 Sina Krokowski
Sina Krokowski Serge Mostowy
Serge Mostowy- Department of Medicine, MRC Centre of Molecular Bacteriology and Infection, Imperial College London, London, UK
Autophagy, an intracellular degradation process, is increasingly recognized as having important roles in host defense. Interactions between Shigella flexneri and the autophagy machinery were first discovered in 2005. Since then, work has shown that multiple autophagy pathways are triggered by S. flexneri, and autophagic responses can have different roles during Shigella infection. Here, we review the interactions between S. flexneri and the autophagy machinery, highlighting that studies using Shigella can reveal the breadth of autophagic responses available to the host.
Introduction
The process of autophagy degrades intracellular material via fusion with lysosomes, and is evolutionarily conserved (de Duve and Wattiaux, 1966; Mizushima et al., 1998; Kabeya et al., 2000; Yang and Klionsky, 2010). It was originally discovered as a non-selective nutrient recycling process in response to starvation (Takeshige et al., 1992). More recently, it has been demonstrated that autophagy can act in a selective manner to maintain cellular homeostasis (Khaminets et al., 2015), and that selective autophagy plays an important role in innate immunity by eliminating intracellular pathogens (Levine et al., 2011; Shibutani et al., 2015). Studies using cellular (in vitro) and animal (in vivo) models have shown that a variety of intracellular bacterial pathogens can interact with the autophagy machinery, and autophagic responses can restrict or promote bacterial replication depending on the infection context. In this review, we discuss canonical and non-canonical autophagy pathways, their interplay with invasive bacteria, and highlight what has been discovered from their interactions with Shigella flexneri.
Canonical and Non-canonical Autophagy Pathways
The process of canonical autophagy is dependent on the systematic recruitment of about 40 autophagy-related (ATG) proteins to an isolation membrane called a phagophore (Feng et al., 2014). First, the Unc-51 like kinase (ULK) complex (ULK1, ULK2, ATG13, ATG101, FIP200), and ATG9L recruit the autophagy-specific class III phosphoinositide 3-kinase [PI(3)K] complex (ATG14L, VPS34, Beclin-1, VPS15) to generate phosphatidylinositol 3-phosphate [PI(3)P] required for phagophore formation. Next, members of the WD-repeat domain phosphoinositide-interacting (WIPI) protein family (WIPI1-WIPI4) bind PI(3)P, ATG2, and ATG16L1, and recruit the ATG16L1 complex (ATG16L1, ATG5, ATG12) to elongate the phagophore. Finally, the accumulation of ATG16L1 initiates the conjugation of microtubule-associated protein light chain 3 (LC3) family members to the phagophore, and closes the autophagosome. Thereafter, autophagosomes mature along the endocytic pathway and fuse with lysosomes to form degradative autolysosomes.
In contrast to canonical autophagy, non-canonical autophagy is a mechanism in which only some ATGs help to form an autophagosome-like vacuole (Codogno et al., 2012; Huang and Brumell, 2014). In this case, a subset of ATG proteins can be recruited to an already-existing (and likely damaged) membrane that is different from a phagophore, e.g., a vacuole containing Salmonella or mycobacteria. LC3-associated phagocytosis (LAP) is the best-studied example of a non-canonical autophagy pathway (Cemma and Brumell, 2012; Huang and Brumell, 2014). Toll-like receptors (TLRs) initiate LAP, then ATG5, Beclin-1, and ATG7 are subsequently recruited to promote phagosome maturation, lysosomal fusion, and killing of phagocytosed bacteria (Sanjuan et al., 2007; Lam et al., 2013; Martinez et al., 2015).
Interplay between Invasive Bacteria and the Autophagy Machinery
In the case of bacterial autophagy (also called xenophagy), autophagy receptors such as p62 (sequestosome 1) and NDP52 (nuclear dot protein, 52 kDa) act as cytosolic sensors, bind ubiquitinated substrates and LC3 family proteins to selectively target bacteria to degradation by canonical autophagy (Levine et al., 2011). Canonical autophagy can control the fate of some intracellular bacteria, such as Listeria monocytogenes (Yoshikawa et al., 2009), Francisella tularensis (Checroun et al., 2006), Salmonella enterica subsp. enterica serovar Typhimurum (Thurston et al., 2009; Zheng et al., 2009), and Mycobacterium tuberculosis (Gutierrez et al., 2004). As a result, canonical autophagy is recognized as a critical component of innate immunity (Levine et al., 2011; Shibutani et al., 2015). On the other hand, some bacteria can replicate inside autophagosome-like structures generated by non-canonical autophagy (Huang and Brumell, 2014). Legionella pneumophila (Choy et al., 2012; Asrat et al., 2014), Coxiella burnettii (Newton et al., 2014), Yersinia pseudotuberculosis (Moreau et al., 2010), Brucella abortus (Starr et al., 2012), Staphylococcus aureus (Fraunholz and Sinha, 2012), and Salmonella Typhimurium (Yu et al., 2014) have been described to benefit from non-canonical autophagy.
To determine the precise role of autophagy in host defense against bacteria, it has been useful to investigate the role of bacterial autophagy in vivo. Animal models used to study bacterial autophagy in vivo include Dictyostelium discoideum (amoeba; Tung et al., 2010), Caenorhabditis elegans (nematode; Jia et al., 2009; Zou et al., 2014), Drosophila melanogaster (fruit fly; Yano et al., 2008), Danio rerio (zebrafish; Mostowy et al., 2013; van der Vaart et al., 2014), and Mus musculus (mouse; Castillo et al., 2012; Wang et al., 2012; Watson et al., 2012; Benjamin et al., 2013; Bonilla et al., 2013; Conway et al., 2013; Marchiando et al., 2013; Kimmey et al., 2015). In agreement with results obtained in vitro, the impact of autophagy in vivo depends on the bacterial pathogen under investigation. These alternative outcomes highlight the molecular complexity of bacterial autophagy. They also suggest difficulties in therapeutic manipulation of autophagy to protect against bacterial infection.
Interactions between S. Flexneri and the Autophagy Machinery
Shigella spp. are Gram-negative enteroinvasive pathogens, causing 163 million illness episodes worldwide per annum (Lima et al., 2015). S. flexneri possess a virulence plasmid which encodes a type III secretion system (T3SS), a needle-like apparatus used to inject bacterial effector proteins into the host cell and enable an intracellular lifestyle (Phalipon and Sansonetti, 2007; Ogawa et al., 2008). Minutes after invasion of host cells, including epithelial cells and macrophages, S. flexneri lyses the phagocytic vacuole and gains access to the host cytosol where it replicates (Ray et al., 2009; Fredlund and Enninga, 2014). To counteract Shigella replication in the cytosol, the host cell employs a variety of antimicrobial responses, including antibacterial autophagy and septin caging (septin caging as a mechanism of host defense will be described later in the text; Ogawa et al., 2005; Mostowy et al., 2010). However, to evade cytosolic immune responses, some cytosolic Shigella can subvert the host actin cytoskeleton to form propulsive actin tails and spread from cell-to-cell (Bernardini et al., 1989; Welch and Way, 2013). Thus, in addition to being an important human pathogen, S. flexneri has emerged as a paradigm to study cell-autonomous immunity and host cell biology during infection (Sansonetti, 2006; Ashida et al., 2011, 2015; Mostowy and Shenoy, 2015).
Autophagy Pathways Induced during Shigella Invasion of Host Cells
S. flexneri invasion of normally non-phagocytic epithelial cells relies upon T3SS effector proteins to induce reorganization of the host cell cytoskeleton, membrane ruffling, and bacterial uptake (Cossart and Sansonetti, 2004). Following entry, the pattern recognition nucleotide-binding oligomerization domain (NOD)- like receptors NOD1 and NOD2 detect bacterial peptidoglycan and trigger pro-inflammatory signaling cascades that restrict bacterial survival (Philpott et al., 2014). Work using the human epithelial cell line HeLa and S. flexneri has demonstrated that NOD proteins interact with ATG16L1, thereby recruiting the autophagy machinery to the bacterial entry site at the plasma membrane (Travassos et al., 2010). These data suggest that NOD proteins link bacterial sensing with autophagosome biogenesis (Figure 1). Interestingly, ATG16L1 also has an autophagy-independent role in the control of Shigella and NOD-mediated inflammatory responses in epithelial cells (Sorbara et al., 2013). In this case, ATG16L1 inhibits NOD1- and NOD2-driven cytokine responses to cytosolic bacteria.
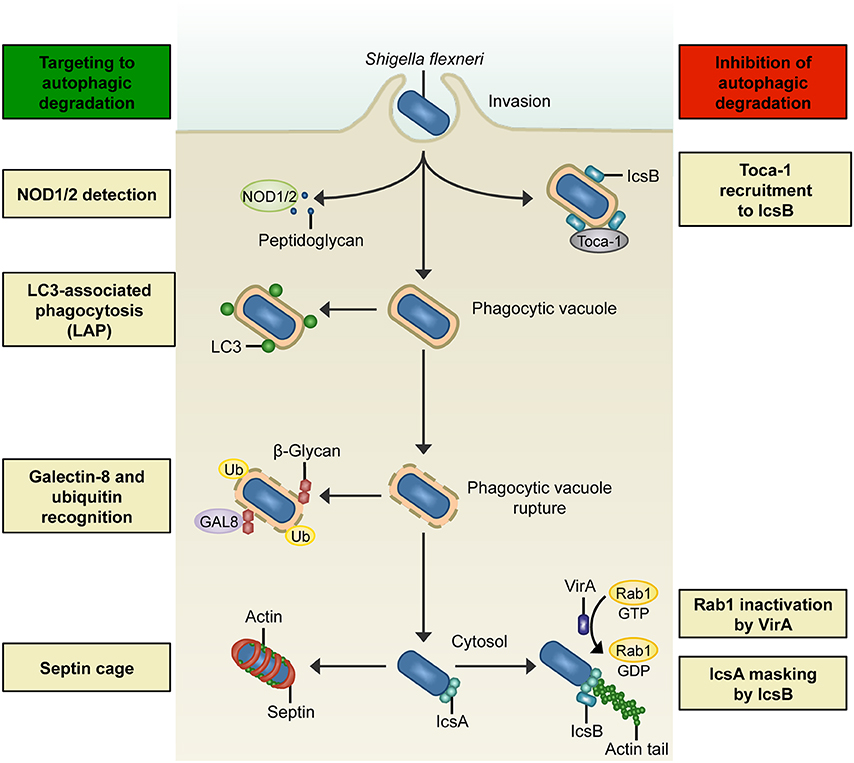
Figure 1. Interactions between S. flexneri and the autophagy machinery. Left: Summary of host mechanisms targeting S. flexneri to degradation by autophagic processes. NOD1/2 detects bacterial peptidoglycan during bacterial entry and recruits ATG16L, thereby triggering an autophagic response. In LC3-associated phagocytosis (LAP), a subset of autophagy proteins (e.g., LC3) is recruited to phagosomal membranes and promotes fusion with lysosomes. Membrane remnants can recruit autophagy components by ubiquitination (Ub), and also by recognition of host cell β-glycans by galectin-8 (GAL8). In the cytosol, actin-polymerizing bacteria can be recognized by ATG5 and entrapped in septin cage-like structures, thereby targeting bacteria to autophagic degradation and preventing their dissemination. Right: Overview of S. flexneri effectors that inhibit degradation by autophagic processes. During invasion, the bacterial effector IcsB recruits Toca-1, which prevents the recruitment of LC3 and other autophagy markers. In the cytosol, Shigella can circumvent Atg5-recognition of IcsA and septin caging by expressing IcsB. Another mechanism of S. flexneri to inhibit autophagosome formation in the cytosol is to secrete VirA to inactivate Rab1.
LAP can also occur during Shigella invasion of epithelial cells (Figure 1), where LAP is dependent on the activity of the T3SS (Campbell-Valois et al., 2015). However, Shigella has a mechanism mediated by the T3SS effector IcsB to counteract LAP. IcsB is a 52 kDa protein that uses the bacterial chaperone IpgA for its stability (Ogawa et al., 2003). Secreted IcsB localizes around the bacterial surface, and though not required for bacterial entry, plays an important role during cell-to-cell spread. Soon after bacterial entry, IcsB recruits transducer of CDC42-dependent actin assembly 1 (Toca-1) to prevent the recruitment of NDP52 and LC3 (Baxt and Goldberg, 2014). In uninfected cells, Toca-1 and neuronal Wiskott-Aldrich syndrome protein (N-WASP) are recruited to the membrane by direct interactions with the Rho guanosine triphosphatase (GTPase) CDC42 (Ho et al., 2004). In contrast, S. flexneri recruits Toca-1 via IcsB (to inhibit recognition by autophagy machinery) and N-WASP via IcsA (to polymerize actin; Leung et al., 2008; Baxt and Goldberg, 2014). Of note, S. flexneri can also be recognized by LC3 after cell-to-cell spreading, when the bacterium is entrapped in a double-membrane vacuole (Campbell-Valois et al., 2015). Here, Shigella can avoid autophagic degradation by secreting IcsB and VirA (the virulence factor VirA will be described later in the text), which act synergistically to promote bacterial escape from LC3-positive vacuoles.
Together, different sensing mechanisms including NOD1/2 and LAP trigger autophagic recognition of invading Shigella. These mechanisms can be viewed as a crucial aspect of host defense to control bacteria at the onset of bacterial interactions with the host cell.
Autophagy Pathways Induced by Shigella Rupture of the Phagocytic Vacuole
Shortly after invasion of host cells, S. flexneri can exit the phagocytic vacuole and gain access to the cytosol. In epithelial cells, rupture of the phagocytic vacuole by Shigella can initiate autophagy by inducing nutrient starvation (Tattoli et al., 2012). In this case, Shigella causes downregulation of the autophagy inhibitor mammalian target of rapamycin complex 1 (mTORC1, which inhibits the ULK1–Atg13–FIP200 complex in the presence of nutrients). From this, it has been proposed that Shigella-induced amino acid starvation can be sensed by the host cell to initiate an immune response.
When Shigella ruptures its phagocytic vacuole in epithelial cells, membrane remnants can be ubiquitinated (Dupont et al., 2009; Figure 1). Here, autophagy receptors p62 and NDP52 recognize ubiquitinated membrane remnants and recruit LC3-positive membrane for autophagosome biogenesis. Moreover, p62 on membrane remnants colocalizes with nuclear factor kappa B (NF-κB) signaling molecules, including tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), thereby serving to dampen the inflammatory response (Dupont et al., 2009). Thus, autophagic degradation has important roles in the control of both infection and inflammation (Shi et al., 2012; Deretic et al., 2013; Shibutani et al., 2015).
Membrane damage exposes host cell glycans to the cytosol, which are damage-associated molecular patterns (DAMPs) recognized by galectins, a family of beta-galactoside-binding proteins (Figure 1). Studies using epithelial cells and macrophages have demonstrated that galectin-3 and galectin-8 accumulate at sites of membrane damage induced by Shigella (Paz et al., 2010; Thurston et al., 2012). While a role for galectin-3 in host defense is not yet clear, galectin-8 initiates the recruitment of NDP52 and LC3 to direct the autophagy machinery to the ruptured phagocytic vacuole (Thurston et al., 2012). The galectin-8-NDP52-LC3 pathway is viewed to occur upstream of the ubiquitin-NDP52-LC3 pathway also targeting the phagocytic vacuole ruptured by Shigella (Paz et al., 2010; Thurston et al., 2012; Boyle and Randow, 2013). In both cases, membrane can act as a danger signal used to direct autophagy components toward invasive bacteria. However, canonical autophagy may also promote repair of damaged vacuolar membranes, as in the case of S. Typhimurium-containing vacuoles (Kreibich et al., 2015). Therefore, the precise role of membrane recognition by the autophagy machinery in the restriction of bacterial proliferation is unknown.
Autophagy Pathways Induced by Shigella in the Cytosol
Once in the cytosol, S. flexneri can subvert the host actin cytoskeleton by expressing the outer membrane autotransporter protein IcsA to recruit N-WASP and the actin related protein 2/3 (ARP2/3) complex, inducing actin-based motility to spread from cell-to-cell (Welch and Way, 2013). IcsA-mediated actin polymerization is also required for recognition of cytosolic Shigella by the autophagy machinery in epithelial cell lines and mouse embryonic fibroblasts (MEFs; Ogawa et al., 2005; Mostowy et al., 2010, 2011). IcsA is recognized by ATG5, triggering the formation of an isolation membrane (Ogawa et al., 2005). Infection of epithelial cell lines and MEFs has shown that this process can be independent of ubiquitin, and is coincident with ATG5 binding to the adaptor protein tectonin beta-propeller repeat containing 1 (TECPR1; Ogawa et al., 2011). TECPR1 interacts with WIPI-2 and PI(3)P, and recruits LC3 to target Shigella to autophagic degradation.
To protect itself from autophagic recognition, cytosolic S. flexneri can inhibit the interaction between IcsA and ATG5 by secreting IcsB, which binds to IcsA and competitively inhibits ATG5 binding (Ogawa et al., 2005; Figure 1). The region of IcsA bound by IcsB/ATG5 overlaps with one of the three N-WASP interacting regions (Ogawa et al., 2005; May and Morona, 2008). It is unknown if N-WASP competes with IcsB/ATG5 in binding to the same region of IcsA, or whether the two other binding regions of IcsA are sufficient to recruit N-WASP. Bacteria without IcsB exhibit an intracellular replication defect in MEFs, highlighting the crucial role of ATG5 recognition in the control of Shigella (Ogawa et al., 2005). Historically, this discovery made Shigella the first example of an intracellular pathogen having an effector that inhibits autophagic recognition for intracellular survival.
Shigella expression of VirA is a separate mechanism to counteract autophagy in epithelial cells (Dong et al., 2012). VirA is localized upstream of IcsA on the Shigella virulence plasmid and is secreted during invasion of host cells (Uchiya et al., 1995), where it acts as a GTPase-activating protein (GAP) that inactivates the GTPase Rab1 (Dong et al., 2012; Figure 1). Rab1, implicated in vesicular transport between the endoplasmic reticulum (ER) and the Golgi, has an important role in autophagosome formation (Zoppino et al., 2010). By inactivating Rab1, VirA disrupts ER-to-Golgi trafficking, and mediates suppression of autophagosome formation against S. flexneri (Dong et al., 2012). VirA is one of 2 bacterial effectors discovered so far, the other being the type 4 secretion system (T4SS) effector RavZ from L. pneumophila (Choy et al., 2012), that can directly interfere with the autophagy machinery (Huang and Brumell, 2014; Mostowy and Shenoy, 2015).
Septins, GTP-binding proteins essential for cell division (Saarikangas and Barral, 2011; Mostowy and Cossart, 2012b), interact with the autophagy machinery and play a crucial role in the control of cytosolic S. flexneri (Mostowy et al., 2010, 2011). Septins are recognized as cytoskeletal components because they form filaments that associate with cellular membranes, actin filaments, and microtubules (Saarikangas and Barral, 2011; Mostowy and Cossart, 2012b; Bezanilla et al., 2015). In epithelial cells, septins are recruited to sites of IcsA-mediated actin polymerization and form cage-like structures necessary for the recruitment of ubiquitin, p62, NDP52, and LC3 to cytosolic Shigella (Mostowy et al., 2010, 2011; Figure 1). Consistent with its role as an autophagy inhibitor, IcsB also masks Shigella from septin caging (Mostowy et al., 2010, 2011). Importantly, septin cages have also been observed in vivo using zebrafish, highlighting septin assembly as an evolutionarily conserved determinant of host defense (Mostowy et al., 2013). From the Shigella-zebrafish infection model, p62 is crucial for septin caging and the restriction of bacterial replication by autophagy in vivo. On the other hand, rapamycin (an inhibitor of mTOR and stimulant of autophagy) can increase Shigella replication and decrease zebrafish survival. Overall, these data indicate that host survival depends on the appropriate autophagic response to control Shigella infection in vivo (Mostowy et al., 2013). In the case of humans, the ATG16L1 T300A polymorphism linked to Crohn's disease, an inflammatory bowel disease, can reduce the process of selective autophagy and host defense against Shigella (Lassen et al., 2014).
Discussion
Originally discovered as a bulk degradation process important for cellular homeostasis (Yang and Klionsky, 2010), autophagy has since been recognized as important for cell-autonomous immunity and host defense (Levine et al., 2011; Mostowy and Shenoy, 2015; Shibutani et al., 2015). On the other hand, some intracellular pathogens have evolved mechanisms to evade or interfere with autophagy processes for intracellular survival (Mostowy and Cossart, 2012a; Choy and Roy, 2013; Huang and Brumell, 2014). Adding to this complexity, it is now clear that ATG proteins also have autophagy-independent roles in immunity and cellular homeostasis (Huang and Brumell, 2014; Mostowy and Shenoy, 2015). In this review, we have highlighted how studies using Shigella can help to investigate the breadth of autophagy responses available to the host.
The S. flexneri infection model can be used to study key issues in cell-autonomous immunity and host cell biology, including the ability of host cells to sense and defend against intracellular bacteria. The host cell has evolved multiple sensing mechanisms that together enable a multi-tiered defense network to counteract Shigella invasion (Table 1). However, Shigella has co-evolved a variety of protection mechanisms, and in some cases can interfere with host cell biology to circumvent autophagic processes (Table 1). Despite the understanding gained from using the Shigella infection model, many outstanding issues remain. For example, what is the role of the host cytoskeleton in autophagy and its ability to restrict or promote bacterial replication? Actin, microtubules, intermediate filaments, and septins are four main cytoskeletal components of mammalian cells, yet their precise roles in autophagy are not understood (Mostowy, 2014). What is the link between autophagy of bacteria and autophagy of mitochondria (also called mitophagy)? Mitochondria can be viewed as bacteria-derived endosymbionts, and recent studies have discovered that autophagy of bacteria and mitochondria use the same machinery (Manzanillo et al., 2013; Randow and Youle, 2014). Finally, to fully determine the role of autophagy in host defense against Shigella will require in depth experimentation to be performed in vivo. Though mammalian models remain poorly suited to image the cell biology of Shigella infection in vivo, the natural translucency of zebrafish larvae enables non-invasive in vivo imaging at high resolution throughout the organism (Kanther and Rawls, 2010; Renshaw and Trede, 2012). Exploiting this, the in vivo role of Shigella interactions with the autophagy machinery can be examined at the subcellular, cellular, and whole-animal level (Mostowy et al., 2013).
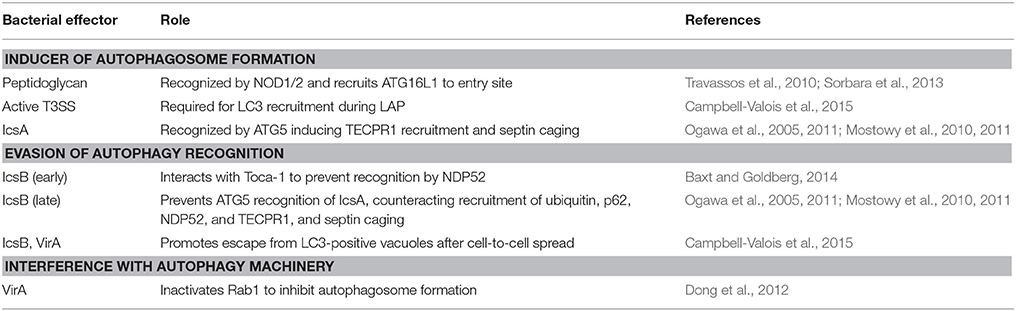
Table 1. S. flexneri mechanisms to induce, evade, or interfere with the autophagy machinery.
Taken together, outcomes generated from studying the interplay between S. flexneri and the autophagy machinery in vitro using tissue culture cells and in vivo using animal models can help to better understand fundamental mechanisms of host defense. They could additionally suggest the development of novel therapies for infectious diseases.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the SM laboratory is supported by a Wellcome Trust Research Career Development Fellowship (WT097411MA) and the Lister Institute of Preventive Medicine.
References
Ashida, H., Mimuro, H., and Sasakawa, C. (2015). Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front. Immunol. 6:219. doi: 10.3389/fimmu.2015.00219
Ashida, H., Ogawa, M., Mimuro, H., Kobayashi, T., Sanada, T., and Sasakawa, C. (2011). Shigella are versatile mucosal pathogens that circumvent the host innate immune system. Curr. Opin. Immunol. 23, 448–455. doi: 10.1016/j.coi.2011.06.001
Asrat, S., de Jesús, D. A., Hempstead, A. D., Ramabhadran, V., and Isberg, R. R. (2014). Bacterial pathogen manipulation of host membrane trafficking. Ann. Rev. Cell Dev. Biol. 30, 79–109. doi: 10.1146/annurev-cellbio-100913-013439
Baxt, L. A., and Goldberg, M. B. (2014). Host and bacterial proteins that repress recruitment of LC3 to Shigella early during infection. PLoS ONE 9:e94653. doi: 10.1371/journal.pone.0094653
Benjamin, J. L., Sumpter, R. Jr, Levine, B., and Hooper, L. V. (2013). Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 13, 723–734. doi: 10.1016/j.chom.2013.05.004
Bernardini, M. L., Mounier, J., d'Hauteville, H., Coquis-Rondon, M., and Sansonetti, P. J. (1989). Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. U.S.A. 86, 3867–3871. doi: 10.1073/pnas.86.10.3867
Bezanilla, M., Gladfelter, A. S., Kovar, D. R., and Lee, W. L. (2015). Cytoskeletal dynamics: a view from the membrane. J. Cell Biol. 209, 329–337. doi: 10.1083/jcb.201502062
Bonilla, D., Bhattacharya, A., Sha, Y., Xu, Y., Xiang, Q., Kan, A., et al. (2013). Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 39, 537–547. doi: 10.1016/j.immuni.2013.08.026
Boyle, K. B., and Randow, F. (2013). The role of ‘eat-me’ signals and autophagy cargo receptors in innate immunity. Curr. Opin. Microbiol. 16, 339–348. doi: 10.1016/j.mib.2013.03.010
Campbell-Valois, F.-X., Sachse, M., Sansonetti, P. J., and Parsot, C. (2015). Escape of actively secreting Shigella flexneri from ATG8/LC3-positive vacuoles formed during cell-to-cell spread is facilitated by IcsB and VirA. mBio 6:e02567–14. doi: 10.1128/mBio.02567-14
Castillo, E. F., Dekonenko, A., Arko-Mensah, J., Mandell, M. A., Dupont, N., Jiang, S., et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. U.S.A. 109, E3168–E3176. doi: 10.1073/pnas.1210500109
Cemma, M., and Brumell, J. H. (2012). Interactions of pathogenic bacteria with autophagy systems. Curr. Biol. 22, R540–R545. doi: 10.1016/j.cub.2012.06.001
Checroun, C., Wehrly, T. D., Fischer, E. R., Hayes, S. F., and Celli, J. (2006). Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U.S.A. 103, 14578–14583. doi: 10.1073/pnas.0601838103
Choy, A., Dancourt, J., Mugo, B., O'Connor, T. J., Isberg, R. R., Melia, T. J., et al. (2012). The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1076. doi: 10.1126/science.1227026
Choy, A., and Roy, C. R. (2013). Autophagy and bacterial infection: an evolving arms race. Trends Microbiol. 21, 451–456. doi: 10.1016/j.tim.2013.06.009
Codogno, P., Mehrpour, M., and Proikas-Cezanne, T. (2012). Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 13, 7–12. doi: 10.1038/nrm3249
Conway, K. L., Kuballa, P., Song, J. H., Patel, K. K., Castoreno, A. B., Yilmaz, O. H., et al. (2013). Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 145, 1347–1357. doi: 10.1053/j.gastro.2013.08.035
Cossart, P., and Sansonetti, P. J. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248. doi: 10.1126/science.1090124
de Duve, C., and Wattiaux, R. (1966). Functions of lysosomes. Ann. Rev. Physiol. 28, 435–492. doi: 10.1146/annurev.ph.28.030166.002251
Deretic, V., Saitoh, T., and Akira, S. (2013). Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737. doi: 10.1038/nri3532
Dong, N., Zhu, Y., Lu, Q., Hu, L., Zheng, Y., and Shao, F. (2012). Structurally distinct bacterial TBC-like GAPs link arf GTPase to Rab1 inactivation to counteract host defenses. Cell 150, 1029–1041. doi: 10.1016/j.cell.2012.06.050
Dupont, N., Lacas-Gervais, S., Bertout, J., Paz, I., Freche, B., Van Nhieu, G. T., et al. (2009). Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6, 137–149. doi: 10.1016/j.chom.2009.07.005
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Fraunholz, M., and Sinha, B. (2012). Intracellular Staphylococcus aureus: live-in and let die. Front. Cell. Infect. Microbiol. 2, 43. doi: 10.3389/fcimb.2012.00043
Fredlund, J., and Enninga, J. (2014). Cytoplasmic access by intracellular bacterial pathogens. Trends Microbiol. 22, 128–137. doi: 10.1016/j.tim.2014.01.003
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. doi: 10.1016/j.cell.2004.11.038
Ho, H. Y., Rohatgi, R., Lebensohn, A. M., Le, M., Li, J., Gygi, S. P., et al. (2004). Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell 118, 203–216. doi: 10.1016/j.cell.2004.06.027
Huang, J., and Brumell, J. H. (2014). Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbol. 12, 101–114. doi: 10.1038/nrmicro3160
Jia, K., Thomas, C., Akbar, M., Sun, Q., Adams-Huet, B., Gilpin, C., et al. (2009). Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc. Natl. Acad. Sci. U.S.A. 106, 14564–14569. doi: 10.1073/pnas.0813319106
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Kanther, M., and Rawls, J. F. (2010). Host–microbe interactions in the developing zebrafish. Curr. Opin. Immunol. 22, 10–19. doi: 10.1016/j.coi.2010.01.006
Khaminets, A., Behl, C., and Dikic, I. (2015). Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 1, 6–16. doi: 10.1016/j.tcb.2015.08.010
Kimmey, J. M., Huynh, J. P., Weiss, L. A., Park, S., Kambal, A., Debnath, J., et al. (2015). Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569. doi: 10.1038/nature16451
Kreibich, S., Emmenlauer, M., Fredlund, J., Rämö, P., Münz, C., Dehio, C., et al. (2015). Autophagy proteins promote repair of endosomal membranes damaged by the Salmonella type three secretion system 1. Cell Host Microbe 18, 527–537. doi: 10.1016/j.chom.2015.10.015
Lam, G. Y., Cemma, M., Muise, A. M., Higgins, D. E., and Brumell, J. H. (2013). Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 9, 985–995. doi: 10.4161/auto.24406
Lassen, K. G., Kuballa, P., Conway, K. L., Patel, K. K., Becker, C. E., Peloquin, J. M., et al. (2014). Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. U.S.A. 111, 7741–7746. doi: 10.1073/pnas.1407001111
Leung, Y., Ally, S., and Goldberg, M. B. (2008). Bacterial actin assembly requires Toca-1 to relieve N-WASP autoinhibition. Cell Host Microbe 3, 39–47. doi: 10.1016/j.chom.2007.10.011
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335. doi: 10.1038/nature09782
Lima, I., Havt, A., and Lima, A. A. (2015). Update on molecular epidemiology of Shigella infection. Curr. Opin. Gastroenterol. 31, 30–37. doi: 10.1097/MOG.0000000000000136
Manzanillo, P. S., Ayres, J. S., Watson, R. O., Collins, A. C., Souza, G., Rae, C. S., et al. (2013). The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501, 512–516. doi: 10.1038/nature12566
Marchiando, A., Ramanan, D., Ding, Y., Gomez, L. E., Hubbard-Lucey, V., Maurer, K., et al. (2013). A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe 14, 216–224. doi: 10.1016/j.chom.2013.07.013
Martinez, J., Malireddi, R. K. S., Lu, Q., Cunha, L. D., Pelletier, S., Gingras, S., et al. (2015). Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906. doi: 10.1038/ncb3192
May, K. L., and Morona, R. (2008). Mutagenesis of the Shigella flexneri autotransporter IcsA reveals novel functional regions involved in IcsA biogenesis and recruitment of host neural Wiscott-Aldrich syndrome protein. J. Bacteriol. 190, 4666–4676. doi: 10.1128/JB.00093-08
Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ishii, T., George, M. D., et al. (1998). A protein conjugation system essential for autophagy. Nature 395, 395–398. doi: 10.1038/26506
Moreau, K., Lacas-Gervais, S., Fujita, N., Sebbane, F., Yoshimori, T., Simonet, M., et al. (2010). Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell. Microbiol. 12, 1108–1123. doi: 10.1111/j.1462-5822.2010.01456.x
Mostowy, S., Bonazzi, M., Hamon, M. A., Tham, T. N., Mallet, A., Lelek, M., et al. (2010). Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8, 433–444. doi: 10.1016/j.chom.2010.10.009
Mostowy, S., Boucontet, L., Mazon Moya, M. J., Sirianni, A., Boudinot, P., Hollinshead, M., et al. (2013). The zebrafish as a new model for the in vivo study of Shigella flexneri interaction with phagocytes and bacterial autophagy. PLoS Pathog. 9:e1003588. doi: 10.1371/journal.ppat.1003588
Mostowy, S., and Cossart, P. (2012a). Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. 22, 283–291. doi: 10.1016/j.tcb.2012.03.006
Mostowy, S., and Cossart, P. (2012b). Septins: the fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 13, 183–194. doi: 10.1038/nrm3284
Mostowy, S., Sancho-Shimizu, V., Hamon, M. A., Simeone, R., Brosch, R., Johansen, T., et al. (2011). p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 286, 26987–26995. doi: 10.1074/jbc.M111.223610
Mostowy, S., and Shenoy, A. R. (2015). The cytoskeleton in cell-autonomous immunity: structural determinants of host defence. Nat. Rev. Immunol. 15, 559–573. doi: 10.1038/nri3877
Mostowy, S. (2014). Multiple roles of the cytoskeleton in bacterial autophagy. PLoS Pathog. 10:e1004409. doi: 10.1371/journal.ppat.1004409
Newton, H. J., Kohler, L. J., McDonough, J. A., Temoche-Diaz, M., Crabill, E., Hartland, E. L., et al. (2014). A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 10:e1004286. doi: 10.1371/journal.ppat.1004286
Ogawa, M., Handa, Y., Ashida, H., Suzuki, M., and Sasakawa, C. (2008). The versatility of Shigella effectors. Nat. Rev. Microbiol. 6, 11–16. doi: 10.1038/nrmicro1814
Ogawa, M., Suzuki, T., Tatsuno, I., Abe, H., and Sasakawa, C. (2003). IcsB, secreted via the type III secretion system, is chaperoned by IpgA and required at the post-invasion stage of Shigella pathogenicity. Mol. Microbiol. 48, 913–931. doi: 10.1046/j.1365-2958.2003.03489.x
Ogawa, M., Yoshikawa, Y., Kobayashi, T., Mimuro, H., Fukumatsu, M., Kiga, K., et al. (2011). A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe 9, 376–389. doi: 10.1016/j.chom.2011.04.010
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731. doi: 10.1126/science.1106036
Paz, I., Sachse, M., Dupont, N., Mounier, J., Cederfur, C., Enninga, J., et al. (2010). Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell. Microbiol. 12, 530–544. doi: 10.1111/j.1462-5822.2009.01415.x
Phalipon, A., and Sansonetti, P. J. (2007). Shigella's ways of manipulating the host intestinal innate and adaptive immune system: a tool box for survival? Immunol. Cell Biol. 85, 119–129. doi: 10.1038/sj.icb.7100025
Philpott, D. J., Sorbara, M. T., Robertson, S. J., Croitoru, K., and Girardin, S. E. (2014). NOD proteins: regulators of inflammation in health and disease. Nat. Rev. Immunol. 14, 9–23. doi: 10.1038/nri3565
Randow, F., and Youle, R. J. (2014). Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15, 403–411. doi: 10.1016/j.chom.2014.03.012
Ray, K., Marteyn, B., Sansonetti, P. J., and Tang, C. M. (2009). Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Micro 7, 333–340. doi: 10.1038/nrmicro2112
Renshaw, S. A., and Trede, N. S. (2012). A model 450 million years in the making: zebrafish and vertebrate immunity. Dis. Model. Mech. 5, 38–47. doi: 10.1242/dmm.007138
Saarikangas, J., and Barral, Y. (2011). The emerging functions of septins in metazoans. EMBO Rep. 12, 1118–1126. doi: 10.1038/embor.2011.193
Sanjuan, M. A., Dillon, C. P., Tait, S. W. G., Moshiach, S., Dorsey, F., Connell, S., et al. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257. doi: 10.1038/nature06421
Sansonetti, P. J. (2006). The bacterial weaponry: lessons from Shigella. Ann. N. Y. Acad. Sci. 1072, 307–312. doi: 10.1196/annals.1326.025
Shi, C. S., Shenderov, K., Huang, N.-N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of autophagy by inflammatory signals limits IL-1B production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. doi: 10.1038/ni.2215
Shibutani, S. T., Saitoh, T., Nowag, H., Münz, C., and Yoshimori, T. (2015). Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 16, 1014–1024. doi: 10.1038/ni.3273
Sorbara, M., Ellison, L., Ramjeet, M., Travassos, L., Jones, N., Girardin, S., et al. (2013). The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 39, 858–873. doi: 10.1016/j.immuni.2013.10.013
Starr, T., Child, R., Wehrly, T. D., Hansen, B., Hwang, S., López-Otin, C., et al. (2012). Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11, 33–45. doi: 10.1016/j.chom.2011.12.002
Takeshige, K., Baba, M., Tsuboi, S., Noda, T., and Ohsumi, Y. (1992). Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311. doi: 10.1083/jcb.119.2.301
Tattoli, I., Sorbara, M. T., Vuckovic, D., Ling, A., Soares, F., Carneiro, L. A. M., et al. (2012). Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575. doi: 10.1016/j.chom.2012.04.012
Thurston, T. L. M., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221. doi: 10.1038/ni.1800
Thurston, T. L. M., Wandel, M. P., von Muhlinen, N., Foeglein, A., and Randow, F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. doi: 10.1038/nature10744
Travassos, L. H., Carneiro, L. A. M., Ramjeet, M., Hussey, S., Kim, Y.-G., Magalhaes, J. G., et al. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62. doi: 10.1038/ni.1823
Tung, S. M., Ünal, C., Ley, A., Peña, C., Tunggal, B., Noegel, A. A., et al. (2010). Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell. Microbiol. 12, 765–780. doi: 10.1111/j.1462-5822.2010.01432.x
Uchiya, K., Tobe, T., Komatsu, K., Suzuki, T., Watarai, M., Fukuda, I., et al. (1995). Identification of a novel virulence gene, virA, on the large plasmid of Shigella, involved in invasion and intercellular spreading. Mol. Microbiol. 17, 241–250. doi: 10.1111/j.1365-2958.1995.mmi_17020241.x
van der Vaart, M., Korbee, C. J., Lamers, G. E., Tengeler, A. C., Hosseini, R., Haks, M. C., et al. (2014). The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense. Cell Host Microbe 15, 753–767. doi: 10.1016/j.chom.2014.05.005
Wang, C., Mendonsa, G. R., Symington, J. W., Zhang, Q., Cadwell, K., Virgin, H. W., et al. (2012). Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc. Natl. Acad. Sci. U.S.A. 109, 11008–11013. doi: 10.1073/pnas.1203952109
Watson, R. O., Manzanillo, P. S., and Cox, J. S. (2012). Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815. doi: 10.1016/j.cell.2012.06.040
Welch, M. D., and Way, M. (2013). Arp2/3-mediated actin-based motility: a tail of pathogen abuse. Cell Host Microbe 14, 242–255. doi: 10.1016/j.chom.2013.08.011
Yang, Z., and Klionsky, D. J. (2010). Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822. doi: 10.1038/ncb0910-814
Yano, T., Mita, S., Ohmori, H., Oshima, Y., Fujimoto, Y., Ueda, R., et al. (2008). Autophagic control of Listeria through intracellular innate immune recognition in Drosophila. Nat. Immunol. 9, 908–916. doi: 10.1038/ni.1634
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., et al. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240. doi: 10.1038/ncb1967
Yu, H. B., Croxen, M. A., Marchiando, A. M., Ferreira, R. B., Cadwell, K., Foster, L. J., et al. (2014). Autophagy facilitates Salmonella replication in HeLa cells. mBio 5:e00865–14. doi: 10.1128/mBio.00865-14
Zheng, Y. T., Shahnazari, S., Brech, A., Lamark, T., Johansen, T., and Brumell, J. H. (2009). The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916. doi: 10.4049/jimmunol.0900441
Zoppino, F. C. M., Militello, R. D., Slavin, I., Álvarez, C., and Colombo, M. I. (2010). Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 11, 1246–1261. doi: 10.1111/j.1600-0854.2010.01086.x
Keywords: autophagy, cytoskeleton, innate immunity, inflammation, Shigella flexneri
Citation: Krokowski S and Mostowy S (2016) Interactions between Shigella flexneri and the Autophagy Machinery. Front. Cell. Infect. Microbiol. 6:17. doi: 10.3389/fcimb.2016.00017
Received: 20 November 2015; Accepted: 25 January 2016;
Published: 10 February 2016.
Edited by:
William D. Picking, University of Kansas, USAReviewed by:
Kenneth G. Campellone, University of Connecticut, USASusu M. Zughaier, Emory University, USA
Thomas A. Kufer, University of Hohenheim, Germany
Copyright © 2016 Krokowski and Mostowy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serge Mostowy, cy5tb3N0b3d5QGltcGVyaWFsLmFjLnVr