 Liljana Petrovska1*
Liljana Petrovska1* Yue Tang1†
Yue Tang1† Melissa J. Jansen van Rensburg2,3†
Melissa J. Jansen van Rensburg2,3† Shaun Cawthraw1Javier Nunez4Samuel K. Sheppard5
Shaun Cawthraw1Javier Nunez4Samuel K. Sheppard5 Richard J. Ellis1
Richard J. Ellis1 Adrian M. Whatmore1
Adrian M. Whatmore1 Tim R. Crawshaw1Richard M. Irvine1
Tim R. Crawshaw1Richard M. Irvine1- 1Bacteriology, Animal and Plant Health Agency Weybridge, Addlestone, United Kingdom
- 2Department of Zoology, University of Oxford, Oxford, United Kingdom
- 3NIHR Health Protection Research Unit in Gastrointestinal Infections, University of Oxford, Oxford, United Kingdom
- 4Veterinary Surveillance, Animal and Plant Health Agency Weybridge, Addlestone, United Kingdom
- 5Department of Biology and Biotechnology, The Milner Centre for Evolution, University of Bath, Bath, United Kingdom
The term “spotty liver disease” (SLD) has been used since the late 1990s for a condition seen in the UK and Australia that primarily affects free range laying hens around peak lay, causing acute mortality and a fall in egg production. A novel thermophilic SLD-associated Campylobacter was reported in the United Kingdom (UK) in 2015. Subsequently, similar isolates occurring in Australia were formally described as a new species, Campylobacter hepaticus. We describe the comparative genomics of 10 C. hepaticus isolates recovered from 5 geographically distinct poultry holdings in the UK between 2010 and 2012. Hierarchical gene-by-gene analyses of the study isolates and representatives of 24 known Campylobacter species indicated that C. hepaticus is most closely related to the major pathogens Campylobacter jejuni and Campylobacter coli. We observed low levels of within-farm variation, even between isolates collected over almost 3 years. With respect to C. hepaticus genome features, we noted that the study isolates had a ~140 Kb reduction in genome size, ~144 fewer genes, and a lower GC content compared to C. jejuni. The most notable reduction was in the subsystem containing genes for iron acquisition and metabolism, supported by reduced growth of C. hepaticus in an iron depletion assay. Genome reduction is common among many pathogens and in C. hepaticus has likely been driven at least in part by specialization following the occupation of a new niche, the chicken liver.
Introduction
Spotty liver disease (SLD) is an important concern for the poultry egg and meat industries. The disease is sporadic in nature and predominantly affects free-range laying hens, causing a drop in egg production, and up to 10% mortality in some flocks (Grimes and Reece, 2011; Jennings et al., 2011; Crawshaw et al., 2015). SLD is characterized by the appearance of 1–2 mm gray/white foci in the liver, described as multifocal fibrinogranulocytic necrotising hepatitis when examined microscopically (Crawshaw et al., 2015). There are similarities between the epidemiology and pathology of SLD and vibrionic hepatitis, with the terms used interchangeably (Jennings et al., 2011). Vibrionic hepatitis was first described in the United States of America in the 1950s (Tudor, 1954) but appears to have declined since the 1960s (Shane and Stern, 2003). A Vibrio-like organism was isolated from cases of vibrionic hepatitis and disease was reproduced (Delaplane et al., 1955; Winterfield and Sevoian, 1957); however, the organism was never fully characterized and questions remain about the nature of this disease and how it relates to the contemporary manifestations of SLD.
Although our understanding of SLD remains limited, recent studies have implicated Campylobacter as an etiological agent. The microscopic pathology of SLD was reproduced in specific pathogen-free (SPF) chicks experimentally infected with a novel thermophilic Campylobacter isolated from SLD cases in the UK (Crawshaw et al., 2015). Analysis of 16S rRNA gene sequences suggested that the SLD-associated Campylobacter isolates represented a novel species within the genus. These organisms grouped with the other thermotolerant Campylobacter species, showing most pairwise identity with type strains of C. jejuni, Campylobacter lari and Campylobacter subantarticus, and Campyloacter insulaenigrae, members of the C. lari group previously isolated from wild birds and marine mammals respectively (Miller et al., 2014a). In 2016, Van et al. formally described a novel thermophilic Campylobacter species, C. hepaticus, which was isolated from cases of SLD in Australia (Van et al., 2016) using the approach pioneered in the UK. C. hepaticus was subsequently confirmed to be the causative agent of SLD, with gross liver lesions typical of clinical cases reproduced in mature layer hens (Van et al., 2017). The results of 16S rRNA gene sequencing, supported by phenotypic and biochemical testing, suggested that the novel SLD-associated Campylobacter previously isolated in the UK was also C. hepaticus. Like other campylobacters, C. hepaticus appears to be fastidious in its growth requirements but is much slower growing than the more commonly isolated thermophilic strains, with some colonies reportedly taking up to 7 days incubation to appear (Crawshaw et al., 2015). This fastidiousness and slow growth may account for the failure of previous attempts to identify the causative agent of SLD. As modified protocols with extended incubation times are not commonly used, the prevalence of C. hepaticus is likely underappreciated.
Among the diverse members of the Campylobacter genus, C. jejuni and C. coli are the best studied, largely because together they are the leading bacterial causes of human gastroenteritis worldwide (Kaakoush et al., 2015). Human disease is strongly associated with the consumption of contaminated poultry (Sheppard et al., 2009a,b), and several genetic factors have been implicated in the survival of Campylobacter outside the host gut (Pascoe et al., 2015; Yahara et al., 2017) and transmission through the food production chain (Yahara et al., 2017). The prevalence and abundance of Campylobacter in the chicken gut and the high levels of carcass contamination at slaughter are thought to contribute to the incidence of human disease (Johnsen et al., 2006; Luber and Bartelt, 2007). Control of Campylobacter in chickens is therefore a potential means for reducing human infection. While human infection is usually thought to cause acute symptoms, Campylobacter has generally been regarded a commensal of chicken. However, there is evidence that C. jejuni does induce humoral and pro-inflammatory responses in chicken (Cawthraw et al., 1994; Smith et al., 2008) and can cause diarrhea (Cogan et al., 2007; Little et al., 2010) and damage to gut mucosa (Stern et al., 1995; Whyte et al., 2001; Line and Bailey, 2006; Wigley, 2015).
Whole-genome sequencing has provided important insights into the genetics and evolution of Campylobacter species that occupy different host niches (Miller et al., 2014a; Gilbert et al., 2016; Graaf-van Bloois et al., 2016; van der Graaf-van Bloois et al., 2016). However, little is known about how variation in species and strains relates to sub-structure within the chicken gut niche, and how this correlates with the emergence of diseases such as SLD. Here, we used a comparative genomics approach to investigate SLD-associated Campylobacter isolates sampled in the UK. By describing genomic features, with reference to published Campylobacter genomes, we identified potential genetic components that contributed to the diversification of the SLD-associated Campylobacter from closely related species and features associated with niche specialization.
Materials and Methods
Bacterial Isolates
Ten putative C. hepaticus isolates from the strain collection of the UK Animal and Plant Health Agency (APHA) (Addlestone, UK) were included in this study (Table 1). Nine of these isolates were previously described in a report of a novel Campylobacter species associated with SLD (Crawshaw et al., 2015). All isolates were cultured from liver samples collected immediately post-mortem from birds showing signs of SLD. Samples were collected from five distinct holdings in the UK between 2010 and 2012 (Table 1). The farms were in different geographic locations with no known epidemiological links between them. All strains were stored at −80°C in 1% (w/v) protease peptone water containing 10% (v/v) glycerol until required.
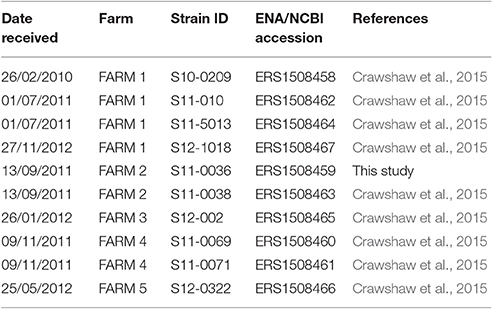
Table 1. Study isolates.
Whole-Genome Sequencing
Isolates were grown from frozen on 10% (v/v) sheep blood agar plates containing Skirrow's antibiotics [vancomycin (10 mg/mL), polymyxin B (2.5 i.u./mL) trimethoprim (5 mg/mL) and actidione (250 mg/mL)] (BASA; Skirrow, 1977) at 42°C in a microaerobic atmosphere (85% (v/v) N2, 7.5% (v/v) CO2, 7.5% (v/v) O2). The bacteria from each plate were harvested into tubes containing 1 mL 0.1 M PBS (pH 7.2) solution. Cells were pelleted by centrifugation and re-suspended in 0.5 mL 0.1 M PBS (pH 7.2) solution. Genomic DNA was extracted using the ArchivePure DNA Cell/Tissue (1 g) kit (5Prime, Gaithersburg, USA). Extracted genomic DNA was fragmented, tagged for multiplexing with the Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced at the APHA on the Illumina MiSeq platform using 150 bp paired-end reads according to the manufacturer's instructions.
Relationships between C. hepaticus Isolates and Other Campylobacter Species
The short-read data were assembled using SPAdes (Bankevich et al., 2012) and the resulting draft assemblies were submitted to the Ribosomal Multilocus Sequence Typing (rMLST) database https://pubmlst.org/rmlst/). Relationships between the study isolates and other Campylobacter species were characterized at the rMLST (Jolley et al., 2012) and core-genome multilocus sequence typing (cgMLST) levels (Maiden et al., 2013). Following annotation of the ribosomal protein genes (rps) using Bacterial Isolate Genome database (BIGSDB) software implemented on the rMLST database (Jolley and Maiden, 2010), the study isolates were compared to the C. hepaticus type strain HV10 (Van et al., 2016) and 76 publicly available genomes comprising 24 Campylobacter species (Supplementary Table S1). The rps genes were concatenated and aligned using MAFFT version 7.037b (Katoh and Standley, 2013) and a Maximum Likelihood tree was generated with MEGA-CC version 7.0 using the general time-reversible model with gamma-distributed rates plus invariant sites with 100 bootstrap replicates. A higher resolution comparison of the study isolates, HV10, and closely related thermophilic Campylobacter species, including C. jejuni, C. coli, C. upsaliensis, C. cuniculorum, C. lari, C. subantarcticus, C. peloridis, C. volucris, and C. ornithocola (Supplementary Table S1), was carried out using the GENOME COMPARATOR module implemented in BIGSDB (Jolley and Maiden, 2010). An ad hoc cgMLST analysis was carried out by comparing the isolates to the annotated genome of C. jejuni NCTC 11168 (GenBank accession AL111168) (Parkhill et al., 2000; Gundogdu et al., 2007), using the default GENOME COMPARATOR settings with the core genome cut-off set to 90%. A Maximum Likelihood tree was reconstructed as described for the rMLST analysis, using the MAFFT alignment of concatenated core gene sequences produced by GENOME COMPARATOR.
Currently there is no multilocus sequence typing (MLST) scheme for C. hepaticus; therefore, we used a read-mapping approach to quantify the variation in the seven gene fragments that comprise the C. jejuni/coli MLST scheme, namely aspA, glnA, gltA, glvA pgm, tkt, and uncA (Dingle et al., 2005). For sequence-read alignment and single nucleotide polymorphism (SNP) detection, paired-end Illumina sequence data were mapped to assembled gene fragments from isolate S12-1018, using BWA (Li and Durbin, 2009). For high-resolution genome-wide SNP detection, sequence data were mapped to the draft genome of HV10 (Van et al., 2016). SNPs were identified using Freebayes (https://github.com/ekg/freebayes) and filtered with a minimum mapping quality of 10 and quality ratio cut-off of 0.9. For phylogenetic analyses, a maximum-likelihood phylogenetic tree was constructed from the SNP alignments after Gubbins was run to remove regions of recombination in the pseudofasta files from SNP calling (Croucher et al., 2015). The phylogenetic trees were built using Figtree as previously described (Petrovska et al., 2016).
Functional Analyses
The draft genomes of HV10 and the UK C. hepaticus isolates were annotated using the RAST server (Aziz et al., 2008; Overbeek et al., 2014), as were the finished genomes of C. jejuni isolates NCTC 11168, M1, PT14, R14, and 4031 (Supplementary Table S1). FIGfam clusters genes based on protein sequence similarity. These genes are then clustered into hierarchical subsystems that display increasing functional breadth (Overbeek et al., 2005). To compare the subsystems generated by RAST, a Student's t-test was performed (two tailed distribution for two-sample populations of unequal variance) using a standard spreadsheet function (Microsoft Excel). P ≤ 0.05 were regarded as significant. Genome comparisons of the study isolates, HV10, and C. jejuni NCTC 11168 were visualized using the BLAST Ring Image Generator (BRIG; Alikhan et al., 2011).
Further analyses were carried out to identify genes involved in antimicrobial resistance, pathogenicity, and iron uptake and metabolism. The SRST2 pipeline (Inouye et al., 2014) was used to search for determinants in the ARG-ANNOT antimicrobial resistance database (Gupta et al., 2014). Putative pathogenicity genes were predicted with the PathogenFinder web-server (Cosentino et al., 2013) using the “All” model. Blastn was used to search for iron uptake related genes in the C. hepaticus isolate genomes after a DNA database was made with genes from iron uptake pathways in C. jejuni NCTC 11168 (Miller et al., 2009). The cut-offs were set at 80% for both identity and coverage so that the genes above these thresholds were recorded as present.
Iron Depletion Assay
The growth of C. hepaticus isolates S11-010, S12-1018, and S12-0322 in regular Brain Heart Infusion broth (BHI) and in iron-depleted BHI was compared to that of C. jejuni NCTC 11168. C. hepaticus isolates were grown for 48 h on 5% sheep blood agar (SBA) plates at 42°C in a microaerobic atmosphere. NCTC 11168 was grown for 24 h under the same conditions. Growth was harvested into PBS at c. 105–106 cfu/ml and 100 μl added to 10 ml BHI in T25 tissue culture flasks. For each isolate, a regular broth and one containing the iron chelator deferoxamine mesylate (Desferal; Sigma) at a final concentration of 20 mM were inoculated (van Vliet et al., 1998). Each C. hepaticus isolate was tested in duplicate on 2 separate occasions, and NCTC 11168 was tested in duplicate on 4 separate occasions. Samples of the broths were removed at 1, 2, 3, and 5 days post-inoculation and quantitative bacteriology performed by plating out serial dilutions on to SBA plates (detection limit = 100 cfu/ml).
Accession Numbers
Nucleotide sequence data were submitted to the European Nucleotide Archive (http://www.ebi.ac.uk/ena) under the primary accession number PRJEB19094. Individual accession numbers are given in Table 1.
Results
C. hepaticus Isolates Form a Distinct Clade Separate from Other Known Campylobacter Species
We used a hierarchical gene-by-gene approach (Maiden et al., 2013) to investigate the relationships between the putative C. hepaticus isolates from the UK, the C. hepaticus type strain (HV10), and 24 other Campylobacter species. Complete nucleotide sequences of the 52 rps genes present in Campylobacter (Cody et al., 2013) were obtained from the 10 SLD-associated Campylobacter isolates sequenced for this study; however, isolate S12-0002 was contaminated and was excluded from further analyses, unless stated otherwise. In the rMLST phylogeny, the study isolates clustered most closely with HV10, confirming that they corresponded to C. hepaticus. The C. hepaticus cluster was positioned between C. jejuni and C. coli (Figure 1A). The ad hoc cgMLST comparison identified 646 genes that were present in ≥90% of 59 isolates including C. hepaticus and 10 other thermophilic Campylobacter species that clustered together in the rMLST phylogeny. The resulting cgMLST phylogeny was consistent with the rMLST tree, with C. hepaticus most closely related to C. coli (Figure 1B). Both phylogenies indicated that the UK C. hepaticus isolates were closely related and segregated according to farm (Figure 1).
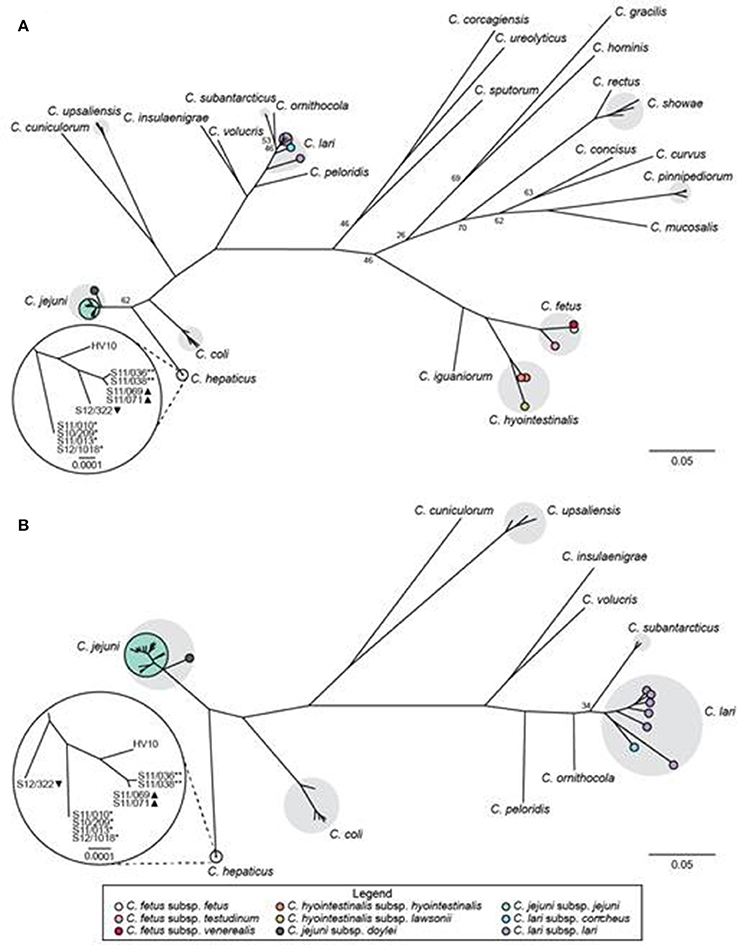
Figure 1. Relationships between C. hepaticus and other Campylobacter species based on gene-by-gene analyses. Maximum likelihood trees based on concatenated nucleotide sequences of (A) 52 rMLST genes from 86 isolates comprising 25 Campylobacter species; and (B) 646 core genes identified by cgMLST in ≥90% of 59 isolates comprising 11 closely related thermophilic Campylobacter species. Bootstrap values <80 (A) or <100 (B) are shown for major nodes. Gray shading indicates taxa with multiple representative isolates. Subspecies are marked with colored circles, as indicated in the legend. The insets provide magnified views of the relationships among C. hepaticus isolates. *, farm 1; **, farm 2; ▴, farm 4; ▾, farm 5.
Local, Farm-Related Phylogenetic Clustering of C. hepaticus Isolates
There was no C. hepaticus MLST scheme at the time of writing; therefore, we used a read-mapping approach to index variation at the 7 loci comprising the MLST scheme of C. jejuni and C. coli, the closest relatives to C. hepaticus. All study isolates, including S12-002, had identical glnA, gltA, and pgm alleles. There were 2 alleles each for aspA, glyA, tkt, and uncA, all of which corresponded to single nucleotide differences, except uncA which had 2 SNPs (Supplementary Figure S1). Although genetic diversity was low, SNPs were associated with sample origin: isolates from the same farm shared the same SNPs in the 7 core MLST genes (Supplementary Figure S1).
To study the relationships among C. hepaticus isolates in more detail, a Maximum Likelihood phylogenetic tree was reconstructed using variable sites within the whole genome sequence with reference to the draft genome of HV10 (Figure 2). The contaminated isolate S12-002 was included in the mapping analyses. The phylogenetic tree also indicated clustering according to farm, with ≤ 11 SNP differences identified between the isolates collected from the same farm. Farm 1 isolates S10-0209, S11-010, S11-5013, and S12-1018 differed by a total of 5 SNPs (Figure 2). Similarly, isolates from farms 2 (S11-0036 and S11-0038) and 4 (S11-0069 and S11-0071) differed by 4 and 11 SNPs, respectively. In contrast, isolates from different farms were separated by at least an order of magnitude more SNPs, with HV10 clustering with isolates from farms 2, 3, and 4. Isolate S12-002 from farm 3 was 113 SNPs apart from S11-069 and S11-0071. Isolate S12-0322 from farm 5 was furthest apart from all other isolates in the phylogenetic tree, with 987 SNP differences to strains S11-0036 and S11-038. HV10 was 1161 SNPs apart from isolate S12-0322 (farm 5), 938 SNPs from S12-1018 (farm 1) and 614 SNPs from isolate S11-036 (farm 2; Figure 2).
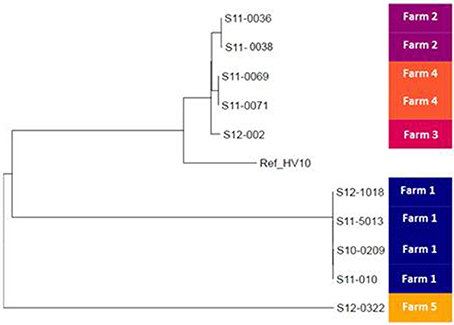
Figure 2. Phylogeny of the UK Campylobacter hepaticus isolates. Maximum likelihood tree constructed by reference to the whole genome sequence of isolate HV10. UK C. hepaticus isolates: S10-0209, S11-010, S11-5013, S12-1018, S11-0036, S11-0038, S12-002, S11-0071, and S12-0322.
C. hepaticus Isolates Have Reduced Genomes
The assembled contigs of the UK C. hepaticus genomes were submitted to RAST, Rapid Annotations based on Subsystem Technology, designed to annotate genes of prokaryotic genomes (Aziz et al., 2008). For comparison, the draft genome of HV10 (Van et al., 2016) and 5 C. jejuni genomes from the public databases, including NCTC 11168, M1, PT14, R14 and 4031 were also submitted to RAST, and pooled data of the UK C. hepaticus isolates were compared with the pooled data of the C. jejuni genomes (Table 1). The RAST results indicated that the UK C. hepaticus isolates were similar in size to each other, but had smaller genomes (1.53 Mb average) than the reference C. jejuni isolates, which were also similar in size (1.67 Mb average; p = 2.6 × E-4; Table 2). The reduction of ~140 Kb resulted in an average of 144 fewer genes (p = 6.1 × E-3). The C. hepaticus isolates had a lower number (average of 44) of RNA coding sequences (average of 52.4) and a lower GC content (average of 28.4%) in comparison to the C. jejuni reference genomes (average of 30.5%). The genome size of the Australian C. hepaticus HV10 isolate was 1.48 Mb with 27.9% GC (Table 2). Genome comparison using BLAST Ring Image Generator (BRIG) indicated multiple deletions in the 10 UK C. hepaticus isolates when compared to the C. jejuni NCTC 11168 genome (Figure 3).
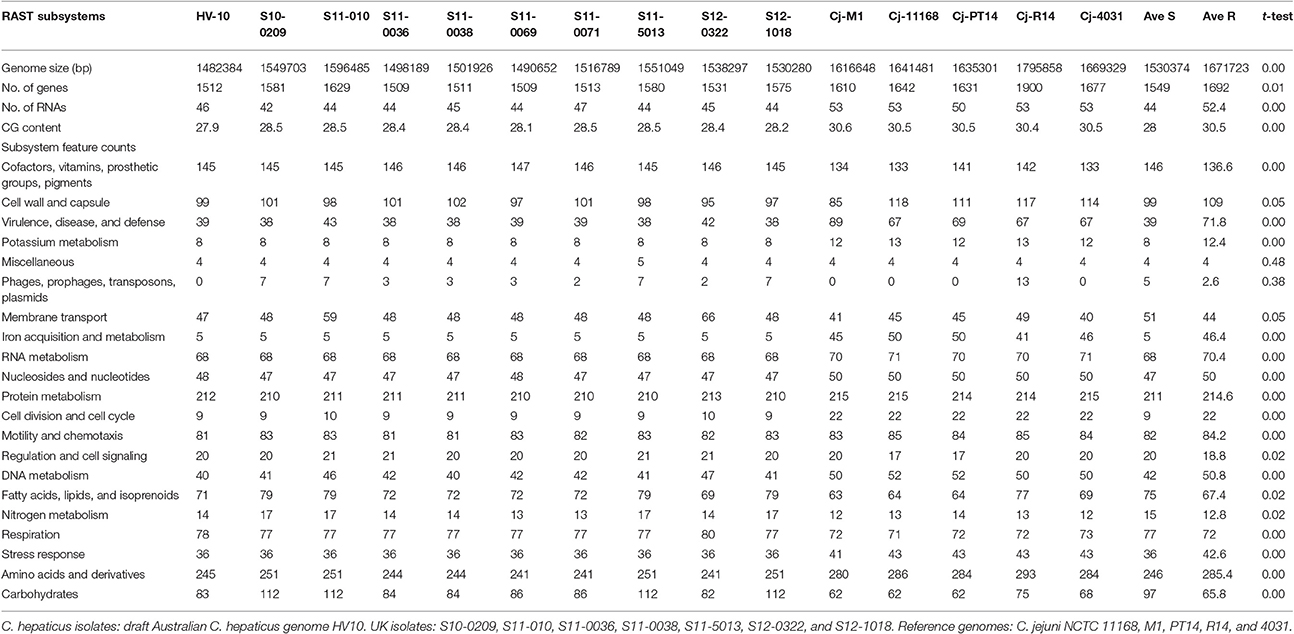
Table 2. Comparison of C. hepaticus with C. jejuni (reference) genomes using RAST.
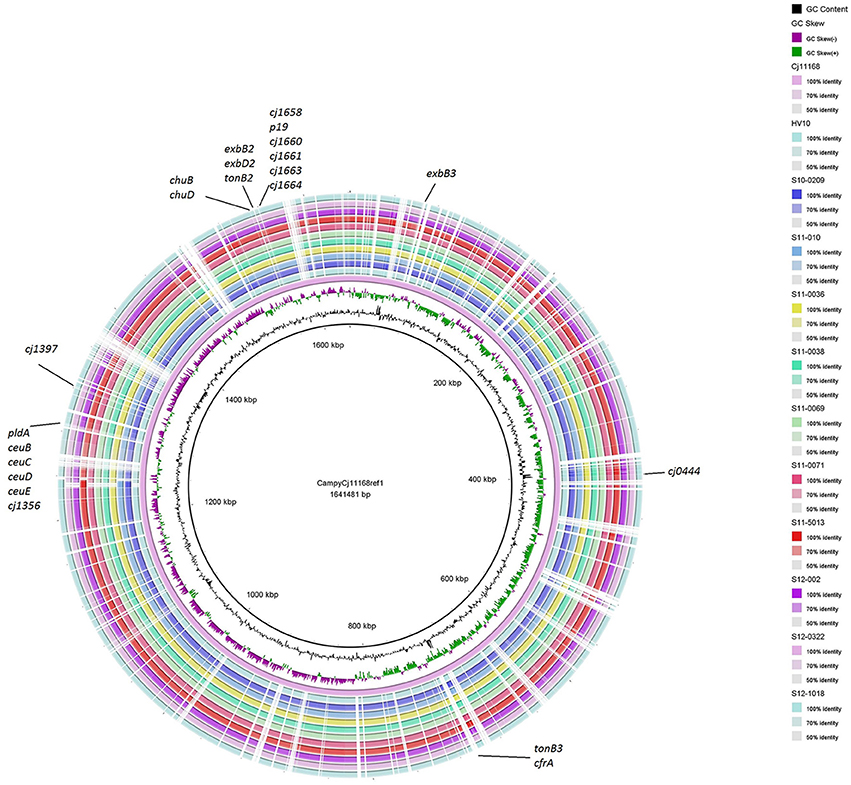
Figure 3. BRIG (BLAST Ring Image Generator) comparison of C. hepaticus isolates with C. jejuni NCTC 11168. Similarities between the reference genome C. jejuni NCTC 11168 and C. hepaticus isolates are presented as concentric rings. Inner circle: C. jejuni NCTC 11168 (purple ring). The 10 UK C. hepaticus isolates and HV10 are presented in the outer concentric rings.
Functional Annotation
RAST uses FIGfam (Aziz et al., 2008) to cluster annotated genomes in subsystems that are further divided into groups, based on protein sequence similarity. Clustered genes are listed as hierarchical subsystems that display increasing functional extensiveness (Overbeek et al., 2005; Table 2). Nineteen of the 21 subsystems reached statistical significance when pooled UK C. hepaticus isolates were compared to pooled C. jejuni reference genomes (Figure 3). C. hepaticus genomes contained significantly fewer genes than the C. jejuni references in 11 of the 21 clustered subsystems, and significantly more in 8 subsystems (Figure 4 and Table 2). The largest decrease was in the subsystem containing genes for iron acquisition and metabolism, with the C. hepaticus isolates containing on average only five genes in comparison to the average of 46.4 (or 11%) present in the reference genomes. Furthermore, within this subsystem, there were no genes identified in the group for iron transport in C. hepaticus, while 8 related genes were identified in C. jejuni. To confirm the absence of iron uptake genes in C. hepaticus, the study genomes were searched using blastn to identify genes from the C. jejuni NCTC 11168 iron uptake pathways (Miller et al., 2009). The cut-offs were set at 80% for both identity and coverage so that the genes above these thresholds were recorded as present. The following loci could not be detected among the UK C. hepaticus isolates: 7/8 genes from the ferri-enterochelin pathway; the entire ferri-rhodotorulic acid pathway; 5/8 genes in the haem pathway; 1/2 genes in the ferrous iron pathway; and cj0444 was missing from the cj0444 pathway (Supplementary Figure S2). The loss of function was tested with an iron depletion assay. There were clear differences between the growth of the C. hepaticus isolates and C. jejuni NCTC 11168 (Figure 5). In regular broth, the C. hepaticus isolates reached peak levels (108–109 cfu/ml) at 3 days post-inoculation, compared to only 1 day for C. jejuni. In the iron-depleted media, the C. jejuni persisted for 5 days at approximately starting levels (103–104 cfu/ml). In contrast, there were no detectable colonies on agar plates at any time point after adding the iron chelator in all tested C. hepaticus isolates.
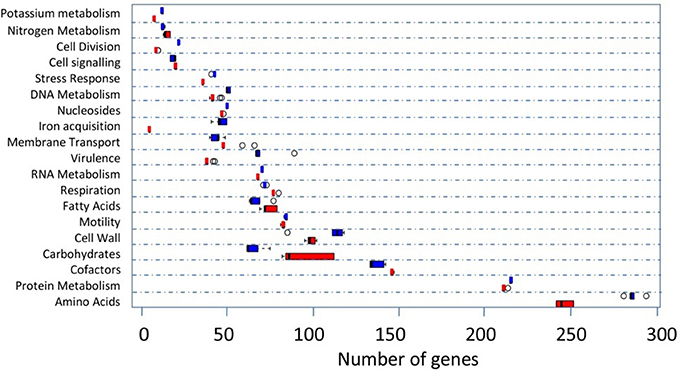
Figure 4. Comparison of the functional subsystems in the UK C. hepaticus (red) and 5 C. jejuni reference genomes (blue) as generated by RAST. References: M1, NCTC 11168, PT14, R14, and 4031. The open circles represent the outliers. Only subsystems with significant differences (student's t-test) are shown. The names of some subsystems are shortened: Cell Division, Cell Division and Cell Cycle; Cell signaling, Regulation and Cell signaling; Nucleosides, Nucleosides and Nucleotides; Iron acquisition, Iron acquisition and metabolism; Virulence, Virulence, Disease and Defense; Fatty Acids, Fatty Acids, Lipids, and Isoprenoids; Motility, Motility and Chemotaxis; Cell Wall, Cell Wall and Capsule; Cofactors, Cofactors, Vitamins, Prosthetic Groups, Pigments; Amino Acids, Amino Acids and Derivatives.
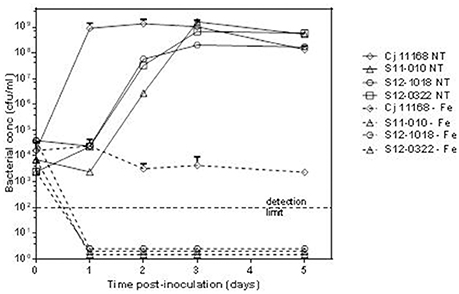
Figure 5. Reduced growth of C. hepaticus isolates in iron depleted conditions. Growth curves for 3 UK C. hepaticus isolates and C. jejuni NCTC 11168 grown in Brain Heart Infusion broth either regular or untreated (NT) or iron-depleted (−Fe).
There were fewer putative virulence, disease, and defense subsystem genes in the UK C. hepaticus isolates, with an average of 39.2 genes identified compared with 71.8 in the C. jejuni genomes. Within this subsystem, on average 13 genes associated with adhesion (adhesion subgroup) were present in the reference genomes, but no known adhesion genes were identified in the C. hepaticus isolates. Similarly, there were six genes in the cytolethal distending toxin (CDT) group in the reference genomes, but no genes of this group were present in the C. hepaticus genomes. In the resistance to antibiotics and toxic compounds subgroup there were 3–5 arsenic resistance genes in the reference genomes, but no resistance genes were present in the C. hepaticus chromosomes.
In the DNA metabolism group, the reference genomes typically contained 5–6 type I restriction-modification pathways; the C. hepaticus isolates contained one of these pathways. On average, 36 genes for stress response were found in the C. hepaticus genomes, which was significantly lower than the 42.6 genes of this group present in the reference genomes. In the oxidative stress pathway, each of the reference genomes contained four genes in the redox-dependent regulation of nucleus processes subsystem and 2 genes in the rubrerythrin subsystem, all of which were absent in the C. hepaticus genomes.
In the amino acids and derivatives group, the pathways of arginine, urea cycle, and polyamines differed between the UK C. hepaticus isolates and reference genomes. The C. hepaticus genomes typically had: putrescine utilization pathways (two genes) that were absent in the reference genomes; a lower number of genes in the arginine deiminase pathway (16 in comparison to 29 in the reference genome); a lower number of polyamine metabolism genes (21 in comparison to 32); and fewer arginine and ornithine degradation genes (20 in comparison to 33 in reference genomes).
The carbohydrates and fatty acids, lipids, and isoprenoids groups were among the 8 subsystems with significantly more genes in the UK C. hepaticus isolates than the C. jejuni reference genomes. The C. hepaticus isolates contained on average 96.7 genes in the carbohydrates group, while an average of only 65.8 genes were present in the reference genomes (Supplementary Table S2).
Genes Related to the Pathogenicity of C. hepaticus
The genomes of HV10 and the UK C. hepaticus isolates, including S12-002, and C. jejuni isolates NCTC 11168, M1 and 4031, were submitted to the PathogenFinder database (v 1.1) (https://cge.cbs.dtu.dk/services/PathogenFinder) to identify the presence of pathogenicity-related genes by comparison of the protein families of all bacterial pathogens present in the database. A total of 511 pathogenicity related factors were identified in the genomes of NCTC 11168, 372 in M1 and 342 in 4031, while none was found in HV10. The UK C. hepaticus isolates contained relatively few genes linked to pathogenesis: 5 were identified in the genomes of S11-0069, S11-0071, S11-0038, S11-0036, and S12-002 (from farms 2, 3, and 4); 6 in S11-0038 (farm 2); 15 in S10-0209, S11-010, S12-1018, S11-5013 (farm1); and 7 in isolate S12-0322 (farm 5; Table 3). The cpp and cmgB3/4 genes, both components of the pTet plasmid (Batchelor et al., 2004), were identified in isolates S11-10, S12-0322, and S12-002 (Table 3). Complete pTet plasmid (Batchelor et al., 2004) sequences were identified in isolates S11-010, S12-002, and S12-0322.
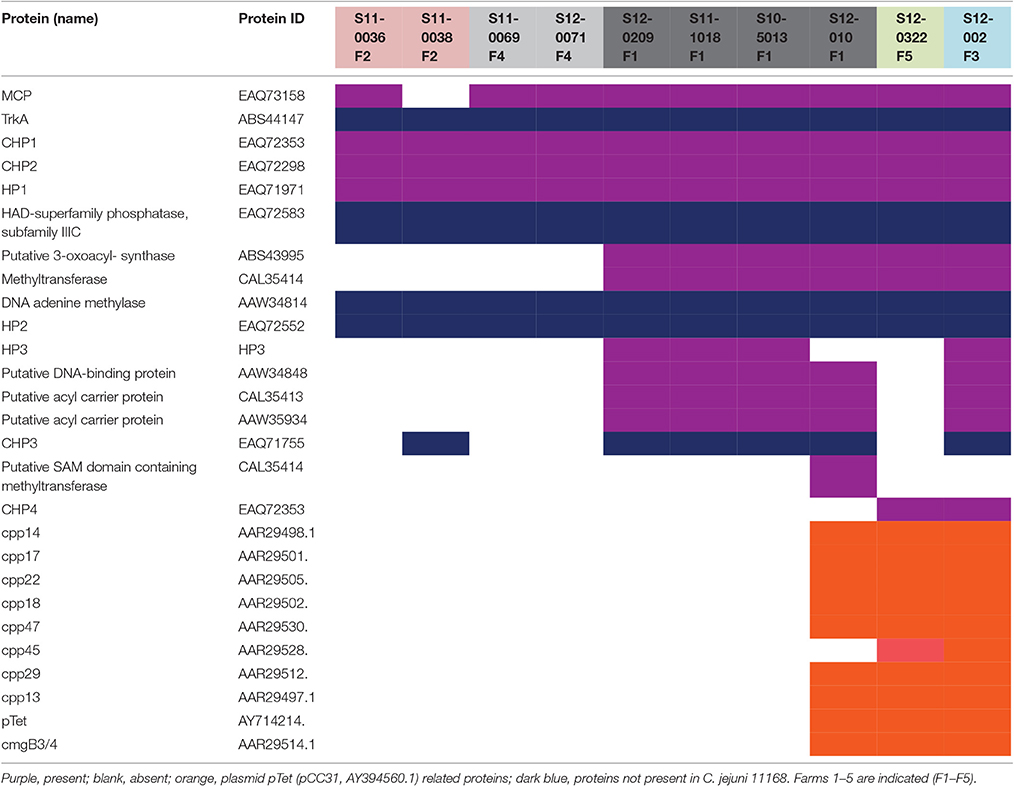
Table 3. Presence of pathogenicity-related genes in C. hepaticus.
Five genes related to pathogenicity in Campylobacter spp. were present in C. hepaticus but not in C. jejuni isolates NCTC 11168, M1 and 4031: (1) putative potassium uptake protein TrkA (ABS44147) identified in C. jejuni subsp. doylei 269.97 (PRJNA17163); (2) HAD-superfamily phosphatase, subfamily IIIC protein EAQ72583; (3) DNA adenine methylase protein AAW34814 identified in C. jejuni RM1221; (4) Hypothetical HP2 protein EAQ72552; and (5) CHP3 conserved hypothetical protein EAQ71755 (Table 3).
Discussion
C. hepaticus has been identified as the cause of SLD (Crawshaw et al., 2015; Van et al., 2016, 2017), and the disease pathology reproduced in SPF birds in the UK and mature layer hens in Australia; however, our understanding of the genomics and evolution of this emerging pathogen remain limited. Hierarchical gene-by-gene analyses of putative C. hepaticus isolates from SLD cases in the UK and representatives of 25 Campylobacter species confirmed that the UK isolates were most closely related to HV10, the C. hepaticus type strain. This verified that C. hepaticus is a cause of SLD in both the UK and Australia, as hypothesized by Van et al. (2016). Previous studies suggested that C. hepaticus was most closely related to members of the C. lari group or C. jejuni and C. coli (Crawshaw et al., 2015; Van et al., 2016); however, these findings were based on phylogenetic analyses of 16S rRNA or heat shock protein 60 gene sequences. The limitations of single gene phylogenies for inferring relationships among species have been acknowledged, particularly 16S rRNA gene sequencing for Campylobacter taxonomy (Gorkiewicz et al., 2003; Miller et al., 2012, 2014b). The higher resolution rMLST and cgMLST analyses carried out in this study confirmed that C. hepaticus was positioned between the major human pathogens C. jejuni and C. coli, which clustered with C. upsaliensis, C. cuniculorum, and members of the C. lari group. These are all thermotolerant spp., many of which have been isolated from birds and some corresponding to emerging human pathogens (Kaakoush et al., 2015).
When analyzed at the MLST, rMLST, and cgMLST levels, the UK C. hepaticus isolates were highly similar to each other. Isolates from farms 2, 3, and 4 all shared the same MLST profile, while those from farms 1 and 5 differed at one and two loci, respectively, including just 5 SNPs in total; none of these profiles appeared in the C. jejuni/coli PubMLST database. At the rMLST and cgMLST levels, the UK isolates remained highly similar, but clustered by farm. High-resolution SNP analysis was used to resolve the relationships among the study isolates, revealing low levels of within-farm diversity. Isolates from the same farm differed by 3–12 SNPs, which contrasted with higher levels of between-farm diversity (173–1,260 SNPs). That the highly similar farm 1 isolates were collected between 2010 and 2012 suggests that these genotypes are stable over time. Overall, the low levels of within-farm diversity were similar to those observed in campylobacteriosis outbreaks (Llarena and Taboada, 2017). Although, the sample size was small, the clustering of isolates suggested a farm-specific subpopulation structure that may reflect ongoing local microevolution, while the between-farm diversity indicated that C. hepaticus is not a newly emerged pathogen. It was of interest that the Australian isolate HV10 was positioned within the diversity of the UK C. hepaticus isolates. Further sampling will be necessary to fully characterize the population structure and global epidemiology of C. hepaticus. When genome sequencing is not feasible, a new MLST scheme based on the C. jejuni/coli scheme may prove beneficial.
Reductive genome evolution has been described in diverse bacteria and is typically associated with specialization, often in an intracellular niche (Georgiades and Raoult, 2010; McCutcheon and Moran, 2011). At 1.48 Mbp, HV10 is the smallest Campylobacter genome sequenced to date (Supplementary Table S1), while the UK C. hepaticus isolates had a slightly larger average genome size of 1.53 Mbp. This represents a reduction of ~171–238 kb compared to their closest relatives C. jejuni and C. coli, which also have relatively small genomes compared to other Campylobacter species (Supplementary Table S1 and references therein). RAST annotation of the UK C. hepaticus genomes indicated a reduction of ~144 genes and 8 RNA coding sequences compared to five C. jejuni reference genomes. Large-scale gene loss and inactivation have been reported in several niche-adapted bacterial pathogens, including: Shigella spp. (Maurelli et al., 1998; Wei et al., 2003); Mycobacterium leprae and Mycobacterium ulcerans (Cole et al., 2001; Rondini et al., 2007); Bordetella pertussis and Bordetella parapertussis (Parkhill et al., 2003); and Rickettsia spp. (Merhej and Raoult, 2011). Likewise, a study of bacteria with different lifestyles identified fewer genes involved in transcription and translation in obligate intracellular bacteria (Merhej et al., 2009). Reduced bacterial genomes also tend to shift toward a higher AT content (McCutcheon and Moran, 2011). The C. hepaticus isolates had a lower average GC content (28.4%) than C. jejuni (30.5%) and most other Campylobacter species (Supplementary Table S1). Likely drivers of the genome reduction observed in C. hepaticus include specialization and genetic isolation following the occupation of a new niche (Georgiades and Raoult, 2010), namely the chicken liver, and perhaps also the transition from a free-living or facultatively parasitic life-cycle to an obligate pathogenic life-cycle (Moran, 2002).
Genome reduction results in gene losses across all functional categories, with biosynthetic pathways commonly eliminated when metabolites are available from the environment (Toft and Andersson, 2010; McCutcheon and Moran, 2011; Hottes et al., 2013; Albalat and Canestro, 2016). In C. hepaticus, 11 out of 21 subsystems defined by RAST were reduced compared to C. jejuni. Gene loss was particularly evident among iron metabolism pathways in C. hepaticus, consistent with adaptation to an iron rich environment such as the chicken liver. The C. hepaticus isolates contained only 10% of the iron metabolism related genes present in C. jejuni isolates (Supplementary Figure S2). Eight subsystems were identified with a higher number of genes in the C. hepaticus isolates than in the reference C. jejuni genomes (Figure 4) with the highest number of gene differences in carbohydrate utilization pathways (average of 96.7 genes in the study isolates and 65.8 in C. jejuni; Supplementary Table S2). This is interesting as Campylobacter is generally considered to be a non-saccharolytic bacterium unable to use glucose and other carbohydrate sources as a growth substrate (Hofreuter, 2014), an observation supported by WGS and BIOLOG studies (Parkhill et al., 2000; Bochner, 2009; Gripp et al., 2011). Carbon source utilization is characteristic for growth of other intracellular gastrointestinal pathogens, for instance Salmonella Typhimurium and Listeria monocytogenes (Dandekar et al., 2012; Fuchs et al., 2012), as well as the close relative of Campylobacter, Helicobacter pylori (Mendz et al., 1993). However, recent studies demonstrated that some C. jejuni strains can metabolize the sugar L-fucose due to the presence of a novel L-fucose pathway including L-fucose permease within a 9 kb genomic island in these strains (Muraoka and Zhang, 2011; Stahl et al., 2011). Furthermore, Stahl and co-workers found that the ability to meatbolise L-fucose in vivo provided C. jejuni with competitive advantage during colonization of the piglet infection model. Similar was not observed in the chick commensal model (Stahl et al., 2011), suggesting potential niche specific advantage for colonization in L-fucose reach environment in the pig small intestine and cecum. It is possible that the C. hepaticus have adopted different carbohydrate utilization mechanisms for opportunistic growth in a carbohydrate rich intracellular environment in the chicken liver.
Reduced genomes can be associated with niche adaptation and increased pathogenicity in some bacteria (Moran, 2002). Niche adaptation requires selection for and against traits to optimize pathogen fitness in the new environment (Bliven and Maurelli, 2012). In C. hepaticus, there was a large reduction in “virulence factors,” with only 5–15 recognized pathogenicity genes detected in these isolates. In contrast, C. jejuni isolates NCTC 11186, M1 and 4031 contained 511, 372, and 342 pathogenicity genes, respectively. With respect to pathogenicity factors identified in C. hepaticus, TrkA, a homolog of the putative potassium uptake protein described as an essential protein for maintenance of ionic homeostasis in response to changes in the environment (Lee et al., 2007), was present in all study isolates but not all C. jejuni reference genomes. The same was true of four other genes: a homolog of the two-component system methyl-accepting chemotaxis proteins (MCPs) that serve as sensors in bacterial chemotactic signaling, detecting attractants, and promoting bacterial movement toward suitable sites for colonization (Li et al., 2014); and 3 conserved hypothetical proteins CHP1, CHP2, and HP1, which have been described in C. jejuni 81–176. Interestingly, a subset of C. hepaticus isolates also contained a homolog protein that is part of the haloacid dehydrogenase (HAD) superfamily, which are involved in a variety of cellular processes ranging from amino acid biosynthesis to detoxification and has only been described in strain 81–176. Similarly, the hypothetical protein HP2 and the conserved hypothetical protein CHP3 present in some C. hepaticus isolates have also only been previously described in strain 81–176. Strain 81–176 displays increased virulence and invades intestinal epithelial cells at levels that are as much as 3 logs higher than other invasive C. jejuni strains (Poly et al., 2005). Furthermore, there was a large reduction in the genes encoding capsular and extracellular polysaccharides (CPS); CPS produced by C. jejuni are known to be important virulence factors that are involved in colonization and invasion (Richards et al., 2013). There were also fewer putative virulence, disease, and defense subsystem genes in the C. hepaticus isolates, including the absence of the CDT group genes encoding a bacterial toxin that initiates a eukaryotic cell cycle block at the G2 stage prior to mitosis (Jinadasa et al., 2011). It is possible that the evolution of attenuated virulence in C. hepaticus could have occurred as a result of immune evasion within the host (Mikonranta et al., 2015) that enables potential establishment of a long term chronic infection (Dennis, 2016) in laying hens with disease manifestation around pick lay. Further analyses of a larger, global C. hepaticus isolate collection are required to robustly infer the pan-genome of C. hepaticus, which in turn will improve our understanding of niche specialization in this organism.
This work highlights the potential importance of C. hepaticus to the poultry industry, especially as infection is likely to be under-detected because isolation requires modifications to the standard C. jejuni/C. coli protocol. Further work is needed to improve the sampling and isolation methods for detection of C. hepaticus on poultry farms. C. hepaticus has not yet been reported in humans; however, consumption of chicken liver is a common source of campylobacteriosis outbreaks (Noormohamed and Fakhr, 2012; Weber et al., 2014; Moffatt et al., 2016). Detection of the C. jejuni pTet tetracycline resistance plasmid in 3 study isolates from 3 separate farms is also a cause of concern. Transfer of genetic material between C. hepaticus and other Campylobacter spp. may mediate exchange of antimicrobial resistance and pathogenicity-related determinants. Further studies of additional isolates are necessary to better understand the population structure and evolution of this important pathogen.
Author Contributions
LP, TC, and RI designed the study; LP, YT, MJvR performed the analyses; JN and RE helped with the analyses; MJvR, TC, AW, SC, and SS revised the manuscript and provided valuable suggestions and LP wrote the manuscript.
Funding
This work was supported by the Department for Environment, Food, and Rural Affairs UK. MJvR is affiliated to the National Institute for Health Research Health Protection Research Unit (NIHR HPRU) in Gastrointestinal Infections at University of Liverpool in partnership with Public Health England (PHE), in collaboration with University of East Anglia, University of Oxford and the Institute of Food Research. MJvR is based at the University of Oxford. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health or Public Health England.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00354/full#supplementary-material
Supplementary Figure S1. MLST diversity among UK C. hepaticus isolates. Nucleotide sequence alignments of the 7 gene fragments comprising the C. jejuni/coli MLST scheme are shown. Consensus sequences were determined by mapping raw reads to the sequence of S12-1018. Variable sites are marked with arrows.
Supplementary Figure S2. Comparison of C. hepaticus and C. jejuni iron uptake pathways. The figure was adapted from a review by Miller et al. (2009). Iron uptake genes in the C. jejuni NCTC 11168 genome were used to make a database and UK C. hepaticus genomes were queried using blastn with 80% cut-offs for both identity and coverage. Blue arrows: iron uptake related genes with arrowheads indicating the direction of transcription; white arrows: flanking genes. Red: genes not detected in C. hepaticus.
Supplementary Table S1. Isolates used in this study.
Supplementary Table S2. Comparison of the Carbohydrates subsystem in C. hepaticus and C. jejuni (reference) genomes using RAST.
References
Albalat, R., and Canestro, C. (2016). Evolution by gene loss. Nat. Rev. Genet. 17, 379–391. doi: 10.1038/nrg.2016.39
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Batchelor, R. A., Pearson, B. M., Friis, L. M., Guerry, P., and Wells, J. M. (2004). Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiology 150(Pt 10), 3507–3517. doi: 10.1099/mic.0.27112-0
Bliven, K. A., and Maurelli, A. T. (2012). Antivirulence genes: insights into pathogen evolution through gene loss. Infect. Immun. 80, 4061–4070. doi: 10.1128/IAI.00740-12.
Bochner, B. R. (2009). Globalphenotypiccharacterizationofbacteria. FEMS Microbiol. Rev. 33, 191–205. doi: 10.1111/j.1574-6976.2008.00149.x.
Cawthraw, S., Ayling, R., Nuijten, P., Wassenaar, T., and Newell, D. G. (1994). Isotype, specificity, and kinetics of systemic and mucosal antibodies to Campylobacter jejuni antigens, including flagellin, during experimental oral infections of chickens. Avian Dis. 38, 341–349. doi: 10.2307/1591960
Cody, A. J., McCarthy, N. D., Jansen van Rensburg, M., Isinkaye, T., Bentley, S. D., Parkhill, J., et al. (2013). Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing. J. Clin. Microbiol. 51, 2526–2534. doi: 10.1128/JCM.00066-13
Cogan, T. A., Thomas, A. O., Rees, L. E., Taylor, A. H., Jepson, M. A., Williams, P. H., et al. (2007). Norepinephrine increases the pathogenic potential of Campylobacter jejuni. Gut 56, 1060–1065. doi: 10.1136/gut.2006.114926
Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., et al. (2001). Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011. doi: 10.1038/35059006
Cosentino, S., Voldby Larsen, M., Moller Aarestrup, F., and Lund, O. (2013). PathogenFinder–distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 8:e77302. doi: 10.1371/annotation/b84e1af7-c127-45c3-be22-76abd977600f
Crawshaw, T. R., Chanter, J. I., Young, S. C., Cawthraw, S., Whatmore, A. M., Koylass, M. S., et al. (2015). Isolation of a novel thermophilic Campylobacter from cases of spotty liver disease in laying hens and experimental reproduction of infection and microscopic pathology. Vet. Microbiol. 179, 315–321. doi: 10.1016/j.vetmic.2015.06.008
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Dandekar, T., Astrid, F., Jasmin, P., and Hensel, M. (2012). Salmonella enterica: a surprisingly well-adapted intracellular lifestyle. Front. Microbiol. 3:164. doi: 10.3389/fmicb.2012.00164
Delaplane, J. P., Smith, H. A., and Moore, R. W. (1955). An unidentified agent causing a hepatitis in chickens. Southwest Vet. 8:356.
Dennis, J. J. (2016). Burkholderia cenocepacia virulence microevolution in the CF lung: variations on a theme. Virulence 1–3. doi: 10.1080/21505594.2016.1253660
Dingle, K. E., Colles, F. M., Falush, D., and Maiden, M. C. (2005). Sequence typing and comparison of population biology of Campylobacter coli and Campylobacter jejuni. J. Clin. Microbiol. 43, 340–347. doi: 10.1128/JCM.43.1.340-347.2005
Fuchs, T. M., Eisenreich, W., Kern, T., and Dandekar, T. (2012). Toward a systemic understanding of Listeria monocytogenes metabolism during infection. Front. Microbiol. 3:23. doi: 10.3389/fmicb.2012.00023
Georgiades, K., and Raoult, D. (2010). Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 1:151. doi: 10.3389/fmicb.2010.00151
Gilbert, M. J., Miller, W. G., Yee, E., Zomer, A. L., van der Graaf-van Bloois, L., Fitzgerald, C., et al. (2016). Comparative genomics of Campylobacter fetus from reptiles and mammals reveals divergent evolution in host-associated lineages. Genome Biol. Evol. 8, 2006–2019. doi: 10.1093/gbe/evw146
Gorkiewicz, G., Feierl, G., Schober, C., Dieber, F., Kofer, J., Zechner, R., et al. (2003). Species-specific identification of campylobacters by partial 16S rRNA gene sequencing. J. Clin. Microbiol. 41, 2537–2546. doi: 10.1128/JCM.41.6.2537-2546.2003
Graaf-van Bloois, L., Miller, W. G., Yee, E., Gorkiewicz, G., Forbes, K. J., Zomer, A. L., et al. (2016). Campylobacter fetus subspecies contain conserved type iv secretion systems on multiple genomic islands and plasmids. PLoS ONE 11:e0152832. doi: 10.1371/journal.pone.0152832
Grimes, T., and Reece, R. (2011). “Spotty liver disease—an emerging disease in free-range layers in Australia,” in Proceedings of the 60th Western Poultry Disease Conference (Sacremento, CA), 53–56.
Gripp, E., Hlahla, D., Didelot, X., Kops, F., Maurischat, S., Tedin, K., et al. (2011). Closely related Campylobacter jejuni strains from different sources reveal a generalist rather than a specialist lifestyle. BMC Genomics 12:584. doi: 10.1186/1471-2164-12-584
Gundogdu, O., Bentley, S. D., Holden, M. T., Parkhill, J., Dorrell, N., and Wren, B. W. (2007). Re-annotation and re-analysis of the Campylobacter jejuni NCTC11168 genome sequence. BMC Genomics 8:162. doi: 10.1186/1471-2164-8-162
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Hofreuter, D. (2014). Defining the metabolic requirements for the growth and colonization capacity of Campylobacter jejuni. Front. Cell. Infect. Microbiol. 4:137. doi: 10.3389/fcimb.2014.00137
Hottes, A. K., Freddolino, P. L., Khare, A., Donnell, Z. N., Liu, J. C., and Tavazoie, S. (2013). Bacterial adaptation through loss of function. PLoS Genet. 9:e1003617. doi: 10.1371/journal.pgen.1003617
Inouye, M., Dashnow, H., Raven, L. A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6, 90. doi: 10.1186/s13073-014-0090-6
Jennings, J. L., Sait, L. C., Perrett, C. A., Foster, C., Williams, L. K., Humphrey, T. J., et al. (2011). Campylobacter jejuni is associated with, but not sufficient to cause vibrionic hepatitis in chickens. Vet. Microbiol. 149, 193–199. doi: 10.1016/j.vetmic.2010.11.005
Jinadasa, R. N., Bloom, S. E., Weiss, R. S., and Duhamel, G. E. (2011). Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 157(Pt 7), 1851–1875. doi: 10.1099/mic.0.049536-0
Johnsen, G., Kruse, H., and Hofshagen, M. (2006). Genotyping of Campylobacter jejuni from broiler carcasses and slaughterhouse environment by amplified fragment length polymorphism. Poult. Sci. 85, 2278–2284. doi: 10.1093/ps/85.12.2278
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C., Colles, F. M., et al. (2012). Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158(Pt 4), 1005–1015. doi: 10.1099/mic.0.055459-0
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 11:595. doi: 10.1186/1471-2105-11-595
Kaakoush, N. O., Castano-Rodriguez, N., Mitchell, H. M., and Man, S. M. (2015). Global Epidemiology of Campylobacter Infection. Clin. Microbiol. Rev. 28, 687–720. doi: 10.1128/cmr.00006-15
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Lee, C. R., Cho, S. H., Yoon, M. J., Peterkofsky, A., and Seok, Y. J. (2007). Escherichia coli enzyme IIANtr regulates the K+ transporter TrkA. Proc. Natl. Acad. Sci. U.S.A. 104, 4124–4129. doi: 10.1073/pnas.0609897104
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Z., Lou, H., Ojcius, D. M., Sun, A., Sun, D., Zhao, J., et al. (2014). Methyl-accepting chemotaxis proteins 3 and 4 are responsible for Campylobacter jejuni chemotaxis and jejuna colonization in mice in response to sodium deoxycholate. J. Med. Microbiol. 63(Pt 3), 343–354. doi: 10.1099/jmm.0.068023-0
Line, J. E., and Bailey, J. S. (2006). Effect of on-farm litter acidification treatments on Campylobacter and Salmonella populations in commercial broiler houses in northeast Georgia. Poult. Sci. 85, 1529–1534. doi: 10.1093/ps/85.9.1529
Little, C. L., Gormley, F. J., Rawal, N., and Richardson, J. F. (2010). A recipe for disaster: outbreaks of Campylobacteriosis associated with poultry liver pate in England and Wales. Epidemiol. Infect. 138, 1691–1694. doi: 10.1017/S0950268810001974
Llarena, A. K., and Taboada, E. (2017). Whole-genome sequencing in epidemiology of Campylobacter jejuni Infections. J. Clin. Microbiol. 55, 1269–1275. doi: 10.1128/JCM.00017-17
Luber, P., and Bartelt, E. (2007). Enumeration of Campylobacter spp. on the surface and within chicken breast fillets. J. Appl. Microbiol. 102, 313–318. doi: 10.1111/j.1365-2672.2006.03105.x
Maiden, M. C., Jansen van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Maurelli, A. T., Fernandez, R. E., Bloch, C. A., Rode, C. K., and Fasano, A. (1998). “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. U.S.A 95, 3943–3948. doi: 10.1073/pnas.95.7.3943
McCutcheon, J. P., and Moran, N. A. (2011). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. doi: 10.1038/nrmicro2670
Mendz, G. L., Hazell, S. L., and Burns, B. P. (1993). Glucose utilization and lactate production by Helicobacter pylori. J. Gen. Microbiol. 139, 3023–3028. doi: 10.1099/00221287-139-12-3023
Merhej, V., and Raoult, D. (2011). Rickettsial evolution in the light of comparative genomics. Biol. Rev. Camb. Philos. Soc. 86, 379–405. doi: 10.1111/j.1469-185XX.2010.00151.x
Merhej, V., Royer-Carenzi, M., Pontarotti, P., and Raoult, D. (2009). Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 4:13. doi: 10.1186/1745-6150-4-13
Mikonranta, L., Mappes, J., Laakso, J., and Ketola, T. (2015). Within-host evolution decreases virulence in an opportunistic bacterial pathogen. BMC Evol. Biol. 15:165. doi: 10.1186/s12862-015-0447-5
Miller, C. E., Williams, P. H., and Ketley, J. M. (2009). Pumping iron: mechanisms for iron uptake by Campylobacter. Microbiology 155(Pt 10), 3157–3165. doi: 10.1099/mic.0.032425-0
Miller, W. G., Chapman, M. H., Yee, E., On, S. L., McNulty, D. K., Lastovica, A. J., et al. (2012). Multilocus sequence typing methods for the emerging Campylobacter Species C. hyointestinalis, C. lanienae, C. sputorum, C. concisus, and C. curvus. Front. Cell Infect. Microbiol. 2:45. doi: 10.3389/fcimb.2012.00045
Miller, W. G., Yee, E., Chapman, M. H., Smith, T. P., Bono, J. L., Huynh, S., et al. (2014a). Comparative genomics of the Campylobacter lari group. Genome Biol. Evol. 6, 3252–3266. doi: 10.1093/gbe/evu249
Miller, W. G., Yee, E., Jolley, K. A., and Chapman, M. H. (2014b). Use of an improved atpA amplification and sequencing method to identify members of the Campylobacteraceae and Helicobacteraceae. Lett. Appl. Microbiol. 58, 582–590. doi: 10.1111/lam.12228
Moffatt, C. R., Greig, A., Valcanis, M., Gao, W., Seemann, T., Howden, B. P., et al. (2016). A large outbreak of Campylobacter jejuni infection in a university college caused by chicken liver pate, Australia, 2013. Epidemiol. Infect. 144, 2971–2978. doi: 10.1017/S0950268816001187
Moran, N. A. (2002). Microbial minimalism: genome reduction in bacterial pathogens. Cell 108, 583–586. doi: 10.1016/S0092-8674(02)00665-7
Muraoka, W. T., and Zhang, Q. (2011). Phenotypic and genotypic evidence for L-fucose utilization by Campylobacter jejuni. J. Bacteriol. 193, 1065–1075. doi: 10.1128/JB.01252-10
Noormohamed, A., and Fakhr, M. K. (2012). Incidence and antimicrobial resistance profiling of Campylobacter in retail chicken livers and gizzards. Foodborne Pathog. Dis. 9, 617–624. doi: 10.1089/fpd.2011.1074
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Parkhill, J., Sebaihia, M., Preston, A., Murphy, L. D., Thomson, N., Harris, D. E., et al. (2003). Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35, 32–40. doi: 10.1038/ng1227
Parkhill, J., Wren, B. W., Mungall, K., Ketley, J. M., Churcher, C., Basham, D., et al. (2000). The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668. doi: 10.1038/35001088
Pascoe, B., Meric, G., Murray, S., Yahara, K., Mageiros, L., Bowen, R., et al. (2015). Enhanced biofilm formation and multi-host transmission evolve from divergent genetic backgrounds in Campylobacter jejuni. Environ. Microbiol. 17, 4779–4789. doi: 10.1111/1462-2920.13051
Petrovska, L., Mather, A. E., AbuOun, M., Branchu, P., Harris, S. R., Connor, T., et al. (2016). Microevolution of monophasic salmonella Typhimurium during epidemic, United Kingdom, 2005-2010. Emerging Infect. Dis. 22, 617–624. doi: 10.3201/eid2204.150531
Poly, F., Threadgill, D., and Stintzi, A. (2005). Genomic diversity in Campylobacter jejuni: identification of C. jejuni 81-176-specific genes. J. Clin. Microbiol. 43, 2330–2338. doi: 10.1128/JCM.43.5.2330-2338.2005
Richards, V. P., Lefebure, T., Pavinski Bitar, P. D., and Stanhope, M. J. (2013). Comparative characterization of the virulence gene clusters (lipooligosaccharide [LOS] and capsular polysaccharide [CPS]) for Campylobacter coli, Campylobacter jejuni subsp. jejuni and related Campylobacter species. Infect. Genet. Evol. 14, 200–213. doi: 10.1016/j.meegid.2012.12.010
Rondini, S., Kaser, M., Stinear, T., Tessier, M., Mangold, C., Dernick, G., et al. (2007). Ongoing genome reduction in Mycobacterium ulcerans. Emerging Infect. Dis. 13, 1008–1015. doi: 10.3201/eid1307.060205
Sheppard, S. K., Dallas, J. F., MacRae, M., McCarthy, N. D., Sproston, E. L., Gormley, F. J., et al. (2009a). Campylobacter genotypes from food animals, environmental sources and clinical disease in Scotland 2005/6. Int. J. Food Microbiol. 134, 96–103. doi: 10.1016/j.ijfoodmicro.2009.02.010
Sheppard, S. K., Dallas, J. F., Strachan, N. J., MacRae, M., McCarthy, N. D., Wilson, D. J., et al. (2009b). Campylobacter genotyping to determine the source of human infection. Clin. Infect. Dis. 48, 1072–1078. doi: 10.1086/597402
Skirrow, M. B. (1977). Campylobacter enteritis: a “new” disease. Br. Med. J. 2, 9–11. doi: 10.1136/bmj.2.6078.9
Smith, C. K., Abuoun, M., Cawthraw, S. A., Humphrey, T. J., Rothwell, L., Kaiser, P., et al. (2008). Campylobacter colonization of the chicken induces a proinflammatory response in mucosal tissues. FEMS Immunol. Med. Microbiol. 54, 114–121. doi: 10.1111/j.1574-695XX.2008.00458.x
Stahl, M., Friis, L. M., Nothaft, H., Liu, X., Li, J., Szymanski, C. M., et al. (2011). L-fucose utilization provides Campylobacter jejuni with a competitive advantage. Proc. Natl. Acad. Sci. U.S.A. 108, 7194–7199. doi: 10.1073/pnas.1014125108
Stern, N. J., Clavero, M. R., Bailey, J. S., Cox, N. A., and Robach, M. C. (1995). Campylobacter spp. in broilers on the farm and after transport. Poult. Sci. 74, 937–941. doi: 10.3382/ps.0740937
Toft, C., and Andersson, S. G. (2010). Evolutionary microbial genomics: insights into bacterial host adaptation. Nat. Rev. Genet. 11, 465–475. doi: 10.1038/nrg2798
Tudor, D. C. (1954). A liver degeneration of unknown origin in chickens. J. Am. Vet. Med. Assoc. 125, 219–220.
van der Graaf-van Bloois, L., Duim, B., Miller, W. G., Forbes, K. J., Wagenaar, J. A., and Zomer, A. (2016). Whole genome sequence analysis indicates recent diversification of mammal-associated Campylobacter fetus and implicates a genetic factor associated with H2S production. BMC Genomics 17:713. doi: 10.1186/s12864-016-3058-7
Van, T. T., Elshagmani, E., Gor, M. C., Anwar, A., Scott, P. C., and Moore, R. J. (2017). Induction of spotty liver disease in layer hens by infection with Campylobacter hepaticus. Vet. Microbiol. 199, 85–90. doi: 10.1016/j.vetmic.2016.12.033
Van, T. T., Elshagmani, E., Gor, M. C., Scott, P. C., and Moore, R. J. (2016). Campylobacter hepaticus sp. nov., isolated from chickens with spotty liver disease. Int. J. Syst. Evol. Microbiol. 66, 4518–4524. doi: 10.1099/ijsem.0.001383
van Vliet, A. H., Wooldridge, K. G., and Ketley, J. M. (1998). Iron-responsive gene regulation in a Campylobacter jejuni fur mutant. J. Bacteriol. 180, 5291–5298.
Weber, R., Auerbach, M., Jung, A., and Glunder, G. (2014). Campylobacter infections in four poultry species in respect of frequency, onset of infection and seasonality. Berl. Munch. Tierarztl. Wochenschr. 127, 257–266.
Wei, J., Goldberg, M. B., Burland, V., Venkatesan, M. M., Deng, W., Fournier, G., et al. (2003). Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 71, 2775–2786. doi: 10.1128/IAI.71.5.2775-2786.2003
Whyte, P., Collins, J. D., McGill, K., Monahan, C., and O'Mahony, H. (2001). The effect of transportation stress on excretion rates of campylobacters in market-age broilers. Poult. Sci. 80, 817–820. doi: 10.1093/ps/80.6.817
Wigley, P. (2015). Blurred lines: pathogens, commensals, and the healthy gut. Front. Vet. Sci. 2:40. doi: 10.3389/fvets.2015.00040
Winterfield, R. W., and Sevoian, M. (1957). Isolation of a causal agent of an avian hepatitis. Vet. Med. 52:273.
Keywords: Campylobacter hepaticus, spotty liver disease, poultry, genome reduction, niche adaptation
Citation: Petrovska L, Tang Y, Jansen van Rensburg MJ, Cawthraw S, Nunez J, Sheppard SK, Ellis RJ, Whatmore AM, Crawshaw TR and Irvine RM (2017) Genome Reduction for Niche Association in Campylobacter Hepaticus, A Cause of Spotty Liver Disease in Poultry. Front. Cell. Infect. Microbiol. 7:354. doi: 10.3389/fcimb.2017.00354
Received: 21 January 2017; Accepted: 21 July 2017;
Published: 11 August 2017.
Edited by:
Alain Stintzi, University of Ottawa, CanadaReviewed by:
Byeonghwa Jeon, University of Alberta, CanadaChristine M. Szymanski, University of Alberta, Canada
Copyright © 2017 Petrovska, Tang, Jansen van Rensburg, Cawthraw, Nunez, Sheppard, Ellis, Whatmore, Crawshaw and Irvine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liljana Petrovska, bGlsamFuYS5wZXRyb3Zza2FAYXBoYS5nc2kuZ292LnVr
†These authors have contributed equally to this work.