 Olivier Dussurget
Olivier Dussurget Hélène Bierne
Hélène Bierne Pascale Cossart1,2,3
Pascale Cossart1,2,3- 1Unité des Interactions Bactéries-Cellules, Institut Pasteur, Paris, France
- 2Inserm, U604, Paris, France
- 3INRA, USC2020, Paris, France
- 4University of Paris Diderot, Sorbonne Paris Cité, Paris, France
Interferons (IFNs) are secreted proteins of the cytokine family that regulate innate and adaptive immune responses to infection. Although the importance of IFNs in the antiviral response has long been appreciated, their role in bacterial infections is more complex and is currently a major focus of investigation. This review summarizes our current knowledge of the role of these cytokines in host defense against the bacterial pathogen Listeria monocytogenes and highlights recent discoveries on the molecular mechanisms evolved by this intracellular bacterium to subvert IFN responses.
Introduction
Listeria monocytogenes is a pathogenic Gram-positive bacillus responsible for a foodborne disease in humans and animals called listeriosis (Vázquez-Boland et al., 2001). This highly versatile bacterium can be isolated from multiple sources such as human and animal feces, soil, water, plants and food. As a common contaminant of fruits, vegetables, seafood, meat and cheese, it represents a major economic problem for the food industry. Infection usually originates from ingestion of contaminated food (Schlech et al., 1983) and may cause febrile gastroenteritis in otherwise healthy persons (Ooi and Lorber, 2005). In contrast, in immunocompromised individuals it leads to a severe invasive disease, which manifests itself as septicemia, meningitis and encephalitis. In the specific case of pregnant women, infection may cause fetal loss or neonatal bacteremia and meningitis. In the United States, incidence of listeriosis ranged from 2.5 to 3.2 cases per million population between 2004 and 2009 (Cartwright et al., 2013). In France, incidence was 3.9 per million between 2001 and 2008, and increased risk of listeriosis was noticed in people with underlying diseases, such as chronic lymphocytic leukemia (Goulet et al., 2012). While relatively rare, listeriosis is among the most deadly foodborne diseases with mortality rates reaching up to 50% depending on the clinical manifestations (Lorber, 1997). In addition to the immunological status of the patient, the clinical outcome of the disease depends on the pathogenic potential of the infecting bacteria. L. monocytogenes strains of serovars 1/2a, 1/2b, and 4b account for 95% of human cases and serovar 4b alone is associated to most outbreaks (Swaminathan and Gernersmidt, 2007).
The capacity of L. monocytogenes to survive and multiply within the gastrointestinal tract is critical for the initial infection, persistence and transmission. L. monocytogenes is well adapted to this environment and produces multiple factors to compete with microbiota and counteract antimicrobial peptides, acidity, hyperosmolarity, hypoxia, bile and iron deprivation (Gahan and Hill, 2005). Crossing of the intestinal epithelium is thought to occur by invasion of enterocytes, in particular goblet cells and M cells of the Peyer's patches. Invasion of enterocytes requires the specific interaction between the Listeria surface protein InlA and its cellular receptor E-cadherin (Lecuit et al., 2001; Disson et al., 2008), which can take place at sites of cell extrusion at the tip and other locations of intestinal villi (Pentecost et al., 2006; Nikitas et al., 2011). Indeed, as recently shown, upon interaction with E-cadherin, Listeria preferentially crosses the intestinal barrier by transcytosis through goblet cells (Nikitas et al., 2011). Entry through ileal Peyer's patches via M cells does not rely on InlA. It has been reported to require Listeria invasion protein InlB (Chiba et al., 2011). After translocation, bacteria reach lymph nodes, the liver and spleen and finally secondary target sites of infection, including the central nervous system and the placenta.
A remarkable feature of L. monocytogenes is its capacity to invade non-professional phagocytic cells such as enterocytes, hepatocytes and trophoblast cells. The exceptional repertoire of virulence factors necessary for entry, survival and multiplication has been extensively studied (Camejo et al., 2011; Cossart, 2011). Expression of many virulence genes relies on the transcriptional activator PrfA, whose role is pivotal for L. monocytogenes transition from saprophytic to intracellular lifestyle (Freitag et al., 2009; Toledo-Arana et al., 2009).
Elimination of L. monocytogenes is mostly based on the capacity of the host to mount an efficient cellular immune response to infection (Mackaness, 1962; Shi and Pamer, 2011). In particular, the fate of infection depends on the level of macrophage activation and on Listeria ability to counteract bactericidal mechanisms of host cells (Shaughnessy and Swanson, 2007; Corr and O'Neill, 2009; Stavru et al., 2011). Bacterial escape from the phagosome and avoidance of autophagy for intracytosolic replication and cell-cell spread have been well characterized. They have been shown to depend on five major virulence factors: the secreted pore-forming toxin listeriolysin O (LLO) and two phospholipases C (PlcA and PlcB) for vacuolar escape, the surface protein ActA for actin-based motility and both ActA and a surface protein of the internalin family, InlK, for autophagy evasion (Cossart, 2011; Dortet et al., 2012). Other strategies of immune escape that lead to the modulation of cytokine expression occur through a variety of mechanisms. Modifications of L. monocytogenes peptidoglycan by the N-deacetylase PgdA and the O-acetyl transferase OatA prevent lysozyme-dependent release of microbe-associated molecular patterns (MAMPs), activation of pathogen-recognition receptors (PRRs) and subsequent production of pro-inflammatory cytokines (Boneca et al., 2007; Aubry et al., 2011; Rae et al., 2011). The toxin LLO induces dephosphorylation of histone H3 and deacetylation of histone H4, which correlate with decreased expression of pro-inflammatory genes, such as the chemokine gene cxcl2 (Hamon et al., 2007). The secreted internalin InlC inhibits inflammation by interacting with IKK-α, a component of the IκB-kinase complex, which is essential for NF-κB activation and expression of pro-inflammatory genes (Gouin et al., 2010). Other evasion mechanisms remain to be characterized, such as the control of the expression of IL-6 by the surface internalin InlH (Personnic et al., 2010). L. monocytogenes also has the capacity to modulate interferon (IFN) production during infection. Type I IFN production by infected cells can be controlled by Listeria multidrug efflux pumps MdrM and MdrT, via the secretion of the second messenger cyclic-di-AMP (Crimmins et al., 2008; Woodward et al., 2010; Schwartz et al., 2012). Synthesis of type III IFN has also recently been shown to be tuned by Listeria nucleomodulin LntA (Lebreton et al., 2011). Our knowledge concerning the role of the IFN cytokine family during listeriosis has rapidly expanded in the last few years and will be the focus of this review.
Interferons
The IFNs Family
IFNs form a family of proteins secreted by many cell types in response to infection. They were originally named for their capacity to interfere with viral proliferation (Isaacs and Lindemann, 1957). This diverse family is composed of three groups of cytokines, namely type I-, type II-, and type III-IFNs, which are important components of innate immune responses (Table 1). Type I-IFNs consist of IFN-α, IFN-β, IFN-δ, IFN-ε, IFN-ζ, IFN-κ, IFN-ν, IFN-τ, and IFN-ω (Levy et al., 2011). Type II-IFN is composed of a single cytokine, IFN-γ (Pestka et al., 2004). Type III-IFNs are IFN-λ 1, IFN-λ 2, and IFN-λ 3 (formerly IL-29, IL-28A, and IL-28B) and IFN-λ 4 (Kotenko, 2011; Prokunina-Olsson et al., 2013). Type I- and type III-IFNs have similar signal transduction systems (see below) and are phylogenetically closer from each other than type II-IFN (Pestka et al., 2004).
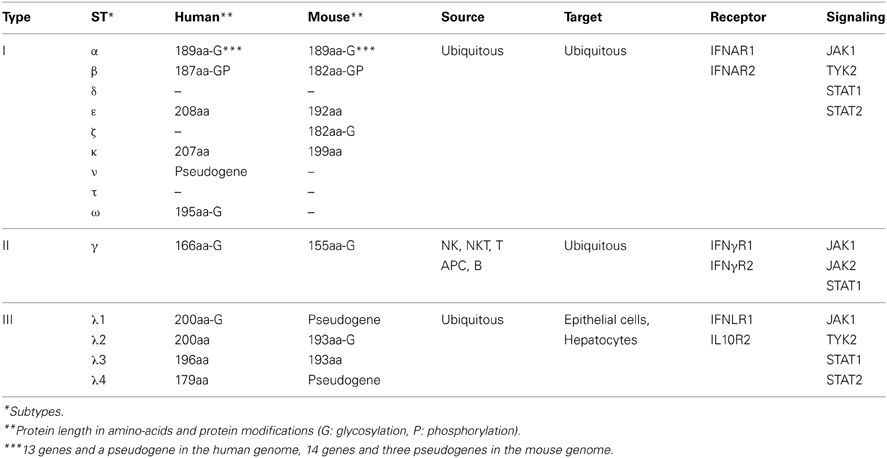
Table 1. The interferon family.
Sequence conservation and chromosome location suggest that type I-IFN genes evolved from a single ancestor through duplication. However, the extent of type I-IFN gene diversification varies greatly depending on the species (Pestka et al., 2004). Generally, a single gene encodes the type I-IFN in fish. In contrast, multiple gene duplications and diversification led to the emergence of sub-types of type I-IFNs in mammals (Table 1). Gene duplication varies also within each sub-type. A single IFN-β gene is found in the human and mouse genomes (Decker et al., 2005; Honda et al., 2006; Durbin et al., 2013). In contrast 13 IFN-α genes and one pseudogene and 14 IFN-α genes and three pseudogenes are found in the human and mouse genomes, respectively (van Pesch et al., 2004; Durbin et al., 2013). A single gene encodes type II-IFN and four genes encode type III-IFNs in human (Decker et al., 2005; Levy et al., 2011). Of note, IFN-λ1 and IFN-λ4 are pseudogenes in mice, which prevents the study of these cytokines in this animal model (Table 1) (Fox et al., 2009).
IFN Activities: General Overview
IFNs are important components of the immune system, which generally trigger cellular protective defenses in response to infection or tumor formation. Type-II IFN (IFN-γ) is a paradigm for this, being an important mediator of innate and adaptive immune responses with a key role in clearance of viral and bacterial pathogens and in tumor control. IFN-γ was first described as an antiviral protein (Wheelock and Sibley, 1965), but is now known to exhibit broader biological activities, non-redundant with that of other types of IFNs. The crucial role of IFN-γ in immunity to infection is reflected by the phenotype of mice lacking the IFN-γ receptor or the IFN-γ gene, which are highly susceptible to Mycobacterium bovis BCG infection (Dalton et al., 1993; Kamijo et al., 1993). Genetic deficiencies resulting in the loss of IFN-γ production or signaling in mice lead to increased susceptibility to infections by other intracellular pathogens, such as L. monocytogenes (see below), Salmonella typhimurium and some viruses (Huang et al., 1993; Harty and Bevan, 1995; Jouanguy et al., 1999). These defects also lead to the loss of tumor control (Kaplan et al., 1998). Patients with deficiencies in the IFN-γ pathway, for instance by mutation in the gene for the IFN-γ receptor 1, are characterized by severe infections with viruses and intracellular bacteria including L. monocytogenes, Salmonella sp. and mycobacteria (Jouanguy et al., 1996; Newport et al., 1996; Roesler et al., 1999; van de Vosse et al., 2009). IFN-γ mediates macrophage activation, i.e., increased phagocytosis and production of pro-inflammatory cytokines, of microbicidal reactive oxygen and nitrogen species, leading to clearance of intracellular pathogens (Schoenborn and Wilson, 2007). In addition, IFN-γ controls differentiation of T cells in Th1 effector cells, antigen processing and presentation by antigen-presenting cells, which participate to cellular immunity against intracellular pathogens (Schroder, 2003; Hu and Ivashkiv, 2009). The immunostimulatory and immunomodulatory properties of IFN-γ have therapeutic implications. Indeed, IFN-γ is used in patients with chronic granulomatous disease to reduce infection and mortality, although the clinical benefit has not been demonstrated in all studies (Holland, 2010).
Type I-IFNs are produced in responses to viruses, many bacteria and parasites. However, in contrast to type II IFNs, these cytokines are not always protective against bacterial infections. Indeed, the role of type I-IFNs in response to bacterial infection is complex and depends on the microorganism (Decker et al., 2005; Monroe et al., 2010; Carrero, 2013). They contribute to resistance of the host against infection by extracellular bacteria, such as Escherichia coli, Helicobacter pylori, Streptococcus agalactiae and S. pneumoniae (Mancuso et al., 2007; Watanabe et al., 2010). In contrast, they are associated with suppression of innate immune responses and increased susceptibility of the host to infection by L. monocytogenes (see below), Brucella abortus, Chlamydia muridarum, Francisella novicida, Salmonella enterica, Staphylococcus aureus, and Yersinia pestis (Auerbuch et al., 2004; Carrero et al., 2004; O'Connell et al., 2004; Qiu et al., 2008; Martin et al., 2009; Henry et al., 2010; de Almeida et al., 2011; Patel et al., 2012; Robinson et al., 2012; Archer et al., 2014). These different effects on infection are likely linked to the wide range of cellular responses induced by their downstream effectors, the products of IFN-stimulated genes (ISGs) (Schoggins et al., 2011). Although Type I-IFNs have long been known to induce antiviral response in the infected host (Isaacs and Lindemann, 1957), they can also induce apoptosis, autophagy, differentiation and migration, inhibit proliferation as well as angiogenesis and mediate cellular damage, inflammation or autoimmunity (Trinchieri, 2010). As a result, type I-IFNs have a therapeutic potential that can be used to treat tumors and viral infections (Pestka, 2007; Heim, 2013; Wilson and Brooks, 2013), while being detrimental for the host in response to a subset of pathogens.
Type III-IFNs have been discovered in 2003 (Kotenko et al., 2003; Sheppard et al., 2003) and their activities have been less extensively characterized than those of type I- and type II-IFNs. However, several studies suggest that type I and type III-IFNs share common biological activities (Levy et al., 2011; Zheng et al., 2013). Although type III-IFNs respond to different stimuli, use different receptors and are not always expressed by the same cells as type I IFNs (see below), engagement of type I- and type III-IFN receptors leads to similar transcriptional responses. Like type I-IFNs, type III-IFNs have been involved in antiproliferative and antiviral responses (Iversen and Paludan, 2010; Mordstein et al., 2010; Durbin et al., 2013; Hamming et al., 2013). Recently, type III-IFNs have been shown to be induced in response to bacterial pathogens, but their downstream effects are not yet characterized (Pietilä et al., 2010; Lebreton et al., 2011; Bierne et al., 2012).
Activation of IFNs
Transcription of IFN genes is induced rapidly in response to microbial infection. Type I-IFNs can be produced by all cells, while type III-IFNs are secreted by specific cell types, including dendritic and epithelial cells. Type I- and type III-IFNs activation is initiated by detection of MAMPs by PRRs such as endosomal transmembrane Toll-like receptors and cytosolic receptors (Stetson and Medzhitov, 2006; Monroe et al., 2010). Upon recognition of MAMPs, PRRs trigger diverse signaling pathways that involve adaptor proteins and cytosolic or organelle-bound protein scaffolds activating kinases converging to phosphorylation of transcription factors and their subsequent translocation into the nucleus (Figure 1). IRF1, IRF3, IRF4, IRF5, IRF7, and IRF8 are important for transcription of the IFN-α genes, with IRF7 considered as the master regulator of IFN-α response (Honda et al., 2005; Tailor et al., 2007; Levy et al., 2011). Regulation of the IFN-β gene is more complex. Activated IRF3, IRF7, AP-1, and NF-κ B bind to the enhancer/promoter regions of the IFN-β gene and participate to the formation of the enhanceosome, which alters chromatin structure and allows transcription (Panne et al., 2007; Panne, 2008). In contrast, IRFs and NF-κ B independently activate transcription of type III-IFN genes (Iversen and Paludan, 2010).
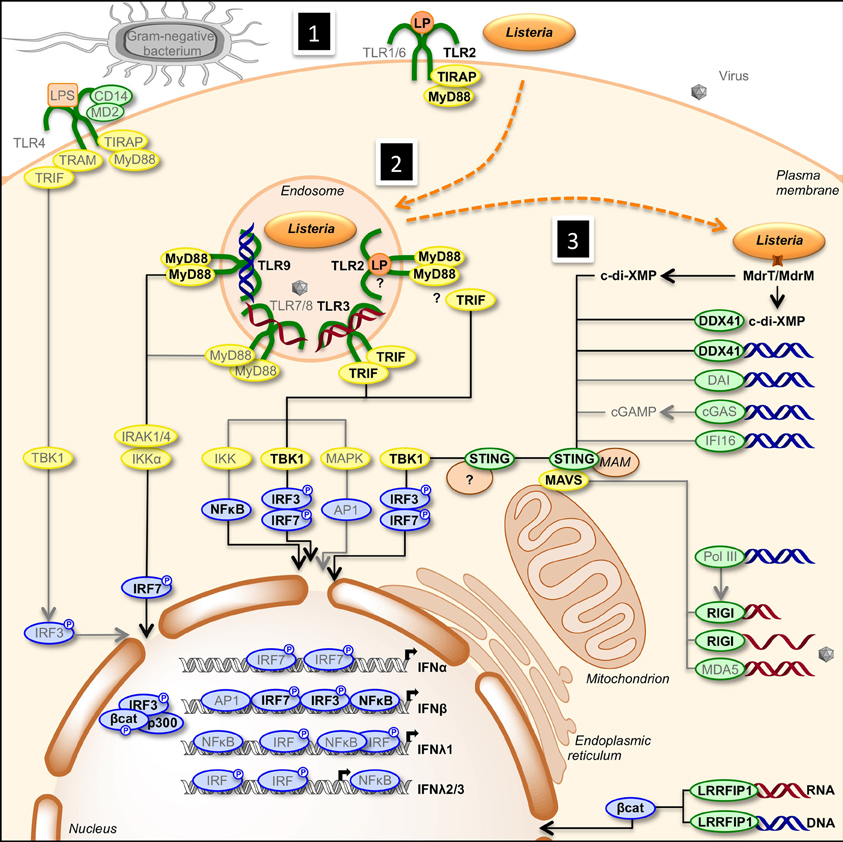
Figure 1. Activation of type I- and type III-IFNs by infection. (1) Cells detect pathogens by membrane-bound PRRs of the TLR family, which trigger the IFN response. Infection of cells with extracellular Gram-negative bacteria leads to activation of TLR4/CD14/MD2 by LPS. Following TLR4 ligation, IRF3 is activated via the adaptors TRAM and TRIF, and TBK1. (2) Intracellular bacteria and viruses are recognized by TLRs located in the endosome. TLR2, TLR3, TLR7/8, and TLR9 detect lipoproteins, dsRNA, ssRNA, and unmethylated CpG DNA, respectively. Engagement of the receptors leads to the induction of MyD88- and TRIF-dependent pathways involving IKK-, TBK1-, and MAPK-mediated activation of NF-κ B, IRF3, IRF7, and AP1, ultimately resulting in transcriptional activation of IFN genes. (3) Infection of cells leads to the release of cyclic-di-nucleotides (c-di-XMP) and nucleic acids (RNA in red, DNA in blue) in the cytosol that signal the presence of microbes and induce expression of interferons. Cyclic-di-nucleotides secreted by bacteria, e.g., through multidrug resistance MdrT and MdrM in Listeria, can activate the protein STING directly and via the cytosolic sensor DDX41. Cytosolic DNA is recognized by DDX41, DAI, cGAS, and IFI16. cGAS converts dsDNA to cGAMP, which stimulates STING. RNA polymerase III (PolIII) can convert cytosolic AT-rich dsDNA to uncapped 5′-triphosphate RNA that, as ssRNA, is sensed by RIGI. Another cytosolic RNA sensor, MDA5, and RIGI signal via the mitochondrial adaptor MAVS and STING. Activated STING recruits TBK1, which phosphorylates transcription factors IRF3 and IRF7. Upon activation, IRFs translocate to the nucleus where they bind to the promoter of type I IFN genes. Bacterial RNA and DNA additionally activate the cytosolic sensor LRRFIP1 that stimulates β-catenin phosphorylation and nuclear translocation where it binds to IRF3, recruits the histone acetyltransferase p300 to the enhanceosome leading to IFNβ transcription. Gray font and arrows: general infection pathways, bold black font and arrows: Listeria infection pathways. Red strands, RNA; blue strands, DNA. AP1, activator protein-1; CD14, cluster of differentiation 14; c-di-XMP, cyclic-di-nucleotide; cGAMP, cyclic-GMP-AMP; cGAS, cGAMP synthase; DAI, DNA-dependent activator of IFN-regulatory factors; DDX41, DEAD (aspartate-glutamate-alanine-aspartate)-box polypeptide 41; ER, endoplasmic reticulum; IFI16, IFN-inducible protein 16; IKK, Iκ B kinases; IRAK, interleukin-1 receptor-associated kinase; LP, lipoprotein; LPS, lipopolysaccharide; LRRFIP1, leucine-rich repeat flightless-interacting protein 1; MAM, mitochondria associated ER membranes; MAPK, mitogen-activated protein kinase; MAVS, mitochondrial antiviral signaling; MD2, myeloid differential protein-2; MDA5, melanoma differentiation-associated gene 5; MyD88, myeloid differentiation factor 88; NF-κ B, nuclear factor of kappa light polypeptide gene enhancer in B-cells; PolIII, RNA polymerase III; RIGI, retinoic acid inducible gene I; STING, stimulator of IFN genes; TBK1, TANK-binding kinase-1; TIRAP, TIR domain-containing adapter protein; TLR, Toll-like receptor; TRAM, TRIF-related adaptor molecule; TRIF, TIR domain-containing adaptor inducing IFN-β.
Regulation of the IFN-γ gene expression is different from that of type I and type III-IFN genes. NK cells and NKT cells are effectors of the innate immune response and primary sources of IFN-γ. Mature NK and NKT cells quickly react to infection by inducing IFN-γ secretion. Upon recognition of ligands expressed on infected cells, NK cell activating-receptors trigger signaling cascades involving adaptor proteins and protein tyrosine kinases leading to activation of Ras/Sos, PLC-γ and MAPK pathways and induction of IFN-γ production (Schoenborn and Wilson, 2007). In addition to receptors, IL-2, IL-15, IL-12, IL-18, and type I-IFNs also contribute to induction of IFN-γ production by NK cells (Newman and Riley, 2007; Schoenborn and Wilson, 2007; Marçais et al., 2013). Similarly, IL-12 and IL-18 induce IFN-γ production by NKT cells (Godfrey and Berzins, 2007). In NK and NKT cells, the IFN-γ gene locus is transcriptionally permissive within accessible chromatin and allows rapid IFN-γ expression upon activation of transcription factors, such as AP-1, NF-κ B, STAT4, and T-bet (Glimcher et al., 2004; Schoenborn and Wilson, 2007; Lazarevic et al., 2013). In addition, naive CD4 and CD8 T cells can differentiate into Th1 CD4 effector T cells and CD8 cytotoxic T lymphocytes capable of IFN-γ secretion (Wilson et al., 2009). IFN-γ production by CD4 and CD8 T cells depends on IL-12, IL-18 and IFN-γ itself and share many signaling pathways with NK cells. Multiple transcription factors act at the IFN-γ promoter, e.g., AP-1, ATF-2/c-Jun, C/EBP, Eomes, Ets-1, NFAT, NF-κB, Runx3, STATs and T-bet (Schoenborn and Wilson, 2007; Samten et al., 2008; Wilson et al., 2009; Lazarevic et al., 2013). Moreover, distal regulatory elements modify the chromatin and remodel the IFN-γ gene locus to facilitate IFN-γ production (Wilson et al., 2009).
IFN Receptors and Signal Transduction
IFNs are rapidly secreted upon infection and then bind to their receptors on the surface of target cells (Table 1). Type I-IFNs bind the ubiquitous IFNAR receptor, which consists of two chains, IFNAR1 and IFNAR2 (Piehler et al., 2012). Type III-IFNs bind and signal through a different receptor complex, made of two chains: IFNLR1 (also known as IL-28Rα) and IL10R2. This receptor is expressed primarily by epithelial cells and hepatocytes (Iversen and Paludan, 2010). Thus, the physiological roles of type I- and type III-IFNs are distinct because of the different distribution of their receptors in tissues, type III-IFNs acting predominantly at mucosal surfaces (Mordstein et al., 2010; Durbin et al., 2013). Type I- and type III-IFNs use different receptors but trigger the same JAK-STAT signal transduction cascade involving TYK2, JAK1, STAT1, and STAT2 albeit with different kinetics (Figure 2) (Marcello et al., 2006). Ultimately, STAT1, STAT2, and IRF9 form a transcription factor complex, referred to as ISGF3, which translocates to the nucleus and binds to IFN-stimulated responsive elements (ISRE) in the promoter of ISGs (Schindler et al., 2007). Type II-IFN, uses a heterodimeric receptor consisting of IFNγR1 and IFNγR2 chains, expressed by many cell types (Bach et al., 1997). IFN-γ activates JAK1, JAK2 and STAT1, leading to transcription of genes bearing a γ-activation sequence (GAS) in their promoter (Figure 2) (Schindler et al., 2007).
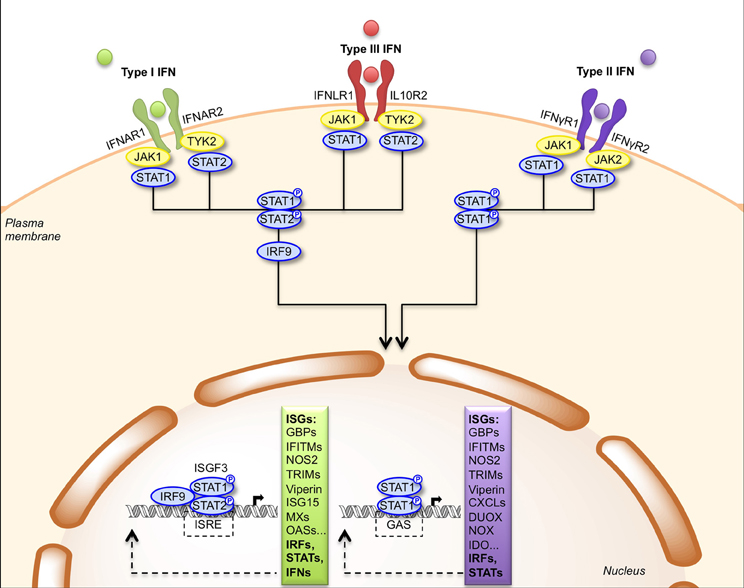
Figure 2. Signal transduction by the type I, type II and type III IFN receptors. Engagement of type I and type III IFNs by their respective receptors, IFNAR1/IFNAR2 and IFNLR1/IL10R2, triggers phosphorylation of JAK1/TYK2 kinases that activate STAT1 and STAT2. Phosphorylated STAT1/STAT2 heterodimers bind IRF9 forming the ISGF3 complex, which translocates to the nucleus where it induces expression of genes with ISRE-dependent promoters. Engagement of type II IFN by the IFNγR1/IFNγR2 receptor induces phosphorylation of JAK1/JAK2 kinases that activate STAT1. Phosphorylated STAT1 homodimers translocate to the nucleus and induces expression of genes with GAS-dependent promoters. CXCL, CXC chemokine ligand; DUOX, dual oxidase; GAS, gamma-IFN activated site; GBP, guanylate-binding protein; IDO, indoleamine 2,3-dioxygenase; IFITM, IFN-induced transmembrane protein; IRF9, IFN-regulatory factor 9; ISG, IFN-stimulated gene; ISGF3, IFN-stimulated gene factor 3; ISRE, IFN-stimulated response element; JAK, Janus activated kinase; MX, myxovirus resistance; NOS2, nitric oxide synthase 2; NOX, NADPH oxidase; OAS, oligoadenylate synthetase; STAT, signal transducer and activator of transcription; TRIM, tripartite motif; TYK2, tyrosine kinase 2.
IFNs and Listeria Infection
Induction of IFNs in Response to Listeria Infection
An initial innate immune response is triggered when Listeria MAMPs activate PRRs of host cells such as epithelial cells and macrophages (Figure 1). Infection induces a robust type I-IFN response. In mice, macrophages have been identified as the major source of IFN-β (Stockinger et al., 2009). In vitro, IFN-β production by bone-marrow-derived murine macrophages has been shown to require bacterial escape from the phagosome and activation of cytosolic surveillance pathways (O'Riordan et al., 2002). Induction of IFN-β depends on the adaptor protein STING and the cytosolic PRR DDX41, which are activated by bacterial secondary messengers c-di-AMP and c-di-GMP and by bacterial DNA (Woodward et al., 2010; Burdette et al., 2011; Sauer et al., 2011; Parvatiyar et al., 2012; Archer et al., 2014). STING is a direct receptor for cyclic-dinucleotides, including the cellular second messenger cyclic GMP-AMP (cGAMP) which is produced by the cytosolic sensor cGAMP synthase (cGAS) upon interaction with microbial DNA (Ablasser et al., 2013; Gao et al., 2013; Wu et al., 2013; Sun et al., 2013; Schoggins et al., 2014). Interestingly, type I-IFN production requires activation of the RIG-I helicase by Listeria RNA in non-immune cells lacking a functional STING signaling pathway (Abdullah et al., 2012; Hagmann et al., 2013). Another cytosolic PRR, the leucine-rich repeat-containing protein LRRFIP1, has also been implicated in IFN-β production by mouse primary peritoneal macrophages in response to Listeria infection, possibly by sensing double stranded DNA and RNA (Yang et al., 2010). While production of IFN-β in response to Listeria infection is independent from TLRs in bone-marrow-derived macrophages (McCaffrey et al., 2004; Stockinger et al., 2004; O'Connell et al., 2005), TLR-2 contributes significantly to IFN-β secretion by peritoneal macrophages, suggesting that specific macrophage populations have evolved different recognition strategies in response to Listeria infection (Aubry et al., 2012).
Listeria infection has recently been shown to induce type III-IFN gene expression in cells of epithelial origin, such as intestinal and trophoblast cells and hepatocytes (Lebreton et al., 2011; Bierne et al., 2012). Similar to type I-IFN, type III-IFN induction is triggered by intracellular Listeria (Bierne et al., 2012).
Listeria infection also triggers a rapid and robust IFN-γ response. After intravenous infection of mice with L. monocytogenes, NK and T cells are the main sources of IFN-γ (Thale and Kiderlen, 2005; Bou Ghanem et al., 2009). IFN-γ producing V1δ+−γδ T cells are other murine immune cells induced at an early stage of Listeria infection in mice inoculated intraperitoneally (Hamada et al., 2008). Using oral infection of mice, the natural route of infection in permissive hosts, L. monocytogenes has been shown to induce IFN-γ production by intraepithelial lymphocytes of the small intestine (Okamoto, 1994). More recently, human E-cadherin (hEcad) expressing mice, a mouse line permissive for Listeria oral infection (Lecuit et al., 2001), were used to study cells involved in intestinal mucosal immunity. Infection induced IFN-γ production in NK cells of the small intestine (Reynders et al., 2011).
Role of IFNs During Listeria Infection
The production of IFN-γ by immune cells promotes bacterial clearance and is thus critical in controlling primary L. monocytogenes infections (Zenewicz and Shen, 2007). Injection of neutralizing monoclonal anti-IFN-γ antibodies in mice infected intraperitoneally with L. monocytogenes inhibits macrophage activation and increases the mortality rate (Buchmeier and Schreiber, 1985). In addition, resistance of IFN-γ gene or IFN-γ receptor knock-out mice infected intravenously with L. monocytogenes is severely impaired (Huang et al., 1993; Harty and Bevan, 1995). Recent work using cell-type specific inactivation of STAT1 in mice elegantly demonstrated the key role of IFN-γ and STAT1 in macrophage activation and clearance of Listeria (Kernbauer et al., 2012). Interestingly, the role of STAT1 was extremely different after infection of immunized mice. STAT1 signaling in T cells and dendritic cells was critical for adaptive immunity to Listeria, while IFN-γ-activated macrophages were not essential anymore once memory cells were produced. Upon oral infection of hEcad mice with Listeria, IFN-γ contributes to the control of bacterial burden in the intestine and of bacterial dissemination to other organs. For instance, blocking IFN-γ with neutralizing antibodies increases Listeria load in the small intestine, the mesenteric lymph nodes and in the spleen of mice infected orally (Reynders et al., 2011).
In contrast to IFN-γ, type I-IFN is beneficial to L. monocytogenes. Mice lacking type I-IFN receptor or IRF3 are more resistant to Listeria intraperitoneal or intravenous infection (Auerbuch et al., 2004; Carrero et al., 2004; O'Connell et al., 2004; Garifulin et al., 2007; Jia et al., 2009). The role of type I-IFNs in increasing host susceptibility could be explained by modulation of components of the immune response involved in controlling bacterial growth such as induction of T cell apoptosis, resulting in greater IL-10 secretion by phagocytic cells, in turn dampening the innate immune response (Carrero and Unanue, 2006), the downregulation of IFN-γR (Rayamajhi et al., 2010; Kearney et al., 2013), or neutrophil recruitment (Brzoza-Lewis et al., 2012). As shown recently, STING-dependent activation of type I-IFN reduces the adaptive immune response to L. monocytogenes (Archer et al., 2014). In contrast, recent studies showed that type I-IFNs can also play a beneficial role for the host during Listeria infection, pointing to the infection route and the timing of type I-IFN production as determinative factors (Pontiroli et al., 2012; Kernbauer et al., 2013).
Interestingly, different strains of L. monocytogenes have been shown to vary greatly in their capacity to induce IFN-β (Reutterer et al., 2008; Schwartz et al., 2012). The LO28 strain hyperinduces IFN-β (Reutterer et al., 2008). This strain bears a non-functional BrtA (also named TetR), the transcriptional repressor of the multidrug efflux pump MdrT (Schwartz et al., 2012; Yamamoto et al., 2012). In Listeria, MdrT allows secretion of c-di-AMP, which triggers IFN-β. Thus, derepression of MdrT in the LO28 strain promotes IFN-β production. Of note, high expression of MdrT in LO28 correlates with both induction of IFN-β and lower virulence. Another Listeria multidrug resistance transporter, MdrM, has been involved in the stimulation of IFN-β production, possibly by secreting c-di-AMP (Crimmins et al., 2008; Woodward et al., 2010; Witte et al., 2012).
The role of type III-IFNs during listeriosis remains to be determined. Since Listeria colonizes several tissues of epithelial origins, such as the liver, intestine and placenta, it is tempting to speculate that IFN-λs play a role in the interaction of Listeria with epithelia. However, a prerequisite to address this question is the establishment of a new animal model, i.e., a mouse line expressing a human E-cadherin, thus permissive for Listeria infection of epithelia (Lecuit et al., 2001) and impaired in type III-IFN responses, such as IL28Rα knockout mice (Mordstein et al., 2010). One should keep in mind that the mouse model is not optimal to address the role of type III-IFN in human listeriosis. Indeed, IFN-λ1 is a pseudogene in mice, while human cells produce this cytokine upon infection with L. monocytogenes. In addition, the type III-IFN receptor is expressed at very low levels in the mouse liver and the IFN-λ response of the mouse liver is very weak (Mordstein et al., 2010). In line with this, it has been recently shown that mouse hepatocytes, in contrast to human hepatocytes, are not responsive to IFN-λ (Hermant et al., 2014).
Role of IFN-Stimulated Genes During Listeria Infection
The beneficial or detrimental effects of IFNs on Listeria infection rely on the functional properties of their downstream effectors. Indeed, IFNs elicit expression of hundreds of interferon-stimulated genes (ISGs), which encode proteins involved in a broad range of cellular functions (reviewed in MacMicking, 2004). However, while about 2,000 ISGs have been identified so far (Rusinova et al., 2012), their functions in immunomodulation remain to be characterized. To date, the contribution of interferon-induced proteins on Listeria infection has mostly been studied in the context of the IFN-γ pathway.
The antilisterial activity of IFN-γ in phagocytic cells involves induction of oxidative and nitrosative defences, via increased expression of enzymes that control production of reactive oxygen and nitrogen species, such as NOX2/CYBB, DUOX2, and iNOS/NOS2 (MacMicking, 2012). These enzymes play an important role in protecting infected cells against Listeria cytoinvasion (Myers et al., 2003; Lipinski et al., 2009). The assembly of these enzymes requires IFN-γ–inducible guanosine triphosphatases (GTPases) of the Gbp (guanylate binding protein) family (Boehm et al., 1998), which not only participate to oxidative pathways but also regulate autophagy (Kim et al., 2011). Several Gbps have been shown to protect cells from Listeria infection by coordinating a potent oxidative and vesicular trafficking program (Kim et al., 2011). IFN-γ also induces the expression of many nuclear genes encoding mitochondrial respiratory chain machinery, via activation of the nuclear receptor ERRα (estrogen-related receptor α). ERRα contributes to mitochondrial ROS production and efficient clearance of L. monocytogenes (Sonoda et al., 2007). A family of IFN-γ-induced chemokines (CXCL9, CXCL10, CXCL11) displays direct antimicrobial activity against L. monocytogenes (Cole et al., 2001). In dendritic cells, one of the IFN-γ-associated ISGs is the immunoregulatory enzyme indoleamine 2,3-dioxygenase (IDO), a key enzyme of the tryptophan metabolism. IDO is proposed to play a role in the containment of Listeria within granulomatous structures, thus avoiding massive T cell activation (Popov et al., 2006).
The function of type I IFN-associated ISGs in Listeria infection is less documented. Zwaferink et al. have observed that upregulation of iNOS/NOS2 by IFN-β promotes necrotic death of macrophages (Zwaferink et al., 2007). Additionally, several interferon-inducible proteins belong to inflammasomes; thus, type I IFN may potentiate inflammasome activation and cell death by pyroptosis (Malireddi and Kanneganti, 2013). Yet, the link between these effectors and the observed harmful effects of type I IFNs on the host is still unclear. Likewise, the role of effectors induced by type III IFNs in Listeria infection of epithelia is not understood.
Of interest, a subset of ISGs is amongst the most induced genes in the intestinal tissue of gnotobiotic humanized mice infected orally with L. monocytogenes (Archambaud et al., 2012). However, which type of IFNs triggers this response and for which function on the intestinal mucosa remain to be explored. In addition, IFN-independent pathways may contribute to expression of these ISGs.
Subversion of IFN Responses by Listeria
Listeria has evolved several mechanisms to avoid immune detection and evade IFN responses. It has been demonstrated that deacetylation of Listeria peptidoglycan by the deacetylase PgdA confers resistance to host lysozyme, thus preventing release of MAMPs, such as DNA, RNA and lipopeptides, that trigger IFN-β production (Boneca et al., 2007). Listeria pgdA mutants are rapidly killed in murine macrophages, which produce lysozyme, and induce a strong secretion of IFN-β compared to wild-type Listeria. The role of PgdA is not limited to the control of type I-IFN production as a pgdA mutant hyperinduces pro-inflammatory cytokines as well. Modification of peptidoglycan by PgdA is an extremely efficient mechanism of immune escape used by Listeria, which correlates with its critical role in virulence.
Remarkably, Listeria has evolved a sophisticated strategy to modulate, either negatively or positively, the expression of ISGs in epithelial cells, by targeting a chromatin-repressive complex, BAHD1 (Bierne et al., 2009; Lebreton et al., 2011, 2012). Indeed, Listeria infection promotes, albeit via an unknown mechanism, the targeting of BAHD1 at the promoter of a set of ISGs, thereby downregulating type I- and type III-IFN responses. On the other hand, Listeria can produce a nucleomodulin, LntA, which when secreted by intracellular bacteria, enters the nucleus of infected cells, binds BAHD1 and inhibits its function (Lebreton et al., 2012, 2014). Thus, LntA stimulates IFN responses. Consistent with the presence of HDAC1/2 in the BAHD1-associated complex, the level of acetylation of lysine 9 on histone H3, which is a mark of active chromatin, increases at the promoters of ISGs in the presence of LntA. When, in which host conditions, and how LntA targets BAHD1 specifically at ISGs remains an open question. The LntA-mediated stimulation of type III-IFN responses might support localized pro-bacterial conditions, as was proposed for IFN-I responses.
Concluding Remarks
We have an extensive knowledge of the molecular and cellular mechanisms involved in Listeria-host interactions. Yet, our understanding of the immune response to Listeria, and more specifically the role IFNs and of their downstream effectors, is far from complete and often relies on studies performed in cultured cells or in mice. However, murine and human listeriosis differ in many aspects (Lecuit, 2007; Hoelzer et al., 2012). For instance, E-cadherin, the major receptor for Listeria in epithelial cells, is not functional for Listeria uptake in the mouse. Thus, the route of entry of Listeria is not strictly the same in mice and humans. Moreover, ISGs induced in response to infection are not identical in mice and humans. Additionally, murine hepatocytes do not respond to type III-IFNs (Hermant et al., 2014), precluding the study of these IFNs during infection by human hepatotropic pathogens, such as L. monocytogenes. Altogether, species-specific differences provide limits to the use of mouse models in characterizing IFN pathways engaged during Listeria infection in humans, especially in key epithelial organs such as the gut, liver and placenta. It will be important to perform future studies using adapted animal models, such as humanized mice permissive to oral infection or transgenic mice with human xenografts (Walters et al., 2006), since the effect of type I-IFN on Listeria infection depends on the route and time of infection (Pontiroli et al., 2012; Kernbauer et al., 2013) and type III-IFN requires bacterial interaction with epithelia (Bierne et al., 2012). Finally, numerous ISGs are induced upon Listeria infection in vitro, but the relevant ISGs and their cellular functions remain to be identified. Validation of ISGs identified in cultured cells in adequate in vivo models or deduced from analyses of patient samples, will be required to address the complex role of IFNs and bacterial subversion strategies and provide new insights into Listeria pathogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors declare no conflict of interest. The authors' work has been supported in part by the Institut Pasteur, Inserm, INRA, Université Paris-Diderot, LabEx IBEID, ANR (Grant EPILIS), French Ligue Nationale contre le Cancer, Fondation Louis-Jeantet, Fondation Le Roch, and European Research Council (Advanced Grant Award 233348 to Pascale Cossart). Pascale Cossart is an international research scholar of the Howard Hughes Medical Institute.
References
Abdullah, Z., Schlee, M., Roth, S., Mraheil, M. A., Barchet, W., Böttcher, J. B. O., et al. (2012). RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J. 31, 4153–4164. doi: 10.1038/emboj.2012.274
Ablasser, A., Goldeck, M., Cavlar, T., Deimling, T., Witte, G., Röhl, I., et al. (2013). cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. doi: 10.1038/nature12306
Archambaud, C., Nahori, M.-A., Soubigou, G., Bécavin, C., Laval, L., Lechat, P., et al. (2012). Impact of lactobacilli on orally acquired listeriosis. Proc. Nat. Acad. Sci. U.S.A. 109, 16684–16689. doi: 10.1073/pnas.1212809109
Archer, K. A., Durack, J., and Portnoy, D. A. (2014). STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog. 10:e1003861. doi: 10.1371/journal.ppat.1003861
Aubry, C., Corr, S. C., Wienerroither, S., Goulard, C., Jones, R., Jamieson, A. M., et al. (2012). Both TLR2 and TRIF contribute to interferon-β production during Listeria infection. PLoS ONE 7:e33299. doi: 10.1371/journal.pone.0033299
Aubry, C., Goulard, C., Nahori, M.-A., Cayet, N., Decalf, J., Sachse, M., et al. (2011). OatA, a peptidoglycan O-acetyltransferase involved in Listeria monocytogenes immune escape, is critical for virulence. J. Infect. Dis. 204, 731–740. doi: 10.1093/infdis/jir396
Auerbuch, V., Brockstedt, D., Meyer-Morse, N., O'Riordan, M., and Portnoy, D. A. (2004). Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 200, 527–533. doi: 10.1084/jem.20040976
Bach, E. A., Aguet, M., and Schreiber, R. D. (1997). The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 15, 563–591. doi: 10.1146/annurev.immunol.15.1.563
Bierne, H., Tham, T. N., Batsche, E., Dumay, A., Leguillou, M., Kerneis-Golsteyn, S., et al. (2009). Human BAHD1 promotes heterochromatic gene silencing. Proc. Natl. Acad. Sci. U.S.A. 106, 13826–13831. doi: 10.1073/pnas.0901259106
Bierne, H., Travier, L., Mahlakõiv, T., Tailleux, L., Subtil, A., Lebreton, A., et al. (2012). Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS ONE 7:e39080. doi: 10.1371/journal.pone.0039080
Boehm, U., Guethlein, L., Klamp, T., Ozbek, K., Schaub, A., Fütterer, A., et al. (1998). Two families of GTPases dominate the complex cellular response to IFN-gamma. J. Immunol. 161, 6715–6723.
Boneca, I. G., Dussurget, O., Cabanes, D., Nahori, M.-A., Sousa, S., Lecuit, M., et al. (2007). A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc. Natl. Acad. Sci. U.S.A. 104, 997–1002. doi: 10.1073/pnas.0609672104
Bou Ghanem, E. N., McElroy, D. S., and D'Orazio, S. E. F. (2009). Multiple mechanisms contribute to the robust rapid gamma interferon response by CD8+ T cells during Listeria monocytogenes infection. Infect. Immun. 77, 1492–1501. doi: 10.1128/IAI.01207-08
Brzoza-Lewis, K. L., Hoth, J. J., and Hiltbold, E. M. (2012). Type I interferon signaling regulates the composition of inflammatory infiltrates upon infection with Listeria monocytogenes. Cell. Immunol. 273, 41–51. doi: 10.1016/j.cellimm.2011.11.008
Buchmeier, N. A., and Schreiber, R. D. (1985). Requirement of endogenous interferon-gamma production for resolution of Listeria monocytogenes infection. Proc. Natl. Acad. Sci. U.S.A. 82, 7404–7408. doi: 10.1073/pnas.82.21.7404
Burdette, D. L., Monroe, K. M., Sotelo-Troha, K., Iwig, J. S., Eckert, B., Hyodo, M., et al. (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. doi: 10.1038/nature10429
Camejo, A., Carvalho, F., Reis, O., Leitão, E., Sousa, S., and Cabanes, D. (2011). The arsenal of virulence factors deployed by Listeria monocytogenes to promote its cell infection cycle. Virulence 2, 379–394. doi: 10.4161/viru.2.5.17703
Carrero, J., and Unanue, E. (2006). Lymphocyte apoptosis as an immune subversion strategy of microbial pathogens. Trends Immunol. 27, 497–503. doi: 10.1016/j.it.2006.09.005
Carrero, J. A. (2013). Confounding roles for type I interferons during bacterial and viral pathogenesis. Int. Immunol. 25, 663–669. doi: 10.1093/intimm/dxt050
Carrero, J. A., Calderon, B., and Unanue, E. R. (2004). Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 200, 535–540. doi: 10.1084/jem.20040769
Cartwright, E. J., Jackson, K. A., Johnson, S. D., Graves, L. M., Silk, B. J., and Mahon, B. E. (2013). Listeriosis outbreaks and associated food vehicles, United States, 1998–2008. Emerging Infect. Dis. 19, 1–9. doi: 10.3201/eid1901.120393
Chiba, S., Nagai, T., Hayashi, T., Baba, Y., Nagai, S., and Koyasu, S. (2011). Listerial invasion protein internalin B promotes entry into ileal Peyer's patches in vivo. Microbiol. Immunol. 55, 123–129. doi: 10.1111/j.1348-0421.2010.00292.x
Cole, A. M., Ganz, T., Liese, A. M., Burdick, M. D., Liu, L., and Strieter, R. M. (2001). Cutting edge: IFN-inducible ELR− CXC chemokines display defensin-like antimicrobial activity. J. Immunol. 167, 623–627.
Corr, S. C., and O'Neill, L. A. J. (2009). Listeria monocytogenes infection in the face of innate immunity. Cell. Microbiol. 11, 703–709. doi: 10.1111/j.1462-5822.2009.01294.x
Cossart, P. (2011). Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc. Natl. Acad. Sci. U.S.A. 108, 19484–19491. doi: 10.1073/pnas.1112371108
Crimmins, G. T., Herskovits, A. A., Rehder, K., Sivick, K. E., Lauer, P., Dubensky, T. W., et al. (2008). Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc. Natl. Acad. Sci. U.S.A. 105, 10191–10196. doi: 10.1073/pnas.0804170105
Dalton, D. K., Pitts-Meek, S., Keshav, S., Figari, I. S., Bradley, A., and Stewart, T. A. (1993). Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 259, 1739–1742. doi: 10.1126/science.8456300
de Almeida, L. A., Carvalho, N. B., Oliveira, F. S., Lacerda, T. L. S., Vasconcelos, A. C., Nogueira, L., et al. (2011). MyD88 and STING signaling pathways are required for IRF3-mediated IFN-β induction in response to Brucella abortus infection. PLoS ONE 6:e23135. doi: 10.1371/journal.pone.0023135
Decker, T., Müller, M., and Stockinger, S. (2005). The Yin and Yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 5, 675–687. doi: 10.1038/nri1684
Disson, O., Grayo, S., Huillet, E., Nikitas, G., Langa-Vives, F., Dussurget, O., et al. (2008). Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455, 1114–1118. doi: 10.1038/nature07303
Dortet, L., Mostowy, S., and Cossart, P. (2012). Listeria and autophagy escape: involvement of InlK, an internalin-like protein. Autophagy 8, 132–134. doi: 10.4161/auto.8.1.18218
Durbin, R. K., Kotenko, S. V., and Durbin, J. E. (2013). Interferon induction and function at the mucosal surface. Immunol. Rev. 255, 25–39. doi: 10.1111/imr.12101
Fox, B. A., Sheppard, P. O., and O'Hara, P. J. (2009). The role of genomic data in the discovery, annotation and evolutionary interpretation of the interferon-lambda family. PLoS ONE 4:e4933. doi: 10.1371/journal.pone.0004933
Freitag, N. E., Port, G. C., and Miner, M. D. (2009). Listeria monocytogenes—from saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 7, 623–628. doi: 10.1038/nrmicro2171
Gahan, C., and Hill, C. (2005). Gastrointestinal phase of Listeria monocytogenes infection. J. Appl. Microbiol. 98, 1345–1353. doi: 10.1111/j.1365-2672.2005.02559.x
Gao, D., Wu, J., Wu, Y. T., Du, F., Aroh, C., Yan, N., et al. (2013). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906. doi: 10.1126/science.1240933
Garifulin, O., Qi, Z., Shen, H., Patnala, S., Green, M. R., and Boyartchuk, V. (2007). Irf3 polymorphism alters induction of interferon beta in response to Listeria monocytogenes infection. PLoS Genet. 3:e152. doi: 10.1371/journal.pgen.0030152
Glimcher, L. H., Townsend, M. J., Sullivan, B. M., and Lord, G. M. (2004). Recent developments in the transcriptional regulation of cytolytic effector cells. Nat. Rev. Immunol. 4, 900–911. doi: 10.1038/nri1490
Godfrey, D. I., and Berzins, S. P. (2007). Control points in NKT-cell development. Nat. Rev. Immunol. 7, 505–518. doi: 10.1038/nri2116
Gouin, E., Adib-Conquy, M., Balestrino, D., Nahori, M.-A., Villiers, V., Colland, F., et al. (2010). The Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the Iκ B kinase subunit IKKα. Proc. Nat. Acad. Sci. U.S.A. 107, 17333–17338. doi: 10.1073/pnas.1007765107
Goulet, V., Hebert, M., Hedberg, C., Laurent, E., Vaillant, V., De Valk, H., et al. (2012). Incidence of listeriosis and related mortality among groups at risk of acquiring listeriosis. Clin. Infect. Dis. 54, 652–660. doi: 10.1093/cid/cir902
Hagmann, C. A., Herzner, A. M., Abdullah, Z., Zillinger, T., Jakobs, C., Schuberth, C., et al. (2013). RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS ONE 8:e62872. doi: 10.1371/journal.pone.0062872
Hamada, S., Umemura, M., Shiono, T., Hara, H., Kishihara, K., Tanaka, K., et al. (2008). Importance of murine Vdelta1 gammadelta T cells expressing interferon-gamma and interleukin-17A in innate protection against Listeria monocytogenes infection. Immunology. 125, 170–177. doi: 10.1111/j.1365-2567.2008.02841.x
Hamming, O. J., Terczynska-Dyla, E. T. N., Vieyres, G., Dijkman, R., Jorgensen, S. E., Akhtar, H., et al. (2013). Interferon lambda 4 signals via the IFN-λ receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J. 32, 3055–3065. doi: 10.1038/emboj.2013.232
Hamon, M. A., Batsche, E., Regnault, B., Tham, T. N., Seveau, S., Muchardt, C., et al. (2007). Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. U.S.A. 104, 13467–13472. doi: 10.1073/pnas.0702729104
Harty, J. T., and Bevan, M. J. (1995). Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity 3, 109–117. doi: 10.1016/1074-7613(95)90163-9
Heim, M. H. (2013). 25 years of interferon-based treatment of chronic hepatitis C: an epoch coming to an end. Nat. Rev. Immunol. 13, 535–542. doi: 10.1038/nri3463
Henry, T., Kirimanjeswara, G. S., Ruby, T., Jones, J. W., Peng, K., Perret, M., et al. (2010). Type I IFN signaling constrains IL-17A/F secretion by gamma-delta T cells during bacterial infections. J. Immunol. 184, 3755–3767. doi: 10.4049/jimmunol.0902065
Hermant, P., Demarez, C., Mahlakõiv, T., Staeheli, P., Meuleman, P., and Michiels, T. (2014). Human but not mouse hepatocytes respond to interferon-lambda in vivo. PLoS ONE 9:e87906. doi: 10.1371/journal.pone.0087906
Hoelzer, K., Pouillot, R., and Dennis, S. (2012). Animal models of listeriosis: a comparative review of the current state of the art and lessons learned. Vet. Res. 43:18. doi: 10.1186/1297-9716-43-18
Holland, S. M. (2010). Chronic granulomatous disease. Clin. Rev. Allerg. Immunol. 38, 3–10. doi: 10.1007/s12016-009-8136-z
Honda, K., Takaoka, A., and Taniguchi, T. (2006). Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360. doi: 10.1016/j.immuni.2006.08.009
Honda, K., Yanai, H., Negishi, H., Asagiri, M., Sato, M., Mizutani, T., et al. (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. doi: 10.1038/nature03464
Hu, X., and Ivashkiv, L. B. (2009). Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31, 539–550. doi: 10.1016/j.immuni.2009.09.002
Huang, S., Hendriks, W., Althage, A., Hemmi, S., Bluethmann, H., Kamijo, R., et al. (1993). Immune response in mice that lack the interferon-gamma receptor. Science 259, 1742–1745. doi: 10.1126/science.8456301
Isaacs, A., and Lindemann, J. (1957). Virus Interference. I. The Interferon. Proc. Royal. Soc. B Biol. Sci. 147, 258–267. doi: 10.1098/rspb.1957.0048
Iversen, M. B., and Paludan, S. R. (2010). Mechanisms of type III interferon expression. J. Interferon Cytokine Res. 30, 573–578. doi: 10.1089/jir.2010.0063
Jia, T., Leiner, I., Dorothee, G., Brandl, K., and Pamer, E. G. (2009). MyD88 and Type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J. Immunol. 183, 1271–1278. doi: 10.4049/jimmunol.0900460
Jouanguy, E., Altare, F., Lamhamedi, S., Revy, P., Emile, J. F., Newport, M., et al. (1996). Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guérin infection. N. Engl. J. Med. 335, 1956–1961. doi: 10.1056/NEJM199612263352604
Jouanguy, E., Döffinger, R., Dupuis, S., Pallier, A., Altare, F., and Casanova, J.-L. (1999). IL-12 and IFN-gamma in host defense against mycobacteria and salmonella in mice and men. Curr. Opin. Immunol. 11, 346–351. doi: 10.1016/S0952-7915(99)80055-7
Kamijo, R., Le, J., Shapiro, D., Havell, E. A., Huang, S., Aguet, M., et al. (1993). Mice that lack the interferon-gamma receptor have profoundly altered responses to infection with Bacillus Calmette-Guérin and subsequent challenge with lipopolysaccharide. J. Exp. Med. 178, 1435–1440. doi: 10.1084/jem.178.4.1435
Kaplan, D. H., Shankaran, V., Dighe, A. S., Stockert, E., Aguet, M., Old, L. J., et al. (1998). Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. U.S.A. 95, 7556–7561. doi: 10.1073/pnas.95.13.7556
Kearney, S. J., Delgado, C., Eshleman, E. M., Hill, K. K., O'Connor, B. P., and Lenz, L. L. (2013). Type I IFNs downregulate myeloid cell IFN-gamma receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences ifngr1 transcription. J. Immunol. 191, 3384–3392. doi: 10.4049/jimmunol.1203510
Kernbauer, E., Maier, V., Rauch, I., Müller, M., and Decker, T. (2013). Route of infection determines the impact of type I interferons on innate immunity to Listeria monocytogenes. PLoS ONE 8:e65007. doi: 10.1371/journal.pone.0065007
Kernbauer, E., Maier, V., Stoiber, D., Strobl, B., Schneckenleithner, C., Sexl, V., et al. (2012). Conditional Stat1 ablation reveals the importance of interferon signaling for immunity to Listeria monocytogenes infection. PLoS Pathog. 8:e1002763. doi: 10.1371/journal.ppat.1002763
Kim, B.-H., Shenoy, A. R., Kumar, P., Das, R., Tiwari, S., and MacMicking, J. D. (2011). A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–721. doi: 10.1126/science.1201711
Kotenko, S. V., Gallagher, G., Baurin, V. V., Lewis-Antes, A., Shen, M., Shah, N. K., et al. (2003). IFN-λ s mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77. doi: 10.1038/ni875
Lazarevic, V., Glimcher, L. H., and Lord, G. M. (2013). T-bet: a bridge between innate and adaptive immunity. Nat. Rev. Immunol. 13, 777–789. doi: 10.1038/nri3536
Lebreton, A., Cossart, P., and Bierne, H. (2012). Bacteria tune interferon responses by playing with chromatin. Virulence 3, 87–91. doi: 10.4161/viru.3.1.18531
Lebreton, A., Job, V., Ragon, M., Le Monnier, A., Dessen, A., Cossart, P., et al. (2014). Structural basis for the inhibition of the chromatin repressor BAHD1 by the bacterial nucleomodulin LntA. MBio. 5:e00775-13. doi:10.1128/mBio.00775-13
Lebreton, A., Lakisic, G., Job, V., Fritsch, L., Tham, T. N., Camejo, A., et al. (2011). A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science 331, 1319–1321. doi: 10.1126/science.1200120
Lecuit, M. (2007). Human listeriosis and animal models. Microbes Infect. 9, 1216–1225. doi: 10.1016/j.micinf.2007.05.009
Lecuit, M., Vandormael-Pournin, S., Lefort, J., Huerre, M., Gounon, P., Dupuy, C., et al. (2001). A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science 292, 1722–1725. doi: 10.1126/science.1059852
Levy, D. E., Marié, I. J., and Durbin, J. E. (2011). Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 1, 476–486. doi: 10.1016/j.coviro.2011.11.001
Lipinski, S., Till, A., Sina, C., Arlt, A., Grasberger, H., Schreiber, S., et al. (2009). DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci. 122, 3522–3530. doi: 10.1242/jcs.050690
Mackaness, G. B. (1962). Cellular resistance to infection. J. Exp. Med. 116, 381–406. doi: 10.1084/jem.116.3.381
MacMicking, J. D. (2004). IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 25, 601–609. doi: 10.1016/j.it.2004.08.010
MacMicking, J. D. (2012). Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 12, 367–382. doi: 10.1038/nri3210
Malireddi, R. K. S., and Kanneganti, T.-D. (2013). Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front. Cell. Infect. Microbiol. 3:77. doi: 10.3389/fcimb.2013.00077
Mancuso, G., Midiri, A., Biondo, C., Beninati, C., Zummo, S., Galbo, R., et al. (2007). Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol. 178, 3126–3133.
Marçais, A., Viel, S., Grau, M., Henry, T., Marvel, J., and Walzer, T. (2013). Regulation of mouse NK cell development and function by cytokines. Front. Immunol. 4:450. doi: 10.3389/fimmu.2013.00450
Marcello, T., Grakoui, A., Barba Spaeth, G., Machlin, E. S., Kotenko, S. V., Macdonald, M. R., et al. (2006). Interferons α and λ inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131, 1887–1898. doi: 10.1053/j.gastro.2006.09.052
Martin, F. J., Gomez, M. I., Wetzel, D. M., Memmi, G., O'Seaghdha, M., Soong, G., et al. (2009). Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Invest. 119, 1931–1939. doi: 10.1172/JCI35879
McCaffrey, R. L., Fawcett, P., O'Riordan, M., Lee, K.-D., Havell, E. A., Brown, P. O., et al. (2004). A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc. Natl. Acad. Sci. U.S.A. 101, 11386–11139. doi: 10.1073/pnas.0403215101
Monroe, K. M., McWhirter, S. M., and Vance, R. E. (2010). Induction of type I interferons by bacteria. Cell. Microbiol. 12, 881–890. doi: 10.1111/j.1462-5822.2010.01478.x
Mordstein, M., Michiels, T., and Staeheli, P. (2010). What have we learned from the IL28 receptor knockout mouse? J. Interferon Cytokine Res. 30, 579–584. doi: 10.1089/jir.2010.0061
Myers, J. T., Tsang, A. W., and Swanson, J. A. (2003). Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J. Immunol. 171, 5447–5453.
Newman, K. C., and Riley, E. M. (2007). Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat. Rev. Immunol. 7, 279–291. doi: 10.1038/nri2057
Newport, M. J., Huxley, C. M., Huston, S., Hawrylowicz, C. M., Oostra, B. A., Williamson, R., et al. (1996). A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med. 335, 1941–1949. doi: 10.1056/NEJM199612263352602
Nikitas, G., Deschamps, C., Disson, O., Niault, T., Cossart, P., and Lecuit, M. (2011). Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J. Exp. Med. 208, 2263–2277. doi: 10.1084/jem.20110560
O'Connell, R. M., Saha, S. K., Vaidya, S. A., Bruhn, K. W., Miranda, G. A., Zarnegar, B., et al. (2004). Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 200, 437–445. doi: 10.1084/jem.20040712
O'Connell, R. M., Vaidya, S. A., Perry, A. K., Saha, S. K., Dempsey, P. W., and Cheng, G. (2005). Immune activation of type I IFNs by Listeria monocytogenes occurs independently of TLR4, TLR2, and receptor interacting protein 2 but involves TNFR-associated NF kappa B kinase-binding kinase 1. J. Immunol. 174, 1602–1607.
Okamoto, M. (1994). Host defense and endogenous interferon-gamma in the intestines during an oral infection with Listeria monocytogenes. Hokkaido Igaku Zasshi 69, 794–809.
Ooi, S. T., and Lorber, B. (2005). Gastroenteritis due to Listeria monocytogenes. Clin. Infect. Dis. 40, 1327–1332. doi: 10.1086/429324
O'Riordan, M., Yi, C. H., Gonzales, R., Lee, K.-D., and Portnoy, D. A. (2002). Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 13861–13866. doi: 10.1073/pnas.202476699
Panne, D. (2008). The enhanceosome. Curr. Opin. Struct. Biol. 18, 236–242. doi: 10.1016/j.sbi.2007.12.002
Panne, D., Maniatis, T., and Harrison, S. C. (2007). An atomic model of the interferon-beta enhanceosome. Cell 129, 1111–1123. doi: 10.1016/j.cell.2007.05.019
Parvatiyar, K., Zhang, Z., Teles, R. M., Ouyang, S., Jiang, Y., Iyer, S. S., et al. (2012). The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 13, 1155–1161. doi: 10.1038/ni.2460
Patel, A. A., Lee-Lewis, H., Hughes-Hanks, J., Lewis, C. A., and Anderson, D. M. (2012). Opposing roles for interferon regulatory factor-3 (IRF-3) and type I interferon signaling during plague. PLoS Pathog. 8:e1002817. doi: 10.1371/journal.ppat.1002817
Pentecost, M., Otto, G., Theriot, J. A., and Amieva, M. R. (2006). Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog. 2:e3. doi: 10.1371/journal.ppat.0020003
Personnic, N., Bruck, S., Nahori, M.-A., Toledo-Arana, A., Nikitas, G., Lecuit, M., et al. (2010). The stress-induced virulence protein InlH controls interleukin-6 production during murine listeriosis. Infect. Immun. 78, 1979–1989. doi: 10.1128/IAI.01096-09
Pestka, S. (2007). The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 282, 20047–20051. doi: 10.1074/jbc.R700004200
Pestka, S., Krause, C. D., and Walter, M. R. (2004). Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32. doi: 10.1111/j.0105-2896.2004.00204.x
Piehler, J., Thomas, C., Garcia, K. C., and Schreiber, G. (2012). Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 250, 317–334. doi: 10.1111/imr.12001
Pietilä, T. E., Latvala, S., Osterlund, P., and Julkunen, I. (2010). Inhibition of dynamin-dependent endocytosis interferes with type III IFN expression in bacteria-infected human monocyte-derived DCs. J. Leukoc. Biol. 88, 665–674. doi: 10.1189/jlb.1009651
Pontiroli, F., Dussurget, O., Zanoni, I., Urbano, M., Beretta, O., Granucci, F., et al. (2012). The timing of IFNβ production affects early innate responses to Listeria monocytogenes and determines the overall outcome of lethal infection. PLoS ONE 7:e43455. doi: 10.1371/journal.pone.0043455
Popov, A., Abdullah, Z., Wickenhauser, C., Saric, T., Driesen, J., Hanisch, F.-G., et al. (2006). Indoleamine 2,3-dioxygenase–expressing dendritic cells form suppurative granulomas following Listeria monocytogenes infection. J. Clin. Invest. 116, 3160–3170. doi: 10.1172/JCI28996
Prokunina-Olsson, L., Muchmore, B., Tang, W., Pfeiffer, R. M., Park, H., Dickensheets, H., et al. (2013). A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 45, 164–171. doi: 10.1038/ng.2521
Qiu, H., Fan, Y., Joyee, A. G., Wang, S., Han, X., Bai, H., et al. (2008). Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J. Immunol. 181, 2092–2102.
Rae, C. S., Geissler, A., Adamson, P. C., and Portnoy, D. A. (2011). Mutations of the Listeria monocytogenes peptidoglycan N-deacetylase and O-acetylase result in enhanced lysozyme sensitivity, bacteriolysis, and hyperinduction of innate immune pathways. Infect. Immun. 79, 3596–3606. doi: 10.1128/IAI.00077-11
Rayamajhi, M., Humann, J., Kearney, S., Hill, K. K., and Lenz, L. L. (2010). Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence 1, 418–422. doi: 10.4161/viru.1.5.12787
Reutterer, B., Stockinger, S., Pilz, A., Soulat, D., Kastner, R., Westermayer, S., et al. (2008). Type I IFN are host modulators of strain-specific Listeria monocytogenes virulence. Cell. Microbiol. 10, 1116–1129. doi: 10.1111/j.1462-5822.2007.01114.x
Reynders, A., Yessaad, N., Vu Manh, T.-P., Dalod, M., Fenis, A., Aubry, C., et al. (2011). Identity, regulation and in vivo function of gut NKp46(+)RORγt(+) and NKp46(+)RORγt(-) lymphoid cells. EMBO J. 30, 2934–2947. doi: 10.1038/emboj.2011.201
Robinson, N., McComb, S., Mulligan, R., Dudani, R., Krishnan, L., and Sad, S. (2012). Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13, 954–962. doi: 10.1038/ni.2397
Roesler, J., Kofink, B., Wendisch, J., Heyden, S., Paul, D., Friedrich, W., et al. (1999). Listeria monocytogenes and recurrent mycobacterial infections in a child with complete interferon-gamma-receptor (IFNgammaR1) deficiency: mutational analysis and evaluation of therapeutic options. Exp. Hematol. 27, 1368–1374. doi: 10.1016/S0301-472X(99)00077-6
Rusinova, I., Forster, S., Yu, S., Kannan, A., Masse, M., Cumming, H., et al. (2012). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, 1040–1046. doi: 10.1093/nar/gks1215
Samten, B., Townsend, J. C., Weis, S. E., Bhoumik, A., Klucar, P., Shams, H., et al. (2008). CREB, ATF, and AP-1 transcription factors regulate IFN-gamma secretion by human T cells in response to mycobacterial antigen. J. Immunol. 181, 2056–2064.
Sauer, J.-D., Sotelo-Troha, K., von Moltke, J., Monroe, K. M., Rae, C. S., Brubaker, S. W., et al. (2011). The ENU-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic-di-nucleotides. Infect. Immun. 79, 688–694. doi: 10.1128/IAI.00999-10
Schindler, C., Levy, D. E., and Decker, T. (2007). JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063. doi: 10.1074/jbc.R700016200
Schlech, W. F., Lavigne, P. M., Bortolussi, R. A., Allen, A. C., Haldane, E. V., Wort, A. J., et al. (1983). Epidemic listeriosis: evidence for transmission by food. N. Engl. J. Med. 308, 203–206. doi: 10.1056/NEJM198301273080407
Schoenborn, J. R., and Wilson, C. B. (2007). Regulation of interferon-γ during innate and adaptive immune responses. Adv. Immunol. 96, 41–101. doi: 10.1016/S0065-2776(07)96002-2
Schoggins, J. W., MacDuff, D. A., Imanaka, N., Gainey, M. D., Shrestha, B., Eitson, J. L., et al. (2014). Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695. doi: 10.1038/nature12862
Schoggins, J. W., Wilson, S. J., Panis, M., Murphy, M. Y., Jones, C. T., Bieniasz, P., et al. (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. doi: 10.1038/nature09907
Schroder, K. (2003). Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189. doi: 10.1189/jlb.0603252
Schwartz, K. T., Carleton, J. D., Quillin, S. J., Rollins, S. D., Portnoy, D. A., and Leber, J. H. (2012). Hyperinduction of host beta interferon by a Listeria monocytogenes strain naturally overexpressing the multidrug efflux pump MdrT. Infect. Immun. 80, 1537–1545. doi: 10.1128/IAI.06286-11
Shaughnessy, L. M., and Swanson, J. A. (2007). The role of the activated macrophage in clearing Listeria monocytogenes infection. Front. Biosci 12, 2683–2692. doi: 10.2741/2364
Sheppard, P., Kindsvogel, W., Xu, W., Henderson, K., Schlutsmeyer, S., Whitmore, T. E., et al. (2003). IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4, 63–68. doi: 10.1038/ni873
Shi, C., and Pamer, E. G. (2011). Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11, 762–774. doi: 10.1038/nri3070
Sonoda, J., Laganiere, J., Mehl, I. R., Barish, G. D., Chong, L.-W., Li, X., et al. (2007). Nuclear receptor ERR-alpha and coactivator PGC-1beta are effectors of IFN-gamma-induced host defense. Genes Dev. 21, 1909–1920. doi: 10.1101/gad.1553007
Stavru, F., Archambaud, C., and Cossart, P. (2011). Cell biology and immunology of Listeria monocytogenes infections: novel insights. Immunol. Rev. 240, 160–184. doi: 10.1111/j.1600-065X.2010.00993.x
Stetson, D., and Medzhitov, R. (2006). Type I interferons in host defense. Immunity 25, 373–381. doi: 10.1016/j.immuni.2006.08.007
Stockinger, S., Kastner, R., Kernbauer, E., Pilz, A., Westermayer, S., Reutterer, B., et al. (2009). Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog. 5:e1000355. doi: 10.1371/journal.ppat.1000355
Stockinger, S., Reutterer, B., Schaljo, B., Schellack, C., Brunner, S., Materna, T., et al. (2004). IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J. Immunol. 173, 7416–7425.
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. doi: 10.1126/science.1232458
Swaminathan, B., and Gernersmidt, P. (2007). The epidemiology of human listeriosis. Microbes Infect. 9, 1236–1243. doi: 10.1016/j.micinf.2007.05.011
Tailor, P., Tamura, T., Kong, H. J., Kubota, T., Kubota, M., Borghi, P., et al. (2007). The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 27, 228–239. doi: 10.1016/j.immuni.2007.06.009
Thale, C., and Kiderlen, A. (2005). Sources of interferon-gamma in early immune response to Listeria monocytogenes. Immunobiol 210, 673–683. doi: 10.1016/j.imbio.2005.07.003
Toledo-Arana, A., Dussurget, O., Nikitas, G., Sesto, N., Guet-Revillet, H., Balestrino, D., et al. (2009). The Listeria transcriptional landscape from saprophytism to virulence. Nature 459, 950–956. doi: 10.1038/nature08080
Trinchieri, G. (2010). Type I interferon: friend or foe? J. Exp. Med. 207, 2053–2063. doi: 10.1084/jem.20101664
van de Vosse, E., van Dissel, J. T., and Ottenhoff, T. H. (2009). Genetic deficiencies of innate immune signalling in human infectious disease. Lancet Infect. Dis. 9, 688–698. doi: 10.1016/S1473-3099(09)70255-5
van Pesch, V., Lanaya, H., Renauld, J. C., and Michiels, T. (2004). Characterization of the murine alpha interferon gene family. J. Virol. 78, 8219–8228. doi: 10.1128/JVI.78.15.8219-8228.2004
Vázquez-Boland, J. A., Kuhn, M., Berche, P., Chakraborty, T., Domínguez-Bernal, G., Goebel, W., et al. (2001). Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14, 584–640. doi: 10.1128/CMR.14.3.584-640.2001
Walters, K.-A., Joyce, M. A., Thompson, J. C., Smith, M. W., Yeh, M. M., Proll, S., et al. (2006). Host-specific response to HCV infection in the chimeric SCID-beige/Alb-uPA mouse model: role of the innate antiviral immune response. PLoS Pathog. 2:e59. doi: 10.1371/journal.ppat.0020059
Watanabe, T., Asano, N., Fichtner-Feigl, S., Gorelick, P. L., Tsuji, Y., Matsumoto, Y., et al. (2010). NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J. Clin. Invest. 120, 1645–1662. doi: 10.1172/JCI39481
Wheelock, E. F., and Sibley, W. A. (1965). Circulating virus, interferon and antibody after vaccination with the 17-D strain of yellow-fever virus. N. Engl. J. Med. 273, 194–198. doi: 10.1056/NEJM196507222730404
Wilson, C. B., and Brooks, D. G. (2013). Decoding the complexity of type I interferon to treat persistent viral infections. Trends Microbiol. 21, 634–640. doi: 10.1016/j.tim.2013.10.003
Wilson, C. B., Rowell, E., and Sekimata, M. (2009). Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 9, 91–105. doi: 10.1038/nri2487
Witte, C. E., Archer, K. A., Rae, C. S., Sauer, J.-D., Woodward, J. J., and Portnoy, D. A. (2012). Innate immune pathways triggered by Listeria monocytogenes and their role in the induction of cell-mediated immunity. Adv. Immunol. 113, 135–156. doi: 10.1016/B978-0-12-394590-7.00002-6
Woodward, J. J., Iavarone, A. T., and Portnoy, D. A. (2010). c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705. doi: 10.1126/science.1189801
Wu, J., Sun, L., Chen, X., Du, F., Shi, H., Chen, C., et al. (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. doi: 10.1126/science.1229963
Yamamoto, T., Hara, H., Tsuchiya, K., Sakai, S., Fang, R., Matsuura, M., et al. (2012). Listeria monocytogenes strain-specific impairment of the TetR regulator underlies the drastic increase in cyclic di-AMP secretion and beta interferon-inducing ability. Infect. Immun. 80, 2323–2332. doi: 10.1128/IAI.06162-11
Yang, P., An, H., Liu, X., Wen, M., Zheng, Y., Rui, Y., et al. (2010). The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a β-catenin-dependent pathway. Nat. Immunol. 11, 487–494. doi: 10.1038/ni.1876
Zenewicz, L., and Shen, H. (2007). Innate and adaptive immune responses to Listeria monocytogenes: a short overview. Microbes Infect. 9, 1208–1215. doi: 10.1016/j.micinf.2007.05.008
Zheng, Y.-W., Li, H., Yu, J.-P., Zhao, H., Wang, S. E., and Ren, X.-B. (2013). Interferon-λ s: special immunomodulatory agents and potential therapeutic targets. J. Innate Immun. 5, 209–218. doi: 10.1159/000345365
Keywords: listeriosis, innate immunity, immune escape, PgdA, LntA, BAHD1
Citation: Dussurget O, Bierne H and Cossart P (2014) The bacterial pathogen Listeria monocytogenes and the interferon family: type I, type II and type III interferons. Front. Cell. Infect. Microbiol. 4:50. doi: 10.3389/fcimb.2014.00050
Received: 05 February 2014; Accepted: 04 April 2014;
Published online: 28 April 2014.
Edited by:
Stephanie M. Seveau, The Ohio State University, USAReviewed by:
Sarah E. F. D'Orazio, University of Kentucky, USAJavier A. Carrero, Washington University School of Medicine, USA
Copyright © 2014 Dussurget, Bierne and Cossart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Dussurget, Unité des Interactions Bactéries-Cellules, Institut Pasteur, 25 rue du Dr Roux, 75015 Paris, France e-mail:b2xpdmllci5kdXNzdXJnZXRAcGFzdGV1ci5mcg==
†Present address: Hélène Bierne, INRA, Institut Micalis, UMR1319 AgroParisTech, Jouy-en Josas, France