 Allison R. Kolbe
Allison R. Kolbe Eduardo Castro-Nallar
Eduardo Castro-Nallar Diego Preciado3
Diego Preciado3- 1Department of Biostatistics and Bioinformatics, Milken Institute School of Public Health, Computational Biology Institute, The George Washington University, Washington, DC, United States
- 2Facultad de Ciencias de la Vida, Center for Bioinformatics and Integrative Biology, Universidad Andrés Bello, Santiago, Chile
- 3Division of Pediatric Otolaryngology, Sheikh Zayed Institute, Children's National Health System, Washington, DC, United States
- 4CIBIO-InBIO, Centro de Investigação em Biodiversidade e Recursos Genéticos, Universidade Do Porto, Vairão, Portugal
Chronic otitis media with effusion (COME) is a common childhood disease characterized by an accumulation of fluid behind the eardrum. COME often requires surgical intervention and can also lead to significant hearing loss and subsequent learning disabilities. Recent characterization of the middle ear fluid (MEF) microbiome in pediatric patients has led to an improved understanding of the microbiota present in the middle ear during COME. However, it is not currently known how the MEF microbiome might vary due to other conditions, particularly respiratory disorders. Here, we apply an amplicon sequence variant (ASV) pipeline to MEF 16S rRNA high-throughput sequencing data from 50 children with COME (ages 3–176 months) undergoing tube placement. We achieve a more detailed taxonomic resolution than previously reported, including species and genus level resolution. Additionally, we provide the first report of the functional roles of the MEF microbiome and demonstrate that despite high taxonomic diversity, the functional capacity of the MEF microbiome remains uniform between patients. Furthermore, we analyze microbiome differences between children with COME with and without a history of lower airway disease (i.e., asthma or bronchiolitis). The MEF microbiome was less diverse in participants with lower airway disease than in patients without, and phylogenetic β-diversity (weighted UniFrac) was significantly different based on lower airway disease status. Differential abundance between patients with lower airway disease and those without was observed for the genera Haemophilus, Moraxella, Staphylococcus, Alloiococcus, and Turicella. These findings support previous suggestions of a link between COME and respiratory illnesses and emphasize the need for future study of the middle ear and respiratory tract microbiomes in diseases such as asthma and bronchiolitis.
Introduction
Chronic otitis media with effusion (COME) is characterized by an accumulation of fluid behind the eardrum which persists for 3 months or more, typically without signs of inflammation (Minovi and Dazert, 2014). COME is a leading cause of hearing loss in children, especially in developing countries (Monasta et al., 2012), and can result in learning disabilities and educational problems (Williams and Jacobs, 2009). Approximately 80% of children worldwide will have experienced an episode of COME by age 10 (Minovi and Dazert, 2014).
More than half of COME cases are preceded by acute otitis media (AOM), which are most commonly caused by bacterial infection with Streptococcus pneumoniae, Moraxella catarrhalis, and Haemophilus influenzae (Minovi and Dazert, 2014; Qureishi et al., 2014). However, the role of these pathogens or other components of the middle ear microbiome in COME is less well-understood. All three of these pathogens have been identified in middle ear fluid (MEF), as well as Alloiococcus otitis, Turicella otitidis, and Staphylococcus sp. (Harimaya et al., 2006; Guvenc et al., 2010; Jervis-Bardy et al., 2015; Chan et al., 2016; Lappan et al., 2018). Interestingly, several of these same species have been linked to the development of asthma and the asthma microbiome, particularly M. catarrhalis and H. influenzae (Bisgaard et al., 2007; Castro-Nallar et al., 2015; Pérez-Losada et al., 2015). However, the relationship between asthma and the middle ear microbiome, particularly in the context of COME, is not known.
Several studies have observed a relationship between otitis media (OM) and respiratory illnesses such as asthma and bronchiolitis. Bronchiolitis is a lung infection that most commonly affects young children at the same age as typical AOM incidence and is associated with airway inflammation and congestion. Asthma is a chronic condition characterized by airway inflammation and increased mucus production, and can result in shortness of breath, coughing, and wheezing, typically at an older age. Bacterial AOM is a common complication associated with bronchiolitis (Andrade et al., 1998; Gomaa et al., 2012). Similarly, large-scale studies in Germany and Mexico have observed that AOM episodes in infancy increases the risk of asthma development later in life (Eldeirawi et al., 2010; MacIntyre et al., 2010). Similar relationships have been observed between COME and asthma. A retrospective study of tube placement patients with COME found a significantly higher rate of asthma diagnosis at follow-up compared to children of the same age, as well as different clinical presentations of COME including mucoid effusion and a higher rate of multiple tube placements (Gamble et al., 1992). Interestingly, COME has also been associated with other atopic diseases such as allergic rhinitis (Alles et al., 2001; Luong and Roland, 2008; Kreiner-Moller et al., 2012). Although the directionality of this relationship is unclear, some have hypothesized that chronic inflammation due to asthma or other atopic diseases can reduce eustachian tube opening and impair mucociliary function (Alles et al., 2001; MacIntyre and Heinrich, 2012). Gamble et al. (1992) suggested that COME in asthmatics is not an isolated disease, but rather the result of atopic disease affecting the mucociliary system of the entire respiratory tract. While the link between bronchiolitis and COME has not been explored as extensively, it is possible that inflammation due to bronchiolitis could have a similar effect. At present, however, the nature of the relationship between COME and respiratory illnesses is poorly understood.
Microbiome dysbiosis has been well-characterized in the respiratory tract associated with asthma and bronchiolitis (Hilty et al., 2010; Huang et al., 2011; Marri et al., 2013; Castro-Nallar et al., 2015; Pérez-Losada et al., 2015; Hasegawa et al., 2016; Mansbach et al., 2016; Kozik and Huang, 2018), but it is not currently known whether this dysbiosis extends to the middle ear, particularly when associated with COME. In this study, we sought to characterize the MEF microbiome associated with COME in patients with and without asthma or bronchiolitis. To accomplish this goal, we re-analyzed the 16S rRNA high-throughput sequencing data previously published by Krueger et al. (2017), incorporating new metadata relating to lower airway disease diagnosis as well as analyzing the metabolic function of the microbial communities. Furthermore, we built upon the previous study by utilizing an amplicon sequence variant (ASV) approach which enhances taxonomic classification and reproducibility (Callahan et al., 2017). This study provides new insight into the MEF microbiome in COME, as well as MEF microbiome changes associated with asthma or bronchiolitis.
Materials and Methods
Sample collection and DNA sequencing methodology for this study were previously described (Krueger et al., 2017). Briefly, middle ear effusion samples were collected from 50 children with chronic otitis media undergoing myringotomy with tympanostomy tube placement at Children's National Health System in Washington, D.C. The cohort ranged from 3 to 176 months of age, with 34 boys and 16 girls. Out of the 50 children, nearly three-fourths suffered from significant hearing loss (36/50). None of these children were treated with antibiotics for 2 weeks prior to sampling. Other (non-antibiotic) medication use was not recorded for this study. Thirteen of the children were diagnosed with lower airway disease such as asthma or bronchiolitis. To fit the categorization of being positive for these, the children needed to meet any of the following criteria: (1) history of pulmonary physician-diagnosed asthma; (2) documented chronic wheezing being treated with a daily respiratory inhaler; or (3) PCR (+) for rhinovirus bronchiolitis diagnosis. Although asthma and bronchiolitis are considered different respiratory illnesses, there often is a spectrum of disease over time and they cannot be reliably distinguished in young children. Therefore, we evaluated them together as lower airway disease in this study.
DNA purification from MEF was performed using the QiaAmp mini kit (Qiagen) and extracted following the MiSeq SOP protocol described in Kozich et al. (2013). The V4 region of the 16S rRNA gene was amplified and libraries were sequenced using the Illumina MiSeq at University of Michigan. Negative controls did not amplify, indicating that bacterial DNA was not present in the reagents, and therefore were not sequenced.
Raw fastq files from Krueger et al. (2017) were re-processed using dada2 version 1.12 (Callahan et al., 2016). This pipeline offers improved taxonomic resolution and reproducibility compared to OTU-based methods (Callahan et al., 2017). Reads were filtered using standard parameters, with no uncalled bases, maximum of two expected errors, and truncating reads at a quality score of two or less. Forward and reverse reads were truncated after 240 and 225 bases, respectively. The standard dada2 pipeline was then applied to perform amplicon sequence variant (ASV) inference, merge paired reads, and identify chimeras. Singleton ASVs are discarded in the dada2 pipeline (Callahan et al., 2017). Taxonomic assignment was performed against the Silva v132 database (Quast et al., 2013) using the dada2-formatted training files for taxonomy and species-level assignment (Callahan, 2018). ASV sequences were aligned using MAFFT (Katoh and Standley, 2013) and used to build a tree with FastTree (Price et al., 2010). The resulting ASV tables and phylogenetic tree were imported into phyloseq (McMurdie and Holmes, 2013) for further analysis.
Functional analysis of microbial communities was performed with PICRUSt2 (Douglas et al., 2019), following the standard pipeline. First, ASVs were aligned to reference sequences using HHMER (Howard Hughes Medical Institute, 2018) and placed into a reference tree with EPA-NG (Barbera et al., 2019) and GAPPA (Czech and Stamatakis, 2019). Hidden-state prediction was performed using castor (Louca and Doebeli, 2018), and along with the ASV abundance table generated by dada2, used to generate metagenome predictions. Finally, KEGG pathway level predictions were performed with MinPath (Ye and Doak, 2009). Exploratory analysis of functional abundances was performed in STAMP (Parks et al., 2014), and visualized with BURRITO (McNally et al., 2018). For visualization purposes, ASVs with <0.1% relative abundance were removed. To test for significant associations between functional profiles and clinical variables, general linear models implemented using MaAsLin, with significance considered at q < 0.25 as recommended by the authors (Morgan et al., 2012).
Alpha diversity indices (ASV richness and Shannon diversity) were calculated on raw counts using the estimate_richness() function in phyloseq and plotted with ggplot2 (Wickham, 2016). Significant differences based on asthma/bronchiolitis status were identified using linear models with clinical metadata (Muc5B +/−, Muc5AC +/−, significant hearing loss +/−, gender, over/under 24 months of age, mucoid/serous fluid) from Krueger et al. (2017) as covariables.
Prior to calculating beta diversity, read counts were normalized for with DESeq2 (Love et al., 2014), using the modified geometric mean (“poscounts”) as implemented in the function estimateSizeFactors(). Beta diversity was calculated using the weighted and unweighted UniFrac distances implemented in the phyloseq package. Significance was determined by PERMANOVA using the R package vegan (Oksanen et al., 2019) with clinical metadata as covariables.
Differential abundance testing was performed with DESeq2 (Love et al., 2014) on normalized data as described before (McMurdie and Holmes, 2013). Using the negative binomial model implemented in DESeq2 accounts for library size differences and biological variability more appropriately than rarefying or using simple proportions (McMurdie and Holmes, 2013). The same variables and covariables listed above were used in the model formula. Significance was determined at α < 0.05. All analyses were performed in R version 3.6 (R Core Team, 2019).
Results
Taxonomic Composition of the Ear Microbiome During Chronic Infection
On average, 98% of sequences were assigned at the genus level (Figure 1), compared to ~84% in Krueger et al. (2017). These included two previously unidentified genera, Achromobacter and Pseudoflavitalea. Furthermore, species level resolution was achieved for some ASVs. These included Turicella otitidis (6.9 ± 2.2%), Alloiococcus otitis (6.0 ± 1.7%), and Stenotrophomonas maltophilia (4.6 ± 1.4%), as well as several low abundance (<1%) ASVs (Supplementary Table 1).
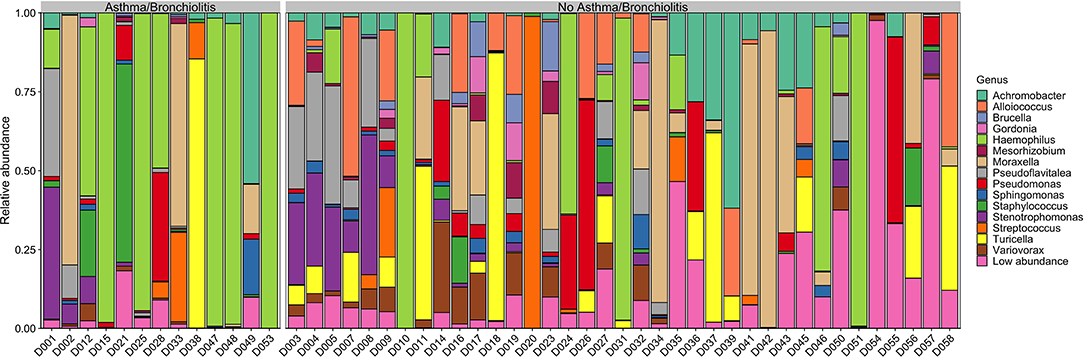
Figure 1. Genus-level relative abundance in patients with or without asthma/bronchiolitis. Genera with mean relative abundance <1% across all samples are plotted as “Low abundance”.
The taxonomic composition of the middle ear effusion is quite diverse, with no core microbiome. Although Haemophilus, Moraxella, and Turicella were the largest genera by mean relative abundance, these genera were only present in approximately half of the studied samples (27/50, 26/50, and 26/50, respectively). Achromobacter and Pseudomonas were the most prevalent genera, present in 39/50 and 38/50 samples, respectively (Supplementary Table 2).
Metabolic Function of the Ear Microbiome During Chronic Infection
The functional roles of the middle ear microbiome were primarily classified into four groups: cellular processes (4.7 ± 0.2%), environmental information processing (20.4 ± 0.3%), genetic information processing (24.8 ± 0.5%), and metabolism (33.8 ± 0.3%). Approximately 16% were unclassified. The sub-pathways with the largest relative abundance were Membrane Transport (19.4 ± 0.3%), Translation (13.4 ± 0.4%), Amino acid metabolism (7.6 ± 0.2%), and Carbohydrate Metabolism (6.3 ± 0.1%). Functional profiles were similar across all patients (Figure 2). No significant associations were identified between the functional profiles and clinical variables using MaAsLin or principal component analysis.
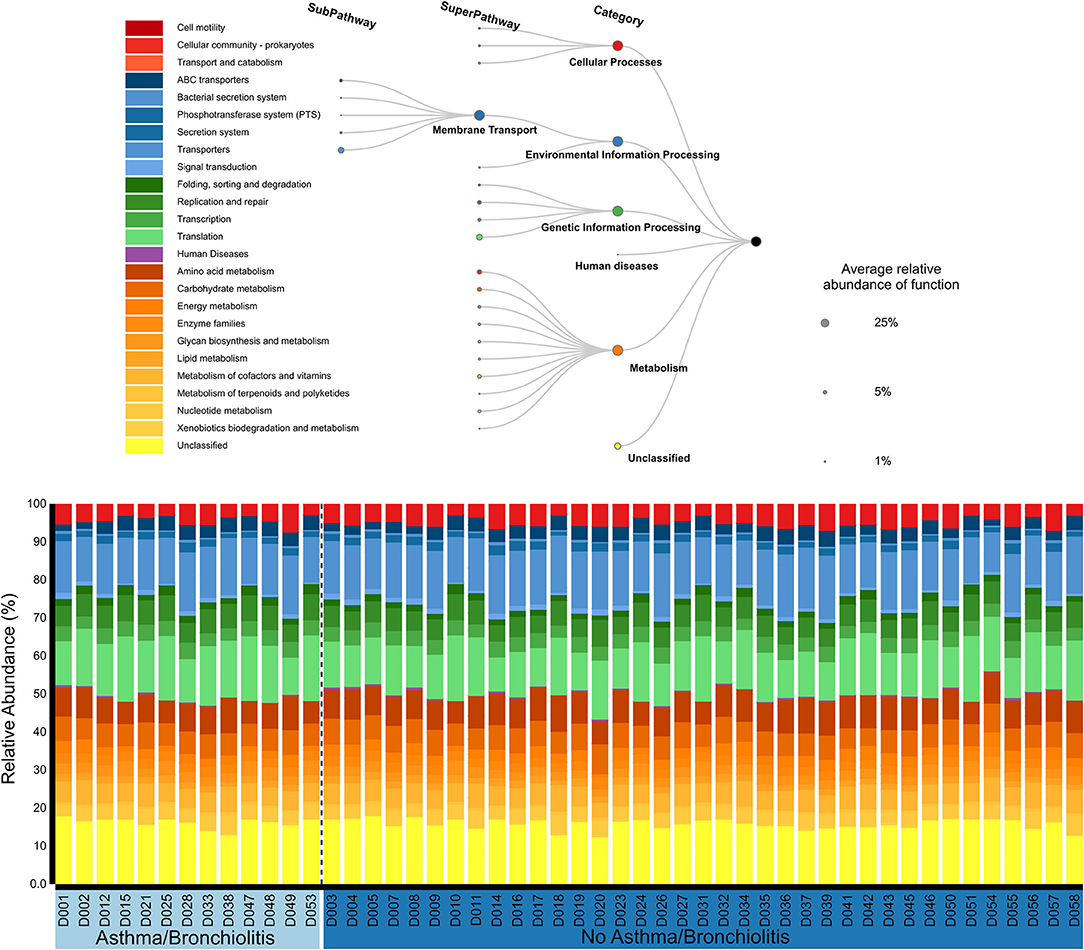
Figure 2. Relative abundances of KEGG functional pathways classified by PICRUSt2 (Douglas et al., 2019) and visualized with BURRITO (McNally et al., 2018).
Variation of Ear Microbiome Composition and Function During Chronic Infection
The α-diversity of the middle ear microbiome estimated by ASV richness and Shannon diversity indices was significantly lower in patients with asthma or bronchiolitis than in patients without (Figure 3; p < 0.05). Mean ASV richness in patients with asthma/bronchiolitis was 16.4 compared to 31.1 in patients without; similarly, mean Shannon diversity in patients with asthma/bronchiolitis was 0.96 compared to 1.72 in patients without. α-diversity did not vary significantly between other clinical variables, including age, gender, significant hearing loss, mucoid/serous effusion, and presence of Muc5B and/or Muc5AC.
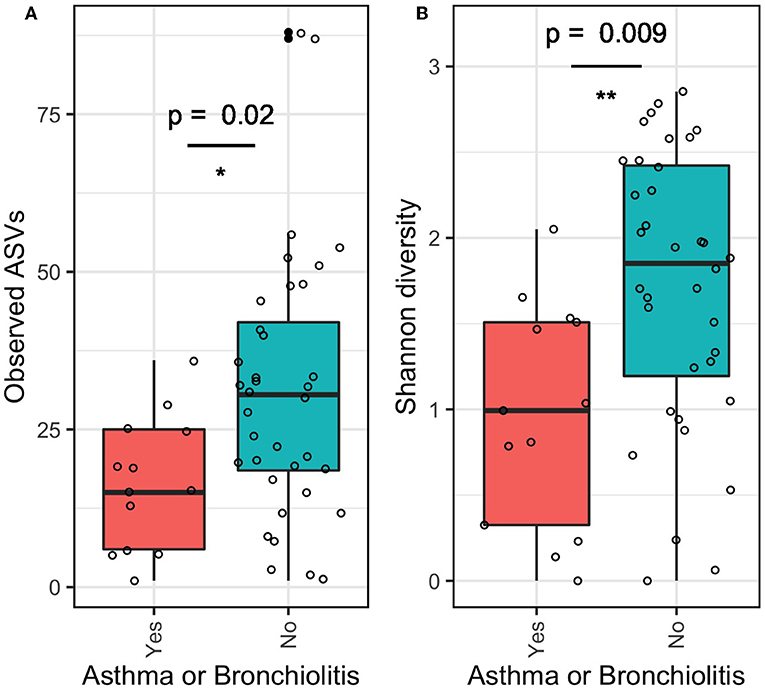
Figure 3. α-diversity of middle ear fluid in patients with asthma or bronchiolitis. Estimates of richness (A; number of observed ASVs) and evenness (B; Shannon diversity) were significantly lower in patients with asthma or bronchiolitis. *p < 0.05, **p < 0.01.
In contrast, principle coordinate analysis with weighted UniFrac distances and analysis with PERMANOVA indicated significant differences in β-diversity between patients with/without significant hearing loss and asthma/bronchiolitis status (Figure 4A). These variables were not significant in principle coordinate analysis with unweighted UniFrac distances (Figure 4B).
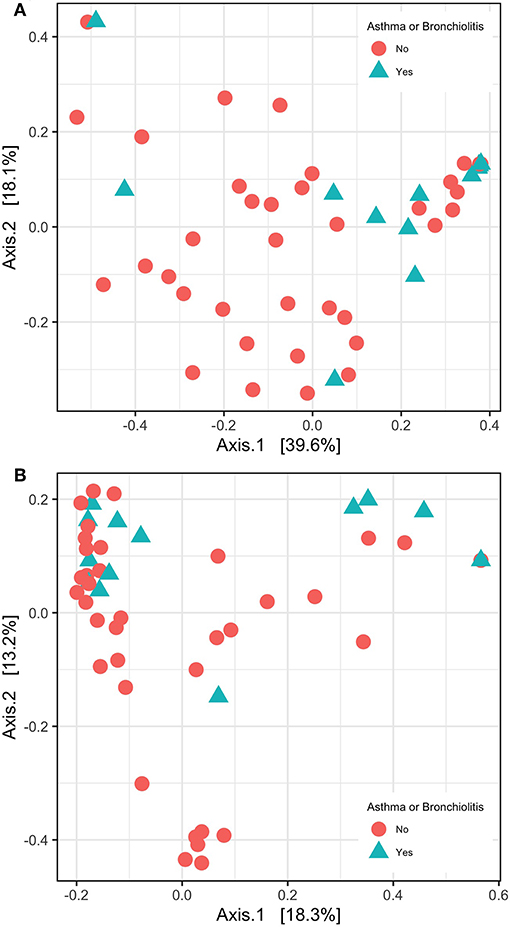
Figure 4. β-diversity shown with principle coordinate analysis of weighted UniFrac distances (A) and unweighted UniFrac distance (B).
Differential abundance testing with DESeq2 identified several ASVs that varied significantly in the relative mean proportions with respect to asthma/bronchiolitis status, after accounting for variation in other clinical and demographic variables. Haemophilus, Staphylococcus, and Moraxella were significantly higher in children with asthma or bronchiolitis, whereas Turicella and Alloiococcus were significantly lower (Figure 5).
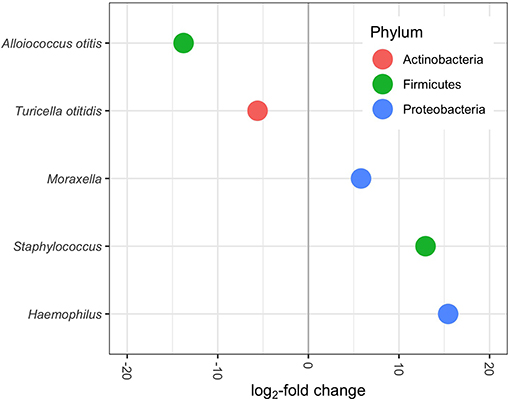
Figure 5. Differentially abundant genera/species (p-adjusted < 0.05) in MEF of patients with lower airway disease diagnosis. Positive log2-fold change indicates increased abundance in patients with asthma or bronchiolitis; negative log2-fold change indicates decreased abundance. Each dot represents one ASV.
Discussion
Chronic otitis media with effusion (COME) is the leading cause of hearing loss among children and affects as many as 80% of children by age 10 (Minovi and Dazert, 2014). Here, we present novel insights from the 16S data presented in Krueger et al. (2017), using amplicon sequence variants (ASVs) instead of OTUs, as well as evaluating links to diagnosed lower airway disease.
Using exact sequence variants instead of OTUs allows for greater precision and reproducibility in taxonomic assignment (Callahan et al., 2017, 2019; Edgar, 2018; Knight et al., 2018; Xue et al., 2018). Previous OTU-based approaches typically functioned by clustering sequences based on a 97% similarity threshold, and then assigning these clusters to reference tree-based OTUs. These approaches did not incorporate sequence quality information or statistical information about the reads into taxonomic assignments, and were therefore losing valuable information (Callahan et al., 2017). Using exact sequence variants improves estimations of diversity and taxonomic predictions, especially for communities which have not been studied extensively (Callahan et al., 2017; Caruso et al., 2019). With the approach implemented in dada2 (Callahan et al., 2016), we were able to achieve species level classifications for multiple ASVs, three of which were present at a mean relative abundance >1%. Of these, Alloiococcus otitis and Turicella otiditis have been previously identified as typical components of the OM microbiome (Tano et al., 2008; von Graevenitz and Funke, 2014; Lappan et al., 2018). In contrast, Stenotrophomonas maltophilia has not been described in many middle ear microbiomes, despite being identified as the source of numerous human diseases including respiratory infections (Brooke, 2012). However, recent work by Kalcioglu et al. (2018) identified S. maltophilia in tympanosclerotic plaques isolated from an individual undergoing surgery due to COME. Therefore, S. maltophilia may be found in a subset of COME patients, particularly those who have developed tympanosclerosis. Interestingly, S. maltophilia was not found in cholesteatomas in the same study, indicating it may serve a specialized role (Kalcioglu et al., 2018).
Furthermore, OTUs that had been previously classified at the family level only were able to be defined at the genus level now using dada2. These were Achromobacter (relative abundance: 6.6%, family: Alcaligenaceae) and Pseudoflavitalea (relative abundance: 5.3%, family: Chitinophagaceae). Pseudoflavitalea is a newly described genus that currently has only been isolated from soil samples (Kim et al., 2016); its potential role in human disease is unknown. On the other hand, Achromobacter xylosoxidans was initially isolated from ear discharge from OM patients (Yabuuchi and Ohyama, 1971) and was recently identified in cholesteatomas of multiple individuals with COME (Kalcioglu et al., 2018). Members of the Achromobacter genus, specifically Achromobacter xylosidans, have been shown to be opportunistic respiratory pathogens, particularly in patients with cystic fibrosis or immune deficiencies (Swenson and Sadikot, 2015). Given that Achromobacter was the most prevalent genus identified (present in 39/50 samples), with a mean relative abundance of 6.6%, further work on the role of Achromobacter in COME is warranted.
The high inter-patient variation and lack of core microbiome indicates that there is no typical middle ear microbiome associated with COME (Figure 1). In contrast to previous reports that found the middle ear microbiome to be dominated by a single genus, typically from Alloicoccus, Moraxella, or Haemophilus (Guvenc et al., 2010; Jervis-Bardy et al., 2015), the middle ear microbiome in this study was highly variable, generally compromising at least two high-abundance genera. This difference may be due to the finer resolution of the ASV pipeline implemented in dada2, and the larger quantity of data retained by not rarefying the counts. Furthermore, even the “typical” agents associated with AOM—namely, Haemophilus, Moraxella, and Streptococcus—were absent in ~50% of the samples from our COME patients. These results highlight the complexity of the middle ear microbiome associated with COME, which is perhaps expected given that a multitude of risk factors and potential etiologies have been identified for COME.
To our knowledge, we present here the first characterization of microbiome function in the middle ear. Although taxonomic composition varied dramatically between patients, microbiome function was remarkably similar across all patients (Figure 2). This supports the idea that microbial community functions often converge despite different taxonomic compositions (Human Microbiome Project, 2012; Louca et al., 2018). These will include core functions that are essential for microbial life, such as translation (Human Microbiome Project, 2012), which was highly abundant in our analysis. Interestingly, one of the top functions of the middle ear microbial community was “Membrane Transport,” which includes a wide range of genes such as transporters, the phosphotransferase system, and the bacterial secretion system. In our case, the majority of genes were subcategorized in the general category “Transporters.” In previous human microbiome studies, membrane transport has been identified as an abundant component of the healthy laryngeal microbiome (Jette et al., 2016) and associated with gut microbiome dysbiosis in irritable bowel disease and obesity (Greenblum et al., 2012). Further work is necessary to understand if these functions represent unique roles of the microbial community during COME, or if overlapping functions are present in AOM and healthy MEF microbiome.
Microbial diversity varied significantly between COME patients with and without asthma/bronchiolitis (Figures 3–5). Measures of α-diversity indicated that both richness and evenness were lower in COME patients with asthma or bronchiolitis. Although α-diversity of the lower respiratory tract has been shown to be higher (Huang et al., 2011; Marri et al., 2013) or unchanged (Goleva et al., 2013) in asthma patients, Castro-Nallar et al. (2015) showed decreased α-diversity in the nasal cavities of asthma patients compared to healthy patients. Decreased α-diversity has also been observed in microbiome profiles associated with increased risk of bronchiolitis (Hasegawa et al., 2017). Therefore, it is possible that the upper and lower respiratory tracts exhibit distinct trends in asthma patients, with decreased α-diversity in the upper respiratory tract extending to the middle ear and increased α-diversity in the lower respiratory tract. This would lend credence to the theory of a unified airway, which suggests that upper and lower airway disease share a pathophysiologic origin, being induced by a commonality in allergic or non-allergic reproducible mechanisms (Giavina-Bianchi et al., 2016). To this point, it is noteworthy that in patients with an allergic asthmatic etiology, a parallel type of allergic inflammatory response is noted in the middle ear (Nguyen et al., 2004). Moreover, patients with asthma are well-known to present with comorbid conditions which contribute to respiratory symptoms, including allergic rhinitis and chronic rhinosinusitis. Epidemiological and pathophysiological links have been described between these conditions (Massoth et al., 2019). This study's microbiome data builds on this point, suggesting an association between asthma and otitis media. However, due to potential bias between different methodologies, further work using paired samples would be necessary to confirm this hypothesis.
Using the weighted UniFrac distance method, we observed significant differences in β-diversity based on two clinical variables: significant hearing loss and asthma/bronchiolitis diagnosis (Figure 4). The association with significant hearing loss was previously observed in Krueger et al. (2017) and was also confirmed here with the present methods; however, the relationship with lower airway disease status was not previously tested. In contrast to other β-diversity methods, UniFrac distances take into account the phylogenetic distance between ASVs. The weighted UniFrac distance additionally considers the abundance of ASVs, whereas the unweighted UniFrac only considers presence/absence (Lozupone et al., 2007). Interestingly, the unweighted UniFrac distance was not significant for any of these three variables. This indicates that similar patterns of presence/absence were evident but ASV abundance varied between patients with asthma/bronchiolitis and significant hearing loss.
Differential abundance analysis with DESeq2 (Love et al., 2014) indicated that four of the five most abundant genera, Haemophilus, Turicella, Alloiococcus, and Moraxella, were differentially abundant between patients with and without asthma/bronchiolitis (Figure 5). Staphylococcus was also significantly higher in patients with lower airway disease. These results highlight a potentially significant difference in the microbiome of COME patients with asthma or bronchiolitis. Both Haemophilus and Moraxella have been studied extensively in the asthma and bronchiolitis-related microbiome (Hilty et al., 2010; Vissing et al., 2013; Castro-Nallar et al., 2015; Pérez-Losada et al., 2015, 2017, 2018; Hasegawa et al., 2016; Mansbach et al., 2016); therefore, it is particularly interesting that these genera were found in greater abundance in asthma patients with COME. Early airway colonization with Haemophilus and Moraxella has been associated with asthma or bronchiolitis development later in childhood (Bisgaard et al., 2007; Vissing et al., 2013), and both have been found in higher abundance in patients with asthma or bronchiolitis (Hilty et al., 2010; Vissing et al., 2013; Castro-Nallar et al., 2015). Staphylococcus-dominated airway microbiomes has also been associated with an increased risk of bronchiolitis (Hasegawa et al., 2017). In contrast, Turicella otiditis and Alloiococcus otitis have been isolated almost exclusively from the human ear and have frequently been attributed to OM (Tano et al., 2008; von Graevenitz and Funke, 2014; Boers et al., 2018). The absence of these species from other parts of the upper respiratory tract has led to a debate about whether they are OM pathogens or part of commensal microbiota. However, it is striking that pathogens associated with asthma and bronchiolitis are increased in COME with lower airway disease, whereas pathogens typically associated with OM are decreased. These results suggest a potential link between the microbiome of the respiratory tract in children with lower airway disease and the microbiome associated with COME. Alternatively, the different microbial communities in patients with comorbid respiratory illnesses could be reflective of disease severity and increased pathogen load in the middle ear. In this study, significant hearing loss served as a proxy for symptom severity and was included in models for differential abundance in DESeq2. It is also possible that medications commonly prescribed for patients with lower airway disease such as corticosteroids could influence the microbiota present in both the lower and upper airways. Denner et al. (2016) showed that corticoid steroid use affects microbiome composition in the lower airways; however, it is not currently known whether this effect would extend to the upper airways. Future work should consider additional metrics of disease severity as well as medication use when evaluating this potential link between the middle ear and respiratory illnesses.
A large body of work suggests a link between atopic disease and OM, and particularly COME. These associated atopic diseases include allergic rhinitis (Alles et al., 2001; Kreiner-Moller et al., 2012; Roditi et al., 2016), eczema (MacIntyre et al., 2010), and asthma (Gamble et al., 1992; Eldeirawi et al., 2010; MacIntyre et al., 2010; Bjur et al., 2012), but the same associations are not always reproduced between studies, leading to significant debate about the nature of these associations (Zernotti et al., 2017). Gamble et al. (1992) hypothesized that COME in asthmatics is not an isolated disease, but instead represents an atopic disease affecting the mucociliary system of entire respiratory tract. Similarly, inflammation from bronchiolitis could result in impaired mucociliary clearance and make the middle ear more susceptible to pathogens. The interesting decrease in specific ear-trophic OM pathogens (Alloiococcus and Turicella) accompanied by an increase in asthma- and bronchiolitis-associated pathogens (Haemophilus, Moraxella, Staphylococcus) in asthma and bronchiolitis patients supports the theory that respiratory disease-associated COME may have a distinct etiology than COME in children without respiratory illnesses (Gamble et al., 1992; Zernotti et al., 2017). Although the directionality of this relationship is unknown, our findings show that COME in children with lower airway disease is characterized by a distinct microbiome, which may also explain the higher frequency of COME complications such as mucoid effusion and COME recurrence observed in asthma patients (Gamble et al., 1992). The relationship between these diseases and their respective microbiomes should be further investigated in future work.
Conclusions
In this study, we performed a detailed characterization of the composition and function of the middle ear microbiome in children with COME. Using an ASV pipeline, we achieved greater resolution at the species and genus levels than using OTUs. Taxonomic profiles varied significantly between children, while microbiome function was remarkably similar across all patients. Furthermore, we identified significant differences in the middle ear microbiome associated with a lower airway disease diagnosis, including α- and β-diversity and relative abundance of Haemophilus, Moraxella, Staphylococcus, Alloiococcus, and Turicella. These results provide novel evidence linking the microbiome of respiratory illnesses with COME and warrant additional work on the link between these important childhood diseases.
Data Availability Statement
Raw data files are available in the NCBI SRA under accession PRJNA555884.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board, Children's National Health System. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
AK, EC-N, DP, and MP-L conceived and designed this study. AK analyzed the data and wrote the manuscript with contributions of all the authors. All authors read and approved this manuscript.
Funding
This study was partially funded the Milken Institute School of Public Health Pilot Fund Program, the Margaret Q. Landenberger Research Foundation and the Fundação para a Ciência e a Tegnologia (T495756868-00032862).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00339/full#supplementary-material
Abbreviations
AOM, acute otitis media; ASV, amplicon sequence variant; COME, chronic otitis media with effusion; OM, otitis media.
References
Alles, R., Parikh, A., Hawk, L., Darby, Y., Romer, J. N., and Scadding, G. (2001). The prevalence of atopic disorders in children with chronic otitis media with effusion. Pediatr. Allergy Immunol. 12, 102–106. doi: 10.1046/j.0905-6157.2000.00008.x
Andrade, M. A., Hoberman, A., Glustein, J., Paradise, J. L., and Wald, E. R. (1998). Acute otitis media in children with bronchiolitis. Pedatrics. 101, 617–619. doi: 10.1542/peds.101.4.617
Barbera, P., Kozlov, A., Czech, L., Morel, B., Darriba, D., Flouri, T., et al. (2019). EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst. Biol. 68, 365–369. doi: 10.1093/sysbio/syy054
Bisgaard, H., Northman Hermansen, M., Buchvald, F., Loland, L., Brydensholt Halkjaer, L., Bonnelykke, K., et al. (2007). Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 357, 1487–1495. doi: 10.1056/NEJMoa052632
Bjur, K. A., Lynch, R. L., Fenta, Y. A., Yoo, K. H., Jacobson, R. M., Li, X., et al. (2012). Assessment of the association between atopic conditions and tympanostomy tube placement in children. Allergy Asthma Proc. 33, 289–296. doi: 10.2500/aap.2012.33.3529
Boers, S. A., de Zeeuw, M., Jansen, R., van der Schroeff, M. P., van Rossum, A. M. C., Hays, J. P., et al. (2018). Characterization of the nasopharyngeal and middle ear microbiota in gastroesophageal reflux-prone versus gastroesophageal reflux non-prone children. Eur. J. Clin. Microbiol. Infect. Dis. 37, 851–857. doi: 10.1007/s10096-017-3178-2
Brooke, J. S. (2012). Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin. Microbiol. Rev. 25, 2–41. doi: 10.1128/CMR.00019-11
Callahan, B. J. (2018). Silva Taxonomic Training Data Formatted for DADA2. Silva version 132. doi: 10.5281/zenodo.1172783
Callahan, B. J., McMurdie, P. J., and Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643. doi: 10.1038/ismej.2017.119
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Callahan, B. J., Wong, J., Heiner, C., Oh, S., Theriot, C. M., Gulati, A. S., et al. (2019). High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 47:e103. doi: 10.1093/nar/gkz569
Caruso, V., Song, X., Asquith, M., and Karstens, L. (2019). Performance of microbiome sequence inference methods in environments with varying biomass. mSystems 4:e00163–18. doi: 10.1128/mSystems.00163-18
Castro-Nallar, E., Shen, Y., Freishtat, R. J., Perez-Losada, M., Manimaran, S., Liu, G., et al. (2015). Integrating microbial and host transcriptomics to characterize asthma-associated microbial communities. BMC Med Genomics 8:50. doi: 10.1186/s12920-015-0121-1
Chan, C. L., Wabnitz, D., Bardy, J. J., Bassiouni, A., Wormald, P. J., Vreugde, S., et al. (2016). The microbiome of otitis media with effusion. Laryngoscope 126, 2844–2851. doi: 10.1002/lary.26128
Czech, L., and Stamatakis, A. (2019). Scalable methods for analyzing and visualizing phylogenetic placement of metagenomic samples. PLoS ONE 14:e0217050. doi: 10.1371/journal.pone.0217050
Denner, D. R., Sangwan, N., Becker, J. B., Hogarth, D. K., Oldham, J., Castillo, J., et al. (2016). Corticosteroid therapy and airflow obstruction influence the bronchial microbiome, which is distinct from that of bronchoalveolar lavage in asthmatic airways. J. Allergy Clin. Immunol. 137, 1398–1405 e1393. doi: 10.1016/j.jaci.2015.10.017
Douglas, G. M., Maffei, V. J., Zaneveld, J., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2019). PICRUSt2: an improved and extensible approach for metagenome inference. bioRxiv. doi: 10.1101/672295
Edgar, R. C. (2018). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 34, 2371–2375. doi: 10.1093/bioinformatics/bty113
Eldeirawi, K., McConnell, R., Furner, S., Freels, S., Stayner, L., Hernandez, E., et al. (2010). Frequent ear infections in infancy and the risk of asthma in Mexican American children. J. Asthma 47, 473–477. doi: 10.3109/02770901003759428
Gamble, J. E., Bizal, J. A., and Daetwyler, E. P. (1992). Otitis media and chronic middle ear effusion in the asthmatic pediatric patient. Ear Nose Throat J. 71, 397–399. doi: 10.1177/014556139207100908
Giavina-Bianchi, P., Aun, M. V., Takejima, P., Kalil, J., and Agondi, R. C. (2016). United airway disease: current perspectives. J. Asthma Allergy 9, 93–100. doi: 10.2147/JAA.S81541
Goleva, E., Jackson, L. P., Harris, J. K., Robertson, C. E., Sutherland, E. R., Hall, C. F., et al. (2013). The effects of airway microbiome on corticosteroid responsiveness in asthma. Am. J. Respir. Crit. Care Med. 188, 1193–1201. doi: 10.1164/rccm.201304-0775OC
Gomaa, M. A., Galal, O., and Mahmoud, M. S. (2012). Risk of acute otitis media in relation to acute bronchiolitis in children. Int. J. Pediatr. Otorhinolaryngol. 76, 49–51. doi: 10.1016/j.ijporl.2011.09.029
Greenblum, S., Turnbaugh, P. J., and Borenstein, E. (2012). Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. U.S.A. 109, 594–599. doi: 10.1073/pnas.1116053109
Guvenc, M. G., Midilli, K., Inci, E., Kuskucu, M., Tahamiler, R., Ozergil, E., et al. (2010). Lack of Chlamydophila pneumoniae and predominance of Alloiococcus otitidis in middle ear fluids of children with otitis media with effusion. Auris Nasus Larynx 37, 269–273. doi: 10.1016/j.anl.2009.09.002
Harimaya, A., Takada, R., Hendolin, P. H., Fujii, N., Ylikoski, J., and Himi, T. (2006). High incidence of Alloiococcus otitidis in children with otitis media, despite treatment with antibiotics. J. Clin. Microbiol. 44, 946–949. doi: 10.1128/JCM.44.3.946-949.2006
Hasegawa, K., Linnemann, R. W., Mansbach, J. M., Ajami, N. J., Espinola, J. A., Petrosino, J. F., et al. (2017). Nasal airway microbiota profile and severe bronchiolitis in infants: a case-control study. Pediatr. Infect. Dis. J. 36, 1044–1051. doi: 10.1097/INF.0000000000001500
Hasegawa, K., Mansbach, J. M., Ajami, N. J., Espinola, J. A., Henke, D. M., Petrosino, J. F., et al. (2016). Association of nasopharyngeal microbiota profiles with bronchiolitis severity in infants hospitalised for bronchiolitis. Eur. Respir. J. 48, 1329–1339. doi: 10.1183/13993003.00152-2016
Hilty, M., Burke, C., Pedro, H., Cardenas, P., Bush, A., Bossley, C., et al. (2010). Disordered microbial communities in asthmatic airways. PLoS ONE 5:e8578. doi: 10.1371/journal.pone.0008578
Howard Hughes Medical Institute (2018). HMMER 3.2.1. Available online at: www.hmmer.org (accessed July 8, 2019).
Huang, Y. J., Nelson, C. E., Brodie, E. L., Desantis, T. Z., Baek, M. S., Liu, J., et al. (2011). Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J. Allergy Clin. Immunol. 127, 372–381 e371–373. doi: 10.1016/j.jaci.2010.10.048
Human Microbiome Project, C. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Jervis-Bardy, J., Rogers, G. B., Morris, P. S., Smith-Vaughan, H. C., Nosworthy, E., Leong, L. E., et al. (2015). The microbiome of otitis media with effusion in Indigenous Australian children. Int. J. Pediatr. Otorhinolaryngol. 79, 1548–1555. doi: 10.1016/j.ijporl.2015.07.013
Jette, M. E., Dill-McFarland, K. A., Hanshew, A. S., Suen, G., and Thibeault, S. L. (2016). The human laryngeal microbiome: effects of cigarette smoke and reflux. Sci. Rep. 6:35882. doi: 10.1038/srep35882
Kalcioglu, M. T., Guldemir, D., Unaldi, O., Egilmez, O. K., Celebi, B., and Durmaz, R. (2018). Metagenomics analysis of bacterial population of tympanosclerotic plaques and cholesteatomas. Otolaryngol. Head Neck Surg. 159, 724–732. doi: 10.1177/0194599818772039
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, S. J., Cho, H., Ahn, J. H., Weon, H. Y., Seok, S. J., Kim, J. S., et al. (2016). Pseudoflavitalea rhizosphaerae gen. nov., sp. nov., isolated from rhizosphere of tomato, and proposal to reclassify Flavitalea soli as Pseudoflavitalea soli comb. nov. Int. J. Syst. Evol. Microbiol. 66, 4167–4171. doi: 10.1099/ijsem.0.001330
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422. doi: 10.1038/s41579-018-0029-9
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Kozik, A. J., and Huang, Y. J. (2018). The microbiome in asthma: role in pathogenesis, phenotype, and response to treatment. Ann. Allergy Asthma Immunol. 122, 270–275. doi: 10.1016/j.anai.2018.12.005.
Kreiner-Moller, E., Chawes, B. L., Caye-Thomasen, P., Bonnelykke, K., and Bisgaard, H. (2012). Allergic rhinitis is associated with otitis media with effusion: a birth cohort study. Clin. Exp. Allergy 42, 1615–1620. doi: 10.1111/j.1365-2222.2012.04038.x
Krueger, A., Val, S., Perez-Losada, M., Panchapakesan, K., Devaney, J., Duah, V., et al. (2017). Relationship of the middle ear effusion microbiome to secretory mucin production in pediatric patients with chronic otitis media. Pediatr. Infect. Dis. J. 36, 635–640. doi: 10.1097/INF.0000000000001493
Lappan, R., Imbrogno, K., Sikazwe, C., Anderson, D., Mok, D., Coates, H., et al. (2018). A microbiome case-control study of recurrent acute otitis media identified potentially protective bacterial genera. BMC Microbiol. 18:13. doi: 10.1186/s12866-018-1154-3
Louca, S., and Doebeli, M. (2018). Efficient comparative phylogenetics on large trees. Bioinformatics 34, 1053–1055. doi: 10.1093/bioinformatics/btx701
Louca, S., Polz, M. F., Mazel, F., Albright, M. B. N., Huber, J. A., O'Connor, M. I., et al. (2018). Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2, 936–943. doi: 10.1038/s41559-018-0519-1
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Luong, A., and Roland, P. S. (2008). The link between allergic rhinitis and chronic otitis media with effusion in atopic patients. Otolaryngol. Clin. North. Am. 41, 311–323, vi. doi: 10.1016/j.otc.2007.11.004
MacIntyre, E. A., Chen, C.-M., Herbarth, O., Borte, M., Schaaf, B., Krämer, U., et al. (2010). Early-life otitis media and incident atopic disease at school age in a birth Cohort. Pediatr. Infect. Dis. J. 29, e96–e99. doi: 10.1097/INF.0b013e3181fcd9e8
MacIntyre, E. A., and Heinrich, J. (2012). Otitis media in infancy and the development of asthma and atopic disease. Curr. Allergy Asthma Rep. 12, 547–550. doi: 10.1007/s11882-012-0308-x
Mansbach, J. M., Hasegawa, K., Henke, D. M., Ajami, N. J., Petrosino, J. F., Shaw, C. A., et al. (2016). Respiratory syncytial virus and rhinovirus severe bronchiolitis are associated with distinct nasopharyngeal microbiota. J. Allergy Clin. Immunol. 137, 1909–1913 e1904. doi: 10.1016/j.jaci.2016.01.036
Marri, P. R., Stern, D. A., Wright, A. L., Billheimer, D., and Martinez, F. D. (2013). Asthma-associated differences in microbial composition of induced sputum. J. Allergy Clin. Immunol. 131, 346-352 e341–343. doi: 10.1016/j.jaci.2012.11.013
Massoth, L., Anderson, C., and McKinney, K. A. (2019). Asthma and chronic rhinosinusitis: diagnosis and medical management. Med. Sci. 7:E53. doi: 10.3390/medsci7040053
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
McNally, C. P., Eng, A., Noecker, C., Gagne-Maynard, W. C., and Borenstein, E. (2018). BURRITO: an interactive multi-omic tool for visualizing taxa-function relationships in microbiome data. Front. Microbiol. 9:365. doi: 10.3389/fmicb.2018.00365
Minovi, A., and Dazert, S. (2014). Diseases of the middle ear in childhood. GMS Curr. Top. Otorhinolaryngol. Head Neck Surg. 13:Doc11. doi: 10.3205/cto000114
Monasta, L., Ronfani, L., Marchetti, F., Montico, M., Brumatti, L., Bavcar, A., et al. (2012). Burden of disease caused by otitis media: systematic review and global estimates. PLoS ONE 7:e36226. doi: 10.1371/journal.pone.0036226
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., Devaney, K. L., Ward, D. V., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. doi: 10.1186/gb-2012-13-9-r79
Nguyen, L. H., Manoukian, J. J., Sobol, S. E., Tewfik, T. L., Mazer, B. D., Schloss, M. D., et al. (2004). Similar allergic inflammation in the middle ear and the upper airway: evidence linking otitis media with effusion to the united airways concept. J. Allergy Clin. Immunol. 114, 1110–1115. doi: 10.1016/j.jaci.2004.07.061
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). “vegan: Community Ecology Package”. R Package Version 2.5–5. Available online at: https://CRAN.R-project.org/package=vegan
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Pérez-Losada, M., Alamri, L., Crandall, K. A., and Freishtat, R. J. (2017). Nasopharyngeal microbiome diversity changes over time in children with asthma. PLoS ONE 12:e0170543. doi: 10.1371/journal.pone.0170543
Pérez-Losada, M., Authelet, K. J., Hoptay, C. E., Kwak, C., Crandall, K. A., and Freishtat, R. J. (2018). Pediatric asthma comprises different phenotypic clusters with unique nasal microbiotas. Microbiome 6:179. doi: 10.1186/s40168-018-0564-7
Pérez-Losada, M., Castro-Nallar, E., Bendall, M. L., Freishtat, R. J., and Crandall, K. A. (2015). Dual transcriptomic profiling of host and microbiota during health and disease in pediatric asthma. PLoS ONE 10:e0131819. doi: 10.1371/journal.pone.0131819
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–596. doi: 10.1093/nar/gks1219
Qureishi, A., Lee, Y., Belfield, K., Birchall, J. P., and Daniel, M. (2014). Update on otitis media - prevention and treatment. Infect. Drug Resist. 7, 15–24. doi: 10.2147/IDR.S39637
Roditi, R. E., Veling, M., and Shin, J. J. (2016). Age: An effect modifier of the association between allergic rhinitis and otitis media with effusion. Laryngoscope 126, 1687–1692. doi: 10.1002/lary.25682
Swenson, C. E., and Sadikot, R. T. (2015). Achromobacter respiratory infections. Ann. Am. Thorac. Soc. 12, 252–258. doi: 10.1513/AnnalsATS.201406-288FR
Tano, K., von Essen, R., Eriksson, P.-O., and Sjöstedt, A. (2008). Alloiococcus otitidis- otitis media pathogen or normal bacterial flora? APMIS 116, 785–790. doi: 10.1111/j.1600-0463.2008.01003.x
Vissing, N. H., Chawes, B. L., and Bisgaard, H. (2013). Increased risk of pneumonia and bronchiolitis after bacterial colonization of the airways as neonates. Am. J. Respir. Crit. Care Med. 188, 1246–1252. doi: 10.1164/rccm.201302-0215OC
von Graevenitz, A., and Funke, G. (2014). Turicella otitidis and Corynebacterium auris: 20 years on. Infection. 42, 1–4. doi: 10.1007/s15010-013-0488-x
Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer-Verlag. doi: 10.1007/978-3-319-24277-4
Williams, C. J., and Jacobs, A. M. (2009). The impact of otitis media on cognitive and educational outcomes. Med. J. Austr. 191, S69–S72. doi: 10.5694/j.1326-5377.2009.tb02931.x
Xue, Z., Kable, M. E., and Marco, M. L. (2018). Impact of DNA sequencing and analysis methods on 16S rRNA gene bacterial community analysis of dairy products. mSphere 3, e00410–00418. doi: 10.1128/mSphere.00410-18
Yabuuchi, E., and Ohyama, A. (1971). Achromobacter xylosoxidans n. sp. from human ear discharge. Japan. J. Microbiol. 15, 477–481. doi: 10.1111/j.1348-0421.1971.tb00607.x
Ye, Y., and Doak, T. G. (2009). A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 5:e1000465. doi: 10.1371/journal.pcbi.1000465
Keywords: otitis media, asthma, bronchiolitis, middle ear microbiome, amplicon sequence variants
Citation: Kolbe AR, Castro-Nallar E, Preciado D and Pérez-Losada M (2019) Altered Middle Ear Microbiome in Children With Chronic Otitis Media With Effusion and Respiratory Illnesses. Front. Cell. Infect. Microbiol. 9:339. doi: 10.3389/fcimb.2019.00339
Received: 23 July 2019; Accepted: 18 September 2019;
Published: 04 October 2019.
Edited by:
Regie Santos-Cortez, University of Colorado Denver School of Medicine, United StatesReviewed by:
W. Edward Swords, University of Alabama at Birmingham, United StatesShujiro Minami, Tokyo Medical Center (NHO), Japan
Rebecca E. Walker, The University of Auckland, New Zealand
Copyright © 2019 Kolbe, Castro-Nallar, Preciado and Pérez-Losada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Allison R. Kolbe, YWtvbGJlQGd3dS5lZHU=