Abstract
We collected 8,700 mosquitoes in three sites in China, which belonged to seven species. Their viromes were tested using metagenomic sequencing and bioinformatic analysis. The abundant viral sequences were detected and annotated belonging to more than 50 viral taxonomic families. The results were verified by PCR, followed by phylogenetic analysis. In the present study, we identified partial viral genes of dengue virus (DENV), a novel circovirus (CCV), densovirus (DNV), Japanese encephalitis virus (JEV), and Wuhan mosquito virus (WMV) in mosquitoes. Metagenomic analysis and PCR amplification revealed three DENV sequences, which were as homologous to the NS3 gene of DENV from Singapore isolated in 2005, with at least 91% nucleotide (nt) identity. Seven fragments of JEV encoding structural proteins were identified belonging to genotype I. They all shared high homology with structural protein genes of JEV isolated from Laos in 2009. The production of infectious virus particles of the newly isolated virus YunnanJEV2017-4 increased after passage from the BHK-21 cell line to the Vero cell line. Novel circovirus-related genes were identified and as being related to an unnamed gene of a mosquito circovirus (MCCV) sequence from the USA isolated in 2011, with at least 41% nt identity: this distant relationship suggests that the parent virus might belong to a novel circovirus genus. Additionally, numerous known viruses and some unknown viruses were also detected in mosquitoes from Yunnan province, China, which will be tested for propagation.
Introduction
Mosquitoes are significant insect vectors for a variety of infectious viruses covering Japanese encephalitis virus (JEV), Circovirus (CCV), Zika virus (ZIKV), Dengue virus (DENV), and Densovirus (DNV), which pose a significant threat to human health and induce an economic burden worldwide (Pham et al., 2005; Klungthong et al., 2007). Mosquitoes are infected with viruses when consuming blood from hosts undergoing viremia. Then the viruses replicate and propagate in the insect host and are introduced into further victims during biting and blood feeding (Ritchie et al., 2007; Motooka et al., 2015). Mosquitoes feed on a wide range of hosts, including humans and other vertebrates, invertebrates, and plants (Shi et al., 2014). Yunnan province in China harbors a diverse range of mosquito-borne viruses (Feng et al., 2015); therefore, regional surveillance is imperative. The detection of viruses in mosquitoes is usually performed by reverse transcription polymerase chain reaction (RT-PCR) and nested PCR approaches (Almeida et al., 2005; Houghton, 2009; Li et al., 2015; Sim et al., 2015). However, compared with Illumina sequencing (Alquezar-Planas et al., 2013; Cholleti et al., 2016; Ergunay et al., 2017), these traditional methods are time-consuming and labor intensive to detect low-level viromes in mosquito vectors (He et al., 2013; Miesen et al., 2016). Thus, metagenomic analysis of mosquitoes is likely to be of great value to avoid missing the detection of viruses with high infectivity and pathogenicity in different collection sites, and the detection of previously unknown viruses.
The present study aimed to build a valid surveillance method to monitor the distribution of viruses from mosquitoes in Yunnan province, China and to provide useful insights into viral isolation, prevention, and control. Diverse and abundant viromes from mosquitoes isolated in Yunnan province were investigated using metagenomic sequencing and PCR. The presence of DENV, JEV, and novel circovirus were confirmed, and JEV was isolated. This preliminary exploration of the metagenomes of mosquito-borne viruses lays the foundation for further research on the territorial distribution, diversity, and surveillance of mosquito-borne viruses in China and other countries.
Materials and methods
Mosquito collection
We collected 8,700 living or freshly dead mosquitoes in Yunnan province, China, during September and October, 2017. The acquired mosquitoes were composed of Culex tritaeniorhynchus (C. tritaeniorhynchus), Armigeres obturbans (Ar. obturbans), Aedes albopictus (Ae. albopictus), Anopheles sinensis (An. sinensis), Anopheles maculatus (An. maculates), Anopheles mininus (An. minimus), and Culex quinquefasciatus (C. quinquefasciatus). The three mosquito samples were grouped based on collection sites (Table 1). The species of mosquitoes in all groups were identified separately and stored at −80°C (Supplementary Table 1). This research was approved by the Ethics Committee and the Research Ethics Committee of Yanbian University Medical College. All experiments involved in active viruses were performed in Biological safety protection third-level laboratory.
Table 1
| Sample | Species | Number | Location | Total reads number | Average (nt) | Viral reads | Viral contigs |
|---|---|---|---|---|---|---|---|
| Sample I | Mosquito Community# | 1000 | Ninger County* | 8,618,693 | 192.20 | 1,032,569 | 4,627 |
| Sample II | Mosquito community# | 1000 | Jinggu county* | 8,797,151 | 194.59 | 763,217 | 3,819 |
| Sample III | Mosquito community# | 6700 | Dali City* | 8,786,675 | 192.34 | 1,263,176 | 6,573 |
| Total/average | 26,202,519 | 193.05 | 2,340,048 | 15,019 |
Mosquito samples employed in metagenomic analysis and data from Illumina sequencing.
Collection sites (Figure 1) were Ninger county (N 23° 03′, E 101° 02′) and Jinggu county (N 23° 50′, E 100° 71′) in Puer city, and Dali city (N 25° 69′, E 100° 19′). #Mosquito species were consist of C. tritaeniorhynchus, Ar. obturbans, Ae. albopictus, An. sinensis, An. maculates, An. Minimus, and C. quinquefasciatus.
Figure 1
Distribution of sample collection sites in Yunnan province, China, 2017. The sample collection sites were labeled with red star.
Preparation of mosquito samples
Briefly, mosquitoes of three samples were mixed with SM buffer (50 mM Tris, 10 mM MgSO4, 0.1 M NaCl, pH 7.5) (MeilunBio, Dalian, China) respectively and ground using a tissue grinder. After removal of mosquito debris and other materials, the mixed samples were centrifuged at 13000 × g for 20 min, and the supernatants were employed to extract the viral nucleic acid.
Extraction of viral nucleic acid and reverse transcription
Clearly, 14 U of Turbo DNase (Ambion, Austin, TX, USA), 25 U of Benzonase Nuclease (Novagen, San Diego, CA, USA), 20 U of RNase I (Fermentas, Ontario, Canada), and 10 × DNase buffer (Ambion) were added to 127 μL of the supernatants to a final volume of 150 μL, followed by digestion at 37°C for 1 h. After removing free nucleic acid and eliminating the contaminating host genomic DNA, the viral nucleic acid in the obtained products was extracted using a Virus Nucleic Acid Kit (Bioer Technology, Hangzhou, China) according to the manufacturer's instructions. The total viral nucleic acids were reverse transcribed using anchored random primers and Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA). Anchored random primers (Table 2) were added separately to the viral nucleic acid and incubated at 75°C for 5 min and placed on ice for 5 min for denaturation. To acquire the reverse transcribed product, 40 U of RNase OUT, 200 U of SuperScript III reverse transcriptase, 1 μL of 0.1 M dithiothreitol (DTT), 1 μL of 10 mM dNTPs, 4 μL of 5 × first strand buffer, and RNase-free H2O were added to a final volume of 20 μL. It was incubated at 25°C for 10 min followed by 50°C for 60 min, and 75°C for 10 min.
Table 2
| Primer type | Primer number | Primers (5′-3′) |
|---|---|---|
| Anchored Random Primers | RT1 | GCCGGAGCTCTGCAGATATCNNNNNN |
| RT 2 | GTATCGCTGGACACTGGACCNNNNNN | |
| RT 3 | ATCGTCGTCGTAGGCTGCTCNNNNNN | |
| RT 4 | CGTAGATAAGCGGTCGGCTCNNNNNN | |
| RT 5 | CATCACATAGGCGTCCGCTGNNNNNN | |
| RT 6 | CGCAGGACCTCTGATACAGGNNNNNN | |
| RT 7 | CGTCCAGGCACAATCCAGTCNNNNNN | |
| RT 8 | CCGAGGTTCAAGCGAGGTTGNNNNNN | |
| RT 9 | ACGGTGTGTTACCGACGTCCNNNNNN | |
| RT 10 | CGACCCTCTTATCGTGACGGNNNNNN | |
| RT 11 | GAGCCCCTAGACACAACGACNNNNNN | |
| RT 12 | GGTGGGCGTGTGAAATCGACNNNNNN | |
| RT 13 | GAAAATGAGAGGGGAGGCGGNNNNNN | |
| Barcode primers | Primer1 | GCCGGAGCTCTGCAGATATC |
| Primer2 | GTATCGCTGGACACTGGACC | |
| Primer3 | ATCGTCGTCGTAGGCTGCTC | |
| Primer4 | CGTAGATAAGCGGTCGGCTC | |
| Primer5 | CATCACATAGGCGTCCGCTG | |
| Primer6 | CGCAGGACCTCTGATACAGG | |
| Primer7 | CGTCCAGGCACAATCCAGTC | |
| Primer8 | CCGAGGTTCAAGCGAGGTTG | |
| Primer9 | ACGGTGTGTTACCGACGTCC | |
| Primer10 | CGACCCTCTTATCGTGACGG | |
| Primer11 | GAGCCCCTAGACACAACGAC | |
| Primer12 | GGTGGGCGTGTGAAATCGAC | |
| Primer13 | GAAAATGAGAGGGGAGGCGG |
Barcode DNA employed in metagenomic analysis (He et al., 2013).
Synthesis of double-strand cDNA (dscDNA)
To degrade the free RNA, 1 μL of RNaseH was added to the above-obtained products. To synthesis dscDNA, anchored random primers were added and incubated at 75°C for 5 min and on ice for 5 min for denaturation. Then 1 μL of Klenow fragment, 1 μL of 10 mM dNTPs, 2 μL of 10 × Klenow buffer, and 6 μL of dd H2O were added. Subsequently, the samples were incubated at 37°C for 60 min, followed by 75°C for 10 min. To eliminate phosphates and free single-strand nucleic acid in the dscDNA reaction, 0.5 μL of Exonuclease I, 1 μL of shrimp alkaline phosphatase (SAP, Takara, Dalian, China), 5 μL of 10 × phosphatase buffer, and 24 μL of DEPC H2O were added and incubated at 37°C for 60 min followed by 75°C for 10 min.
Sequence-independent single primer amplification (SISPA) and purification of PCR products
To obtain large quantity of the viral nucleic acids, the dscDNA was amplified using SISPA. The 50-μL reaction comprised 10 μL of dscDNA, 2 μL of barcode primer (Table 2), 1 μL of Accuprime Taq DNA polymerase, 5 μL of 10 × Accuprime buffer I, and 32 μL of dd H2O. The PCR condition comprised 95°C for 3 min; 40 cycles of 95°C for 20 s, 54°C for 20 s, and 68°C for 70 s, with a final extension at 68°C for 7 min. The acquired PCR products were purified using a PCR purification kit (QIAGEN, Hilden, Germany) and eluted in 30 μL of TE buffer (100 mM Tris-HCl, 10 mM EDTA, pH8.0).
Metaviral sequencing
The purified PCR products were sent to the Wuhan Genome Institute (BGI, Shenzhen, China) for Illumina sequencing. Concisely, to obtain ~180 bp DNA fragments, the purified PCR products were ultrasonicated and dATP and Klenow fragment were added to produce 3′-dA overhangs. To establish genomic DNA libraries, DNA fragments were bound to Illumina adaptors and amplified using PCR with adaptor primers. Amplicons were ligated to flow cells to which fluorescently labeled dNTPs were added. Identification of DNA sequences was performed by the Sequencing-by-Synthesis method (SBS, Illumina). Base calling was monitored by the program GAPipeline (BGI) with default settings. No-calling reads and adaptor sequences were eliminated. The remaining sequences were assembled into contigs using SOAPdenovo software (BGI). Contigs and sequences longer than 100 bp were defined as significant data for further in silico analysis.
Computational analysis
Alignments of contigs and sequences were operated using BLASTx and BLASTn with the non-redundant and viral reference databases in GenBank (https://www.ncbi.nlm.nih.gov/genbank/). BLAST hits with an E value ≤ 10e−5 were considered significant. After elimination of bacterial and eukaryotic sequences, virus-like sequences were analyzed.
Identification of detected viruses
In accordance with the alignment results of viral contigs and the match position of viral contigs with the corresponding viruses in GenBank, specific primers (Table 3) were designed and synthesized to identify the detected viruses. Viral nucleic acids were extracted using a Virus Nucleic Acid Kit (Bioer Technology) and amplified using the designed primers and a PCR Master Mix (Tiangen, Beijing, China).
Table 3
| Primer Name | Primers (5′-3′) | Product (bp) |
|---|---|---|
| YunnanWMV8-2017-1/2/3/4/5-F | GTGAAGAAAGATGAAGGAT | 561 |
| YunnanWMV8-2017-1/2/3/4/5-R | CTATCCAGGGCCTCCCATCA | |
| YunnanDENV2017-1/2/3-F | AGGAGCCCTGTGGGACGTCCC | 1836 |
| YunnanDENV2017-1/2/3-R | TTTTCTTCCACTGGCAAACTCCTT | |
| YunnanDNV2017-1/2/3-F | ACTGGACCAACCGTTGGTG | 387 |
| YunnanDNV2017-1/2/3-R | AGGGCTATGTGCGTTAACAAT | |
| YunnanDNV2017-4/5-F | ACAAAACAAACTCATCAGTCGGC | 429 |
| YunnanDNV2017-4/5-R | TTAGATGATGTAAGGGTTTTGGTTG | |
| YunnanDNV2017-6/7/8-F | CTACTAGCAATGGTTAAACTGG | 801 |
| YunnanDNV2017-6/7/8-R | TCAAGAATCCGGCTGTTTGGTA | |
| YunnanMCCV2017-1/2-F | GCCGGAGCTCTGCAGATAT | 645 |
| YunnanMCCV2017-1/2-R | CCGGAGCTCAGACGTGTGCT | |
| YunnanJEV2017-1/2-F | AGCCGGAGCTCTGCAGATAT | 870 |
| YunnanJEV2017-1/2-R | CTACAGACGTGTGCTCTTC | |
| YunnanJEV2017-3-F | TTTAACTGTCTGGGAATGG | 1064 |
| YunnanJEV2017-3-R | ACCAACCTCCCCACAGGGG | |
| YunnanJEV2017-4/5/6/7-F | ATGAAGCTATCAAACTTTCAAG | 2001 |
| YunnanJEV2017-4/5/6/7-R | GGCATGCACATTGGTCGCT | |
| JEV-F* | CTATTGGTCGCTCCGGCTTACAGT | 1500 |
| JEV-R* | TGTCAATGGCGCAGCCAGTGTC |
Primer pairs used in PCR identification.
The primers used in JEV identification after viral isolation.
Phylogenetic analysis
The acquired PCR products were sequenced, and the sequences were aligned against sequences of representative viruses using Clustal W version 2.0. (Accession numbers are shown in the phylogenetic trees). Maximum-likelihood phylogenetic trees were produced using MEGA 7 with 1,000 bootstrap replicates.
Cell culture
The hamster cell line BHK-21 and Vero cell line (both available in our laboratory) were employed in this study. BHK-21 cells and Vero cells were cultured in Dulbecco's modified Eagle's medium (DMEM. HyClone, Logan, UT, USA) with 10% fetal bovine serum (FBS. HyClone) and 1% penicillin and streptomycin (Pen Strep. HyClone), and incubated at 37°C in 5% CO2.
Isolation of viruses
Viral isolation was conducted for the positive samples. Briefly, the mosquito samples were diluted 7-fold with DMEM containing 2% FBS and the infected BHK-21 cells were cultured for 5–7 days. Cultures were examined daily for evidence of a virally induced cytopathic effect (CPE). Cultures without a CPE were blind passaged three times.
Identification of the isolated virus by PCR and western blot
The isolated virus YunnanJEV2017-4 was identified by PCR technique using the specific primers (Table 3) of the envelope (E) protein genes. Moreover, the further verification was performed by Western blot method with an anti-E monoclonal antibody (Abcam, Cambridge, UK) as primary antibody and a HRP-conjugated goat anti-mouse antibody (ZSGB-Bio, Beijing, China) as second antibody.
Observation of negative-stain electron microscopy
BHK-21 cells with suspected JEV-induced CPE were collected by repeated freezing and thawing three times, followed by centrifugation at 10,000 × g for 15 min. The obtained virus suspension was centrifuged at 60,000 × g for 4 h and the supernatant was removed gently and discarded. Subsequently, the viral precipitate was resuspended with an isometric mixture of 6.1% (v / v) pH 7.2 glutaraldehyde (HEDEBIO, Beijing, China) fixative and DMEM, of which 25 μL were added to the copper grid. After desiccation, one drop of 3% phosphotungstic acid (JINDU, Shanghai, China) was added for negative staining. Before observation under an electron microscopic(FEI, Hillsboro, USA), the grid was placed in an incubator (SANYO, Osaka, Japan) at 37°C for desiccation.
Detection of the viral titer of different passages
The viral titers of different passages of JEV (passage 5 and 10) were evaluated in BHK-21 cells in triplicate. Cells were infected with serial tenfold dilutions of JEV and incubated for 120 h at 37°C. The cytopathic effect of JEV at each dilution was evaluated in eight replicates. The TCID50 (50% tissue culture infective dose) was calculated by the Reed-Muench method (Krah, 1991).
The variability of envelope (E) genes of different passage viruses
The obtained JEV (P5 and P10) from BHK-21 cells line were passaged in Vero cells line respectively. Viral RNA of JEV (P5 and P10) from infected BHK-21 and Vero cells was extracted from the supernatant using an RNA viral kit (Bioer Technology). The cDNA was synthesized with the purified RNA using a SuperScript™ III first-strand synthesis system (Invitrogen) with the reverse primer JWR1 5′-AGATCCTGTGTTCTTCCTCACCACCA-3′, according to the manufacturer's instructions. Envelope genes of JEV (P5 and P10) from infected BHK-21 and Vero cells were amplified using the paired primers shown in Table 3 and sequenced. The alignment of the four envelope genes was performed using MEGA 7.0.
Statistical analysis
The data analysis was performed using SPSS 17.0 software. Group comparisons for viral titers were done by t-test for independent means. Experiments were performed in triplicate and a minimum of three independent experiments were evaluated. The value of P < 0.05 was considered statistically significant.
GenBank accession numbers
Amplified sequences were deposited in the GenBank under the following accession numbers: YunnanWMV8-2017-1/2/3/4/5 (MH193505–MH193509), YunnanDENV2017-1/2/3 (MH193510–MH193512), YunnanDNV2017-1/2/3/4/5/6/7/8 (MH193513–MH193520), YunnanMCCV2017-1/2 (MH193521–MH193522), YunnanJEV2017-1/2/3/4/5/6/7 (MH193523–MH193529), respectively.
Results
Metaviral sequencing and the virome of mosquitoes
To obtain the clean data of the virome in mosquitoes, contaminating host sequences and barcode DNA were eliminated. A total of 26,202,519 reads were acquired by Illumina sequencing, with averaging read lengths of 193.05 nt (Table 1). The amount of viral sequences was 11.98, 8.68, and 14.38% in sample I, II, and III respectively, of which the relative abundance of viral families was shown in Figure 2A. The intersection among the three samples was displayed in Figure 2B. There were 45, 42, and 53 viral families in sample I, II, and III respectively. Remarkably, the three samples all possessed representative sequences from 38 common families: Luteoviridae, Marseilleviridae, Tombusviridae, Parvoviridae, Adenoviridae, Baculoviridae, Nudiviridae, Podoviridae, Reoviridae, Secoviridae, Bunyaviridae, Iridoviridae, Iflaviridae, Dicistroviridae, Ascoviridae, Inoviridae, Orthomyxoviridae, Asfarviridae, Caulimoviridae, Nodaviridae, Herpesviridae, Arteriviridae, Circoviridae, Rhabdoviridae, Mimiviridae, Closteroviridae, Polydnaviridae, Phycodnaviridae, Picornaviridae, Partitiviridae, Retroviridae, Myoviridae, Nyamiviridae, Mesoniviridae, Permutotetraviridae, Siphoviridae, Flaviviridae, Tymoviridae (Supplementary Table 2).
Figure 2
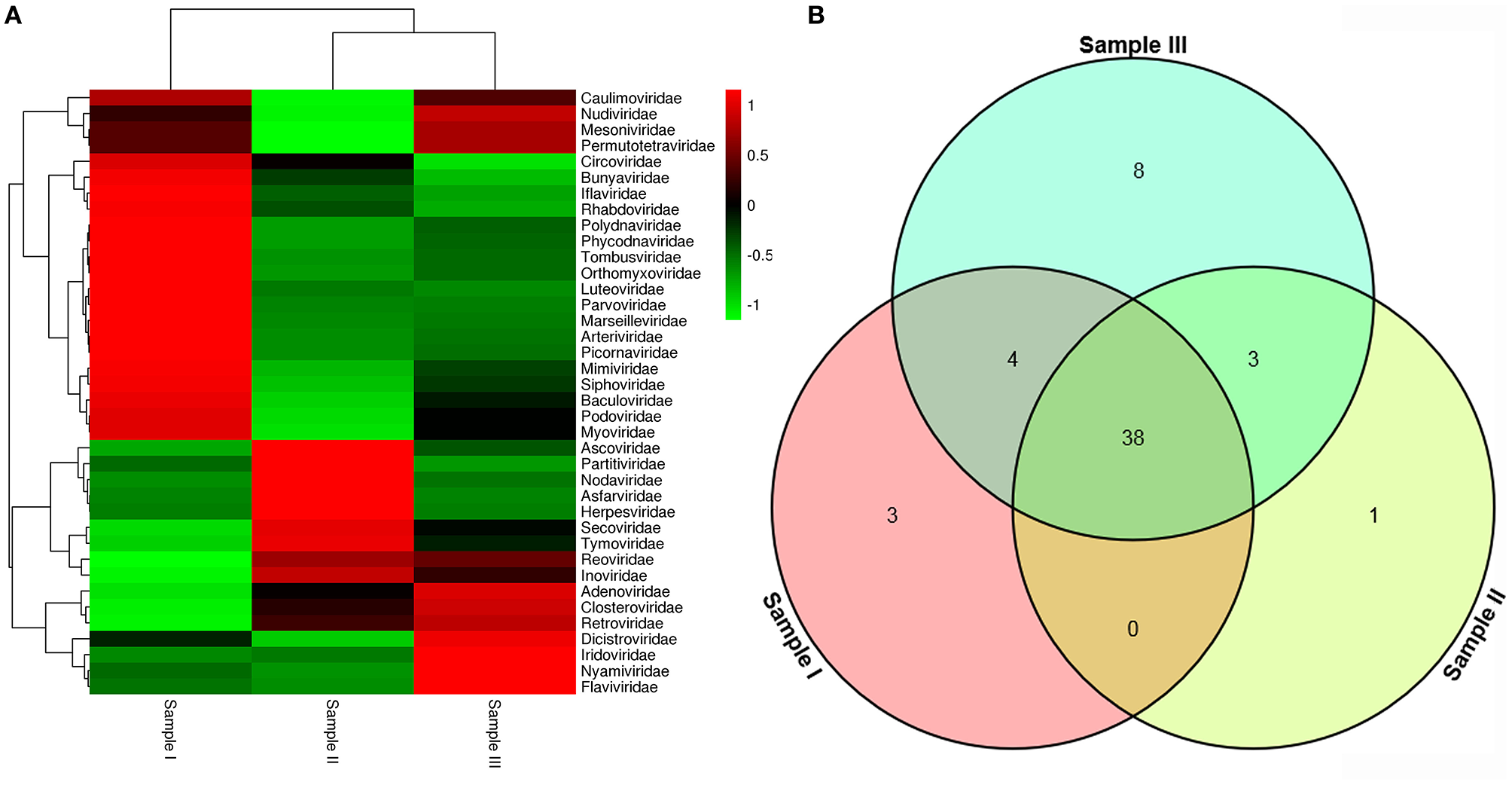
Abundance of viral families and their relationship in three samples. Viral sequences were sorted according to viral families and the relative abundance were displayed in Heat Map (A). The amount of viral families in each sample and their intersection among the three samples were shown in Venn diagram (B).
PCR verification of the metavirome results
Viral sequences were assembled into contigs using SOAPdenovo. From the 15,019 assembled viral contigs, it was detected that 46 WMV8-like contigs with a read coverage of 43 × (278–836 nt), 83 DENV-like contigs with a read coverage of 34 × (207–2356 nt), 164 DNV-like contigs with a read coverage of 127 × (257–936 nt), 26 MCCV-like contigs with a read coverage of 17 × (219–1053 nt), and 182 JEV-like contigs with a read coverage of 139 × (317–3285 nt). The WMV8-like contigs shared 91.3–93.3% nt identity with known WMV8 sequences. The DENV-like contigs shared 91.9–92.8% nt identity with known DENV sequences. The DNV-like contigs shared 80.6–90.0% nt identity with known DNV sequences. The MCCV-like contigs shared 41.6–42.3% nt identity with known MCCV sequences. The JEV-like contigs shared 86.6–97.7% nt identity with known JEV sequences. To confirm the outcome of metavirome sequencing, specific primers were designed and synthesized to amplify the identified viruses (Table 3). The following were the results of PCR identification of detected viruses in mosquito samples.
PCR amplification of wuhan mosquito virus
Based on the alignment results of viral contigs, viral PCR amplicons from sample I shared high identity with Wuhan mosquito virus (WMV) genes. Five 561-nucleotides-long segments (YunnanWMV8-2017-1/2/3/4/5) that shared ~96–98% nt identity and ~89–95% amino acid (aa) identity with each other were isolated. BLASTN against the GenBank identified these segments as homologous to the L gene of WMV, with at least 91% nt identity and 87% aa identity suggesting that viruses were from a WMV8 variant (Figure 3).
Figure 3
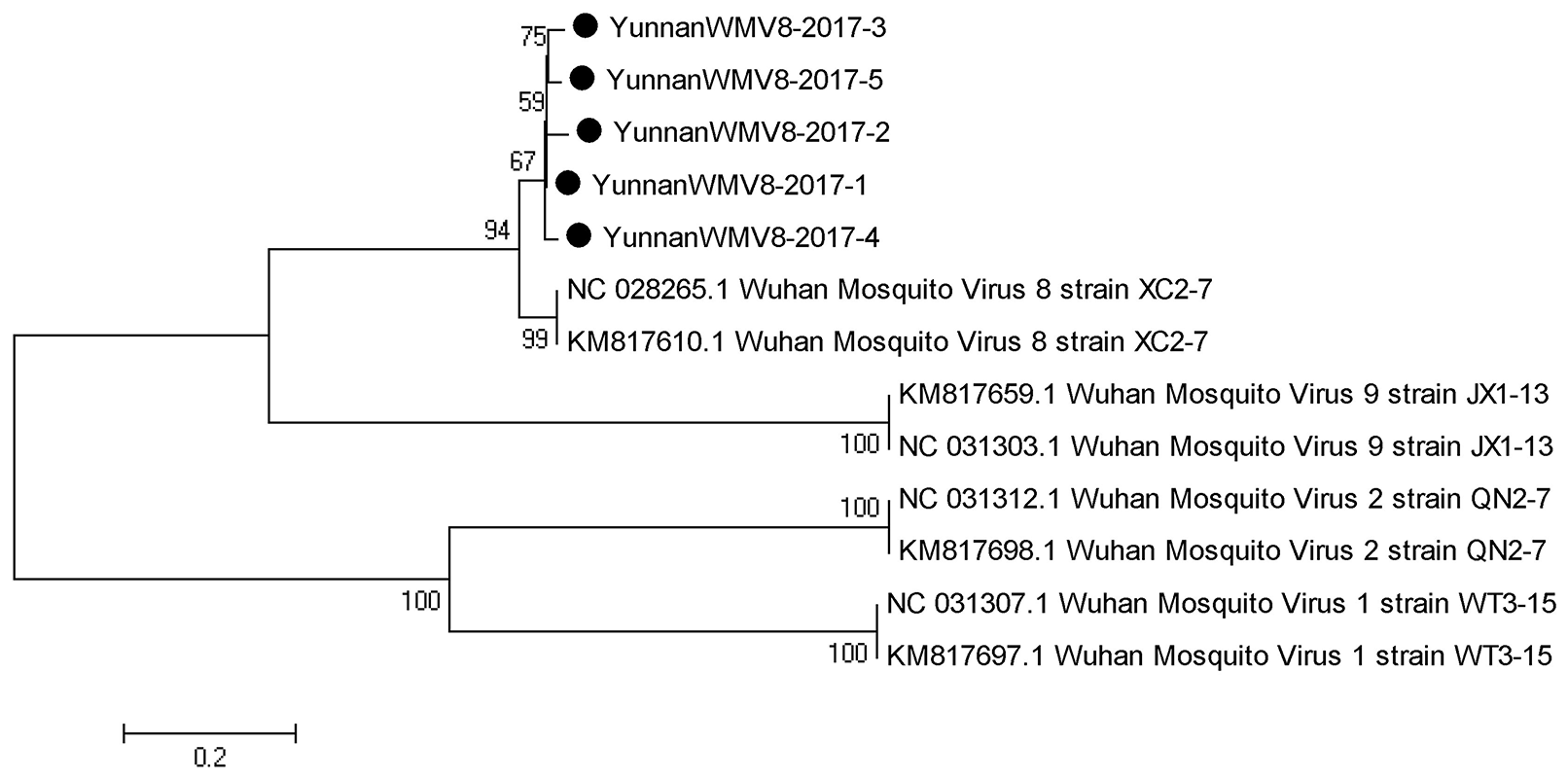
Phylogenetic tree of Wuhan mosquito virus. Phylogenetic trees based on RNA-dependent RNA polymerase (L) gene of Wuhan mosquito virus. Using the Maximum-likelihood method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the genes identified in this study.
PCR amplification of dengue virus
Based on the alignment results of viral contigs, viral PCR amplicons from samples III shared high identity with dengue virus (DENV, genus Flavivirus, family Flaviviridae). Three 1836-nucleotides-long segments (YunnanDENV2017-1/2/3) that shared ~98–99% nt identity and ~95–97% aa identity with each other were isolated. BLASTN against the GenBank identified these segments as homologous to the non-structural protein 3 (NS3) gene of DENV sequence from Singapore isolated in 2005, with at least 91% nt identity and 88% aa identity suggesting that viruses were from its variant (Figure 4). Phylogenetic analysis showed that the newly identified DENV sequences were genotype IV genes.
Figure 4
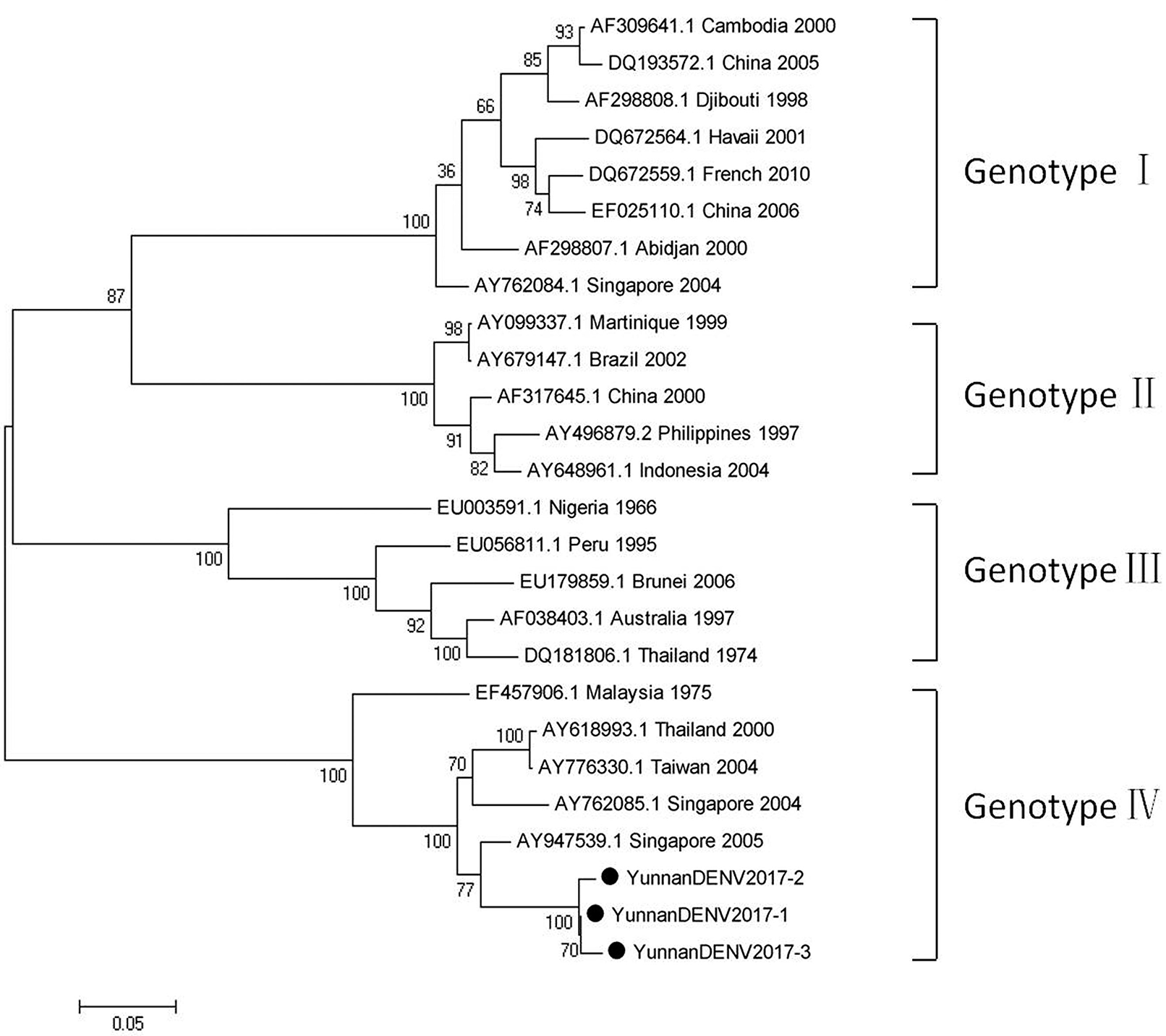
Phylogenetic tree of DENV. Phylogenetic tree based on Non-structural protein 3 (NS3) gene of DENV. Using the Maximum-likelihood method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the genes identified in this study.
PCR amplification of mosquito densovirus
In line with the alignment results of viral contigs, viral PCR amplicons from sample II shared high identity with Aedes albopictus densovirus (DNV, genus Brevidensovirus, family Parvoviridae). Eight viral segments were isolated, among which three 387-nucleotides-long segments (YunnanDNV2017-1/2/3), two 429-nucleotides-long segments (YunnanDNV2017-4/5) and three 801-nucleotides-long segments (YunnanDNV2017-6/7/8) that shared ~96–97%, ~97% and ~96–98% nt identity and ~89–93%, ~93% and ~91–95% aa identity respectively with each other. BLASTN against the GenBank identified these segments as homologous to the non-structural protein 2 (NS2) gene of DNV from China isolated in 2006 (Figure 5), with at least 87, 80, and 83% nt identity and 85, 79, and 85% aa identity respectively.
Figure 5
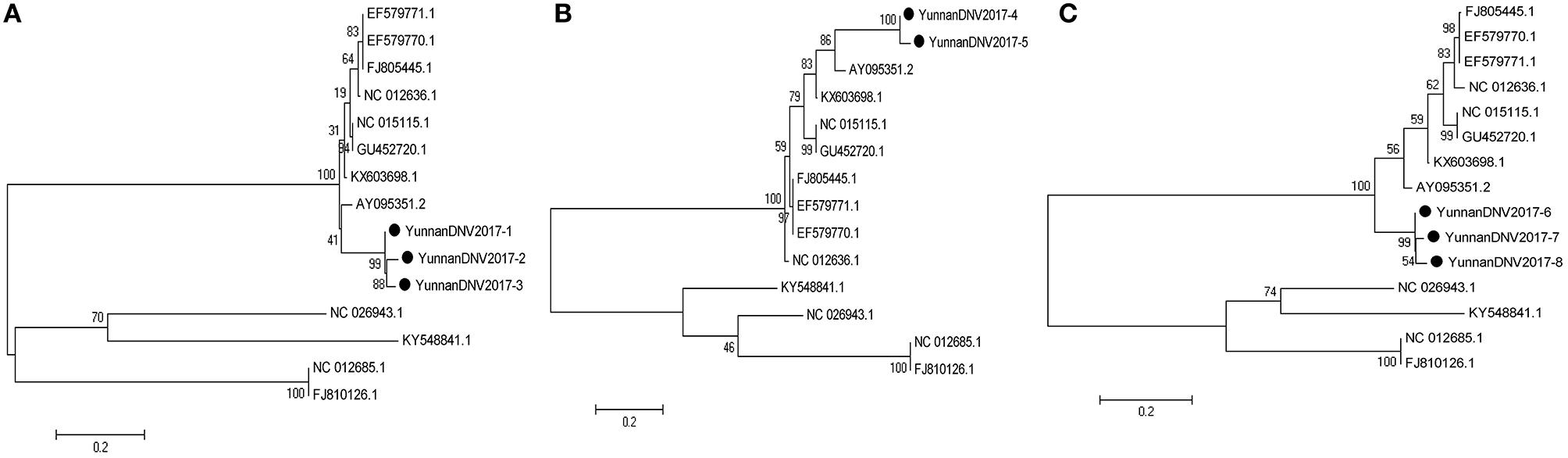
Phylogenetic trees of Mosquito densovirus. Phylogenetic tree based on Non-structural protein 2 (NS2) gene of Mosquito densovirus together with YunnanDNV2017-1/2/3 (A), YunnanDNV2017-4/5 (B), and YunnanDNV2017-6/7/8 (C). Using the Maximum-likelihood method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the genes identified in this study.
PCR amplification of mosquito circovirus
Based on the alignment results of viral contigs, viral PCR amplicons from samples III shared high identity with mosquito circovirus (MCCV, family Circoviridae). Two 645-nucleotides-long segments (YunnanMCCV2017-1/2) that shared ~97% nt identity and ~93% aa identity with each other were isolated. BLASTN against the GenBank identified these segments as homologous to an unnamed gene of MCCV, with at least 41% nt identity and 40% aa identity with MCCV sequence from America isolated in 2011 suggesting that viruses may be novel circovirus (Figure 6).
Figure 6
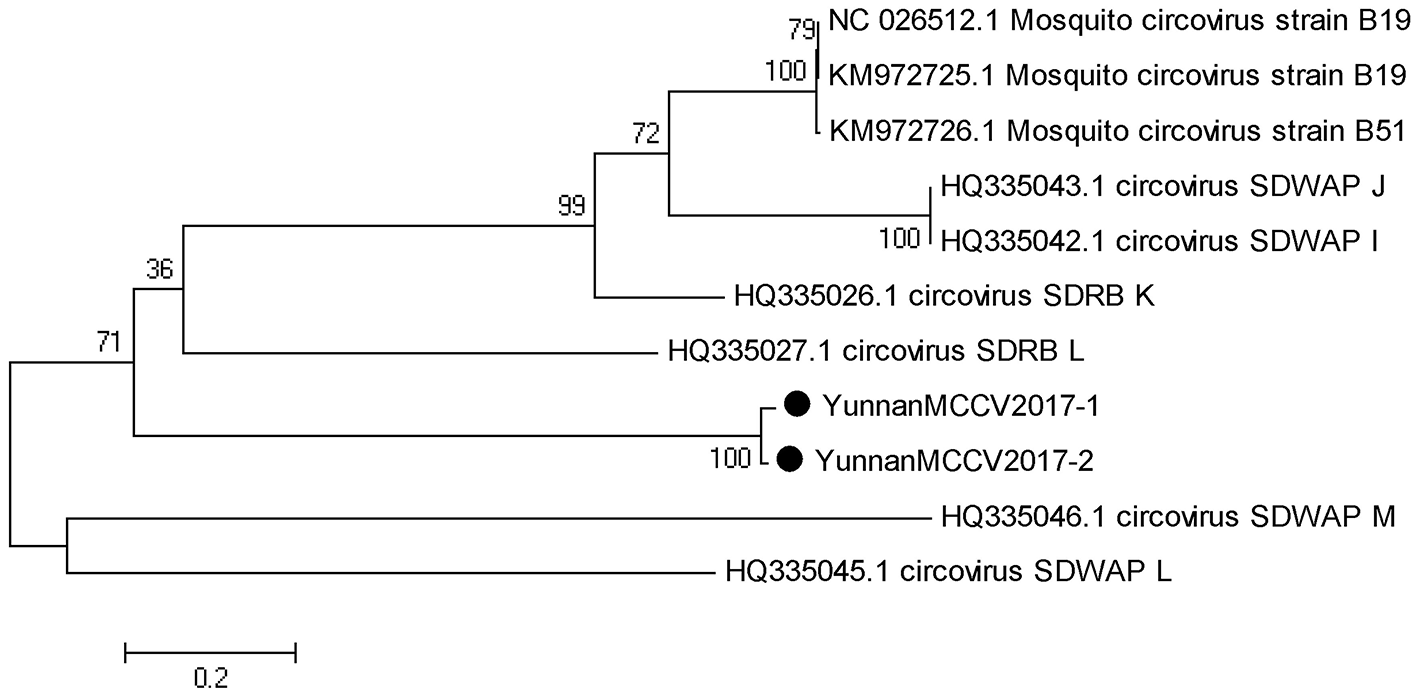
Phylogenetic tree of Mosquito circovirus. Phylogenetic tree based on Mosquito circovirus. Using the Maximum-likelihood method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the genes identified in this study.
PCR amplification of japanese encephalitis virus
According to the alignment results of viral contigs, viral PCR amplicons from sample III shared high identity with Japanese encephalitis virus (JEV, genus Flavivirus, family Flaviviridae). Two 870-nucleotides-long segments (YunnanJEV2017-1/2), one 1064-nucleotides-long segments (YunnanJEV2017-3) and four 2001-nucleotides-long segments (YunnanJEV2017-4/5/6/7) that all shared ~98% nt identity and shared ~95% and ~95–97% aa identity respectively with each other were isolated. BLASTN against the GenBank identified these segments as homologous to the C+prM, E and M+E genes of JEV sequence from Laos isolated in 2009, with at least 86, 95, and 96% nt identity and 83, 91, and 94% aa identity respectively. Phylogenetic analysis showed that the newly identified JEV genes all belonged to genotype I (Figure 7).
Figure 7
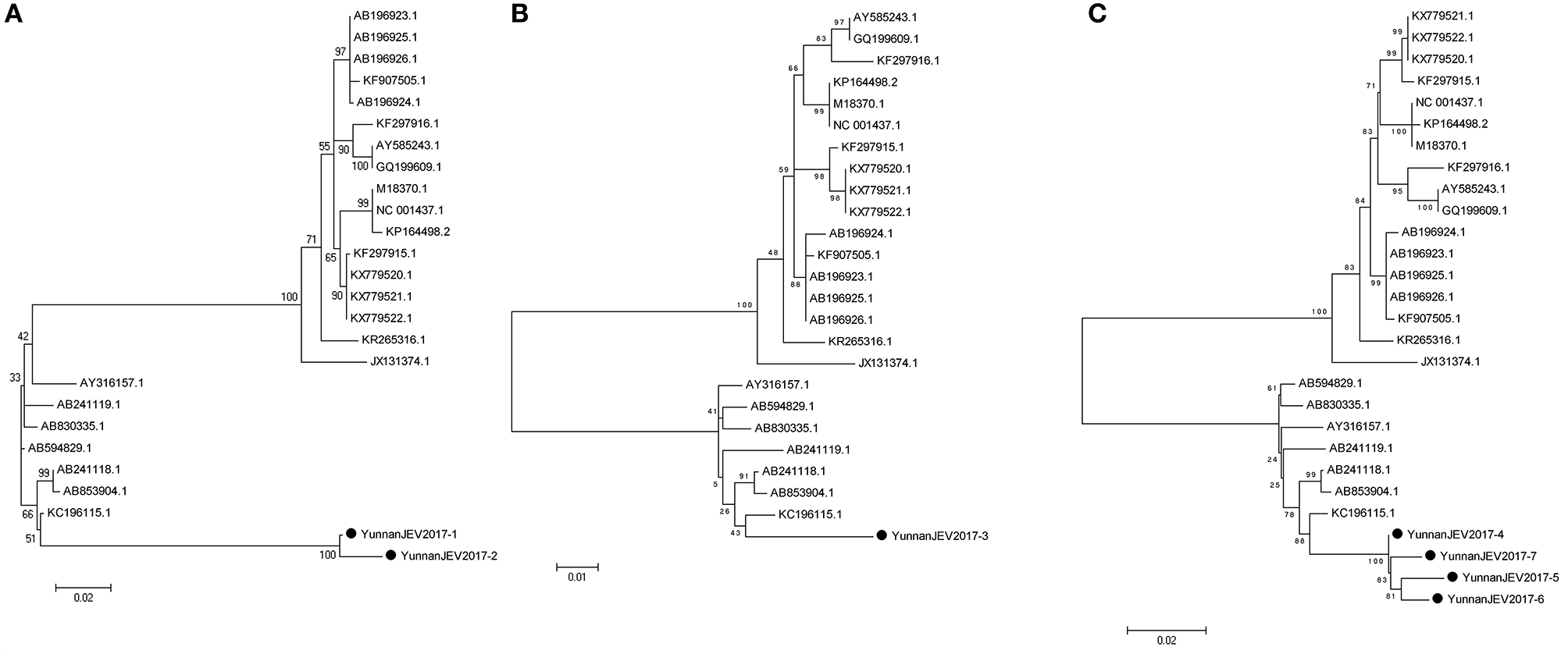
Phylogenetic trees of JEV. Phylogenetic tree based on C+prM gene of JEV together with YunnanJEV2017-1/2 (A), E gene of JEV together with YunnanJEV2017-3 (B), and prM+E gene with YunnanJEV2017-4/5/6/7 (C). Using the Maximum-likelihood method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the genes identified in this study.
Viral identification of japanese encephalitis virus
After inoculation into BHK-21 cells, the positive JEV strain was verified by PCR technique. Levels of intracellular JEV were measured by Western blot method using an E-specific monoclonal antibody. YunnanJEV2017-4-infected cells were positive for E protein expression. Viral particles were observed using negative-stain electron microscopy. The particles of YunnanJEV2017-4 were rounded with a diameter of approximately 30–40 nm. Moreover, there were tiny protrusions on the surface, which appeared similar to JEV (Figure 8).
Figure 8
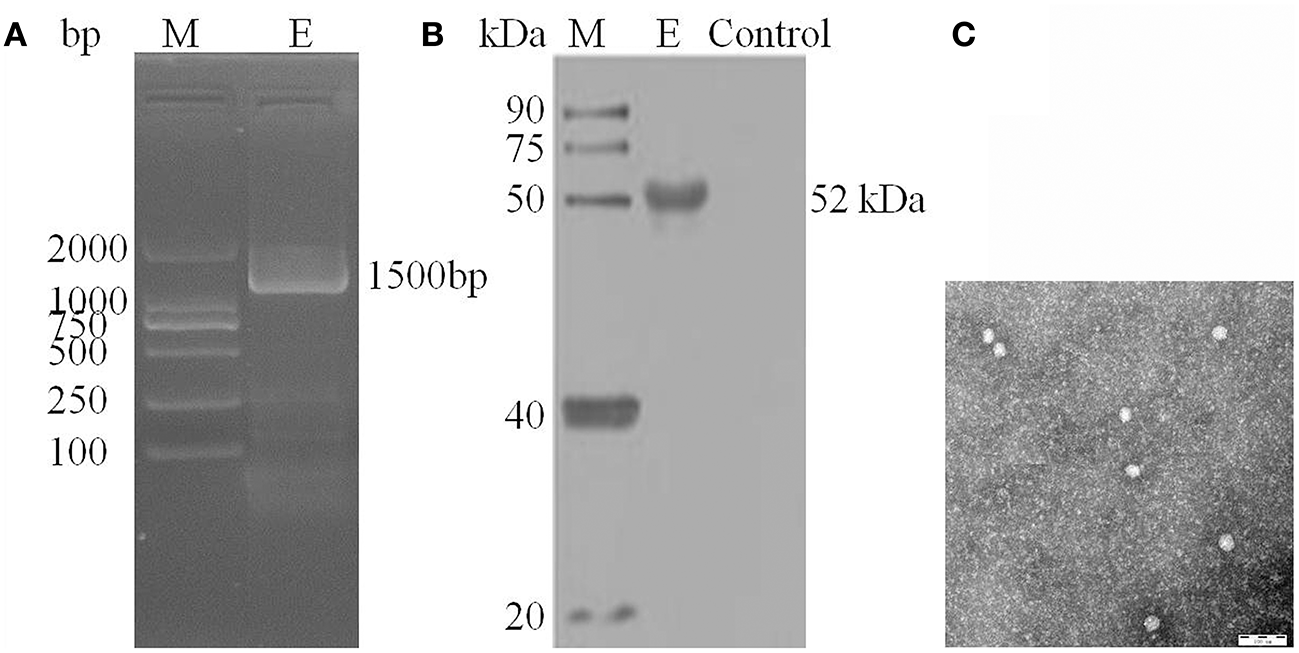
Identification of JEV-China/YN2017-4 isolation in Dali city of Yunnan province by PCR, Western blot, and negative-stain electron microscopy. PCR identification of JEV-China/YN2017-4 after BHK-21 cells infection (A). Western blot identification of JEV-China/YN2017-4 isolation strain was assessed with an anti-E monoclonal antibody (Abcam, Cambridge, UK) and a HRP-conjugated goat anti-mouse antibody (ZSGB-Bio, Beijing, China) (B). Negative-stain electron microscopy of JEV-China/YN2017-4 particles (C).
The viral titers of the newly isolated JEV in cells
In BHK-21 cells, the mean viral titers of YunnanJEV2017-4 of different passage viruses were 5.36 × 104 TCID50/0.1 mL (P5) and 6.42 × 104 TCID50/0.1 mL (P10), respectively. It implied that multiple passages might result in various viral titers The YunnanJEV2017-4 E genes of P5 and P10 were aligned using MegAlign after sequencing. Nucleotide identity analysis showed that P5 shared 98.9% nt identity with P10. After passage in Vero cells using YunnanJEV2017-4 from infected BHK-21 cells, the E genes of YunnanJEV2017-4 (P5 and P10) from BHK-21 and Vero cells were sequenced. By nucleotide alignment, 11 and 12 nucleotide sites in E genes of JEV (P5 and P10) appeared polymorphic respectively. By means of amino acid sequences alignment, one amino acid site (position: 138 aa) in E proteins of P5 and P10 was both mutated, with amino acid lysine (K) changing to glutamic (E). The viral titers were detected in Vero cells in triplicate, and mean of those of YunnanJEV2017-4 were 2.84 × 105 TCID50/0.1 mL (P5) and 3.57 × 105 TCID50/0.1 mL (P10), respectively. It showed a significant increase (P < 0.05) compared to BHK-21 cells.
Discussion
Mosquitoes are the intermediate host of numerous viruses that infect humans, animals, insects, plants, and other species, and play a significant role in the prevalence of many infectious diseases (Zhang et al., 2013; Fisher et al., 2015; Shi et al., 2015). Virus identification by traditional virological methods was very laborious and time-consuming before 2007 (Krah, 1991). Compared with the traditional method, metagenomic sequencing by Illumina sequencing cooperated with high throughput analysis is more efficiency, resulting in identification of many viruses from different species in recent years by several laboratories (Wilson et al., 2015). Moreover, metagenomic sequencing can reveal the composition of the virome in the mosquito samples. Metagenomic sequencing revealed the high abundance of viruses in mosquitoes (Bolling et al., 2015; Joseph et al., 2016; Shi et al., 2017). It can also contributed to the discovery of novel viruses and their identification and characterization (Carissimo et al., 2016; Frey et al., 2016). In the present study, more than 50 viral families were detected by Illumina sequencing in mosquito samples. However, there was a difference in the percentage of viral sequences obtained from the three mosquito groups. It could be related to the geographic location of the vectors (Atoni et al., 2018; Xia et al., 2018; Zakrzewski et al., 2018). Interestingly, the results show that these (and many other) viruses can be (collectively) found in non-Ae. aegypti mosquito species (Ae. albopictus was included in the samples) (Dodson and Rasgon, 2017; Dodson et al., 2018). Moreover, new findings of viral distribution and evolution in Yunnan province, China were obtained.
RT-PCR and PCR were used in confirming the results of Illumina sequencing. The detection of DENV in the mosquito samples shows that DENV still exist in Yunnan province, China. Many strains of DENV were uncovered by previous studies (Shi et al., 2014). The detection of new DENV strains is important, as it suggests a new prevalence of DENV in this area. In addition, the identification of novel circovirus is significant, suggesting that increased environmental surveillance is required because of its severe infectivity to animals, especially pigs, which are significant food animals. Meanwhile, the presence of unidentified viruses in the Illumina sequencing results may be ascribed to inadequate mosquito sampling and limited collection sites.
JEV and DENV were detected simultaneously, indicating that these viruses co-circulate in Yunnan province and implying that co-infection by the viruses is possible. Though the detection of the virus is not sufficient to prove transmission, detection of co-circulation of DENV and JEV in the same geographic location is important (Dodson and Rasgon, 2017; Dodson et al., 2018). The simultaneous detection of the genes of two viruses may bring a new challenge to the control of DENV.
The detection and isolation of JEV indicate that the virus still exists in Yunnan province. After passage in Vero cells using YunnanJEV2017-4 from infected BHK-21 cells, one amino acid site (position: 138 aa) in E proteins of JEV (P5 and P10) was both mutated, with lysine changing to glutamic. It was reported that, at the position of 138 in the E protein, lysine in that could attenuate viral infectivity, whereas glutamic could enhance viral infectivity (Ni et al., 1994). Subsequently, the viral titers were detected, and we found that there was an increase compared to BHK cells. The results show that viral titer of YunnanJEV2017-4 increased when passaged in Vero cells, suggesting the possible increased threat to human health induced by JEV transmission crossing species barrier.
Clearly, our results show that the distribution of viruses in mosquitoes collected in Yunnan province is dependent on the geographical location of mosquitoes. However, the present work explored only a small portion of the virome of mosquitoes in this region. More study in different areas and countries might be needed to assess the diversity of mosquito-borne viruses. In conclusion, our findings provide a useful insight into viral isolation and characterization of DENV and novel circovirus, which could be applied in the future.
Statements
Author contributions
PX, HJL, and NJ conceived and designed the experiments. PX, JH, CL, YL, MW, and NJ performed the experiments. PX, HL, JR, and NJ analyzed the data. YZ, XG, LX, HZ, HL, and MT contributed reagents, materials, analysis tools. PX and NJ wrote the paper. All authors read and approved the final version of the manuscript.
Acknowledgments
This work was supported by the National Program on Key Research Project of China [grant number 2016YFD0500401 and 2017YFD0501803], the Special Fund for Agro-scientific Research in the Public Interest [grant number 201203082], and the National Natural Science Foundation of China [grant number 31272573], the Yunnan Academician Workstation [grant number 2018IC151], and the National Natural Science Foundation [grant number 3180130343].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00364/full#supplementary-material
References
1
AlmeidaR. S.SpilkiF. R.RoeheP. M.ArnsC. W. (2005). Detection of Brazilian bovine respiratory syncytial virus strain by a reverse transcriptase-nested-polymerase chain reaction in experimentally infected calves. Vet. Microbiol.105, 131–135. 10.1016/j.vetmic.2004.11.004
2
Alquezar-PlanasD. E.MourierT.BruhnC. A.HansenA. J.VitcetzS. N.MorkS.et al. (2013). Discovery of a divergent HPIV4 from respiratory secretions using second and third generation metagenomic sequencing. Sci. Rep. 3:2468. 10.1038/srep02468
3
AtoniE.WangY.KarunguS.WaruhiuC.ZohaibA.ObandaV.et al. (2018). Metagenomic virome analysis of Culex mosquitoes from Kenya and China. Viruses10:30. 10.3390/v10010030
4
BollingB. G.WeaverS. C.TeshR. B.VasilakisN. (2015). Insect-specific virus discovery: significance for the arbovirus community. Viruses7, 4911–4928. 10.3390/v7092851
5
CarissimoG.EiglmeierK„ Reveillaud, J.HolmI.DialloM.DialloD.et al. (2016). Identification and characterization of two novel RNA viruses from Anopheles gambiae species complex mosquitoes. PLoS ONE11:e0153881. 10.1371/journal.pone.0153881
6
CholletiH.HayerJ.AbilioA. P.MulandaneF. C.Verner-CarlssonJ.FalkK. I.et al. (2016). Discovery of novel viruses in mosquitoes from the zambezi valley of mozambique. PLoS ONE11:e0162751. 10.1371/journal.pone.0162751
7
DodsonB. L.PujhariS.RasgonJ. L. (2018). Vector competence of selected North American Anopheles and Culex mosquitoes for Zika virus. Peer J. 6:e4324. 10.7717/peerj.4324
8
DodsonB. L.RasgonJ. L. (2017). Vector competence of Anopheles and Culex mosquitoes for Zika virus. Peer J. 5:e3096. 10.7717/peerj.3096
9
ErgunayK.BrinkmannA.LitzbaN.GunayF.KarS.OterK.et al. (2017). A novel rhabdovirus, related to Merida virus, in field-collected mosquitoes from Anatolia and Thrace. Arch. Virol. 162, 1903–1911. 10.1007/s00705-017-3314-4
10
FengY.HeB.FuS.YangW.ZhangY.TuC.et al. (2015). Isolation and identification of the Akabane virus from mosquitoes in Yunnan Province, China. Bing. Du. Xue. Bao. 31, 51–57.
11
FisherR. G.SmithD. M.MurrellB.SlabbertR.KirbyB. M.EdsonC.et al. (2015). Next generation sequencing improves detection of drug resistance mutations in infants after PMTCT failure. J. Clin. Virol. 62, 48–53. 10.1016/j.jcv.2014.11.014
12
FreyK. G.BiserT.HamiltonT.SantosC. J.PimentelG.MokashiV. P.et al. (2016). Bioinformatic characterization of mosquito viromes within the Eastern United States and Puerto Rico: discovery of novel viruses. Evol. Bioinform. Online12, 1–12. 10.4137/EBO.S38518
13
HeB.LiZ.YangF.ZhengJ.FengY.GuoH.et al. (2013). Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLoS ONE8:e61950. 10.1371/journal.pone.0061950
14
HoughtonM. (2009). The long and winding road leading to the identification of the hepatitis C virus. J. Hepatol. 51, 939–948. 10.1016/j.jhep.2009.08.004
15
JosephR. F.NathanD. G.BenjaminJ. K.JamesW. L.StevenM. L.LawrenceS. F.et al. (2016). WestAfrican Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses,and totiviruses. Virology498, 288–299. 10.1016/j.virol.2016.07.031
16
KlungthongC.GibbonsR. V.ThaisomboonsukB.NisalakA.KalayanaroojS.ThirawuthV.et al. (2007). Dengue virus detection using whole blood for reverse transcriptase PCR and virus isolation. J. Clin. Microbiol. 45, 2480–2485. 10.1128/JCM.00305-07
17
KrahD. L. (1991). A simplified multiwell plate assay for the measurement of hepatitis A virus infectivity. Biologicals19, 223–227.
18
LiH.CaoY. X.HeX. X.FuS. H.LyuZ.HeY.et al. (2015). Real-time RT-PCR Assay for the detection of Tahyna Virus. Biomed. Environ. Sci. 28, 374–377. 10.3967/bes2015.052
19
MiesenP.IvensA.BuckA. H.van RijR. P. (2016). Small RNA profiling in dengue virus 2-infected aedes mosquito cells reveals viral piRNAs and novel host miRNAs. PLoS. Negl. Trop. Dis. 10:e0004452. 10.1371/journal.pntd.0004452
20
MotookaD.NakamuraS.HagiwaraK.NakayaT. (2015). Viral detection by high-throughput sequencing. Methods Mol. Biol. 1236, 125–134. 10.1007/978-1-4939-1743-3_11
21
NiH.BurnsN. J.ChangG. J.ZhangM. J.WillsM. R.TrentD. W.et al. (1994). Comparison of nucleotide and deduced amino acid sequence of the 5' non-coding region and structural protein genes of the wild-type Japanese encephalitis virus strain SA14 and its attenuated vaccine derivatives. J. Gen. Virol. 75, 1505–1510. 10.1099/0022-1317-75-6-1505
22
PhamT. N.MacparlandS. A.CoffinC. S.LeeS. S.BurseyF. R.MichalakT. I. (2005). Mitogen-induced upregulation of hepatitis C virus expression in human lymphoid cells. J. Gen. Virol. 86, 657–666. 10.1099/vir.0.80624-0
23
RitchieS. A.van den HurkA. F.ZborowskiP.KerlinT. J.BanksD.WalkerJ. A.et al. (2007). Operational trials of remote mosquito trap systems for Japanese encephalitis virus surveillance in the Torres Strait, Australia. Vector. Borne. Zoonotic. Dis. 7, 497–506. 10.1089/vbz.2006.0643
24
ShiC.LiuY.HuX.XiongJ.ZhangB.YuanZ. (2015). A metagenomic survey of viral abundance and diversity in mosquitoes from Hubei province. PLoS ONE10:e0129845. 10.1371/journal.pone.0129845
25
ShiJ.DuanZ.SunJ.WuM.WangB.ZhangJ.et al. (2014). Identification and validation of a novel microRNA-like molecule derived from a cytoplasmic RNA virus antigenome by bioinformatics and experimental approaches. Virol. J. 11:121. 10.1186/1743-422X-11-121
26
ShiM.NevilleP.NicholsonJ.EdenJ. S.ImrieA.HolmesE. C.et al. (2017). High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in western Australia. J. Virol. 91, e00680–e00617. 10.1128/JVI.00680-17
27
SimS.AwP. P.WilmA.TeohG.HueK. D.NguyenN. M.et al. (2015). Tracking dengue virus intra-host genetic diversity during human-to-mosquito transmission. PLoS Negl. Trop. Dis. 9:e0004052. 10.1371/journal.pntd.0004052
28
WilsonM. R.ShanbhagN. M.ReidM. J.SinghalN. S.GelfandJ. M.SampleH. A.et al. (2015). Diagnosing Balamuthia mandrillaris encephalitis with metagenomic deep sequencing. Ann. Neurol. 78, 722–730. 10.1002/ana.24499
29
XiaH.WangY.ShiC.AtoniE.ZhaoL.YuanZ.et al. (2018). Comparative metagenomic profiling of viromes associated with four common mosquito species in China. Virol. Sin. 33, 59–66. 10.1007/s12250-018-0015-4
30
ZakrzewskiM.RašićG.DarbroJ.KrauseL.PooY. S.FilipovićI.et al. (2018). Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep. 8:4690. 10.1038/s41598-018-22945-y
31
ZhangX.ShengJ.PlevkaP.KuhnR. J.DiamondM. S.RossmannM. G. (2013). Dengue structure differs at the temperatures of its human and mosquito hosts. Proc. Natl. Acad. Sci. U.S.A. 110, 6795–6799. 10.1073/pnas.1304300110
Summary
Keywords
metagenomic analysis, mosquito, virome, virus detection, phylogenetic analysis
Citation
Xiao P, Li C, Zhang Y, Han J, Guo X, Xie L, Tian M, Li Y, Wang M, Liu H, Ren J, Zhou H, Lu H and Jin N (2018) Metagenomic Sequencing From Mosquitoes in China Reveals a Variety of Insect and Human Viruses. Front. Cell. Infect. Microbiol. 8:364. doi: 10.3389/fcimb.2018.00364
Received
13 April 2018
Accepted
01 October 2018
Published
19 October 2018
Volume
8 - 2018
Edited by
Rachel L. Roper, The Brody School of Medicine at East Carolina University, United States
Reviewed by
Helen Piontkivska, Kent State University, United States; Edward Rybicki, University of Cape Town, South Africa
Updates
Copyright
© 2018 Xiao, Li, Zhang, Han, Guo, Xie, Tian, Li, Wang, Liu, Ren, Zhou, Lu and Jin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Zhang zhangying636@126.comHuijun Lu huijun_lu@126.comNingyi Jin jinningyi2000@126.com
This article was submitted to Virus and Host, a section of the journal Frontiers in Cellular and Infection Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.