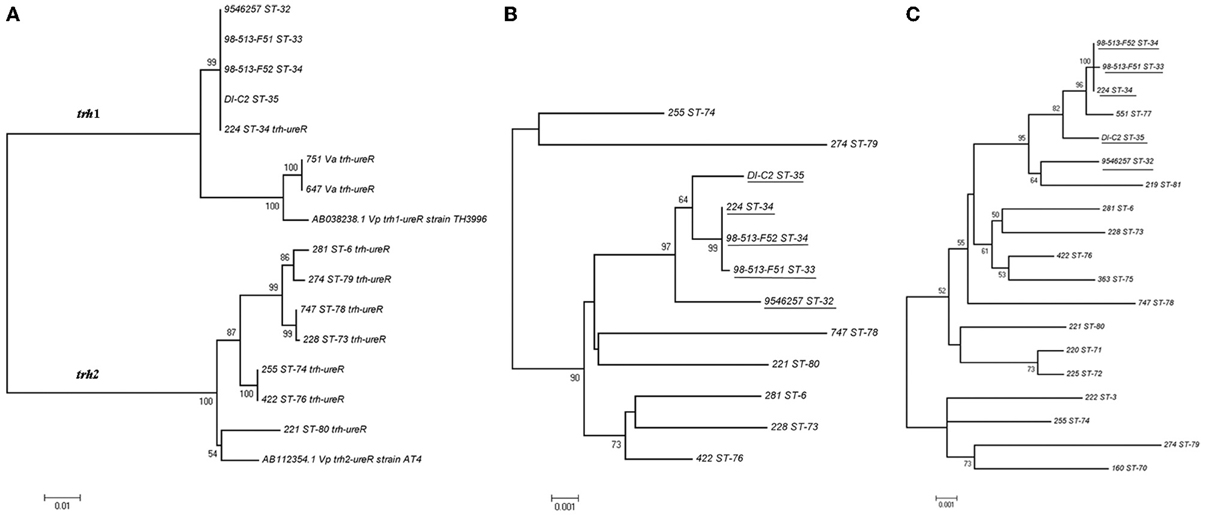
Figure 5. Minimum evolution trees (ME) trees constructed from either the concatenated sequences of the seven loci (MLST) or ureR-trh region of each of the sequence types (STs) obtained in this study. (A) ME tree of the ureR-trh region obtained for the trh+ isolates together with representative sequences of either trh1 or trh2 retrieved from GenBank. (B) ME tree of the concatenated sequences of the seven loci of the trh+ isolates. (C) ME tree of concatenated sequences of all the V. parahaemolyticus isolates from this study. Underlined are the isolates carrying ureR-trh1 region. Scale represents the evolutionary distance and boot-strap values over 50% are shown in the branches.ureR-trh region sequences of V. alginolyticus isolates are indicated in the figure as Va.
Comparison of the minimum evolution (ME) trees generated from the concatenated sequences of the seven genes (MLST) of all trh+ V. parahaemolyticus isolates and their 1417 bp ureR-trh region, revealed different evolutionary stories between the housekeeping genes and this virulence region (Figures 5A,B). As expected, the ureR-trh1 region was identical among the members of the clonal complex 34 (CC34), which all belong to closely related STs (Figure 5B). In the case of the V. alginolyticus isolates, this region was more similar (99%) to the one in V. parahaemolyticus (TH3996) than to the one present in CC34 isolates (96%), indicating that this region of V. alginolyticus was acquired from a different genetic background that the one in CC34 isolates. On the other hand, even though strains 225 and 422 contained identical ureR-trh2 region, they belonged to very distant STs, (ST-74 and ST-76, respectively), both located at distant part of the ME MLST tree.
These results also demonstrate how MLST, in conjunction with ureR-trh cluster analysis, is a powerful technique for revealing evolutionary events inside the V. parahaemolyticus species. It has been suggested that Vibrio spp. are more vulnerable to horizontal gene transfer than the other prokaryotic species (Dryselius et al., 2007), and that frequent recombination events further drive clonal diversification (Gonzalez-Escalona et al., 2008). In this study, the conservation of the ureR-trh1 among isolates belonging to CC34 (Gonzalez-Escalona et al., 2008) suggests that this region was acquired before those isolates started to diverge.
MLST analyses indicate that V. parahaemolyticus isolates 225 (ST-74) and 422 (ST-76), both containing an identical ureR-trh2 region, have diverged in the past (Figure 5B), and suggest that this region was acquired recently. Another possible explanation could be that this region evolved or changed more slowly in comparison with the other housekeeping genes (MLST). However, since MLST analyzes housekeeping genes, presuming a recent acquisition of that region seems a more parsimonious explanation. Whether this ureR-trh2 region was acquired recently by only one or both of these isolates, remains unknown.
Furthermore, analysis of the ME tree generated with the concatenated sequences of the seven genes of all the V. parahaemolyticus isolates in this study, including tdh+/trh– and tdh–/trh– isolates, showed that some isolates were highly related despite their tdh or trh content (Figure 5C). These findings support previous reports that these elements could be laterally acquired (Nishibuchi and Kaper, 1995; Park et al., 2000). An example is strain 551 (ST-77), which, although not carrying either determinant, is nonetheless closely related to isolates belonging to the ancestral type of CC34 (ST-34) which contains ureR-trh1 region (Figure 5C). Isolate 551 is a SLV of isolates belonging to ST-34 and differed by six nucleotide changes on locus pyrC. This amount of changes in this locus, compared to the other six, indicates that these different variants of the locus have arisen by recombination rather than mutation (Gonzalez-Escalona et al., 2008). The lack of ureR-trh1 region in this isolate (551) could be due to a loss of this region or that this isolate did not acquired it. Whole genome sequencing of these strains will provide some insights into the loss/acquisition of the ureR-trh1 region by these isolates.
Geographical distribution of trh+ V. parahaemolyticus STs in norway
The isolation of multiple trh+ V. parahaemolyticus strains from all four sampling sites shows that these types of isolates are prevalent on the coast of Norway (Figure 1, Table 1). MLST analysis showed that most areas have unique V. parahaemolyticus populations, as evidenced by their different STs; exceptions were ST-76 and ST-77, which were isolated from 3 (Hvaler, Vallø, and Oslo) and 2 (Oslo and Flødevigen) of the four areas tested, respectively. The presence of the same ST across those different areas has no easy explanation although this could be due to either ocean currents or interchange of blue mussels among different Norwegian coastal areas.
One area of special interest was Hvaler, where V. parahaemolyticus strains belonged to three different ST with different degrees of similitude. For example ST-79 (trh2-2) and ST-74 (trh2-4) were located closely in the concatenated sequence ME tree (Figure 5B), and carried different trh gene variant. While strain 438 ST-76 (trh2-4), was identical to other strains isolated from Oslo and Vallø, it also carried the same trh gene variant than ST-74. This detail indicates that there are different populations of trh+ V. parahaemolyticus in the same area (Hvaler), carrying the same trh variant, suggesting horizontal gene transfer of this gene among them. Interestingly, the ST-76 strains in Hvaler and Vallø were isolated from blue mussels while the ST-76 strains from Oslo were all isolated from water.
Also, it appears to be a high prevalence of trh2 vs. trh1 gene in environmental V. parahaemolyticus in Norway, but in order to offer a more conclusive answer to this phenomenon more strains from different areas, seasons, and years, should be analyzed. trh2+ was observed in all environmental trh+ V. parahaemolyticus and two of the 3 Norwegian clinical cases. In the other hand, trh1 gene was observed exclusively in the V. alginolyticus analyzed, and in one of the 3 Norwegian clinical cases. trh1+ V. alginolyticus were isolated in two distant regions Flødevigen and Oslo, Nordstrand bad, indicating that there appears to be strains in those regions that act as a reservoir of trh genes in the Norwegian aquatic environment.
Conclusion
Our study presents new information regarding the genetic relationship among trh+ isolates, and introduces a PCR screening method demonstrating that the organization of the trh-ureR-nik-ure gene cluster found in the Norwegian trh+ V. parahaemolyticus isolates is identical to the one previously described for strain TH3996 (Park et al., 2000). Overall, these results showed that the genetic background of trh+ V. parahaemolyticus in Norwegian coasts is highly complex and there appears to be a movement of the same, or highly similar, strains between coastal regions. Additionally, the current study is (to the author's knowledge) the first to indicate a genetic relationship between trh+ and/or tdh+ V. parahaemolyticus and non-pathogenic environmental isolates. These intriguing results warrant closer examination of the trh+ and/or tdh+ isolates [224 (ST-34) and 228 (ST-73)] and their non-pathogenic “relatives” [551 (ST-77) and 281 (ST-6)], which may provide unique information regarding the transfer of genetic elements carrying trh and/or tdh. In conclusion, MLST and ureR-trh sequence analysis generated two different evolutionary trees, suggesting that ureR-trh genes have been acquired or lost at different times by Norwegian V. parahaemolyticus isolates.
Author Contributions
Anette B. Ellingsen, Jaran S. Olsen, Per E. Granum, and Liv M. Rørvik designed parts of the study. Anette B. Ellingsen and Liv M. Rørvik organized the collection of Vibrios. Anette B. Ellingsen, Jaran S. Olsen, and Narjol González-Escalona performed laboratory and phylogentic analyses. Anette B. Ellingsen, Liv M. Rørvik and Narjol González-Escalona analyzed the data obtained from the whole study. Anette B. Ellingsen, Jaran S. Olsen, Per E. Granum, Liv M. Rørvik and Narjol González-Escalona wrote the final manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are deeply grateful to Tone Aarskaug (Norwegian Defense Research Establishment) for technical support and Jon Bohlin for assistance in the phylogenetic analysis of trh and ureR sequence data. The authors would also like to thank Dr. Angelo DePaola for his helpful comments. The authors thank Dr. Lili Fox Vélez for her editorial assistance on this manuscript.
References
Alam, M. J., Tomochika, K. I., Miyoshi, S. I., and Shinoda, S. (2002). Environmental investigation of potentially pathogenic Vibrio parahaemolyticus in the Seto-Inland Sea, Japan. FEMS Microbiol. Lett. 208, 83–87. doi: 10.1111/j.1574-6968.2002.tb11064.x
Ansaruzzaman, M., Lucas, M., Deen, J. L., Bhuiyan, N. A., Wang, X. Y., Safa, A., et al. (2005). Pandemic serovars (O3:K6 and O4:K68) of Vibrio parahaemolyticus associated with diarrhea in mozambique: spread of the pandemic into the African continent. J. Clin. Microbiol. 43, 2559–2562. doi: 10.1128/JCM.43.6.2559-2562.2005
Bauer, A., Ostensvik, O., Florvag, M., Ormen, O., and Rorvik, L. M. (2006). Occurrence of Vibrio parahaemolyticus, V. cholerae, and V. vulnificus in norwegian blue mussels (Mytilus edulis). Appl. Environ. Microbiol. 72, 3058–3061. doi: 10.1128/AEM.72.4.3058-3061.2006
Bauer, A., and Rorvik, L. M. (2007). A novel multiplex PCR for the identification of Vibrio parahaemolyticus, Vibrio cholerae and Vibrio vulnificus. Lett. Appl. Microbiol. 45, 371–375. doi: 10.1111/j.1472-765X.2007.02195.x
Bej, A. K., Patterson, D. P., Brasher, C. W., Vickery, M. C., Jones, D. D., and Kaysner, C. A. (1999). Detection of total and hemolysin-producing Vibrio parahaemolyticus in shellfish using multiplex PCR amplification of tl, tdh and trh. J. Microbiol. Methods 36, 215–225. doi: 10.1016/S0167-7012(99)00037-8
Caburlotto, G., Ghidini, V., Gennari, M., Tafi, M. C., and Lleo, M. M. (2008). Isolation of a Vibrio parahaemolyticus pandemic strain from a marine water sample obtained in the northern adriatic. Euro. Surveill. 13, 1–3.
Cai, S. H., Wu, Z. H., Jian, J. C., and Lu, Y. S. (2007). Cloning and expression of gene encoding the thermostable direct hemolysin from Vibrio alginolyticus strain HY9901, the causative agent of vibriosis of crimson snapper (Lutjanus erythopterus). J. Appl. Microbiol. 103, 289–296. doi: 10.1111/j.1365-2672.2006.03250.x
Chowdhury, A., Ishibashi, M., Thiem, V. D., Tuyet, D. T., Tung, T. V., Chien, B. T., et al. (2004a). Emergence and serovar transition of Vibrio parahaemolyticus pandemic strains isolated during a diarrhea outbreak in vietnam between 1997 and 1999. Microbiol. Immunol. 48, 319–327. doi: 10.1111/j.1348-0421.2004.tb03513.x
Chowdhury, N. R., Stine, O. C., Morris, J. G., and Nair, G. B. (2004b). Assessment of evolution of pandemic Vibrio parahaemolyticus by multilocus sequence typing. J. Clin. Microbiol. 42, 1280–1282. doi: 10.1128/JCM.42.3.1280-1282.2004
Chowdhury, N. R., Chakraborty, S., Ramamurthy, T., Nishibuchi, M., Yamasaki, S., Takeda, Y., et al. (2000). Molecular evidence of clonal Vibrio parahaemolyticus pandemic strains. Emerg. Infect. Dis. 6, 631–636. doi: 10.3201/eid0606.000612
Cook, D. W., Bowers, J. C., and DePaola, A. (2002). Density of total and pathogenic (tdh+) Vibrio parahaemolyticus in Atlantic and Gulf coast molluscan shellfish at harvest. J. Food Prot. 65, 1873–1880.
DePaola, A., Nordstrom, J. L., Bowers, J. C., Wells, J. G., and Cook, D. W. (2003). Seasonal abundance of total and pathogenic Vibrio parahaemolyticus in Alabama oysters. Appl. Environ. Microbiol. 69, 1521–1526. doi: 10.1128/AEM.69.3.1521-1526.2003
Dryselius, R., Kurokawa, K., and Iida, T. (2007). Vibrionaceae, a versatile bacterial family with evolutionarily conserved variability. Res. Microbiol. 158, 479–486. doi: 10.1016/j.resmic.2007.04.007
Ellingsen, A. B., Jorgensen, H., Wagley, S., Monshaugen, M., and Rorvik, L. M. (2008). Genetic diversity among norwegian Vibrio parahaemolyticus. J. Appl. Microbiol. 105, 2195–2202. doi: 10.1111/j.1365-2672.2008.03964.x
Foxman, B., Zhang, L., Koopman, J. S., Manning, S. D., and Marrs, C. F. (2005). Choosing an appropriate bacterial typing technique for epidemiologic studies. Epidemiol. Perspect. Innov. 2, 10. doi: 10.1186/1742-5573-2-10
Fuenzalida, L., Hernandez, C., Toro, J., Rioseco, M. L., Romero, J., and Espejo, R. T. (2006). Vibrio parahaemolyticus in shellfish and clinical samples during two large epidemics of diarrhoea in southern Chile. Environ. Microbiol. 8, 675–683. doi: 10.1111/j.1462-2920.2005.00946.x
Gonzalez-Escalona, N., Blackstone, G. M., and DePaola, A. (2006). Characterization of a Vibrio alginolyticus strain, isolated from Alaskan oysters, carrying a hemolysin gene similar to the thermostable direct hemolysin-related hemolysin gene (trh) of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 72, 7925–7929. doi: 10.1128/AEM.01548-06
Gonzalez-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L. A., and DePaola, A. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
Hervio-Heath, D., Colwell, R. R., Derrien, A., Robert-Pillot, A., Fournier, J. M., and Pommepuy, M. (2002). Occurrence of pathogenic vibrios in coastal areas of France. J. Appl. Microbiol. 92, 1123–1135. doi: 10.1046/j.1365-2672.2002.01663.x
Honda, T., Abad-Lapuebla, M. A., Ni, Y. X., Yamamoto, K., and Miwatani, T. (1991). Characterization of a new thermostable direct haemolysin produced by a Kanagawa-phenomenon-negative clinical isolate of Vibrio parahaemolyticus. J. Gen. Microbiol. 137, 253–259. doi: 10.1099/00221287-137-2-253
Honda, T., and Iida, T. (1993). The pathogenicity of Vibrio parahaemolyticus and the role of the thermostable direct haemolysin and related haemolysins. Rev. Med. Microbiol. 4, 106–113. doi: 10.1097/00013542-199304000-00006
Hurley, C. C., Quirke, A., Reen, F. J., and Boyd, E. F. (2006). Four genomic islands that mark post-1995 pandemic Vibrio parahaemolyticus isolates. BMC Genomics 7:104. doi: 10.1186/1471-2164-7-104
Iida, T., and Honda, T. (2006). “Vibrio parahaemolyticus,” in The Biology of Vibrios, eds F. L. Thompson, B. Austin, and J., Swings (Washington, DC: ASM Press), 340–348
Iida, T., Park, K. S., Suthienkul, O., Kozawa, J., Yamaichi, Y., Yamamoto, K., et al. (1998). Close proximity of the tdh, trh and ure genes on the chromosome of Vibrio parahaemolyticus. Microbiology 144(Pt 9), 2517–2523.
Izutsu, K., Kurokawa, K., Tashiro, K., Kuhara, S., Hayashi, T., Honda, T., et al. (2008). Comparative genomic analysis using microarray demonstrates a strong correlation between the presence of the 80-kilobase pathogenicity island and pathogenicity in Kanagawa phenomenon-positive Vibrio parahaemolyticus strains. Infect. Immun. 76, 1016–1023. doi: 10.1128/IAI.01535-07
Johnson, C. N., Flowers, A. R., Young, V. C., Gonzalez-Escalona, N., DePaola, A., Noriea, N. F., et al. (2009). Genetic relatedness among tdh+ and trh+ Vibrio parahaemolyticus cultured from Gulf of Mexico oysters (Crassostrea virginica) and surrounding water and sediment. Microb. Ecol. 57, 437–443. doi: 10.1007/s00248-008-9418-3
Kaysner, C. A., Abeyta, C. Jr. Stott, R. F., Lilja, J. L., and Wekell, M. M. (1990). Incidence of urea-hydrolyzing Vibrio parahaemolyticus in Willapa Bay, Washington. Appl. Environ. Microbiol. 56, 904–907.
Kaysner, C. A., Abeyta, C. Jr. Trost, P. A., Wetherington, J. H., Jinneman, K. C., Hill, W. E., et al. (1994). Urea hydrolysis can predict the potential pathogenicity of Vibrio parahaemolyticus strains isolated in the Pacific Northwest. Appl. Environ. Microbiol. 60, 3020–3022.
Kishishita, M., Matsuoka, N., Kumagai, K., Yamasaki, S., Takeda, Y., and Nishibuchi, M. (1992). Sequence variation in the thermostable direct hemolysin-related hemolysin (trh) gene of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 58, 2449–2457.
Kumar, S., Tamura, K., and Nei, M. (2004). MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5, 150–163. doi: 10.1093/bib/5.2.150
Lantagne, D., Balakrish, N. G., Lanata, C. F., and Cravioto, A. (2013). The cholera outbreak in haiti: where and how did it begin? Curr. Top. Microbiol. Immunol. doi: 10.1007/82_2013_331. [Epub ahead of print].
Lu, P. L., Chang, S. C., Pan, H. J., Chen, M. L., and Luh, K. T. (2000). Application of pulsed-field gel electrophoresis to the investigation of a nosocomial outbreak of Vibrio parahaemolyticus. J. Microbiol. Immunol. Infect. 33, 29–33.
Makino, K., Oshima, K., Kurokawa, K., Yokoyama, K., Uda, T., Tagomori, K., et al. (2003). Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361, 743–749. doi: 10.1016/S0140-6736(03)12659-1
Marshall, S., Clark, C. G., Wang, G., Mulvey, M., Kelly, M. T., and Johnson, W. M. (1999). Comparison of molecular methods for typing Vibrio parahaemolyticus. J. Clin. Microbiol. 37, 2473–2478.
Martinez-Urtaza, J., Huapaya, B., Gavilan, R. G., Blanco-Abad, V., Ansede-Bermejo, J., Cadarso-Suarez, C., et al. (2008a). Emergence of Asiatic Vibrio diseases in South America in phase with El Nino. Epidemiology 19, 829–837. doi: 10.1097/EDE.0b013e3181883d43
Martinez-Urtaza, J., Lozano-Leon, A., Varela-Pet, J., Trinanes, J., Pazos, Y., and Garcia-Martin, O. (2008b). Environmental determinants of the occurrence and distribution of Vibrio parahaemolyticus in the rias of Galicia, Spain. Appl. Environ. Microbiol. 74, 265–274. doi: 10.1128/AEM.01307-07
Martinez-Urtaza, J., Lozano-Leon, A., DePaola, A., Ishibashi, M., Shimada, K., Nishibuchi, M., et al. (2004). Characterization of pathogenic Vibrio parahaemolyticus isolates from clinical sources in Spain and comparison with Asian and North American pandemic isolates. J. Clin. Microbiol. 42, 4672–4678. doi: 10.1128/JCM.42.10.4672-4678.2004
Martinez-Urtaza, J., Simental, L., Velasco, D., DePaola, A., Ishibashi, M., Nakaguchi, Y., et al. (2005). Pandemic Vibrio parahalemolyticus O3: K6, Europe. Emerg. Infect. Dis. 11, 1319–1320. doi: 10.3201/eid1108.050322
Masini, L., De Grandis, G., Principi, F., Mengarelli, C., and Ottaviani, D. (2007). Research and characterization of pathogenic vibrios from bathing water along the Conero Riviera (Central Italy). Water Res. 41, 4031–4040. doi: 10.1016/j.watres.2007.05.003
Matsumoto, C., Okuda, J., Ishibashi, M., Iwanaga, M., Garg, P., Rammamurthy, T., et al. (2000). Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J. Clin. Microbiol. 38, 578–585.
McCarthy, S. A., and Khambaty, F. M. (1994). International dissemination of epidemic Vibrio cholerae by cargo ship ballast and other nonpotable waters. Appl. Environ. Microbiol. 60, 2597–2601.
McCarthy, S. A., McPhearson, R. M., Guarino, A. M., and Gaines, J. L. (1992). Toxigenic Vibrio cholerae O1 and cargo ships entering Gulf of Mexico. Lancet 339, 624–625. doi: 10.1016/0140-6736(92)90918-S
McLaughlin, J. B., DePaola, A., Bopp, C. A., Martinek, K. A., Napolilli, N. P., Allison, C. G., et al. (2005). Outbreak of Vibrio parahaemolyticus gastroenteritis associated with Alaskan oysters. N. Engl. J. Med. 353, 1463–1470. doi: 10.1056/NEJMoa051594
Nair, G. B., Ramamurthy, T., Bhattacharya, S. K., Dutta, B., Takeda, Y., and Sack, D. A. (2007). Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin. Microbiol. Rev. 20, 39–48. doi: 10.1128/CMR.00025-06
Nishibuchi, M., and Kaper, J. B. (1995). Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect. Immun. 63, 2093–2099.
Nishibuchi, M., Taniguchi, T., Misawa, T., Khaeomanee-Iam, V., Honda, T., and Miwatani, T. (1989). Cloning and nucleotide sequence of the gene (trh) encoding the hemolysin related to the thermostable direct hemolysin of Vibrio parahaemolyticus. Infect. Immun. 57, 2691–2697.
Okura, M., Osawa, R., Iguchi, A., Arakawa, E., Terajima, J., and Watanabe, H. (2003). Genotypic analyses of Vibrio parahaemolyticus and development of a pandemic group-specific multiplex PCR assay. J. Clin. Microbiol. 41, 4676–4682. doi: 10.1128/JCM.41.10.4676-4682.2003
Okura, M., Osawa, R., Tokunaga, A., Morita, M., Arakawa, E., and Watanabe, H. (2008). Genetic analyses of the putative O and K antigen gene clusters of pandemic Vibrio parahaemolyticus. Microbiol. Immunol. 52, 251–264. doi: 10.1111/j.1348-0421.2008.00027.x
Park, K. S., Iida, T., Yamaichi, Y., Oyagi, T., Yamamoto, K., and Honda, T. (2000). Genetic characterization of DNA region containing the trh and ure genes of Vibrio parahaemolyticus. Infect.Immun. 68, 5742–5748. doi: 10.1128/IAI.68.10.5742-5748.2000
Quilici, M. L., Robert-Pillot, A., Picart, J., and Fournier, J. M. (2005). Pandemic Vibrio parahaemolyticus O3:K6 spread, France. Emerg. Infect. Dis. 11, 1148–1149. doi: 10.3201/eid1107.041008
Rodriguez-Castro, A., Ansede-Bermejo, J., Blanco-Abad, V., Varela-Pet, J., Garcia-Martin, O., and Martinez-Urtaza, J. (2009). Prevalence and genetic diversity of pathogenic populations of Vibrio parahaemolyticus in coastal waters of Galicia, Spain. Environ. Microbiol. Rep. 2, 58–66. doi: 10.1111/j.1758-2229.2009.00064.x
Su, Y. C., and Liu, C. (2007). Vibrio parahaemolyticus: a concern of seafood safety. Food Microbiol. 24, 549–558. doi: 10.1016/j.fm.2007.01.005
Wong, H.-C., Chen, M.-C., Liu, S.-H., and Liu, D.-P. (1999). Incidence of highly genetically diversified Vibrio parahaemolyticus in seafood imported from Asian countries. Int. J. Food Microbiol. 52, 181–188. doi: 10.1016/S0168-1605(99)00143-9
Wong, H. C., Liu, S. H., Ku, L. W., Lee, I. Y., Wang, T. K., Lee, Y. S., et al. (2000). Characterization of Vibrio parahaemolyticus isolates obtained from foodborne illness outbreaks during 1992 through 1995 in taiwan. J. Food Prot. 63, 900–906.
Xu, M., Iida, T., Yamamoto, K., Takarada, Y., Miwatani, T., and Honda, T. (1994). Demonstration and characterization of simultaneous production of a thermostable direct hemolysin (TDH/I) and a TDH-related hemolysin (TRHx) by a clinically isolated Vibrio parahaemolyticus strain, TH3766. Infect. Immun. 62, 166–171.
Keywords: tdh, trh, V. parahaemolyticus, V. alginolyticus, MLST, urease, Vibrio, PCR
Citation: Ellingsen AB, Olsen JS, Granum PE, Rørvik LM and González-Escalona N (2013) Genetic characterization of trh positive Vibrio spp. isolated from Norway. Front. Cell. Infect. Microbiol. 3:107. doi: 10.3389/fcimb.2013.00107
Received: 27 August 2013; Paper pending published: 15 September 2013;
Accepted: 11 December 2013; Published online: 25 December 2013.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Maria M. Lleo, Università di Verona, ItalyJaime Martinez-Urtaza, European Centre for Disease Prevention and Control, Sweden
Copyright © 2013 Ellingsen, Olsen, Granum, Rørvik and González-Escalona. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Narjol González-Escalona, Center for Food Safety and Applied Nutrition, Food and Drug Administration, 5100 Paint Branch Parkway HFS-712, College Park, MD 20740, USA e-mail:bmFyam9sLmdvbnphbGV6LWVzY2Fsb25hQGZkYS5oaHMuZ292