 Pan Tao
Pan Tao Zhangyong Ning1
Zhangyong Ning1 Xiangqi Hao
Xiangqi Hao Xi Lin
Xi Lin Shoujun Li
Shoujun Li- 1College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
- 2Guangdong Provincial Key Laboratory of Prevention and Control for Severe Clinical Animal Diseases, Guangzhou, China
- 3Guangdong Technological Engineering Research Center for Pet, Guangzhou, China
This study aimed to detect changes in the complete transcriptome of MDCK cells after infection with the H5N1 and H3N2 canine influenza viruses using high-throughput sequencing, search for differentially expressed RNAs in the transcriptome of MDCK cells infected with H5N1 and H3N2 using comparative analysis, and explain the differences in the pathogenicity of H5N1 and H3N2 at the transcriptome level. Based on the results of our comparative analysis, significantly different levels of expression were found for 2,464 mRNAs, 16 miRNAs, 181 lncRNAs, and 262 circRNAs in the H3N2 infection group and 448 mRNAs, 12 miRNAs, 77 lncRNAs, and 189 circRNAs in the H5N1 infection group. Potential functions were predicted by performing Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of the target genes of miRNAs, lncRNAs and circRNAs, and the ncRNA-mRNA regulatory network was constructed based on differentially expressed RNAs. A greater number of pathways regulating immune metabolism were altered in the H3N2 infection group than in the H5N1 infection group, which may be one reason why the H3N2 virus is less pathogenic than is the H5N1 virus. This study provides detailed data on the production of ncRNAs during infection of MDCK cells by the canine influenza viruses H3N2 and H5N1, analyzed differences in the total transcriptomes between H3N2- and H5N1-infected MDCK cells, and explained these differences with regard to the pathogenicity of H3N2 and H5N1 at the transcriptional level.
Introduction
Influenza viruses belong to the Orthomyxoviridae family of RNA viruses that cause influenza in humans or animals, and the viral genome is composed of a single negative strand of segmented RNA. To date, four types of influenza viruses have been identified: influenza A virus (IAV), influenza B virus (IBV), influenza C virus (ICV), and most recently influenza D virus (IDV) (Hause et al., 2013, 2014).
IAV, which causes acute respiratory diseases in many hosts such as birds, humans and pigs, is an important pathogenic microorganism worldwide. Moreover, IAV has a wide host specificity and infects a variety of hosts. Influenza has been divided into avian influenza, swine influenza, human influenza and canine influenza. In the early days, IAVs were believed to be unable to infect dogs under natural conditions, but several subtypes were subsequently isolated from dogs with respiratory symptoms. Currently, two main subtypes of canine influenza virus (CIV) have been identified: equine-origin H3N8 (Crawford et al., 2005; Daly et al., 2008; Kruth et al., 2008; Kirkland et al., 2010) and avian-origin H3N2 (Song et al., 2008; Lee et al., 2009; Li et al., 2010). In addition, various subtypes of IAVs are reported to be able to infect dogs, including influenza A H1N1 pdm09 viruses, H5N1 avian influenza viruses (AIVs), H5N2 subtypes of AIVs, reassortants of wild-type H3N1 IAVs and H9N2 subtypes of AIVs (Songserm et al., 2006; Dundon et al., 2010; Lin et al., 2011; Guang-jian et al., 2012; Song et al., 2012; Sun et al., 2013).
Avian-origin H3N2 CIV, which was initially circulating only in Asian countries (Song et al., 2009; Li et al., 2010; Li G. et al., 2018), has now spread to the United States and the rest of the world (Pulit-Penaloza et al., 2017; Voorhees et al., 2017). H3N2 CIV causes sneezing and clinical symptoms of typical respiratory diseases, such as runny nose, cough and fever, as well as damage to many other organs outside the respiratory tract, in dogs (Luo et al., 2018; Zheng et al., 2018).
The highly pathogenic avian influenza (HPAI) H5N1 virus was first reported in Thailand in October 2004 in dogs with severe pulmonary congestion and edema and a bloody nose (Songserm et al., 2006). An epidemiological survey of 629 village dogs in Thailand found that approximately one-quarter had antibodies against H5N1, indicating that they were infected with the virus or had been infected in the past (Butler, 2006). Our laboratory also isolated an avian influenza H5N1 virus from a dog in 2013. The dogs that were infected with the highly pathogenic AIV H5N1 subtype showed anorexia, dyspnea, cough, conjunctivitis and a brief increase in body temperature within 2 days, but the virus did not spread between dogs (Maas et al., 2007; Giese et al., 2008). The highest viral replication titer of the nose swab was 6.3 log10TCID50/mL on average, and lung lesions in the H5N1 infection group were more severe than those in the H3N2 infection group (Zheng et al., 2018).
Why is the H5N1 influenza virus more pathogenic than H3N2, and why does it lead to a more severe inflammatory response? Current studies on H3N2 and H5N1 avian-origin CIV transcriptomes are limited to miRNAs and mRNAs (Fu et al., 2018; Zheng et al., 2018), whereas no studies have examined changes in mRNAs, miRNAs, lncRNAs, and circRNAs or performed a detailed analysis of the correlations between ncRNA and mRNA levels. In the present study, we analyzed the entire transcriptome to obtain an understanding of the mechanism of pathogenicity and differences in inflammation between the viruses and to provide guidance for future treatment.
Dogs are one of the most numerous domesticated animals and often serve as companion animals to humans. Because dogs can be infected with both avian and human influenza viruses, they are likely to be a “mixing vessel” for genetic rearrangement of influenza viruses, making the study of canine influenza important for public and human health (Gonzalez et al., 2014; Zhu et al., 2015).
Materials and Methods
Viruses and Cells
The H3N2 (A/canine/Guangdong/B/2013) and H5N1 (A/canine/Guangdong/01/2013) CIVs were isolated in 2013 from dogs with severe respiratory symptoms in Guangdong, China, and preserved in our laboratory. Madin-Darby canine kidney (MDCK) cells were obtained from American Type Culture Collection (ATCC) and propagated in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 atmosphere. Viruses were propagated in the MDCK cells at 37°C and 5% CO2 for 48 h. All experiments with live viruses were performed in an enhanced animal biosafety level 3 facility at the South China Agricultural University. The protocol number was SYXK (YUE) 2016-0136.
Sample Collection and RNA Isolation
MDCK cells were cultured with DMEM containing 10% FBS in a 37°C incubator with a 5% CO2 atmosphere. Upon reaching 90% confluence, MDCK cells were infected with the H3N2 (MOI = 0.1) and H5N1 (MOI = 0.1) influenza viruses, and the viruses used in these infections were purified using a sucrose gradient. Then, the cells were cultured with DMEM containing 2% FBS in a 37°C incubator with a 5% CO2 atmosphere for 24 h before being harvested. The control group was cultured without virus under the same conditions (independent triplicate experiments). Total RNA was isolated from MDCK cells using TRIzol (Takara) according to the manufacturer's protocol. RNA concentrations were detected using a Qubit2.0 fluorometer (Invitrogen), and the RNA integrity and genomic DNA contamination were detected by separating the samples on an agarose gel. RNA concentrations and purity were determined by measuring the OD, A260/A280 (>1.8) and A260/A230 (>1.6). RNA samples were stored at −80°C until further use.
RNA Sequencing and Data Analysis
Small RNA library construction: T4 RNA ligase 2 (New England Biolabs) was used to connect the 3′-end connector to the RNA. Reverse transcription primer hybridization: T4 RNA ligase 1 (New England Biolabs) was used to connect the 5'-end connector to the RNA. Reverse transcription reaction: The final library product was obtained by PCR amplification of the reverse transcription product. Construction of a chain-specific library for ribosome removal: Sequencing libraries were generated using RNase R digestion and rRNA-depleted RNAs. The library preparations were sequenced on an Illumina Hiseq™ platform (repeated 3 times). The raw sequencing data were analyzed with FastQC using cutadapt to remove joints and trimmomatic to remove low-quality bases and reads at both ends.
Screen of Differentially Expressed mRNAs, miRNAs, lncRNAs, and circRNAs and Clustering Analysis
The differences in expression among mRNAs, lncRNAs and circRNAs were analyzed using DESeq2, and the differential expression of miRNA was analyzed using edgeR (Anders and Huber, 2010). The results of the differential analyses were visualized, with a p < 0.05 and multiple differences >2 as screening conditions. A Venn diagram and a heat map were constructed, and a clustering analysis was performed based on the results of the differential expression analysis. A network diagram was drawn based on the correlations between miRNA and mRNA, lncRNA and mRNA, and circRNA and mRNA expression.
Annotation of GO Terms and Analysis of KEGG Signaling Pathways
The differentially expressed miRNA, lncRNA, and circRNA target genes and mRNAs were annotated using GO and analyzed for enriched KEGG signaling pathways.
Analysis of the ceRNA Network (ceRNET)
The circRNA-miRNA-mRNA ceRNET and the lncRNA-miRNA-mRNA ceRNET were constructed after determining the negative regulatory relationship between the differentially expressed miRNAs and their differentially expressed target genes (mRNAs/lncRNAs and mRNAs/circRNAs).
Real-Time qPCR
Significant regulatory pathways were selected from the ceRNET for RT-qPCR verification of differential expression. The cDNA templates were synthesized using PrimeScriptTM RT Master Mix (Perfect Real Time) (Takara, Otsu, Japan, Product no: RR036A). RT-qPCR was performed using the SYBR Premix Ex TaqTM (Tli RNaseH Plus) (Takara, Otsu, Japan, Product no: TRR820A) and an LC480 Real-Time PCR System (Roche, Basel, Switzerland) in accordance with the manufacturer's specifications. Small RNA samples were isolated using the E.Z.N.A.TM miRNA Kit (OMEGA BIO-TEK. Product no: R6842-01). The miRcute Plus miRNA First Chain cDNA Synthesis Kit (Tiangen, Beijing, China. Product no: KP211) was used for cDNA synthesis. RT-qPCR was performed using the miRcute Plus miRNA qPCR Detection Kit (Tiangen, Beijing, China, Product no: FP411) and an LC480 instrument (Roche, Basel, Switzerland). GAPDH was used as the endogenous control for mRNAs and lncRNAs, and U6 was used as the endogenous control for miRNAs. We used the 2−ΔΔCt method to analyze the data. All samples were analyzed in triplicate, and the data are presented as means ± standard deviations (n = 3).
Results
Replication Kinetics of H3N2 and H5N1 Strains in MDCK Cells
The kinetics of H3N2 and H5N1 CIV replication were measured and analyzed based on the TCID50 at 3, 12, 24, 36, 48, and 72 hpi (Figure 1) in Reed-Muench. H3N2 and H5N1 CIVs replicated well in MDCK cells. H5N1 replicated significantly more vigorously than H3N2 (p < 0.01) at every assessed time point throughout the replication kinetics experiment. Viral replication was distinctly dose dependent for both H3N2 and H5N1 CIVs during the first 24 hpi, whereas the dose dependency was negligible from 24 to 72 hpi. The peak titers of the H5N1 CIV reached up to 109.6 of the TCID50/mL at 24 hpi, whereas H3N2 CIV replicated significantly less rapidly, with peak virus titers reaching only 107.7 of the TCID50/mL.
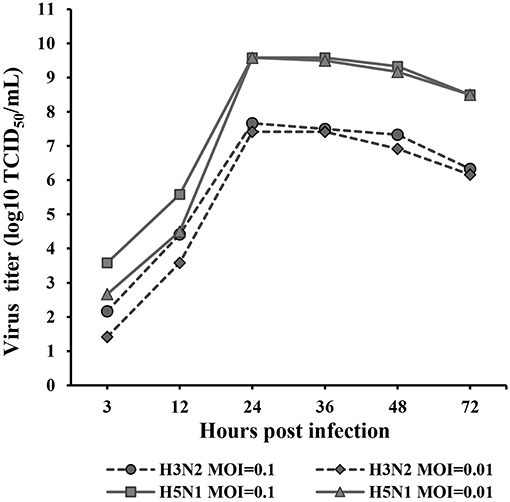
Figure 1. Replication kinetics of H3N2 and H5N1 in MDCK cells. Monolayers of MDCK cells were infected with each virus at MOIs of 0.1 and 0.01. Virus titers were determined using the TCID50 assay at 3, 12, 24, 36, 48, and 72 hpi. TPCK-trypsin (0.25 mg/mL) was added to the medium to promote the propagation of the H3N2 influenza A virus. The data were analyzed using one-way ANOVA (p < 0.05). Data are presented as the means ± SD of independent triplicate experiments.
Analysis of Differentially Expressed mRNAs, miRNA, lncRNAs, and circRNAs
Through comparative analysis, we found that 2,464 mRNAs, 16 miRNAs, 181 lncRNAs, and 262 circRNAs were significantly differentially expressed in the H3N2 group compared with the control group and that 448 mRNAs, 12 miRNAs, 77 lncRNAs and 189 circRNAs were differentially expressed in the H5N1 group compared with the control group. Moreover, 1,950 mRNAs, 20 miRNAs, 162 lncRNAs, and 75 circRNAs were differentially expressed in the H3N2 group compared with the H5N1 group (Table 1, Figure 2 and Supplementary Material).
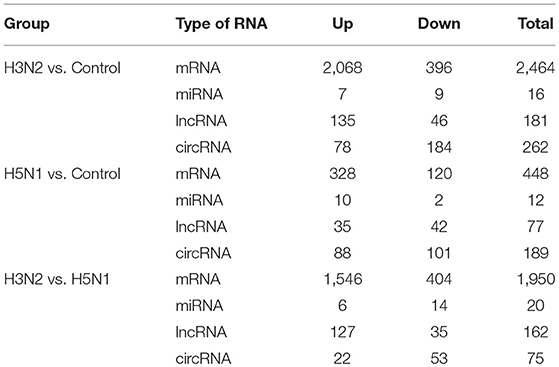
Table 1. Differential expression profiles of mRNAs, miRNAs, lncRNAs, and circRNAs.
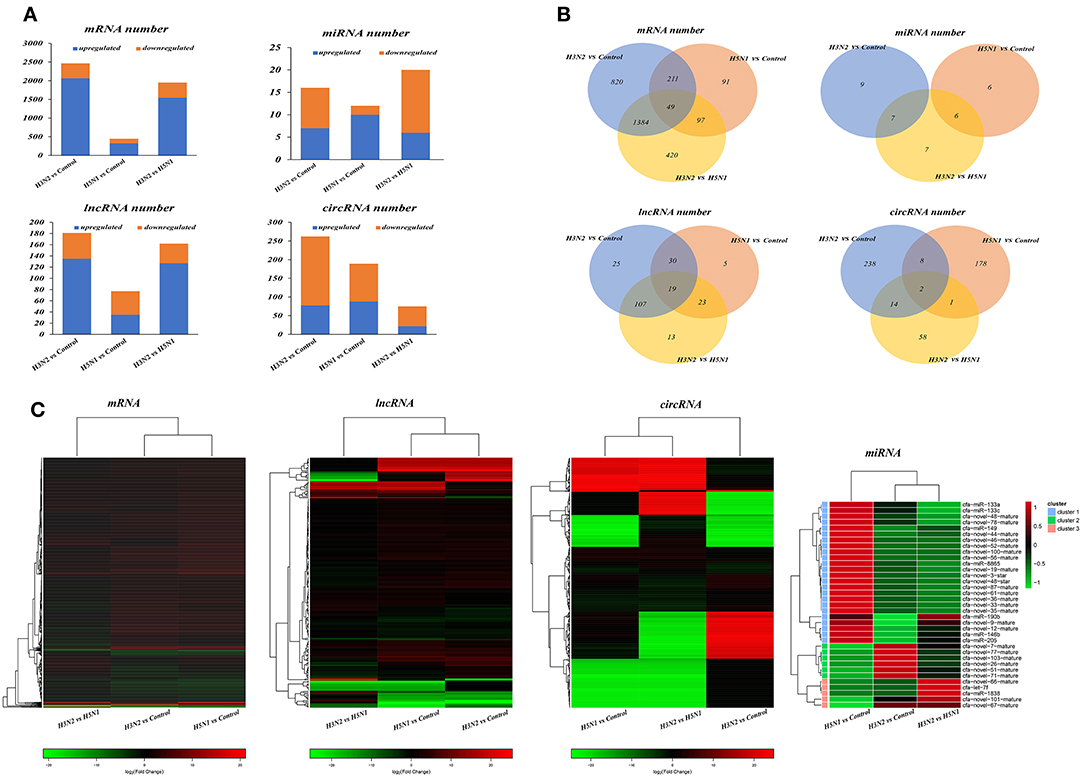
Figure 2. (A) Statistical histogram of the differential expression analysis. The horizontal axis represents the comparison and the vertical axis shows the number of differentially expressed transcripts. Orange represents downregulated transcripts, and blue represents upregulated transcripts. (B) Differentially expressed transcripts. In the Venn diagram, different comparison groups are represented by different colors. The numbers in the figure represent specific or common differences in the number of transcripts. The overlapping areas represent the number of differentially expressed transcripts shared by different comparison groups; the non-overlapping areas represent the number of differentially expressed transcripts that were unique to the different comparison groups. (C) Heat map of differentially expressed transcripts based on fold changes. Each row represents a transcript and each column represents a comparison group. Red indicates upregulated expression, and green indicates downregulated expression.
Correlation Analysis of mRNAs and ncRNAs
We analyzed the targeting relationship between differentially expressed ncRNAs and mRNAs to further study the correlation between the ncRNA-mRNA regulatory network in MDCK cells infected with CIVs. The analysis identified 706 differentially expressed miRNA-mRNA, 66 lncRNA-mRNA and 86 circRNA-mRNA pairs in the H3N2 group compared with the control group and 50 differentially expressed miRNA-mRNA, 13 lncRNA-mRNA, and 6 circRNA-mRNA pairs in the H5N1 group compared with the control group (Figure 3).
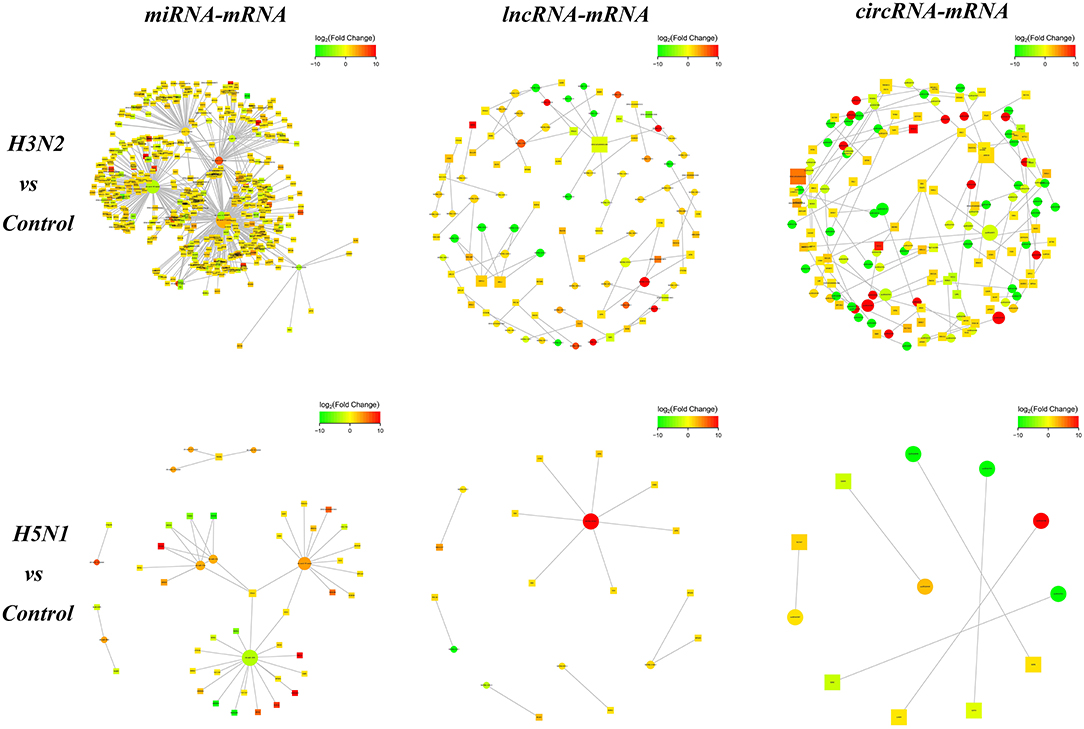
Figure 3. The network diagram of ncRNA and mRNA interactions. Square nodes indicate ncRNAs, round nodes indicate mRNAs, and the edges indicate the interactions between the two genes. The size of the node is proportional to the connectivity (degree) of the node; namely, the more edges are connected to the node, the larger the degree of the node, indicating that the gene is more important in the network. The color of the node represents the difference in gene expression in this group of samples, namely, the logFC value; green represents downregulation, red represents upregulation, and the color depth represents differences in the altered expression.
Gene Ontology (GO) Functional Enrichment Analysis of Differentially Expressed Genes
We conducted a GO enrichment analysis and statistical analysis of biological processes to determine the functional classifications of these differentially expressed mRNAs and target genes of ncRNAs. A large number of genes in the H3N2 and H5N1 infection groups are involved in cellular process, single-organism process and metabolic process. However, many genes in biological processes were enriched in the H3N2 infection group, and each enriched functional classification also contained a large number genes. Based on the results of the GO enrichment analysis, miRNAs were more abundant than lncRNAs and circRNAs in terms of the number of target genes and functional classifications (Figure 4).
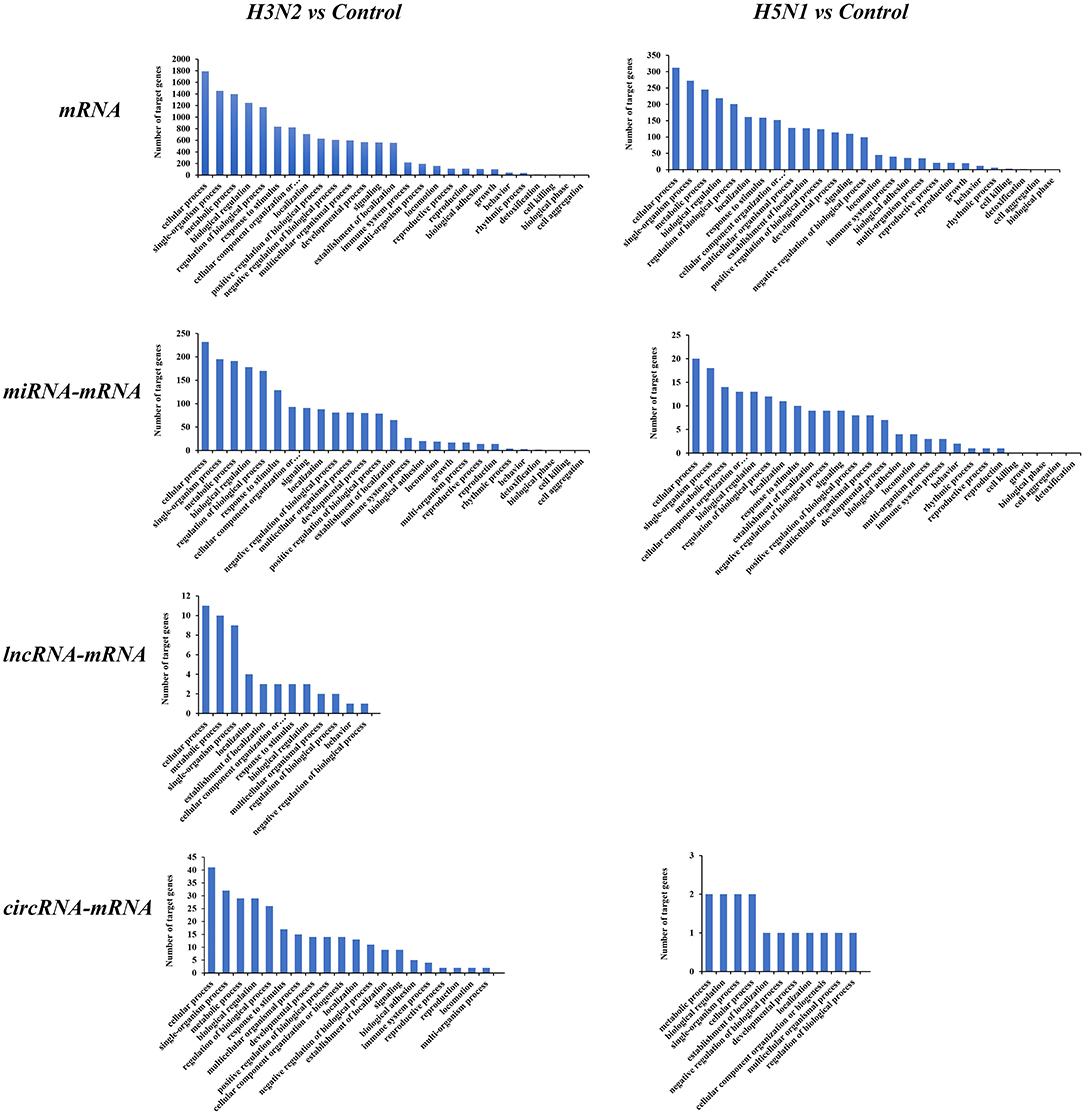
Figure 4. GO pathways of differentially expressed mRNAs and target genes of differentially expressed miRNAs, lncRNAs, and circRNAs. The horizontal axis shows the functional classification, and the vertical axis shows the number of genes in the classification.
Analysis of the Enrichment of Differentially Expressed Genes in Various Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways
KEGG analysis was also performed for mRNAs and target genes of miRNAs, lncRNAs and circRNAs that were differentially expressed in cells infected with H3N2 and H5N1 to explore canine host resistance mechanisms to H3N2 and H5N1 infection and their differences. Then, we conducted a statistical analysis of cytokine and pathogen-host interaction pathways and found that a large number regulatory pathways and genes were enriched in the H3N2 infection group, particularly the MAPK signaling pathway, the endocytosis pathway, the p53 signaling pathway and other pathways with more obvious advantages. Notably, miRNAs can target more than one mRNA to regulate gene expression, indicating that miRNA-mediated targeted regulation of mRNA expression is more important than are the regulatory pathways involving lncRNAs and circRNAs (Figure 5).
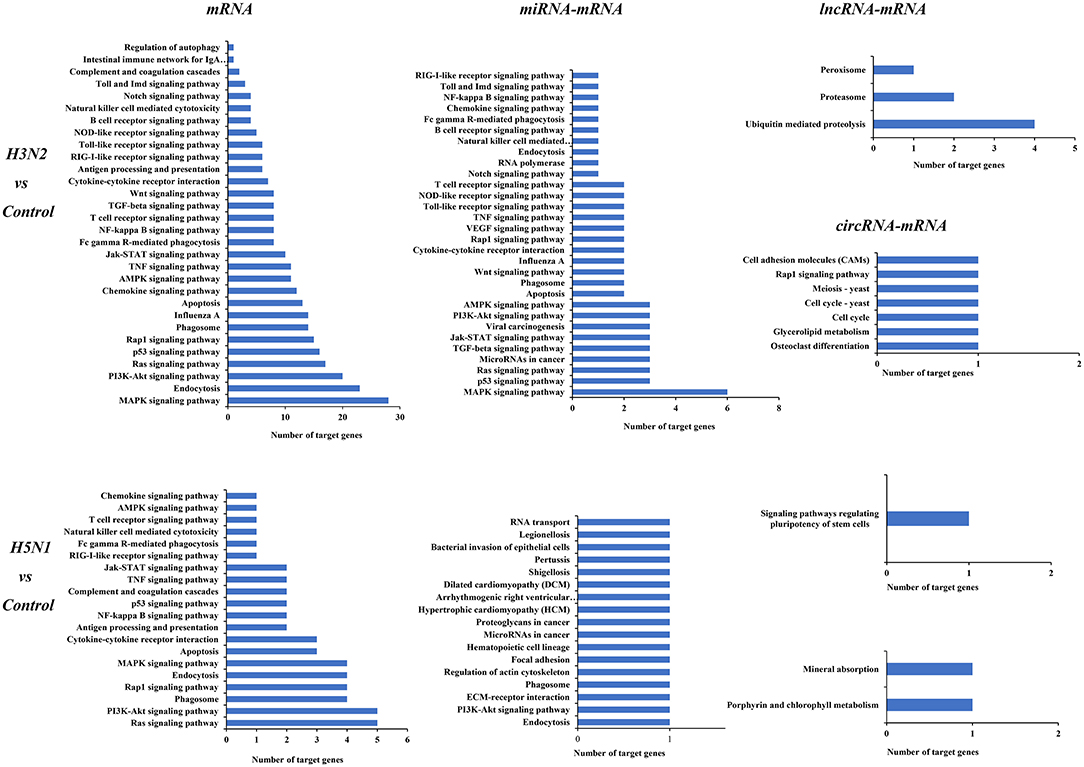
Figure 5. KEGG pathways of differentially expressed mRNAs and target genes of differentially expressed miRNAs, lncRNAs, and circRNAs. The horizontal axis shows the number of genes, and the vertical axis shows each pathway.
The Competing Endogenous RNA (ceRNA) Regulatory Network
Notably, lncRNAs and circRNAs limit miRNA-mediated regulation of target gene expression and act as miRNA sponges to indirectly regulate gene expression. According to the theory of ceRNAs, we constructed lncRNA-miRNA-mRNA and circRNA-miRNA-mRNA regulatory networks. In the comparison of the H3N2 and control groups, the lncRNA-miRNA-mRNA network contained 15 lncRNAs, 6 miRNAs and 237 mRNAs, and the circRNA-miRNA-mRNA network contained 3 circRNAs, 1 miRNA and 9 mRNAs. In the comparison of the H5N1 and control groups, the lncRNA-miRNA-mRNA network comprised 6 lncRNAs, 4 miRNAs and 18 mRNAs, and a circRNA-miRNA-mRNA regulatory network was not identified. Enrichment of mRNAs in the regulatory network was assessed via analysis of KEGG pathways (Figure 6).
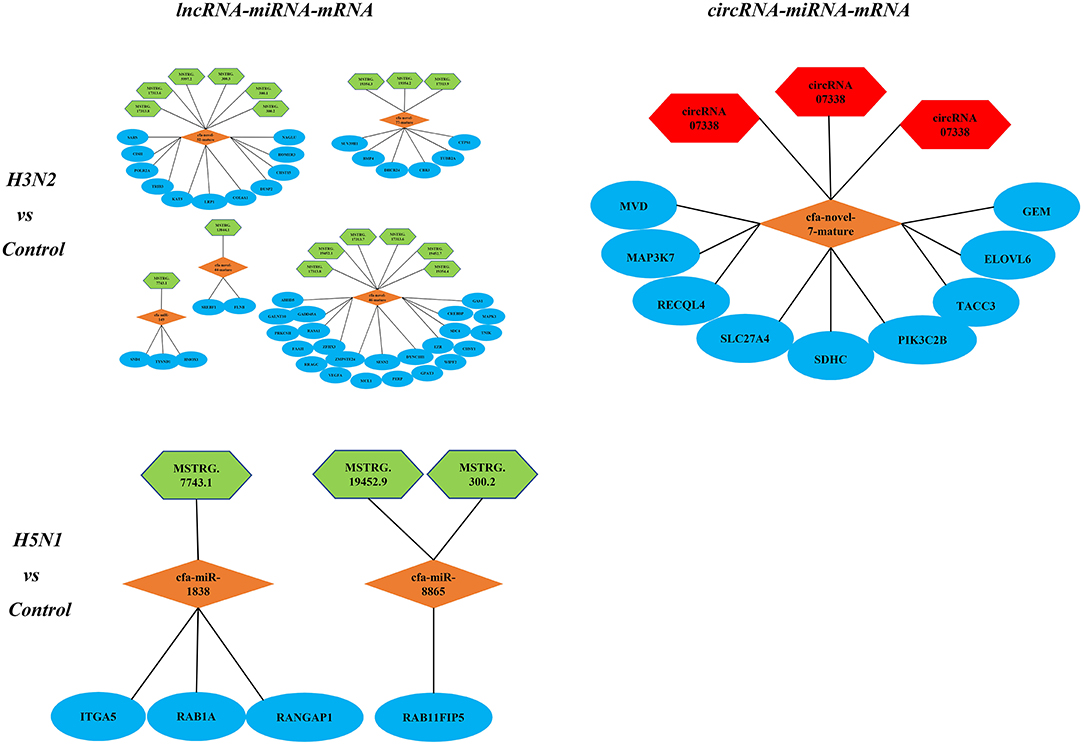
Figure 6. KEGG pathways associated with the lncRNA-miRNA-mRNA and circRNA-miRNA-mRNA regulatory networks; green represents lncRNAs, red represents circRNAs, orange represents miRNAs, and blue represents mRNAs.
Differential Expression of ncRNAs and mRNAs Confirmed by RT-qPCR
In the ceRNA network, mRNAs in the Jak-STAT signaling pathway, p53 signaling pathway and viral carcinogenesis regulatory network were enriched and verified by RT-qPCR (primer sequences are shown in Table 2). The sequencing results were consistent with the trends in the qPCR verification results (Table 3 and Figure 7).

Table 2. Primer sequences used for real-time PCR.
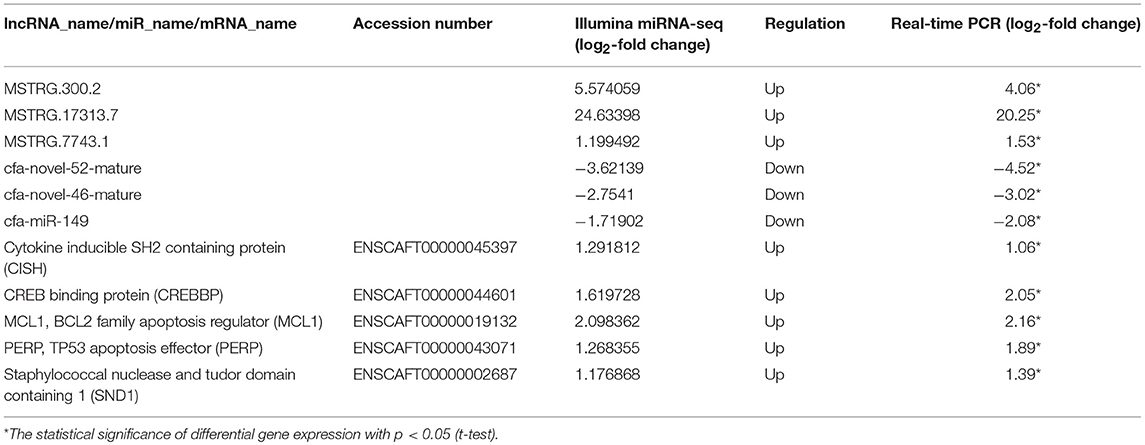
Table 3. Relative RNA expression of selected differentially expressed genes (DEGs) determined using RNA-seq and quantitative real-time PCR analyses.
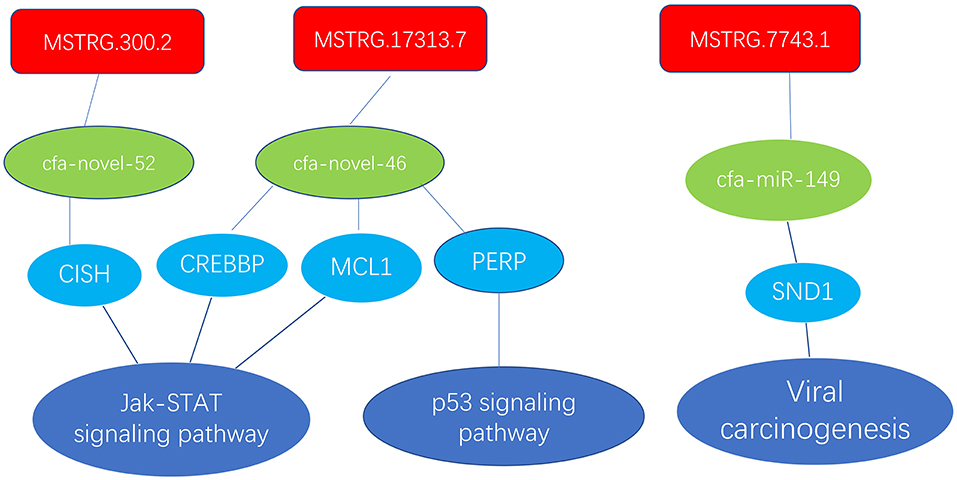
Figure 7. Enrichment of mRNAs in the Jak-STAT signaling pathway, p53 signaling pathway and viral carcinogenesis regulation network; lncRNAs are depicted in red, green represents miRNAs, and blue represents mRNAs.
Discussion
In this study, a high-throughput sequencing technique was used to compare the entire transcriptomes of MDCK cells infected with the H5N1 and H3N2 viruses. Although fewer differentially expressed mRNAs were identified in the H5N1 infection group than in the H3N2 infection group, a higher proportion of mRNAs exhibited a more significant differential expression pattern in the H5N1 infection group. In the H5N1 group, 24 (log2-fold change > 10) of 448 mRNAs were differentially expressed mRNAs, while only 27 (log2-fold change > 10) of 2,464 mRNAs were differentially expressed in the H3N2 group, 10 of which were shared by the two groups. In the ncRNA-mRNA regulatory network, the H3N2 group had more regulatory pairs than did the H5N1 group. Thus, H3N2-infected MDCK cells may be able to resist the viral infection through more pathways to reach a balanced state. This finding also illustrates why H5N1 is more virulent than H3N2 at the RNA level.
In the circRNA-miRNA-mRNA regulatory network, we identified 3 circRNAs in the H3N2 infection group. One miRNA and 9 mRNAs had a targeted relationship, while the H5N1 infection group did not possess this regulatory network. In the lncRNA-miRNA-mRNA regulatory network, 15 lncRNAs, 6 miRNAs, and 237 mRNAs were identified in the H3N2 infection group. Six lncRNAs, 4 miRNAs, and 18 mRNAs were identified in the H5N1 infection group. Based on this result, H3N2 infection may induce broad regulation in MDCK cells, and the circRNA-miRNA-mRNA regulatory network is not common in influenza infection. Regarding GO and KEGG enrichment analyses, we also intuitively observed a greater number of GO annotations and enriched KEGG pathways in the H3N2 infection group than in the H5N1 infection group. In conclusion, we postulate that H5N1 is more virulent than H3N2 for several reasons. (1). H5N1 infects cells and causes intracellular cytokine storms, resulting in rapid cell death (Li X. et al., 2018). (2). Cells infected with H3N2 utilize more resistance pathways to eliminate the virus, while H5N1 infection activates relatively few pathways in cells, which showed the some differences in MDCK in antiviral responses between H3N2 and H5N1 at the RNA level of the transcriptome.
Notably, lncRNAs play important roles in many cellular activities, such as the regulation of epigenetics, the cell cycle and cell differentiation, and has become a hot topic in genetic research (Wapinski and Chang, 2011; Kwok and Tay, 2017). MiRNAs and their target genes have a variety of relationships (Perez et al., 2009; Fan and Wang, 2016; Nakamura et al., 2016), and misaligned miRNAs can be used as diagnostic and prognostic biomarkers (Okkenhaug and Vanhaesebroeck, 2003; Hale et al., 2010; Haneklaus et al., 2013). In the present study, we examined the lncRNA-miRNA-mRNA regulatory networks of the Jak-STAT signaling pathway, the p53 signaling pathway and the viral carcinogenesis pathway for RT-qPCR verification. The Jak-STAT signaling pathway, which is closely related to the type I interferon (IFN)-mediated innate immune response, is an important regulator of cell proliferation, differentiation, survival, motility, apoptosis, development and the immune response (Bartunek et al., 1999; Liu et al., 2010). Currently, p53 is the most widely analyzed functional transcription factor. The aggregation and induced activation of p53 are the core cell signaling events in a variety of stress-induced injury responses and have important regulatory roles in inhibiting cell cycle progression and inducing DNA damage repair, cell autophagy and apoptosis (Crighton et al., 2006; Duan et al., 2015; Sui et al., 2015). In the next step, we further explored the mechanisms of these pathways.
In this study, the total transcriptome of the MDCK cell model infected with CIVs was analyzed for the first time. The results further revealed the differences in pathogenicity between H3N2 and H5N1 at the RNA level of the transcriptome. However, this study has only analyzed the differences in the RNA levels at 24 hpi in canine cells and further studies are needed to analyze both earlier time points as well as the differences in the RNA transcriptome in dogs infected with the two types of influenza viruses.
Author Contributions
PT, XH, XL, and QZ conducted the experiments. PT and ZN analyzed the data and wrote the paper. SL designed the experiments and revised the paper.
Funding
This project was supported in part by the National Natural Science Foundation of China (31872454, 31672563), The National Key Research and Development Program of China (2016YFD0501004), The Natural Science Foundation Guangdong province (2018B030311037, 2018A030313633), The Guangdong Provincial Key Laboratory of Prevention and Control for Severe Clinical Animal Diseases (2017B030314142).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00076/full#supplementary-material
Supplementary Data Sheet 1. The original data.
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Bartunek, P., Koritschoner, N. P., Brett, D., and Zenke, M. (1999). Molecular cloning, expression and evolutionary analysis of the avian tyrosine kinase JAK1. Gene 230, 129–136. doi: 10.1016/S0378-1119(99)00080-3
Butler, D. (2006). Thai dogs carry bird-flu virus, but will they spread it? Nature 439:773. doi: 10.1038/439773a
Crawford, P. C., Dubovi, E. J., Castleman, W. L., Stephenson, I., Gibbs, E. P., Chen, L., et al. (2005). Transmission of equine influenza virus to dogs. Science 310, 482–485. doi: 10.1126/science.1117950
Crighton, D., Wilkinson, S., O'Prey, J., Syed, N., Smith, P., Harrison, P. R., et al. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134. doi: 10.1016/j.cell.2006.05.034
Daly, J. M., Blunden, A. S., Macrae, S., Miller, J., Bowman, S. J., Kolodziejek, J., et al. (2008). Transmission of equine influenza virus to English foxhounds. Emerg. Infect. Dis. 14, 461–464. doi: 10.3201/eid1403.070643
Duan, L., Perez, R. E., Davaadelger, B., Dedkova, E. N., Blatter, L. A., and Maki, C. G. (2015). p53-regulated autophagy is controlled by glycolysis and determines cell fate. Oncotarget 6, 23135–23156. doi: 10.18632/oncotarget.5218
Dundon, W. G., De Benedictis, P., Viale, E., and Capua, I. (2010). Serologic evidence of pandemic (H1N1) 2009 infection in dogs, Italy. Emerg. Infect. Dis. 16, 2019–2021. doi: 10.3201/eid1612.100514
Fan, N., and Wang, J. (2016). MicroRNA 34a contributes to virus-mediated apoptosis through binding to its target gene Bax in influenza A virus infection. Biomed. Pharmacother. 83, 1464–1470. doi: 10.1016/j.biopha.2016.08.049
Fu, C., Luo, J., Ye, S., Yuan, Z., and Li, S. (2018). Integrated lung and tracheal mRNA-Seq and miRNA-Seq analysis of dogs with an avian-like H5N1 canine influenza virus infection. Front. Microbiol. 9:303. doi: 10.3389/fmicb.2018.00303
Giese, M., Harder, T., Teifke, J., Klopfleisch, R., Breithaupt, A., Mettenleiter, T., et al. (2008). Experimental infection and natural contact exposure of dogs with avian influenza virus (H5N1). Emerg. Infect. Dis. 14, 308–310. doi: 10.3201/eid1402.070864
Gonzalez, G., Marshall, J. F., Morrell, J., Robb, D., McCauley, J. W., Perez, D. R., et al. (2014). Infection and pathogenesis of canine, equine, and human influenza viruses in canine tracheas. J. Virol. 88, 9208–9219. doi: 10.1128/JVI.00887-14
Guang-jian, Z., Zong-shuai, L., Yan-li, Z., Shi-jin, J., and Zhi-jing, X. (2012). Genetic characterization of a novel influenza A virus H5N2 isolated from a dog in China. Vet. Microbiol. 155, 409–416. doi: 10.1016/j.vetmic.2011.08.017
Hale, B. G., Albrecht, R. A., and García-Sastre, A. (2010). Innate immune evasion strategies of influenza viruses. Future Microbiol. 5, 23–41. doi: 10.2217/fmb.09.108
Haneklaus, M., Gerlic, M., O'Neill, L. A. J., and Masters, S. L. (2013). miR-223: infection, inflammation and cancer. J. Intern. Med. 274, 215–226. doi: 10.1111/joim.12099
Hause, B. M., Collin, E. A., Liu, R., Huang, B., Sheng, Z., Lu, W., et al. (2014). Characterization of a novel influenza virus in cattle and swine: proposal for a new genus in the Orthomyxoviridae family. MBio 5:e00031-14. doi: 10.1128/mBio.00031-14
Hause, B. M., Ducatez, M., Collin, E. A., Ran, Z., Liu, R., Sheng, Z., et al. (2013). Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathog. 9:e1003176. doi: 10.1371/journal.ppat.1003176
Kirkland, P. D., Finlaison, D. S., Crispe, E., and Hurt, A. C. (2010). Influenza virus transmission from horses to dogs, Australia. Emerg. Infect. Dis. 16, 699–702. doi: 10.3201/eid1604.091489
Kruth, S. A., Carman, S., and Weese, J. (2008). Seroprevalence of antibodies to canine influenza virus in dogs in Ontario. Can. Vet. J. 49, 800–802.
Kwok, Z. H., and Tay, Y. (2017). Long noncoding RNAs: lincs between human health and disease. Biochem. Soc. T. 45, 805–812. doi: 10.1042/BST20160376
Lee, C., Song, D., Kang, B., Kang, D., Yoo, J., Jung, K., et al. (2009). A serological survey of avian origin canine H3N2 influenza virus in dogs in Korea. Vet. Microbiol. 137, 359–362. doi: 10.1016/j.vetmic.2009.01.019
Li, G., Wang, R., Zhang, C., Wang, S., He, W., Zhang, J., et al. (2018). Genetic and evolutionary analysis of emerging H3N2 canine influenza virus. Emerg. Microbes Infect. 7:73. doi: 10.1038/s41426-018-0079-0
Li, S., Shi, Z., Jiao, P., Zhang, G., Zhong, Z., Tian, W., et al. (2010). Avian-origin H3N2 canine influenza A viruses in Southern China. Infect. Genetics Evol. 10, 1286–1288. doi: 10.1016/j.meegid.2010.08.010
Li, X., Fu, Z., Liang, H., Wang, Y., Qi, X., Ding, M., et al. (2018). H5N1 influenza virus-specific miRNA-like small RNA increases cytokine production and mouse mortality via targeting poly(rC)-binding protein 2. Cell Res. 28, 157–171. doi: 10.1038/cr.2018.3
Lin, D., Sun, S., Du, L., Ma, J., Fan, L., Pu, J., et al. (2011). Natural and experimental infection of dogs with pandemic H1N1/2009 influenza virus. J. Gen. Virol. 93, 119–123. doi: 10.1099/vir.0.037358-0
Liu, W. J., Sun, D. X., Yu, Y., Li, G., Tang, S. Q., Zhang, Y., et al. (2010). Association of Janus kinase 2 polymorphisms with growth and reproduction traits in chickens. Poult. Sci. 89, 2573–2579. doi: 10.3382/ps.2010-00988
Luo, J., Lu, G., Ye, S., Ou, J., Fu, C., Zhang, X., et al. (2018). Comparative pathogenesis of H3N2 canine influenza virus in beagle dogs challenged by intranasal and intratracheal inoculation. Virus Res. 255, 147–153. doi: 10.1016/j.virusres.2018.05.023
Maas, R., Tacken, M., Ruuls, L., Koch, G., and Rooij, E. (2007). Avian influenza (H5N1) susceptibility and receptors in dogs. Emerg. Infect. Dis. 13, 1219–1221. doi: 10.3201/eid1308.070393
Nakamura, S., Horie, M., Daidoji, T., Honda, T., Yasugi, M., Kuno, A., et al. (2016). Influenza A virus-induced expression of a GalNAc transferase, GALNT3, via MicroRNAs is required for enhanced viral replication. J. Virol. 90, 1788–1801. doi: 10.1128/JVI.02246-15
Okkenhaug, K., and Vanhaesebroeck, B. (2003). PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 3, 317–330. doi: 10.1038/nri1056
Perez, J. T., Pham, A. M., Lorini, M. H., Chua, M. A., Steel, J., and TenOever, B. R. (2009). MicroRNA-mediated species-specific attenuation of influenza A virus. Nat. Biotechnol. 27, 572–576. doi: 10.1038/nbt.1542
Pulit-Penaloza, J. A., Simpson, N., Yang, H., Creager, H. M., Jones, J., Carney, P., et al. (2017). Assessment of molecular, antigenic, and pathological features of canine influenza A(H3N2) viruses that emerged in the United States. J. Infect. Dis. 216, S499–S507. doi: 10.1093/infdis/jiw620
Song, D., Kang, B., Lee, C., Jung, K., Park, B., and Oh, J. (2008). Transmission of avian influenza virus (H3N2) to dogs. Emerg. Infect. Dis. 14, 741–746. doi: 10.3201/eid1405.071471
Song, D., Lee, C., Kang, B., Jung, K., Oh, T., Kim, H., et al. (2009). Experimental infection of dogs with avian-origin canine influenza a virus (H3N2). Emerg. Infect. Dis. 15, 56–58. doi: 10.3201/eid1501.080755
Song, D., Moon, H. J., An, D. J., Jeoung, H. Y., Kim, H., Yeom, M. J., et al. (2012). A novel reassortant canine H3N1 influenza virus between pandemic H1N1 and canine H3N2 influenza viruses in Korea. J. Gen. Virol. 93, 551–554. doi: 10.1099/vir.0.037739-0
Songserm, T., Amonsin, A., Jam-on, R., Sae-Heng, N., Pariyothorn, N., Payungporn, S., et al. (2006). Fatal avian influenza A H5N1 in a dog. Emerg. Infect. Dis.12:1744. doi: 10.3201/eid1211.060542
Sui, X., Han, W., and Pan, H. (2015). p53-induced autophagy and senescence. Oncotarget 6, 11723–11724. doi: 10.18632/oncotarget.4170
Sun, X., Xu, X., Liu, Q., Liang, D., Li, C., He, Q., et al. (2013). Evidence of avian-like H9N2 influenza A virus among dogs in Guangxi, China. Infect. Genetics Evol. 20, 471–475. doi: 10.1016/j.meegid.2013.10.012
Voorhees, I. E. H., Glaser, A. L., Toohey-Kurth, K., Newbury, S., Dalziel, B. D., Dubovi, E. J., et al. (2017). Spread of canine influenza A(H3N2) virus, United States. Emerg. Infect. Dis. 23, 1950–1957. doi: 10.3201/eid2312.170246
Wapinski, O., and Chang, H. Y. (2011). Long noncoding RNAs and human disease. Trends Cell Biol. 21, 354–361. doi: 10.1016/j.tcb.2011.04.001
Zheng, Y., Fu, X., Wang, L., Zhang, W., Zhou, P., Zhang, X., et al. (2018). Comparative analysis of MicroRNA expression in dog lungs infected with the H3N2 and H5N1 canine influenza viruses. Microb. Pathog. 121, 252–261. doi: 10.1016/j.micpath.2018.05.015
Keywords: canine influenza, H3N2, H5N1, deep sequencing, total transcriptome
Citation: Tao P, Ning Z, Hao X, Lin X, Zheng Q and Li S (2019) Comparative Analysis of Whole-Transcriptome RNA Expression in MDCK Cells Infected With the H3N2 and H5N1 Canine Influenza Viruses. Front. Cell. Infect. Microbiol. 9:76. doi: 10.3389/fcimb.2019.00076
Received: 10 January 2019; Accepted: 05 March 2019;
Published: 26 March 2019.
Edited by:
Ricardo Martin Gomez, CONICET Institute of Biotechnology and Molecular Biology (IBBM), ArgentinaReviewed by:
Christian Nelson, SUNY Cortland, United StatesSoledad Negrotto, CONICET Instituto de Medicina Experimental, Academia Nacional de Medicina, Argentina
Copyright © 2019 Tao, Ning, Hao, Lin, Zheng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shoujun Li, c2hvdWp1bmxpQHNjYXUuZWR1LmNu