 Kamal El Bissati1*
Kamal El Bissati1* Henry Redel2†Li-Min Ting2
Henry Redel2†Li-Min Ting2 Joseph D. Lykins3
Joseph D. Lykins3 Martin J. McPhillie4Rajendra Upadhya2
Martin J. McPhillie4Rajendra Upadhya2 Patrick M. Woster5
Patrick M. Woster5 Nigel Yarlett6Kami Kim2,7*†
Nigel Yarlett6Kami Kim2,7*† Louis M. Weiss2,7*
Louis M. Weiss2,7*- 1Department of Ophthalmology and Visual Science, The University of Chicago, Chicago, IL, United States
- 2Department of Medicine, Albert Einstein College of Medicine and Montefiore Medical Center, Bronx, NY, United States
- 3Department of Internal Medicine and Department of Emergency Medicine, Virginia Commonwealth University Health System, Richmond, VA, United States
- 4School of Chemistry, University of Leeds, Leeds, United Kingdom
- 5Department of Drug Discovery and Biomedical Sciences, Medical University of South Carolina, Charleston, SC, United States
- 6Haskins Laboratories, Department of Chemistry and Physical Sciences, Pace University, New York, NY, United States
- 7Department of Pathology, Albert Einstein College of Medicine and Montefiore Medical Center, Bronx, NY, United States
Twenty-two compounds belonging to several classes of polyamine analogs have been examined for their ability to inhibit the growth of the human malaria parasite Plasmodium falciparum in vitro and in vivo. Four lead compounds from the thiourea sub-series and one compound from the urea-based analogs were found to be potent inhibitors of both chloroquine-resistant (Dd2) and chloroquine-sensitive (3D7) strains of Plasmodium with IC50 values ranging from 150 to 460 nM. In addition, the compound RHW, N1,N7-bis (3-(cyclohexylmethylamino) propyl) heptane-1,7-diamine tetrabromide was found to inhibit Dd2 with an IC50 of 200 nM. When RHW was administered to P. yoelii-infected mice at 35 mg/kg for 4 days, it significantly reduced parasitemia. RHW was also assayed in combination with the ornithine decarboxylase inhibitor difluoromethylornithine, and the two drugs were found not to have synergistic antimalarial activity. Furthermore, these inhibitors led to decreased cellular spermidine and spermine levels in P. falciparum, suggesting that they exert their antimalarial activities by inhibition of spermidine synthase.
Introduction
Malaria is a global health threat, especially in the developing world. Plasmodium falciparum causes the most lethal form of the disease. It is responsible for a high number of clinical cases and deaths annually. About 3.2 billion people remain at risk of malaria. In 2015 alone, there was an estimated 214 million new cases of malaria and 438,000 deaths (World Health Organization, 2015). The number of malaria cases fell from an estimated 262 million in 2000 to 214 million in 2015 (World Health Organization, 2015), due to employment of artemisinin and drug-impregnated bed nets (White et al., 2014; World Health Organization, 2015). Artemisinin-based combination therapies ACTs have become widely adopted as first-line treatment in almost all countries where malaria is endemic (White, 2008). However, recent studies report decreases in parasite clearance rates following artesunate monotherapy or artesunate-mefloquine combination therapy in Thailand and Cambodia, suggesting increasing resistance to this therapeutic approach (Dondorp et al., 2009; Amaratunga et al., 2012; Phyo et al., 2012; Ferreira et al., 2013). In light of the challenge posed by resistant strains of P. falciparum, development of new drugs to combat this infection is increasingly necessary and will save lives.
The intraerythrocytic life cycle of P. falciparum is dynamic, with the parasite undergoing numerous morphological and physiological transformations throughout the course of infection. Whilst actively dividing, the parasite is capable of generating 36 daughter parasites within 2 days. Innumerable parasitic cellular processes offer diverse chemotherapeutic targets for the inhibition of parasite replication and, thereby, abrogation of disease. Among these targets, is the biosynthesis of polyamines. There is a 10 to 20-fold increase in polyamine levels when the parasite transitions from ring stage to schizonts in infected erythrocytes and there is evidence of parasitic dependence on the polyamine pathway for intraerythrocytic development (Assaraf et al., 1984; Gupta et al., 2005). Moreover, inhibitors of this pathway cause decreased polyamine levels, which, in turn, results in transcriptional arrest (van Brummelen et al., 2009). Consequently, the polyamine pathway appears to be a valid target for further exploration and small molecular development, given its essential role in parasite survival. Polyamines, which include putrescine, spermidine, spermine, and cadaverine (an analog of putrescine), are amine-containing, cationic, low molecular mass compounds, and ubiquitous in eukaryotes and prokaryotes. These polycation compounds interact electrostatically with anionic macromolecules like RNA, DNA, ATP, proteins, and phospholipids (Igarashi and Kashiwagi, 2000; Wallace et al., 2003). These interactions regulate replication, transcription, membrane biogenesis, maintenance of chromatin conformation, specific gene expression, ion channels, and confer protection of nucleic acids against oxidative stress (Igarashi and Kashiwagi, 2000; Wallace et al., 2003). Synthesis of polyamines in most cells is initiated by the production of putrescine from ornithine through the activity of ornithine decarboxylase (ODC). The addition of either one or two aminopropyl groups to the terminal amino groups of putrescine form spermidine and spermine, respectively via S-adenosylmethionine (AdoMet) decarboxylase. Most animal and yeast cells can take up polyamines and convert them back to spermidine and putrescine via spermidine/spermine-N-acetyltransferase and polyamine oxidase (Pegg and McCann, 1982; Marton and Pegg, 1995). P. falciparum lacks the capacity for polyamine interconversion, and the parasite controls polyamine levels exclusively through de novo spermidine synthase, which determines levels of spermidine (Müller et al., 2001; Clark et al., 2010). This enzyme has the additional unique function of producing low levels of spermine (Haider et al., 2005).
Many inhibitors of both ODC and AdoMetDC have been synthesized, with the goal of interfering with polyamine metabolism in tumor cells as anti-cancer therapy and prevention (Marton and Pegg, 1995; Casero and Marton, 2007). DFMO (alpha-difluoromethylornithine) was successfully exploited against West African human sleeping sickness (Trypanosoma brucei gambiense) (Bacchi et al., 1980; Burri and Brun, 2003). Interestingly, the functions of AdoMetDC and ODC are combined into a single unique bi-functional protein (Pf AdoMet/ODC) in P. falciparum (Müller et al., 2000). This enzyme has formed the basis of multiple studies assessing polyamine metabolism as a chemotherapeutic target in this organism. Inhibitors of this enzyme, however, have only cytostatic effects in vitro with cure achieved only with co-administration with polyamine analogs in murine malaria models (Bitonti et al., 1989).
Other polyamine analogs interfering with polyamine functions and metabolism have been synthesized and tested in different organisms. The tetraamines, homologs of spermine in which the external aminopropyl groups present in spermine were replaced by aminobutyl groups, are shown along with oligoamines to be effective in the treatment of microsporidiosis, an opportunistic infection associated with severe HIV infection (Bacchi et al., 2002). Spermine analogs were found to condense DNA and are indeed powerful inhibitors of human cell proliferation (Osland and Kleppe, 1977).
Based on these findings, we tested the oligoamines (SL-11158 and SL-11144) and the tetraamine SL-11093, with demonstrated efficacy against microsporidia, for their activities against malaria parasites. Here we report the identification of four lead compounds from the thiourea sub-series and one compound from the urea-based analogs from the list of twenty-two polyamine analogs effective against chloroquine-sensitive and -resistant parasites, at nanomolar levels. Moreover, these compounds were tested in vivo in a murine model with Plasmodium yoelii and found to significantly reduce levels of parasitemia. These effects are correlated with an observed decrease in spermidine and spermine levels and highlight the importance of this polyamine to the parasite.
Methods
Strains
The P. falciparum strains 3D7 and Dd2 used in this study were obtained from the Malaria Research and Reference Reagent Resource Center (MR4). P. yoelii clones were obtained from the WHO Registry Standard Malaria Parasites, University of Edinburgh.
Chemicals
Hypoxanthine radiolabelled chemicals were purchased from NEN Life Science Products. Compounds SL-11144, SL-11158, SL-11091, and SL-11093 were synthesized by the Frydman group as described elsewhere (Reddy et al., 1998; Valasinas et al., 2003). All of the remaining compounds were synthesized by Dr. Patrick M. Woster, Medical University of South Carolina, as previously described (Zou et al., 2001; Bi et al., 2006; Verlinden et al., 2011; Verlinden et al., 2015).
Cell Culture and Materials
P. falciparum was cultured in vitro with erythrocytes using a gas mixture of 3% O2, 3% CO2, and 94% N2 as described by the method of Trager and Jensen (1976). Briefly, we used RPMI medium 1,640 supplemented with 30 mg/liter hypoxanthine (Sigma), 25 mM Hepes (Sigma), 0.225% NaHCO3 (Sigma), 0.5% Albumax I (Life Technologies, Grand Island, NY) and 10 μg/ml of gentamycin (Life Technologies) for the parasite growth. Synchronization of the parasites was obtained by incubation with 5% sorbitol treatments (Lambros and Vanderberg, 1979). The stage of the parasite was confirmed by fixed smears of the infected erythrocytes using Giemsa staining and observation by bright-field microscopy.
Hypoxanthine Incorporation Assay
The susceptibility of parasites to different compounds was assessed by tritiated hypoxanthine uptake as described by Desjardins et al. (1979). Briefly, infected erythrocytes with 3% at the ring stages were incubated with the compounds from the list of twenty-two polyamine analogs at 0, 0.01, 0.05, 0.1, 1, 5, 10, 50, 100, 250, 500, and 750 μM in a medium free of hypoxanthine for 48 h. Two hundred microliter of the mixture was then added to a 96-well plate with 3H-hypoxanthine at a concentration of 0.5 μCi/well. The cells were incubated for 24 h, washed on an ultrafilter and radioactivity was counted using a scintillation counter. IC50 values are calculated from the sigmoidal inhibition curves using Prism and are represented in nM. Values are means of three independent experiments each performed in triplicate.
Infection of Mice With Blood-Stage Parasites and Drug Treatments
Naive 8-weeks-old female Swiss Webster mice were intravenously infected with 2 × 105 infected red blood cells (iRBCs) of P. yoelii YM parasites; 3 mice were included in each infection group. Two groups per treatment were performed. Drugs were dissolved in dimethyl sulfoxide (DMSO). For example, to inject 35 mg/kg, we first dissolved 35 mg of the drug in 1 mL of DMSO and then diluted it in 0.05% Tween 80 H2O, for a total of 10 mL. We then injected 200 μL of this solution into mice with a body weight of 20 g. The polyamine compounds and pyrimethamine were administered intraperitoneally. All mice were treated for 4 days. Parasitemia was monitored by Giemsa staining of blood smears obtained after the 4 days of treatment and on a daily basis. Parasites were harvested by collecting blood samples from the tail vein of infected mice. The mice tolerated (without observable toxicity) up to 35 mg/kg when the schedule described above was used. All mice studies were performed with the Institutional Animal Care and Use Committee at the Albert Einstein College of Medicine approval and oversight.
Polyamine Quantification
Polyamine content was quantified as described previously (Bacchi et al., 2004). Briefly, P. falciparum Dd2 strain infected red blood cells were treated in the late schizont stage (42 h post invasion) with 1 μM of RHW (IC50 = 200 nM), 750 nM of compound 13 (IC50 = 150 nM), and with 5 mM DFMO (IC50 = 1 mM), to ensure complete parasite arrest. Treated and untreated cultures, at 10% parasitemia, were harvested at trophozoite phase (18–24 h). Polyamine interconversion was assayed by incubation infected red blood cells to 0.25 μCi (2.1 nmol) [1-14C]spermine as described previously by Bacchi et al. (2001). After incubation, mixtures were centrifuged and the supernatant was discarded. Pellets were extracted with 10% TCA overnight and frozen. Separation of polyamines was performed by HPLC equipped with a 5 μm C-18 reversed-phase column, samples and standards were detected by a UV detector (Perkin Elmer), and signals were integrated using a β-ram (IN/US Systems) Version 3.1 software package.
Molecular Modeling
The graphical user interface Maestro (version 10.3, Schrodinger LLC, New York, NY, 2018) was used to visualize the Pf SpdS protein (PDB IDs: 2I7C and 4CWA), which was prepared using the Protein Preparation Wizard. Only chain A was used in both crystal structures for further modeling. The ligand RHW was prepared using the Maestro user interface. MacroModel force-field based molecular modeling (MacroModel v10.3, Schrodinger LLC, New York, NY, 2018) was used to predict the binding pose of the ligand RHW (Harder et al., 2015). The obtained molecular conformations were visualized using PyMoL (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
Results
Effects of Polyamine Analogs on Growth of 3D7 and Dd2 of P. falciparum Strains
Previous studies have shown that polyamine analogs with a backbone of repeating N-butyl subunits, such as pentamines, oligoamines or bis-(aryl)-substituted 3-7-3 analogs (Figure 1A), sterilized Encephalitozoon cuniculi-infected monolayer cells and cured two murine model infections (Bacchi et al., 2002). The IC50 of SL-11158 and SL-11144 against microsporidia was 8.2 and 0.62 μM (Bacchi et al., 2002). In order to examine the antimalarial activity of polyamine analogs, we have tested the effect of increasing concentrations of these compounds on the intraerythrocytic life cycle of P. falciparum in culture, by following the incorporation of radiolabeled hypoxanthine into parasite nucleic acids. We then determined the 50% inhibitory concentration (IC50) that blocks the replication of P. falciparum inside the red cells. The study was performed with two strains of P. falciparum (3D7 and Dd2) with different levels of sensitivity to the antimalarial drugs pyrimethamine and chloroquine (Figure 2 and Table 1). As a control, we confirmed the IC50 of chloroquine in the two strains. As expected, the strain 3D7 was sensitive to chloroquine with IC50 of 5 ± 0.2 nM and Dd2 was highly resistant to chloroquine (IC50 300 ± 21 nM) (Supplementary Figure 1).
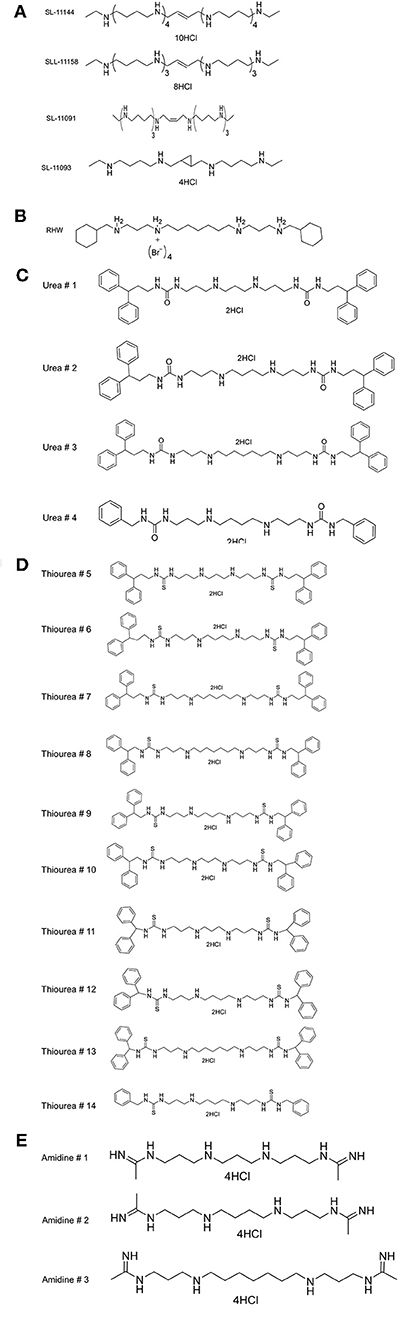
Figure 1. Structures of (A) various oligoamine and tetraamine analogs of polyamines, (B) N1, N7-bis (3-(cyclohexylmethylamino) propyl) heptane-1,7-diamine tetrabromide (RHW), (C) Urea, (D) Thiourea, (E) Amidine.
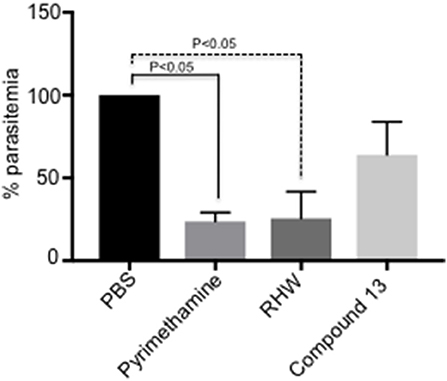
Figure 2. In vivo study of the effect on P. yoelii-infected mice (n = 3 animals per group) of polyamine analogs administered ip at 35 mg/kg/day as indicated in Methods. Pyrimethamine was administered ip as a positive control at 10 mg/kg/day. A control untreated group received PBS. Activity was determined by percentage of parasitemia at day 6-post infection relative to untreated control. The results are shown as the means of three independent experiments ± standard deviation. Significant differences relative to untreated control are determined by student's t-test.
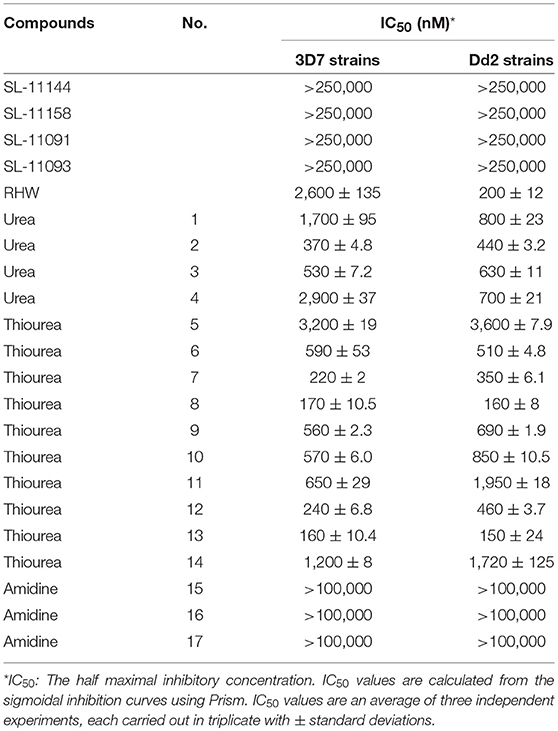
Table 1. Structure of various analogs of polyamines and their affect on the growth of 3D7 and Dd2 strains of P. falciparum under in vitro conditions.
As shown in Table 1 and Supplementary Figure 1, RHW exhibited antimalarial activity, with an IC50 of 2.6 μM against 3D7 and 0.2 μM against Dd2 parasite strains. This prompted the testing of a variety of polyamine analogs for antiparasitic effects. We tested 17 compounds belonging to three different classes, namely ureas, thioureas, and amidines (Figures 1C–E). The polyamines of the thiourea group were found to be the most potent in inhibiting parasite growth, with IC50 ranging from 160 to 3,200 nM for 3D7 strain and 150 to 3,600 nM for Dd2 strain (Table 1). Compound 13 was found to be more effective in inhibiting parasitic growth of both 3D7 and Dd2 strains with IC50 values of 160 nM and 150 nM, respectively. Both 3D7 and Dd2 parasite strains exhibited varied sensitivity toward the urea class of polyamine analogs, with compound 2 exhibiting the strongest growth inhibition of the class, with an IC50 of 370 nM for 3D7 and 440 nM for Dd2 strains (Table 1). The amidine class of analogs showed the least effect on growth of both strains of P. falciparum with an IC50 > 100 μM (Table 1). To further confirm the inhibitory effects of these classes of polyamine analogs on P. falciparum growth, we employed the pLDH colorimetric assay (data not shown), which measures parasite-specific lactate dehydrogenase activity (Makler and Hinrichs, 1993; Makler et al., 1993).
Efficacies of Polyamine Analogs in vivo
We tested the efficacy of the most potent compounds on the growth inhibitory potential of Plasmodium employing a murine model. We infected mice with P. yoelii, as described in materials and methods, and the infected mice were treated with RHW and compound 13, the compounds with the most efficacy in vitro. PBS infected mice served as control and pyrimethamine drug treated animals were used as a positive control group. As shown in Figure 2, treatment of animals with RHW resulted in a significant (p < 0.05) decrease in parasitemia compared to the untreated control. Even though compound 13 decreased parasitemia in vivo, the level of inhibition was not significantly different (p > 0.05) to the PBS control group (Figure 2 and Supplementary Figure 2). In conclusion, these in vivo results show the efficacy of RHW on the inhibition of parasitemia in the murine model.
Polyamine Quantification
We examined the effects of polyamine analogs on overall uptake of arginine and putrescine, and production of spermidine and spermine. Table 2 shows the results of polyamine contents in red blood cells [experiment was performed twice, each in five replicate experiments, where the polyamine content of Plasmodium infected red blood cells was compared to treated cells with RHW, compound 13, and ornithine decarboxylase inhibitor difluoromethylornithine (DFMO) as a control]. We monitored the intracellular content of arginine, putrescine, spermidine, and spermine. Data show that DFMO substantially decreases the amount of spermidine. However, spermine levels were similar for both untreated and DFMO treated. These results were in concordance with previous data that show the same effect of DFMO (Sugiura et al., 1984). In contrast, infected red blood cells treated with RHW showed a significant decrease in spermidine (p < 0.05) and spermine levels (p < 0.01). These data suggest an effect of RHW on polyamine metabolism in Plasmodium, including an effect on enzymes involved on spermidine and spermine synthesis. In addition, when RHW or compound 13 was assayed in combination with DFMO in both Dd2 and 3D7 strains, the two drugs were found not to have synergistic antimalarial activity (Figure 3).
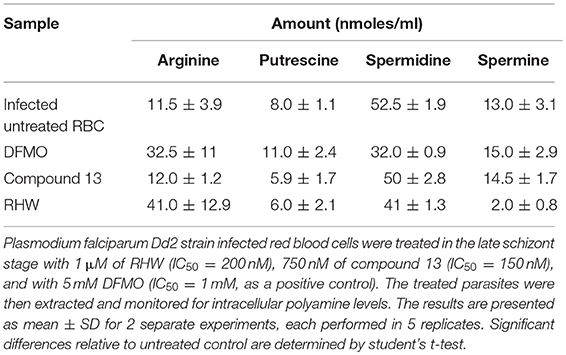
Table 2. Effect of treatment of RHW and compound 13 on the polyamine contents of Plasmodium falciparum.
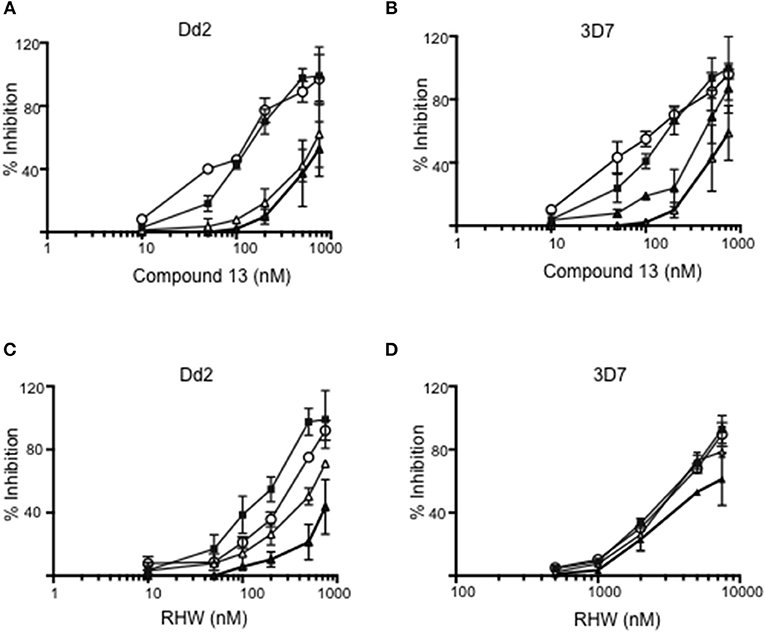
Figure 3. Inhibition of Dd2 and 3D7 strains by different concentrations of compound 13 (A,B) and RHW (C,D) in the absence of DFMO (closed squares), or in presence of 500 mM (open triangles), 1 mM (open circles), and 3 mM (closed triangles) of DFMO.
Molecular Modeling of Compound RHW
In order to understand the putative binding conformations of RHW to P. falciparum spermidine synthase (Pf SpdS) and the observed biological activity, we utilized in silico molecular modeling. Sprenger et al. proposed three areas of the active site: the distal aminopropyl cavity, the putrescine site and the larger dcAdoMet site (composed of the central aminopropyl cavity and the MTA cavity) within Pf SpdS (Sprenger et al., 2016). Because RHW showed potent activity in vitro and in vivo, all our molecular modeling was performed with this inhibitor. RHW is a long linear molecule consisting of 20 rotatable bonds (Figure 1B). Attempts to dock and predict the binding mode of RHW using Schrodinger Glide XP mode were unsuccessful. Instead, we modified an existing ligand (5-(1H-benzimidazol-2-yl) pentan-1-amine) within the Pf SpdS active site of PBD ID: 4CWA (Sprenger et al., 2015), and subjected it to MacroModel minimization using force-field OPLS_2005, keeping the protein backbone rigid (locked) and allowing the side chains to move and generate a lower energy ligand:protein complex (Figure 4). RHW is predicted to bind across all three areas of the active site (Figure 4A), and when overlaid with inhibitor AdoDATO from crystal structure 2I7C (Dufe et al., 2007), RHW showed a similar mode of binding (Figure 4B). Some movement of the amino acid side chains (W234, C266, I235, H236, E231, M50, W51, and D178) was observed to accommodate the terminal cyclohexylmethyl groups. Of these, only H236 and D178 displayed significant movement (2.0 Å) of their side chain positions.
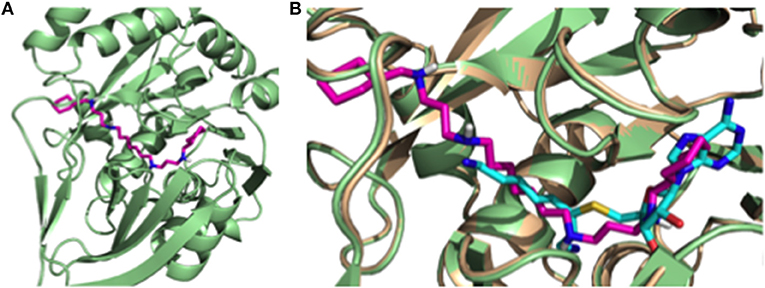
Figure 4. (A) Putative binding conformation of RHW (pink) in PfSpdS (green) traversing the three areas of the active site. (B) Overlay of RHW modeled conformation (pink) with 217C (wheat) and it's co-crystal inhibitor AdoDATO (sky blue).
Discussion
Multiple polyamine analogs in this study have demonstrated potent antimalarial properties, with IC50 at the nanomolar range for P. falciparum strains, both sensitive and resistant to chloroquine. Drug-resistance continues to present challenges in treating P. falciparum infection, and medications previously useful for treating drug-resistant isolates are increasingly inadequate, as illustrated by the emergence of artemisinin-resistance.
The utilization of polyamine analogs to treat infection has been previously demonstrated in experimental murine models of microsporidiosis, with several of the analogs demonstrating significant efficacy (Bacchi et al., 2004). The compounds with the most efficacies were shown to be tetraamines and oligoamines. These tetraamine compounds, which are structural homologs of spermine, have been studied extensively, and have been shown to interrupt and modify DNA conformational structure, inducing bends and kinks in the double helix (Hsieh et al., 1994). The oligoamine compounds have been demonstrated previously to contribute to the collapse of DNA, promoting condensation of DNA. These compounds have also documented anti-tumor activity (Frydman et al., 2003; Huang et al., 2003).
Given the importance of polyamine metabolism for transcriptional activity and cellular replication microsporidia and Plasmodium, testing analogs effective against microsporidia formed the basis of our initial approach toward P. falciparum. Surprisingly, these compounds showed no or minimal antimalarial activity. The lack of effect of the tetraamine compounds on P. falciparum prompted further synthetic efforts.
The addition of cyclohexylmethyl side groups to the tetraamine backbone resulted in the compound abbreviated as RHW. This compound showed substantial promise, with efficacy at the nanomolar range against chloroquine-resistant P. falciparum, and further medicinal chemistry allowed for additional modifications to these compounds. Some of these modifications proved beneficial, enhancing antimalarial activity, such as the urea and thiourea examples, while others were detrimental (amidine analogs). It has also been noted previously (Verlinden et al., 2011) that the compounds with aromatic rings demonstrated enhanced efficacy when compared to matched-pair compounds without these groups. The length of the carbon chain between the two central amines appears to also be important (see compounds 1–3). While the exact, mechanistic underpinning of this difference requires further exploration, this observation should inform any medicinal chemistry efforts with these polyamine analogs. We present a putative model of RHW bound to P. falciparum spermidine synthase (Pf SpdS) (Figure 4).
The observed differences in responsiveness to these compounds between microsporidia and P. falciparum highlight differences in the metabolic handling of polyamines by these two pathogens. It has been noted previously that microsporidia, including E. cuniculi, have an operative polyamine synthesis and interconversion pathway, the latter of which is lacking in Plasmodium (Müller et al., 2001). This difference in capacity could, in part, explain the differences observed in response to the tetraamine compounds. An alternative explanation could be structural differences in particular enzymes targeted by these compounds. The data presented here demonstrate decreased levels of spermine in infected cells and argue in favor of the polyamine analogs inhibiting spermidine synthase. While functional studies demonstrating this relationship are still needed, this seems a probable hypothesis for the mechanism by which the action of these compounds is exerted. Another potential target to inhibit polyamine synthesis is through blocking the transport of these molecules. Polyamine transporters have not yet been identified in the P. falciparum genome, but a drug combination selectively inhibiting both polyamine biosynthesis and transport may provide a promising anti-malarial strategy (van Brummelen et al., 2009).
The polyamine analogs investigated in this study have been demonstrated to hold promise as antimalarial agents and should be further characterized with respect to kinetics and impact on spermidine synthase. Moreover, following optimization, future studies with respect to efficacy, toxicity, and delivery to infected patients will be of value. Malaria is an enormous threat to the health of populations globally, responsible for millions of deaths annually. Novel therapeutic strategies are desperately needed in the continued fight against Plasmodium, and these polyamine analogs have the potential to represent one of these strategies, with the goal of more effectively treating infections and preventing the severe morbidity and mortality caused by this parasite worldwide.
Author Contributions
KE, LW, and KK designed research. KE, HR, L-MT, RU, and NY performed research. KE, HR, L-MT, RU, NY, PW, JL, MM, LW, and KK analyzed data. KE, JL, KK, and LW wrote the paper. All authors read and approved the final manuscript version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Rima McLeod and Dr. Gang Cheng for critical reading of the manuscript. This research was supported by grants R01AI087625 (KK), RC4AI092801 (KK), R01AI031788 (LW) from the National Institute of Allergy and Infectious Disease and RO1 CA149095 (PW). This work was also supported by Knights Templar Eye Foundation (to KE).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00009/full#supplementary-material
References
Amaratunga, C., Sreng, S., Suon, S., Phelps, E. S., Stepniewska, K., Lim, P., et al. (2012). Artemisinin-resistant Plasmodium falciparum in Pursat province, Western Cambodia: a parasite clearance rate study. Lancet Infect. Dis. 12, 851–858. doi: 10.1016/S1473-3099(12)70181-0
Assaraf, Y. G., Golenser, J., Spira, D. T., and Bachrach, U. (1984). Polyamine levels and the activity of their biosynthetic enzymes in human erythrocytes infected with the malarial parasite, Plasmodium falciparum. Biochem. J. 222, 815–819. doi: 10.1042/bj2220815
Bacchi, C. J., Lane, S., Weiss, L. M., Yarlett, N., Takvorian, P., and Wittner, M. (2001). Polyamine synthesis and interconversion by the microsporidian Encephalitozoon cuniculi. J. Eukaryot. Microbiol. 48, 374–381. doi: 10.1111/j.1550-7408.2001.tb00327.x
Bacchi, C. J., Nathan, H. C., Hutner, S. H., McCann, P. P., and Sjoerdsma, A. (1980). Polyamine metabolism: a potential therapeutic target in trypanosomes. Science 210, 332–334. doi: 10.1126/science.6775372
Bacchi, C. J., Rattendi, D., Faciane, E., Yarlett, N., Weiss, L. M., Frydman, B., et al. (2004). Polyamine metabolism in a member of the phylum Microspora (Encephalitozoon cuniculi): effects of polyamine analogues. Microbiology 150, 1215–1224. doi: 10.1099/mic.0.26889-0
Bacchi, C. J., Weiss, L. M., Lane, S., Frydman, B., Valasinas, A., Reddy, V., et al. (2002). Novel synthetic polyamines are effective in the treatment of experimental microsporidiosis, an opportunistic AIDS-associated infection. Antimicrob. Agents Chemother. 46, 55–61. doi: 10.1128/AAC.46.1.55-61.2002
Bi, X., Lopez, C., Bacchi, C. J., Rattendi, D., and Woster, P. M. (2006). Novel alkylpolyaminoguanidines and alkylpolyaminobiguanides with potent antitrypanosomal activity. Bioorg. Med. Chem. Lett. 16, 3229–3232. doi: 10.1016/j.bmcl.2006.03.048
Bitonti, A. J., Dumont, J. A., Bush, T. L., Edwards, M. L., Stemerick, D. M., McCann, P. P., et al. (1989). Bis(benzyl)polyamine analogs inhibit the growth of chloroquine-resistant human malaria parasites (Plasmodium falciparum) in vitro and in combination with alpha-difluoromethylornithine cure murine malaria. Proc. Natl. Acad. Sci. U.S.A. 86, 651–655. doi: 10.1073/pnas.86.2.651
Burri, C., and Brun, R. (2003). Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 90, S49–S52. doi: 10.1007/s00436-002-0766-5
Casero, R. A., and Marton, L. J. (2007). Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 6, 373–390. doi: 10.1038/nrd2243
Clark, K., Niemand, J., Reeksting, S., Smit, S., van Brummelen, A. C., Williams, M., et al. (2010). Functional consequences of perturbing polyamine metabolism in the malaria parasite, Plasmodium falciparum. Amino Acids 38, 633–644. doi: 10.1007/s00726-009-0424-7
Desjardins, R. E., Canfield, C. J., Haynes, J. D., and Chulay, J. D. (1979). Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16, 710–718. doi: 10.1128/AAC.16.6.710
Dondorp, A. M., Nosten, F., Yi, P., Das, D., Phyo, A. P., Tarning, J., et al. (2009). Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361, 455–467. doi: 10.1056/NEJMoa0808859
Dufe, V. T., Qiu, W., Müller, I. B., Hui, R., Walter, R. D., and Al-Karadaghi, S. (2007). Crystal structure of Plasmodium falciparum spermidine synthase in complex with the Substrate decarboxylated S-adenosylmethionine and the potent inhibitors 4MCHA and AdoDATO. J. Mol. Biol. 373, 167–177. doi: 10.1016/j.jmb.2007.07.053
Ferreira, P. E., Culleton, R., Gil, J. P., and Meshnick, S. R. (2013). Artemisinin resistance in Plasmodium falciparum: what is it really? Trends Parasitol. 29, 318–320. doi: 10.1016/j.pt.2013.05.002
Frydman, B., Blokhin, A. V., Brummel, S., Wilding, G., Maxuitenko, Y., Sarkar, A., et al. (2003). Cyclopropane-containing polyamine analogues are efficient growth inhibitors of a human prostate tumor xenograft in nude mice. J. Med. Chem. 46, 4586–4600. doi: 10.1021/jm030175u
Gupta, R. D., Krause-Ihle, T., Bergmann, B., Müller, I. B., Khomutov, A. R., Müller, S., et al. (2005). 3-Aminooxy-1-Aminopropane and derivatives have an antiproliferative effect on cultured Plasmodium falciparum by decreasing intracellular polyamine concentrations. Antimicrob. Agents Chemother. 49, 2857–2864. doi: 10.1128/AAC.49.7.2857-2864.2005
Haider, N., Eschbach, M.-L., Dias, S., de, S., Gilberger, T.-W., Walter, R. D., et al. (2005). The spermidine synthase of the malaria parasite Plasmodium falciparum: molecular and biochemical characterisation of the polyamine synthesis enzyme. Mol. Biochem. Parasitol. 142, 224–236. doi: 10.1016/j.molbiopara.2005.04.004
Harder, E., Damm, W., Maple, J., Wu, C., Reboul, M., Xiang, J. Y., et al. (2015). OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12:281–96. doi: 10.1021/acs.jctc.5b00864
Hsieh, H.-P., Muller, J. G., and Burrows, C. J. (1994). Structural effects in novel steroidal polyamine-DNA binding. J. Am. Chem. Soc. 116, 12077–12078. doi: 10.1021/ja00105a068
Huang, Y., Hager, E. R., Phillips, D. L., Dunn, V. R., Hacker, A., Frydman, B., et al. (2003). A novel polyamine analog inhibits growth and induces apoptosis in human breast cancer cells. Clin. Cancer Res. 9, 2769–2777. doi: 10.3389/fmicb.2019.00009
Igarashi, K., and Kashiwagi, K. (2000). Polyamines: mysterious modulators of cellular functions. Biochem. Biophys. Res. Commun. 271, 559–564. doi: 10.1006/bbrc.2000.2601
Lambros, C., and Vanderberg, J. P. (1979). Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420. doi: 10.2307/3280287
Makler, M. T., and Hinrichs, D. J. (1993). Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 48, 205–210. doi: 10.4269/ajtmh.1993.48.205
Makler, M. T., Ries, J. M., Williams, J. A., Bancroft, J. E., Piper, R. C., Gibbins, B. L., et al. (1993). Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 48, 739–741. doi: 10.4269/ajtmh.1993.48.739
Marton, L. J., and Pegg, A. E. (1995). Polyamines as targets for therapeutic intervention. Annu. Rev. Pharmacol. Toxicol. 35, 55–91. doi: 10.1146/annurev.pa.35.040195.000415
Müller, S., Coombs, G. H., and Walter, R. D. (2001). Targeting polyamines of parasitic protozoa in chemotherapy. Trends Parasitol. 17, 242–249. doi: 10.1016/S1471-4922(01)01908-0
Müller, S., Da'dara, A., Lüersen, K., Wrenger, C., Gupta, R. D., Madhubala, R., et al. (2000). In the human malaria parasite Plasmodium falciparum, polyamines are synthesized by a bifunctional ornithine decarboxylase, S-adenosylmethionine decarboxylase. J. Biol. Chem. 275, 8097–8102. doi: 10.1074/jbc.275.11.8097
Osland, A., and Kleppe, K. (1977). Polyamine induced aggregation of DNA. Nucleic Acids Res. 4, 685–695. doi: 10.1093/nar/4.3.685
Pegg, A. E., and McCann, P. P. (1982). Polyamine metabolism and function. Am. J. Physiol. Cell Physiol. 243, C212–C221. doi: 10.1152/ajpcell.1982.243.5.C212
Phyo, A. P., Nkhoma, S., Stepniewska, K., Ashley, E. A., Nair, S., McGready, R., et al. (2012). Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet Lond. Engl. 379, 1960–1966. doi: 10.1016/S0140-6736(12)60484-X
Reddy, V. K., Valasinas, A., Sarkar, A., Basu, H. S., Marton, L. J., and Frydman, B. (1998). Conformationally restricted analogues of 1N,12N-bisethylspermine: synthesis and growth inhibitory effects on human tumor cell lines. J. Med. Chem. 41, 4723–4732. doi: 10.1021/jm980172v
Sprenger, J., Carey, J., Svensson, B., Wengel, V., and Persson, L. (2016). Binding and inhibition of spermidine synthase from Plasmodium falciparum and implications for in vitro inhibitor testing. PLoS ONE 11:e0163442. doi: 10.1371/journal.pone.0163442
Sprenger, J., Svensson, B., Hålander, J., Carey, J., Persson, L., and Al-Karadaghi, S. (2015). Three-dimensional structures of Plasmodium falciparum spermidine synthase with bound inhibitors suggest new strategies for drug design. Acta Crystallogr. D Biol. Crystallogr. 71, 484–493. doi: 10.1107/S1399004714027011
Sugiura, M., Shafman, T., and Kufe, D. (1984). Effects of polyamine depletion on proliferation and differentiation of murine erythroleukemia cells. Cancer Res. 44, 1440–1444.
Trager, W., and Jensen, J. B. (1976). Human malaria parasites in continuous culture. Science 193, 673–675. doi: 10.1126/science.781840
Valasinas, A., Reddy, V. K., Blokhin, A. V., Basu, H. S., Bhattacharya, S., Sarkar, A., et al. (2003). Long-chain polyamines (oligoamines) exhibit strong cytotoxicities against human prostate cancer cells. Bioorg. Med. Chem. 11, 4121–4131. doi: 10.1016/S0968-0896(03)00453-X
van Brummelen, A. C., Olszewski, K. L., Wilinski, D., Llinás, M., Louw, A. I., and Birkholtz, L.-M. (2009). Co-inhibition of Plasmodium falciparum S-adenosylmethionine decarboxylase/ornithine decarboxylase reveals perturbation-specific compensatory mechanisms by transcriptome, proteome, and metabolome analyses. J. Biol. Chem. 284, 4635–4646. doi: 10.1074/jbc.M807085200
Verlinden, B. K., de Beer, M., Pachaiyappan, B., Besaans, E., Andayi, W. A., Reader, J., et al. (2015). Interrogating alkyl and arylalkylpolyamino (bis)urea and (bis)thiourea isosteres as potent antimalarial chemotypes against multiple lifecycle forms of Plasmodium falciparum parasites. Bioorg. Med. Chem. 23, 5131–5143. doi: 10.1016/j.bmc.2015.01.036
Verlinden, B. K., Niemand, J., Snyman, J., Sharma, S. K., Beattie, R. J., Woster, P. M., et al. (2011). Discovery of novel alkylated (bis)urea and (bis)thiourea polyamine analogues with potent antimalarial activities. J. Med. Chem. 54, 6624–6633. doi: 10.1021/jm200463z
Wallace, H. M., Fraser, A. V., and Hughes, A. (2003). A perspective of polyamine metabolism. Biochem. J. 376, 1–14. doi: 10.1042/bj20031327
White, N. J. (2008). Qinghaosu (artemisinin): the price of success. Science 320, 330–334. doi: 10.1126/science.1155165
White, N. J., Pukrittayakamee, S., Hien, T. T., Faiz, M. A., Mokuou, O. A., and Dondorp, A. M. (2014). Malaria. Lancet 383, 22–28. doi: 10.1016/B.978-0-7020-5101-2.00044-3
World Health Organization (2015) World Health Organization Malaria Report. Geneva: World Health Organization.
Keywords: polyamine, Plasmodium, malaria, spermidine, spermine, spermidine synthase, thiourea
Citation: El Bissati K, Redel H, Ting L-M, Lykins JD, McPhillie MJ, Upadhya R, Woster PM, Yarlett N, Kim K and Weiss LM (2019) Novel Synthetic Polyamines Have Potent Antimalarial Activities in vitro and in vivo by Decreasing Intracellular Spermidine and Spermine Concentrations. Front. Cell. Infect. Microbiol. 9:9. doi: 10.3389/fcimb.2019.00009
Received: 20 June 2018; Accepted: 14 January 2019;
Published: 14 February 2019.
Edited by:
Alexis Kaushansky, Seattle Children's Research Institute, United StatesReviewed by:
Paras Jain, Intellectual Ventures, United StatesRapatbhorn Patrapuvich, Mahidol University, Thailand
Copyright © 2019 El Bissati, Redel, Ting, Lykins, McPhillie, Upadhya, Woster, Yarlett, Kim and Weiss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamal El Bissati, a2VsYmlzc2F0aUB1Y2hpY2Fnby5lZHU=
Kami Kim, a2FtaWtpbUBoZWFsdGgudXNmLmVkdQ==
Louis M. Weiss, bG91aXMud2Vpc3NAZWluc3RlaW4ueXUuZWR1
†Present Address: Henry Redel, Rutgers Robert Wood Johnson Medical School, Department of Medicine, East Brunswick, NJ, United States; NYU School of Medicine, Department of Medicine, New York, NY, United States
Kami Kim, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, FL, United States