 Tze Guan Tan
Tze Guan Tan Ying Shiang Lim
Ying Shiang Lim Alrina Tan
Alrina Tan Royston Leong†
Royston Leong† Norman Pavelka
Norman Pavelka- Singapore Immunology Network (SIgN), Agency for Science, Technology and Research (A*STAR), Singapore, Singapore
Candida albicans is a ubiquitous fungal symbiont that resides on diverse human barrier surfaces. Both mammalian and fungal cells can convert arachidonic acid into the lipid mediator, prostaglandin E2 (PGE2), but the physiological significance of fungus-derived PGE2 remains elusive. Here we report that a C. albicans mutant deficient in PGE2 production suffered a loss of competitive fitness in the murine gastrointestinal (GI) tract and that PGE2 supplementation mitigated this fitness defect. Impaired fungal PGE2 production affected neither the in vitro fitness of C. albicans nor hyphal morphogenesis and virulence in either systemic or mucosal infection models. Instead, fungal production of PGE2 was associated with enhanced fungal survival within phagocytes. Consequently, ablation of colonic phagocytes abrogated the intra-GI fitness boost conferred by fungal PGE2. These observations suggest that C. albicans has evolved the capacity to produce PGE2 from arachidonic acid, a host-derived precursor, to promote its own colonization of the host gut. Analogous mechanisms might undergird host-microbe interactions of other symbiont fungi.
Introduction
Candida albicans is one of the most successful fungal symbionts in humans, colonizing 40–80% of individuals in industrialized nations and typically representing the predominant species within the fungal microbiota (Odds, 1987; MacCallum, 2010). In healthy people, C. albicans dwells on diverse barrier sites of the body, including the oral cavity, skin, female reproductive tract, and the intestines, where it does not cause symptomatic disease (Odds, 1987; MacCallum, 2010). However, C. albicans can turn pathogenic and result in mucosal or life-threatening invasive bloodstream infections under a variety of conditions that compromise host immunity, damage barrier surfaces, or disrupt the microbiota (Brown et al., 2012; Fan et al., 2015). Molecular profiling studies have identified C. albicans strains in the gastrointestinal (GI) tract as the provenance of hematogenous isolates during systemic infection (Nucci and Anaissie, 2001; Odds et al., 2006). Understanding the fungal and host factors that modulate the ability of C. albicans to thrive in different host niches as a pathogen or symbiont could therefore inform our development of new methods of combatting human fungal infections.
Screens of C. albicans mutants in mice have revealed several fungal pathways that mediate colonization of the host gut. Salient pathways include the regulation of the acquisition of iron and nutrients, in particular carbon and nitrogen, cell wall remodeling that augments fungal adhesion to intestinal mucins, and morphogenetic changes (White et al., 2007; Rosenbach et al., 2010; Chen et al., 2011; Pande et al., 2013; Perez et al., 2013; Sem et al., 2016; Bohm et al., 2017; Mamouei et al., 2017). Fungal colonization is, in turn, restricted by the host immune system. Intestinal epithelial cells (IECs) produce antimicrobial peptides (AMPs) that kill C. albicans (Fan et al., 2015), and gut-resident CX3CR1+ macrophages control the growth of C. albicans and other symbiont fungi in mice (Leonardi et al., 2018). Accordingly, some of the aforementioned genetic programs in C. albicans might also promote intestinal symbiosis by calibrating the host immune response toward the fungus or vice versa. For instance, cell wall remodeling can mask the exposure of β-glucan, an essential component of fungal cell walls, to host immunological sensing via dectin-1 (Ballou et al., 2016; Pradhan et al., 2018), and recognition of surface β-glucan by dectin-1 is strongly correlated with the competitive fitness of Candida species in the murine intestine (Sem et al., 2016). In addition, C. albicans upregulates the key morphogenetic regulator, EFG1, upon colonizing the gut, and EFG1 is required for C. albicans to overcome host immunological pressures to persist in the GI tract long-term (Pierce and Kumamoto, 2012).
Prostaglandin E2 (PGE2) is a lipid mediator produced by mammalian cells that exerts pleiotropic effects on physiology, in particular the modulation of immunological responses (Kalinski, 2012). For instance, PGE2 suppresses the maturation of dendritic cells and restrains the phagocytic activity of monocytes, macrophages, and neutrophils (Kalinski, 2012; Medeiros et al., 2012). Depending on the tissue context, PGE2 can either promote (Chizzolini et al., 2008; Yao et al., 2009; Gagliardi et al., 2010; Smeekens et al., 2010) or suppress (Betz and Fox, 1991; Valdez et al., 2012; Ma et al., 2016) pro-inflammatory CD4+ T helper (Th)-1 and−17 responses, which help rein in C. albicans infections in the bloodstream and at barrier surfaces, respectively (Kashem et al., 2015).
Of note, many fungal species, including C. albicans, also produce PGE2 in vitro when supplied exogenously with the host-derived precursor, arachidonic acid (AA) (Noverr et al., 2002). C. albicans and most fungal species lack AA, which is derived from mammalian membrane phospholipids—but C. albicans can induce host cells to release AA (Castro et al., 1994). Moreover, fungus-derived PGE2 exhibits immunomodulatory properties analogous to those of mammalian PGE2 in vitro (Noverr et al., 2001) and in vivo (Kim et al., 2014). PGE2 has also been reported to promote yeast-hyphae morphological switching (Kalo-Klein and Witkin, 1990; Noverr et al., 2001) and biofilm formation in vitro (Alem and Douglas, 2004)—traits commonly associated with fungal virulence. Given that the fungus is more typically associated with the host as a benign symbiont and inter-individual transmission often occurs in the absence of overt infection (Russell and Lay, 1973; Odds et al., 2006), it seems unlikely, however, that C. albicans has evolved the cellular machinery for synthesizing PGE2 from an exogenous substrate for the sole purpose to increase its pathogenic potential or to cause immune-related disorders in the host. We thus posit that fungus-derived PGE2 might mediate asymptomatic colonization by C. albicans of the mammalian intestinal environment. Indeed, we found that a C. albicans mutant with impaired PGE2 production, while not exhibiting any defects in in vitro fitness or in vivo virulence, suffered a loss of competitive fitness relative to the wildtype (WT) strain in the murine intestine. We further report evidence suggesting that fungal PGE2 promotes intestinal symbiosis by C. albicans in part by reducing its killing by phagocytes.
Materials and Methods
Fungal Strains
Fungal strains used in this study are listed in Table S1. All fungal strains were grown in yeast peptone dextrose (YPD) media [1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) glucose] overnight at 30°C prior to use in experiments unless otherwise stated.
Construction of Fungal Strains
Fungal mutants were generated using the SAT1-flipping strategy as previously described (Sasse and Morschhauser, 2012; Sem et al., 2016). Briefly, the plasmid pSFS2 containing the SAT1 flipper cassette was used to disrupt the open reading frame of the OLE2, FET3, and FET31 genes of C. albicans. Sequences (500–600 bp) flanking the targeted genes were amplified using the primers listed in Table S2. Gene deletion was verified by quantitative polymerase chain reaction (qPCR) using the primers listed in Table S2.
Mice and Treatments
C57BL/6J and Ccr2-KO (B6.129S4-Ccr2tm1Ifc/J) mice were maintained under specific-pathogen-free conditions at the A*STAR Biological Resource Centre (BRC) in Biopolis, Singapore. Adult mice (6–8 weeks of age) of either sex were used in all experiments. All animal experiments were approved by the BRC Institutional Animal Care and Use Committee (IACUC) in accordance with the guidelines of the Agri-Food & Veterinary Authority (AVA) and the National Advisory Committee for Laboratory Animal Research (NACLAR), Singapore. To deplete phagocytes, mice were injected intraperitoneally (i.p.) with control or clodronate-containing liposomes (Liposoma B.V.) every 2 days starting from 2 days prior to the inoculation of mice with fungi. To perform PGE2 supplementation, mice were injected i.p. with 16.7 μg/kg of 16,16-dimethyl-PGE2 (Cayman Chemical) or vehicle [methyl acetate (Sigma)] daily, starting from day 0, throughout the course of fungal colonization.
Measurement of Fungal PGE2 Production
Fungal PGE2 production was measured by enzyme-linked immunosorbent assay (ELISA) as previously described (Yano and Fidel, 2011). Briefly, fungi were inoculated into fresh YPD to an optical density (OD) at 600 nm of 0.2 and grown at 37°C with shaking at 250 rpm for 2–2.5 h before peroxide-free arachidonic acid (Cayman Chemical) was added to a final concentration of 0.5 mM. Fungal growth was continued for another 48 h before supernatants were harvested. Levels of PGE2 metabolite (PGEM), a stable derivative of PGE2, in fungal supernatants were assayed using the PGEM EIA kit (Cayman Chemical) as per manufacturer instructions.
Quantification of Fungal Titers
Fungal titers for all experiments were determined by plating fungal cultures, tissue homogenates, or lavage fluids on YPD agar containing 50 U/ml penicillin (Gibco) and 50 μg/ml streptomycin (Gibco). Plates were incubated at 37°C for 24–48 h and colony-forming-units (CFUs) enumerated.
Murine Infection With C. albicans
To elicit systemic candidiasis, mice were injected intravenously via the tail vein with a sublethal dose of C. albicans (2 × 105 cells). Oropharyngeal candidiasis (OPC) was established as previously described (Solis and Filler, 2012). Briefly, mice were injected subcutaneously (s.c.) with 100 mg/kg cortisone 21-acetate (Sigma) in 0.05% (v/v) Tween®-80 (Sigma)/phosphate buffered saline (PBS) 1 day prior to infection to induce transient immunosuppression. Mice were subsequently sedated with 150 mg/kg ketamine and 10 mg/kg xylazine before sublingual inoculation with a small cotton ball saturated with C. albicans cells (2 × 107 cells/ml) for 75 min. Vulvovaginal candidiasis (VVC) was established as previously described (Yano and Fidel, 2011). Briefly, female mice were treated s.c. with 0.1 mg of β-estradiol 17-valerate (Sigma) in sesame oil (Sigma) 3 days prior to infection. β-estradiol injections were maintained weekly thereafter. Mice were intravaginally inoculated with 5 × 104 C. albicans cells in 20 μl of PBS. Infected mice were monitored daily (systemic candidiasis and OPC) for survival and weight loss. Tongues from mice with OPC were homogenized by pressing the tissues through a 40 μm cell strainer. Homogenates were plated to assess fungal burden. Mice with VVC were intravaginally lavaged with 100 μl of PBS. Vaginal lavage fluid was plated to determine fungal burden or used for neutrophil quantification by flow cytometry as described below under “Antibodies and flow cytometry.”
Intestinal Colonization With C. albicans
Three to four days prior to inoculation, mice were placed on drinking water containing 1 g/l of penicillin G (Sigma) and 2 g/l of streptomycin (Sigma) (Sem et al., 2016). Antibiotics were changed twice weekly and maintained throughout the course of the experiment. Mice were gavaged with 107 C. albicans cells to establish intestinal colonization. To determine the intestinal fungal load, stools were collected, weighed, homogenized in PBS, and plated.
In vivo Intestinal Competition
Intestinal fungal competition experiments were performed as previously described (Sem et al., 2016). Briefly, mice were gavaged with 2 × 107 cells comprising the SC5314-dTomato strain and the test strain (WT or ole2/ole2) at a 1:1 ratio. The relative frequency of the test strain at each time point was determined by counting the number of fluorescent vs. non-fluorescent colonies under a fluorescence stereomicroscope (Olympus). The fitness coefficient of each test strain in each mouse was calculated as previously described (Sem et al., 2016). To control for inter-experimental variability, fitness coefficients were normalized to the mean of the coefficients for the WT strain within each treatment group (e.g., clodronate-treated group) for each experiment. Normalization was performed by calculating the difference between each fitness coefficient value and the mean for the WT strain.
In vitro Competition
In vitro competition experiments were performed as previously described (Sem et al., 2016). Briefly, 1.25 × 106 cells comprising the SC5314-dTomato strain and the test strain (WT or ole2/ole2) at a 1:1 ratio were inoculated into 50 ml of media [YPD or yeast nitrogen base (YNB) (BD Difco) with amino acids] containing 0.5 mM of arachidonic acid or not and grown at 37°C. Every 8 or 16 h, fungal cultures were harvested, plated, and 1.25 × 106 cells re-inoculated into 50 ml of fresh media as described above. Relative proportions and fitness coefficients of each test strain were derived as described above.
Infection of Macrophages With C. albicans
Fungal resistance to macrophages at 24 h was determined as previously described (Sem et al., 2016). Briefly, confluent J774A.1 macrophages (ATCC) were infected with C. albicans cells at low multiplicities of infection (MOIs) ranging from 0.005 to 0.0003125 to ensure that essentially 100% of fungi were engulfed by the macrophages at 3 h post-infection. Infected macrophages were incubated at 37°C and fungal colonies enumerated after 24 h. Fungi were also seeded in wells without macrophages at varying densities to assess the number of CFUs plated in each experiment. The percentage of fungal cells that escaped killing by macrophages was calculated as previously described (Noble and Johnson, 2005). For short-term fungal killing assays, J774A.1 macrophages were infected at a MOI of 0.1. Extracellular fungi were removed 1 h post-infection by gently washing the macrophages with PBS. Macrophages were lysed with 0.02% Triton X-100 after an additional 2 h of incubation at 37 °C and cell lysates plated on YPD to enumerate the intra-macrophage fungal load. Where indicated, 100 nM of PGE2 (Cayman Chemical) or the corresponding volume of vehicle (ethanol) was added to the cultures.
Filamentation Assays
Filamentation of C. albicans strains was assessed as previously described (Sem et al., 2016). Briefly, C. albicans cells were incubated in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum (FCS) at 37°C in 96-well plates for 24 h, or plated on Spider agar (1% nutrient broth, 1.35% agar, 0.4% potassium phosphate, 2% mannitol) at 37°C for 4 days before colonies were scored for their morphology, i.e., wrinkled vs. smooth, indicative of filamentous vs. non-filamentous cells, respectively. Arachidonic acid was added at 0.5 mM where indicated.
In vitro Cytotoxicity Assay
Confluent J774A.1 macrophages were infected with C. albicans cells at a MOI of 1 for 6 h. LDH released into the culture media was measured using the CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega) as per kit instructions and normalized to LDH levels in uninfected cells lysed with 1% Triton X-100 (Sigma).
Measurement of LL-37 Production From IECs
Confluent human colonic epithelial cell lines HT-29 and Caco-2 were infected with C. albicans cells at a MOI of 0.5 for 24 h. LL-37 secreted into the culture media was measured by the LL-37 ELISA kit (Cusabio) as per manufacturer instructions.
Assessment of Fungal Resistance to mCRAMP
Fungal resistance to mCRAMP was evaluated as previously described (Sem et al., 2016). Briefly, overnight cultures of C. albicans were inoculated into fresh YPD in the presence of 400 nM PGE2 (Cayman Chemical) or vehicle (ethanol) for 3 h. Cells were washed, resuspended in the appropriate concentration of mCRAMP1−39 (Synpeptide), and incubated at 37°C for 2 h. Cells were washed extensively after mCRAMP exposure and stained with 50 μM of propidium iodide (PI) (Thermo Fisher Scientific) for 15 min. The proportion of live PI− fungal cells was determined by flow cytometry.
Measurement of Transcript Levels by Quantitative PCR
RNA was extracted from a small segment of the distal colon harvested from C. albicans-colonized mice 7 days after inoculation. For in vitro infection experiments, J774A.1 macrophages were infected with fungi at the indicated MOIs and the macrophages lysed with TRIzol™ (Invitrogen) 6 h post-infection. Both colon and macrophage samples were homogenized in TRIzol™ using 0.5 mm glass beads (Sigma) and RNA extracted as per manufacturer instructions. cDNA was prepared from RNA using SuperScript™ III reverse transcriptase (Invitrogen) as per manufacturer instructions. Levels of transcripts of interest in cDNA were measured by qPCR using PerfeCTa SYBR® Green FastMix (Quantabio) and normalized to those of Rpl13a.
Isolation of Colonic Lamina Propria Cells
To remove the epithelial layer, colons were sliced open longitudinally to expose the luminal surface and shaken in DMEM containing 1 mM dithiothreitol, 5 mM EDTA, and 3% (v/v) FCS for 20 min at 37°C. Remaining tissue was washed in cold DMEM, minced, and digested in DMEM containing 1.5 mg/ml collagenase D (Roche), 50 μg/ml DNase I (Roche), and 2% (v/v) FCS for 45 min at 37 °C. Digested tissue was washed and filtered twice to obtain a single-cell suspension.
Antibodies and Flow Cytometry
Mammalian cells were stained with antibodies against CD3ε (145-2C11), CD4 (RM4-5), CD8α (53-6.7), CD11b (M1/70), CD11c (N418), CD19 (6D5), CD45 (30F11), CD64 (X54-5/7.1), I-A/E (M5/114.15.2), Ly6G (1A8) from Biolegend and 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific) to exclude dead cells. All immunocyte subsets were gated on live leukocytes (DAPI−CD45+). Macrophages, DCs, B cells, CD4+ T cells, and CD8+ T cells from the colonic lamina propria were defined as CD64+CD11b+CD11c+I-A/E+, CD64−CD11c+I-A/E+, CD3ε−CD19+, CD3ε+CD19−CD4+, and CD3ε+CD19−CD8α+, respectively. Intravaginal neutrophils were defined as CD11b+Ly6G+ cells. Samples were acquired on the MACSQuant® VYB (Miltenyi Biotec) and analyzed using FlowJo v10.2 (Tree Star).
Statistics
Pairwise comparisons were performed using the Student's t-test or the paired Student's t-test, as appropriate, while multi-group comparisons were performed using the Two-way Analysis of Variance (ANOVA) with Sidak's multiple comparisons test (Prism 7; Graph-Pad), unless otherwise stated. P-values were deemed significant if <0.05. Data routinely presented as mean ± standard deviation (s.d.) unless otherwise stated.
Results
PGE2-Defective C. albicans Mutant Exhibits No Fitness Defects in vitro
PGE2 production has been described for C. albicans and other pathogenic fungi such as Cryptococcus neoformans and Aspergillus species (Noverr et al., 2002) as well as for some gut-dwelling symbiont Candida species (Shiraki et al., 2008; Ells et al., 2011; Mishra et al., 2014; Grozer et al., 2015). We measured in vitro PGE2 production across an array of symbiont fungi and confirmed that all species tested, including Saccharomyces cerevisiae, secreted PGE2 when supplied with exogenous AA (Figure 1A), implying a PGE2 biosynthetic pathway conserved across the majority of fungal species.
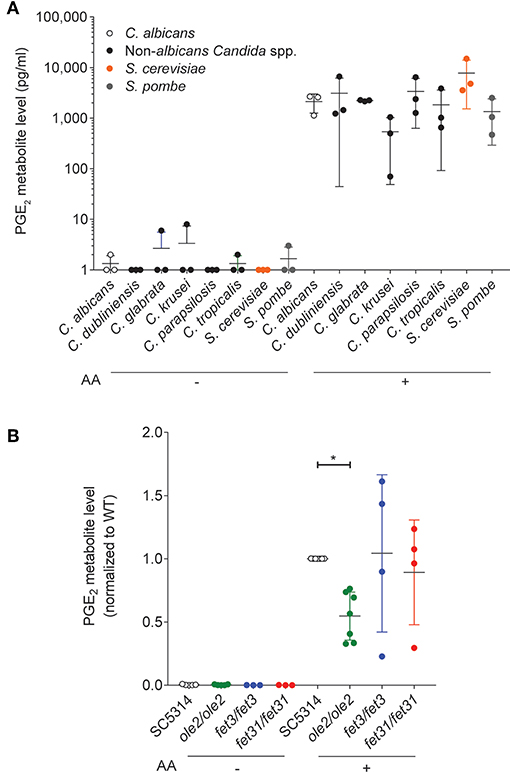
Figure 1. In vitro PGE2 production by various C. albicans strains. Levels of PGE2 metabolite, a stable derivative of PGE2, in the supernatants of in vitro cultures of various fungal species (A) or C. albicans strains (B), were assayed by ELISA. Arachidonic acid (AA) was added (+) or not (–) to the cultures. Data pooled from 3 to 7 independent experiments. Mean ± s.d. *p < 0.05; One-way ANOVA with Dunnett's multiple comparisons test.
We next sought a C. albicans mutant with altered PGE2 production which we could use to study the physiological function of fungus-derived PGE2. Although the PGE2 biosynthetic pathway in C. albicans and other fungi remains largely uncharacterized, three genes, OLE2, FET3, and FET31, have been described to promote PGE2 production in vitro (Erb-Downward and Noverr, 2007; Krause et al., 2015). We generated knockout (KO) mutants for each of these genes and assessed the mutants' capacities for PGE2 secretion in vitro at 37°C. As expected, PGE2 production by all fungal strains was strictly dependent on the provision of exogenous AA (Figure 1B). However, only the ole2/ole2 mutant exhibited reproducibly diminished PGE2 secretion. Concordant with a previous report (Erb-Downward and Noverr, 2007), PGE2 production was not completely abrogated by OLE2 ablation, implying the existence of alternative pathways to PGE2 generation by C. albicans. We thus focused on the ole2/ole2 mutant for all subsequent experiments.
To determine whether fungal PGE2 modifies fungal fitness in vitro, we maintained either the WT or the ole2/ole2 strain in competition with a fluorescently tagged WT strain (SC5314-dTomato) in serially passaged liquid batch cultures. The abundance of each strain relative to that of SC5314-dTomato was determined at regular intervals by plating the cultures on yeast peptone dextrose (YPD) agar followed by scoring of fluorescent and non-fluorescent colonies. The fitness of each strain can in turn be rigorously quantified by deriving a fitness coefficient from the rate of change of the relative abundance of each strain (Sem et al., 2016). Even after 80 h [equivalent to ~44 and 63 generations for growth in YNB media and YPD, respectively) of serial subculturing, we detected no differences in competitive fitness between the ole2/ole2 and WT strains in both the nutrient-rich YPD broth and the nutrient-poor YNB media, whether or not exogenous AA was supplied (Figure 2, Figure S1). Therefore, fungal PGE2 does not appear to be important for fitness in vitro.
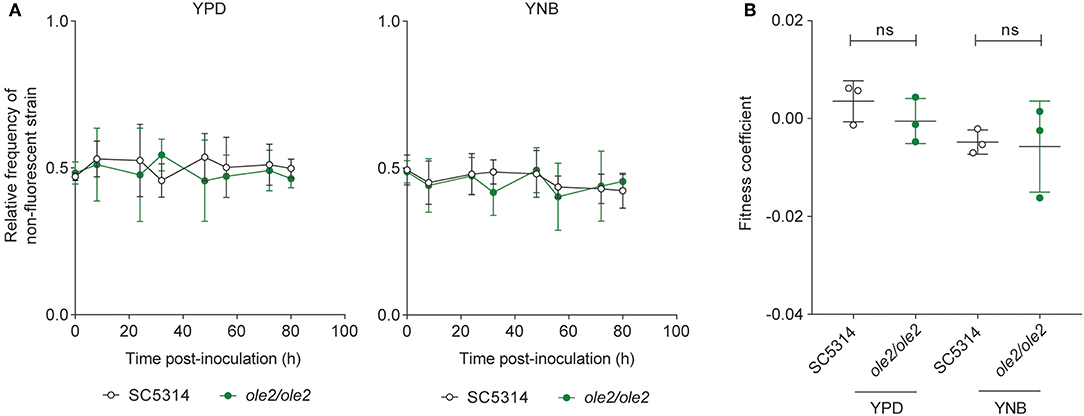
Figure 2. Fungal PGE2 is not required for the in vitro competitive fitness of C. albicans. WT C. albicans or the ole2/ole2 mutant was grown in rich (YPD) or minimal (YNB) media in competition with a fluorescently-tagged WT strain at a 1:1 ratio in serial batch cultures for 80 h. (A) Relative frequencies of the WT or ole2/ole2 strain throughout the serial passages. (B) Fitness coefficients of the WT or ole2/ole2 strain, determined as outlined in Materials and Methods. Data pooled from 3 independent experiments. Mean ± s.d. ns, not significant. Student's t-test.
Fungal PGE2 Is Not Required for Virulence
Since PGE2 was reported to increase filamentation and biofilm formation in vitro (Noverr et al., 2001; Alem and Douglas, 2004; Kim et al., 2014), we asked whether defective PGE2 synthesis by the ole2/ole2 mutant influences its virulence. Surprisingly, the ole2/ole2 mutant was fully capable of hyphal formation in vitro, both in response to fetal calf serum (FCS) and when plated on Spider agar, regardless of the presence of AA (Figures 3A,B). Moreover, the ability of C. albicans to damage host cells—whose cell membranes are an abundant source of endogenous AA (Martin et al., 2016)—evidenced by the release of lactate dehydrogenase from infected murine macrophages, was not significantly altered by fungal PGE2 (Figure 3C). Nonetheless, there exist occasional discrepancies between in vivo virulence and in vitro pathogenic traits (Noble et al., 2010). To clarify the role of fungus-derived PGE2 during in vivo infections, we adopted murine models of hematogenously disseminated candidiasis and mucosal candidiasis of the oropharyngeal cavity and vaginal tract, all of which engage distinct anti-fungal immunological responses (Fidel, 2005; Conti et al., 2009; Kashem et al., 2015). The ole2/ole2 mutant manifested comparable virulence as the WT strain in all tissues tested, across a variety of pathological parameters, including overall survival, fungal burden, weight loss, and neutrophil recruitment (Figures 3D–F, Figure S2). Our in vitro and in vivo infection data are hence not consistent with a role for fungal PGE2 in mediating C. albicans virulence.
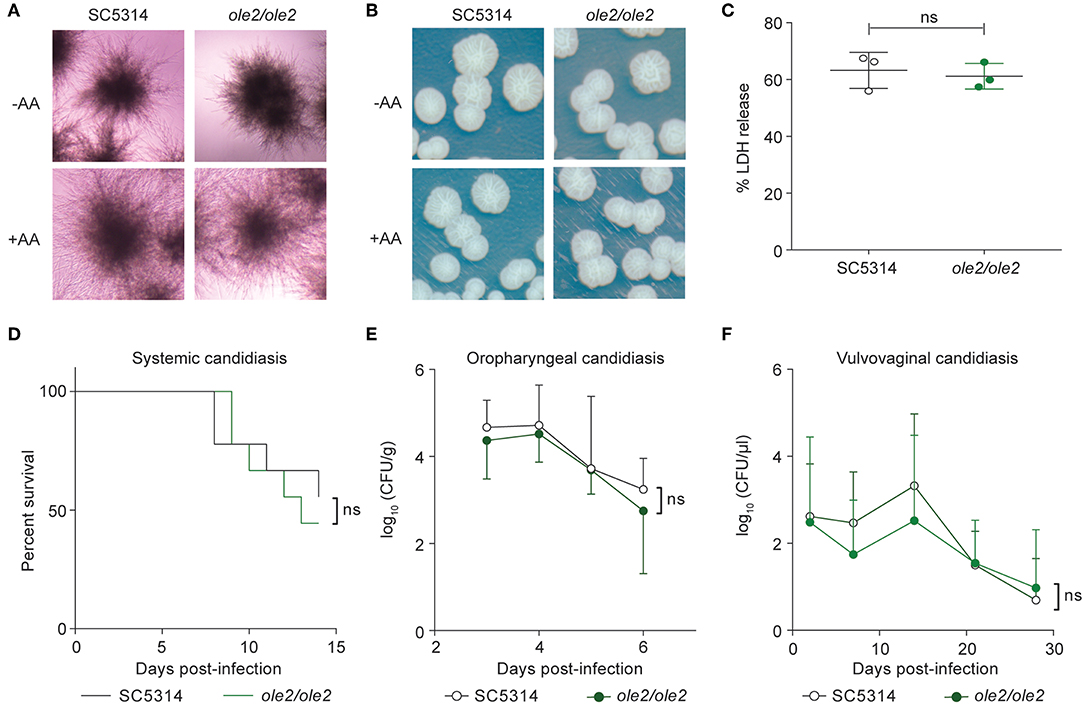
Figure 3. Fungal PGE2 is not required for virulence. (A,B) Hyphae formation of the WT and ole2/ole2 C. albicans strains in response to fetal calf serum (FCS) (A) or when plated on Spider agar. (B) AA was added where indicated. (C) In vitro cytotoxicity of the indicated fungal strains to J774A.1 murine macrophages, determined by quantifying the release of lactate dehydrogenase (LDH) from the macrophages. Data presented as a percentage of the maximal amount of LDH released by completely lysed cells. (D) Survival of mice infected intravenously with 2 × 105 cells of the indicated C. albicans strains. (E,F) Kinetics of the fungal CFUs in the tongue (E) and vaginal lavage fluid (F) of mice infected sublingually (E) or intravaginally (F) with the indicated fungal strains. Data pooled from 2 independent experiments. Mean ± s.d. (C,E,F). n = 8–10 mice/group. ns, not significant. Student's t-test (C,E,F); Log-rank test (D).
Fungal PGE2 Promotes Intestinal Colonization
C. albicans is most commonly associated with the host as a symbiont in the gut. We therefore sought to evaluate whether fungal PGE2 contributes to the fitness of C. albicans in the intestines. Mice are usually refractory to colonization by C. albicans but can be induced to harbor high titers of fungi for sustained durations if treated with antibiotics to deplete their indigenous microbiota (Koh et al., 2008; Fan et al., 2015). Colonization of the murine GI tract with C. albicans represents a bona fide model for fungal symbiosis with the mammalian host because it does not lead to symptomatic disease and invasive hyphae are rarely detected within the intestinal fungal population (White et al., 2007; Koh et al., 2008; Bohm et al., 2017).
To assess the intestinal fitness of the ole2/ole2 mutant, we inoculated antibiotics-treated mice with either ole2/ole2 or WT C. albicans in competition with SC5314-dTomato and tracked the relative frequency of each strain over time. The ole2/ole2 mutant was significantly compromised in its competitive fitness relative to the WT strain, starting as early as 3 days post-inoculation, as evidenced by its lower relative frequency in the fecal fungal load and its lower fitness coefficient, determined as per our in vitro competitive fitness experiments (Figure 4A, Figure S3A). Although we were not able to test a reconstituted ole2/ole2::OLE2 mutant, colonization of mice with another independently-derived ole2/ole2 strain recapitulated the inferior competitive fitness of C. albicans in the GI tract in the absence of OLE2 (Figures S3B,C). While we consistently observed a lower fitness coefficient for the ole2/ole2 mutant relative to WT C. albicans across independent experiments, the fitness coefficient differential between the two strains varied in magnitude between experiments (Figure S3), possibly due to variation across different cohorts of mice. To facilitate a comparison of fungal fitness across pooled groups of mice, we normalized fitness coefficients to the mean coefficient of the WT strain within each experiment, thereby removing the batch effects on fungal fitness. This analysis confirmed a significant impairment in the competitive fitness of the PGE2-deficient ole2/ole2 mutant in the murine gut (Figure 4B).
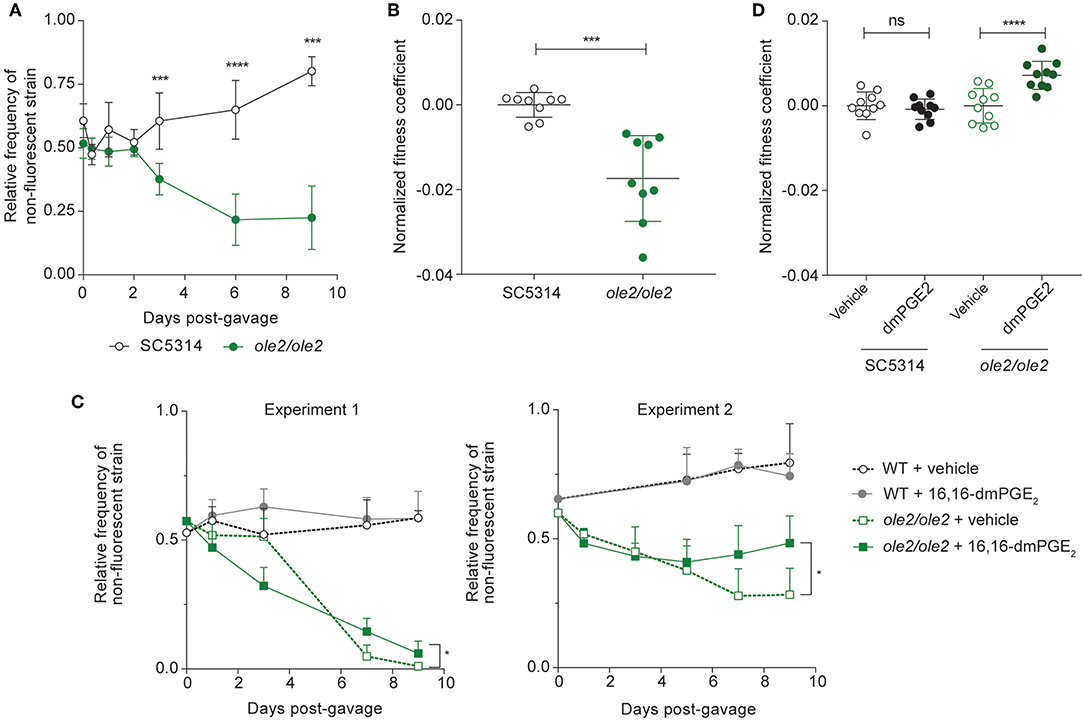
Figure 4. Fungal PGE2 promotes intestinal colonization by C. albicans. Antibiotics-treated mice were gavaged with the indicated C. albicans strains and stools were plated on YPD agar at regular intervals over 9 days. (A,B) Relative frequencies (A) and fitness coefficients, normalized to the mean of the WT coefficients (B), of the indicated fungal strains in the intestines of gavaged mice. (C,D) Relative frequencies (C) and fitness coefficients (D) of the indicated fungal strains in mice supplemented with vehicle or 16,16-dimethyl-PGE2 (dmPGE2). Coefficients for each fungal strain were normalized to the mean of the respective vehicle coefficients. Fitness coefficients were determined as described in Materials and Methods. Data pooled from 2 independent experiments. Mean ± s.d. n = 9−10 mice/group. ****p < 0.0001; ***p < 0.001. Student's t-test (A,B); Two-way ANOVA with Sidak's multiple comparisons test (C,D).
To determine if the loss of competitive fitness of the ole2/ole2 strain in the gut is PGE2-dependent, we supplemented C. albicans-colonized mice daily with 16,16-dimethyl-PGE2 (dmPGE2), a stable analog of PGE2. Indeed, dmPGE2 treatment partially restored the competitive fitness of the ole2/ole2 mutant (Figures 4C,D, Figure S3D), in support of a role for fungal PGE2 in mediating intestinal fitness of C. albicans under steady-state conditions. In contrast, dmPGE2 provision did not further enhance the competitive fitness of the WT strain. Taken together, these data indicate that fungal PGE2 exerts a specific function for C. albicans: promote its colonization of the host GI tract.
Fungal PGE2 Does Not Affect AMP Production or Fungal Resistance to AMPs
AMPs such as mouse cathelicidin-related antimicrobial peptide (mCRAMP), encoded by Camp, have been demonstrated to mediate intestinal resistance to C. albicans colonization (Fan et al., 2015). We postulated that fungal PGE2 might promote intestinal fitness of C. albicans by influencing AMP production from IECs or fungal resistance to AMPs. To address the former hypothesis, we stimulated human colonic IEC lines with WT C. albicans or the ole2/ole2 mutant but observed no difference in the secretion of LL-37, the human ortholog of mCRAMP, by the IEC lines (Figure S4A). Indeed, infection of the IEC lines by fungi in vitro did not induce appreciable production of LL-37 above the background level. We also assessed Camp transcript levels in the colons of mice colonized with either WT or ole2/ole2 C. albicans but found no difference in the quantities of Camp mRNA induced by the two fungal strains in vivo (Figure S4B). Finally, we exposed fungi grown in the presence or absence of PGE2 to mCRAMP in vitro to evaluate the effects of PGE2 on fungal resistance to AMPs. Both the WT and ole2/ole2 strains manifested comparable susceptibility to mCRAMP, and the addition of PGE2 did not significantly alter fungal resistance to mCRAMP (Figure S4C). Hence the collective evidence does not support a role for fungus-derived PGE2 in altering AMP production from IECs or fungal resistance to AMPs.
Fungus-Derived PGE2 Reduces Killing of C. albicans by Intestinal Phagocytes
Since intestinal macrophages limit fungal colonization of the gut (Leonardi et al., 2018) and tissue-resident phagocytes are required for control of fungal infection in various tissues (Kashem et al., 2015; Dominguez-Andres et al., 2017; Sparber et al., 2018), we postulated that fungal PGE2 might potentiate fungal fitness by acting on the fungi themselves and/or host tissue phagocytes to improve the ability of C. albicans to evade killing by phagocytes. To elucidate the relationship between fungal PGE2, tissular phagocytes, and fungal fitness in the host, we assessed the competitive fitness of the WT and ole2/ole2 strains in mice depleted of phagocytes. We first treated mice with clodronate-containing liposomes, which is toxic to a variety of phagocytic leukocytes, including macrophages and dendritic cells (DCs) (Weisser et al., 2012). In our experiments, clodronate treatment depleted colonic CD11b+ dendritic cells (DCs) but not macrophages, CD11b− DCs, or non-phagocytic leukocytes (Figures S5A,B). The poor efficiency of macrophage ablation in clodronate-treated mice could possibly arise from differential accessibility and susceptibility of macrophages in the colon to intraperitoneally-administered clodronate relative to those from other tissue sites. Nonetheless, clodronate treatment was sufficient to abrogate the fitness defect of the ole2/ole2 mutant vis-à-vis the WT strain that was observed in control mice, thereby implicating CD11b+ DCs as another tissue-resident phagocytic population that restricts C. albicans in the gut (Figures 5A,B, Figure S6A). We further validated the preceding results by repeating the fungal competitive colonization experiment in a genetic model of phagocyte depletion. CCR2 is a chemokine receptor required for efficient reconstitution of the intestinal macrophage and CD11b+ DC compartments (Bain et al., 2014; Scott et al., 2015). Consequently, Ccr2-KO mice display a paucity of both macrophages and CD11b+ DCs but not CD11b− DCs or lymphocytes in the colon (Figures S5C,D). In agreement with our findings in clodronate-treated mice, Ccr2 deficiency abolished the fitness difference between the WT and ole2/ole2 strains observed in WT mice (Figures 5C,D, Figure S6B). Our results collectively support a role for fungal PGE2 in reducing the eradication of C. albicans by host intestinal phagocytes, including macrophages and CD11b+ DCs. We thus sought to compare the ex vivo fungicidal activity of host phagocytes toward WT and ole2/ole2 C. albicans. Owing to difficulties in isolating sufficient numbers of primary macrophages or DCs from the murine gut for this purpose, we turned to a well-described murine macrophage line J774A.1 and measured fungal killing by J774A.1 cells upon C. albicans infection in vitro using a previously reported method (Sem et al., 2016). Indeed, the ole2/ole2 mutant exhibited reduced resistance to macrophages relative to the WT strain at 24 h post-infection but not at 3 h (Figure 5E, Figures S7A,B), and this defect in resistance to host killing can be reversed by the provision of exogenous PGE2 (Figure 5F). We reasoned that the effects of fungus-derived PGE2 on fungal killing at 24 h post-infection might require the transcriptional reprogramming of macrophages, as previously described (Suram et al., 2013; Kim et al., 2014; Yun et al., 2016). However, we did not observe a difference in the levels of transcripts associated with pro-inflammatory (Il1b) and immunosuppressive (Arg1, Chil3, Retnla, Il10) macrophage subsets between macrophages infected with WT or ole2/ole2 C. albicans (Figure S7C). Overall, these results suggest that fungal PGE2 confers C. albicans with a competitive advantage in the mammalian GI tract by protecting the fungi from the microbicidal activity of intestinal phagocytes.
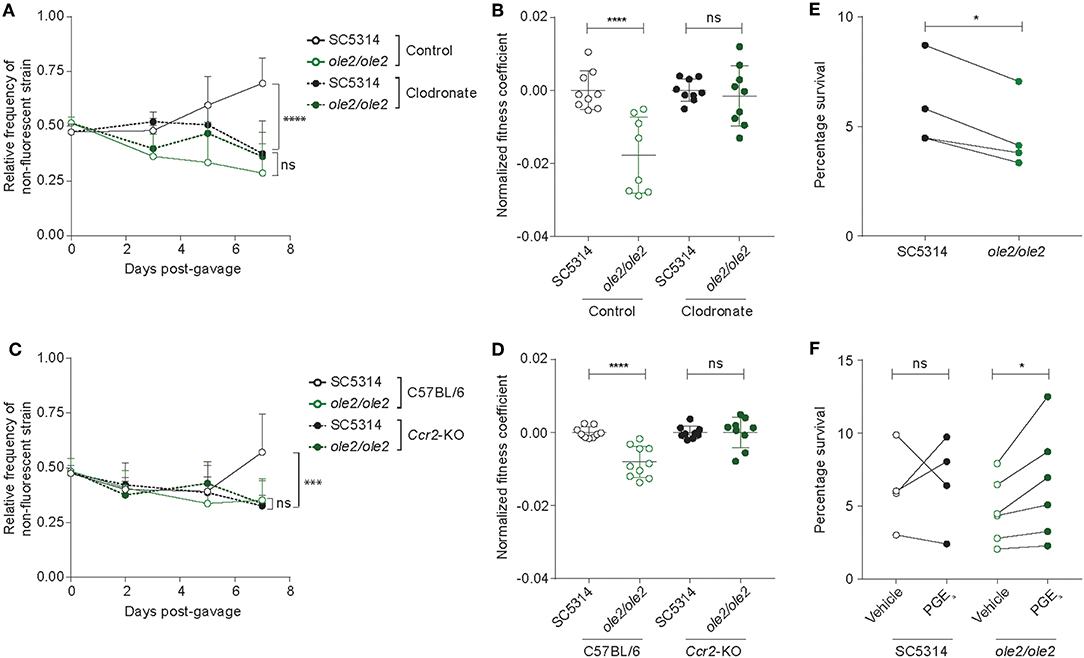
Figure 5. Ablation of colonic phagocytes abrogates the fitness defect of the ole2/ole2 mutant. In vivo intestinal colonization experiments were performed as in Figure 4. (A,B) Mice were treated with control or clodronate-containing liposomes. (C,D) WT or Ccr2-KO mice were used for intestinal colonization. (A,C) Kinetics of the relative frequencies of the indicated fungal strains. P-values indicate comparisons of relative frequencies of the two fungal strains within each treatment (A) or genetic (C) group. (B,D) Fitness coefficients of the indicated fungal strains determined as described in Materials and Methods. Coefficients for each treatment group were normalized to the mean of the respective WT coefficients. (E) J774A.1 murine macrophages were infected with the indicated C. albicans strains for 24 h and the percentage survival of each strain was determined as outlined in Materials and Methods. (F) Infection of macrophages as in (A), but with addition of exogenous PGE2 or vehicle to the cultures. Data pooled from 2 (A–D) or 4–6 (E,F) independent experiments. Mean ± s.d. (A–D). n = 9–10 mice/group (A–D). ****p < 0.0001; ***p < 0.001; *p < 0.05; ns, not significant. Two-way ANOVA with Sidak's multiple comparisons test (A–D); paired student's t-test (E,F).
Discussion
In this study, we sought to clarify the physiological function of PGE2 production by C. albicans, a phenomenon long described (Noverr et al., 2001) but of unclear importance to either host or fungus. Using a PGE2-deficient fungal mutant, we found no contribution of fungal PGE2 to the in vitro and in vivo virulence of C. albicans across disparate murine models of candidiasis that affect different organs and engage distinct antifungal immunological responses. Instead, we uncovered a specific role for PGE2 production by C. albicans in potentiating fungal colonization of the murine intestine. Interestingly, the effects of fungal PGE2 appear to be linked to protection from killing by host phagocytes. Our results raise several questions that merit further elaboration.
How has C. albicans evolved the ability to synthesize PGE2 from AA, a substrate that it lacks? No fungal orthologs of the mammalian biosynthetic enzymes for PGE2–the cyclooxygenases, COX-1 and COX-2, and the PGE2 synthase, PTGES—have been identified to date (Erb-Downward and Noverr, 2007; Erb-Downward et al., 2008). Even using a sensitive method based on profile hidden Markov models (HMMER v.3) failed to identify distant homologs of the mammalian PGE2 biosynthetic enzymes (F. L. Sirota & S. Maurer-Stroh, unpublished observation). In addition, the three genes identified to contribute to PGE2 synthesis in C. albicans—OLE2, FET3, and FET31—do not harbor enzymatic domains that are known to bind to or act on AA or downstream prostaglandins. Indeed, FET3 and FET31 are putative iron transporters in C. albicans and might instead modulate fungal PGE2 production indirectly, possibly by acting as cofactors or regulating cellular levels of iron that in turn impact PGE2 synthesis. Genetic deficiency of OLE2 in C. albicans did not completely eliminate PGE2 production in vitro (Erb-Downward and Noverr, 2007; Figure 1), implying the existence of additional components of the fungal PGE2 synthetic pathway. It is possible that the impact of OLE2 on fungal PGE2 production in the gut might be more profound than that observed in vitro, though we have not been able to address this empirically due to difficulties in precisely measuring PGE2 levels in intestinal contents, which contain high levels of substances that interfere with ELISA data linearity (data not shown). Thus, C. albicans has convergently evolved a mechanism to generate PGE2 from a host-derived precursor, presumably in response to evolutionary pressures exerted by its lifestyle as an obligate symbiont in warm-blooded organisms.
Why does C. albicans goes through the effort of producing PGE2 at all? Earlier studies have implicated fungus-derived PGE2 in pathogenesis, since PGE2 promotes virulence traits such as germ tube and biofilm formation in vitro (Noverr et al., 2001; Alem and Douglas, 2004). Moreover, fungal PGE2 can suppress host immunity, which might in turn favor fungal growth. For instance, PGE2 emanating from a fungal bloom in the gut induced an immunoregulatory M2 phenotype in alveolar macrophages, which in turn exacerbated allergic airway inflammation in mice (Kim et al., 2014), and exposure to fungal PGE2 diminished the protective immune response generated in response to immunization with DCs pulsed with C. albicans yeasts (Kundu and Noverr, 2011). However, the preceding findings do not point to an overt benefit for C. albicans, for which symbiosis is the status quo and infection is adventitious and often a byproduct of sustained immunodeficiency and/or microbial dysbiosis. Accordingly, we observed no role for fungal PGE2 in mediating the virulence of C. albicans under both in vitro and in vivo settings. Conversely, fungal PGE2 production appeared to be important for promoting intestinal colonization by C. albicans. C. albicans is located at high titers in the gut and acquisition of the fungus often occurs early during ontogeny via transmission of maternal intestinal strains to the infant (Russell and Lay, 1973; Odds et al., 2006). It would thus be evolutionarily advantageous for C. albicans to evolve an independent means of generating PGE2 to facilitate its own entrenchment as a symbiont in the gut.
How does fungal PGE2 mediate intestinal colonization by C. albicans? Here we provide evidence in support of gut phagocytes, in particular macrophages and CD11b+ DCs, as a target of fungal PGE2, since depletion of colonic phagocytes by two orthogonal approaches abrogated the fitness defect of the ole2/ole2 mutant vis-à-vis the WT strain. The ole2/ole2 mutant also sustained increased killing by a murine macrophage line in vitro, a phenomenon that was reversed by the addition of exogenous PGE2. PGE2-dependent fungal resistance to phagocytes could occur via the paracrine activity of fungal PGE2 on proximal phagocytes, and/or an autocrine effect of fungal PGE2 on the fungus itself. PGE2 has wide-ranging effects on phagocytes, most of which converge on immunosuppression (Kalinski, 2012; Medeiros et al., 2012). For instance, PGE2 reduces the phagocytosis of fungi by macrophages (Serezani et al., 2012) and polarizes both fungi-exposed macrophages and DCs toward an immunoregulatory phenotype, characterized by an increased expression of suppressive factors such as arginase-1 (Suram et al., 2013; Kim et al., 2014; Yun et al., 2016). Hence, PGE2 secreted by C. albicans proximal to or engulfed by intestinal phagocytes might dampen their uptake of more fungi, phagosome maturation, and/or production of inflammatory, antifungal factors beyond the ones we had tested (Figure S7C). The effects of fungal PGE2 can also radiate beyond those of phagocytes to T cell responses primed by fungi-experienced phagocytes (Gagliardi et al., 2010; Smeekens et al., 2010), which in turn control fungal growth in the gut. PGE2 is labile and likely has short-range activity, possibly even at the level of individual fungi-containing phagosomes in phagocytes (Hoffmann et al., 2012). This might account for the apparent cell-autonomous effect of fungal PGE2 on intestinal fitness of C. albicans.
In addition, PGE2 might act directly on C. albicans itself to augment its resistance to host immunity. Mucosal microbial communities, including those of Candida, have been likened to biofilms (de Vos, 2015), and certain genes crucial for the intestinal fitness of C. albicans are also required for biofilm formation (Ganguly and Mitchell, 2011; Bohm et al., 2017; Lagree and Mitchell, 2017), indicating common mechanisms underlying fungal fitness in both biofilms and the gut. Fungus-derived PGE2 might therefore elicit biofilm-like transcriptional programs in gut fungi that enhance fitness via various means, such as the induction of stress response pathways that improve fungal resistance to immunological assaults from the host.
Our study focused on the role of fungus-derived PGE2 but does not rule out a role for host-derived PGE2 or other AA-derived lipid mediators in modulating intestinal symbiosis by C. albicans. Host phagocytes secrete PGE2 when stimulated with C. albicans (Smeekens et al., 2010; Suram et al., 2013; Yun et al., 2016), though it is plausible that host- and fungus-derived PGE2 exert distinct activities in a tissue- and cell-type-specific fashion, especially in light of the differential accessibility of immunocyte populations to fungi and the instability of PGE2. Nevertheless, we predict that the effects of fungal PGE2 on the intestinal fitness of C. albicans would be amplified in the absence of host-derived PGE2. Notably, C. albicans, as well as other fungi and parasites, produces immunomodulatory lipid mediators from AA aside from PGE2 in vitro, including prostaglandin D2 and leukotrienes (Szkudlinski, 2000; Herve et al., 2003; Tsitsigiannis et al., 2005; Valdez et al., 2012), some of which have been implicated in modulating microbial fitness during pathogenesis. It is tempting to speculate that non-PGE2 lipids can, akin to PGE2, modulate the fitness of C. albicans and other symbiont microbes in the intestines and other barrier surfaces.
We report here a novel mechanism used by C. albicans to colonize the gut that is distinct from previously delineated pathways, such as the suppression of filamentation and changes in nutrient metabolism and iron acquisition (Nguyen et al., 2009). Since fungal colonization is a prerequisite for disseminated or mucosal diseases, this work provides new potential targets for therapeutic control of intestinal fungal populations to prevent human fungal infections.
Data Availability Statement
All datasets generated for this study are included in the manuscript/Supplementary Files.
Ethics Statement
The animal study was reviewed and approved by A*STAR Biological Resource Centre (BRC) Institutional Animal Care and Use Committee (IACUC).
Author Contributions
TT, YL, AT, and RL performed experiments and data analysis. TT wrote the manuscript. NP edited the manuscript and supervised the study.
Funding
This study was supported by A*STAR Investigatorship award no. JCO/1437a00117 and by SIgN core funding to NP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This manuscript has been released as a Pre-Print at BioRxiv (Tan et al., 2018). We thank G. T. T. Le for technical assistance; S. Maurer-Stroh and F. L. Sirota for sharing unpublished bioinformatics analyses; Singapore Immunology Network (SIgN) Mouse Core for providing mice. This paper is dedicated to the memory of RL.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00359/full#supplementary-material
References
Alem, M. A., and Douglas, L. J. (2004). Effects of aspirin and other nonsteroidal anti-inflammatory drugs on biofilms and planktonic cells of Candida albicans. Antimicrob. Agents Chemother. 48, 41–47. doi: 10.1128/AAC.48.1.41-47.2004
Bain, C. C., Bravo-Blas, A., Scott, C. L., Perdiguero, E. G., Geissmann, F., Henri, S., et al. (2014). Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 15, 929–937. doi: 10.1038/ni.2967
Ballou, E. R., Avelar, G. M., Childers, D. S., Mackie, J., Bain, J. M., Wagener, J., et al. (2016). Lactate signalling regulates fungal beta-glucan masking and immune evasion. Nat. Microbiol. 2:16238. doi: 10.1038/nmicrobiol.2016.238
Betz, M., and Fox, B. S. (1991). Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 146, 108–113.
Bohm, L., Torsin, S., Tint, S. H., Eckstein, M. T., Ludwig, T., and Perez, J. C. (2017). The yeast form of the fungus Candida albicans promotes persistence in the gut of gnotobiotic mice. PLoS Pathog. 13:e1006699. doi: 10.1371/journal.ppat.1006699
Brown, G. D., Denning, D. W., Gow, N. A., Levitz, S. M., Netea, M. G., and White, T. C. (2012). Hidden killers: human fungal infections. Sci. Transl. Med. 4:165rv113. doi: 10.1126/scitranslmed.3004404
Castro, M., Ralston, N. V., Morgenthaler, T. I., Rohrbach, M. S., and Limper, A. H. (1994). Candida albicans stimulates arachidonic acid liberation from alveolar macrophages through alpha-mannan and beta-glucan cell wall components. Infect. Immun. 62, 3138–3145.
Chen, C., Pande, K., French, S. D., Tuch, B. B., and Noble, S. M. (2011). An iron homeostasis regulatory circuit with reciprocal roles in Candida albicans commensalism and pathogenesis. Cell Host Microbe 10, 118–135. doi: 10.1016/j.chom.2011.07.005
Chizzolini, C., Chicheportiche, R., Alvarez, M., de Rham, C., Roux-Lombard, P., Ferrari-Lacraz, S., et al. (2008). Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 112, 3696–3703. doi: 10.1182/blood-2008-05-155408
Conti, H. R., Shen, F., Nayyar, N., Stocum, E., Sun, J. N., Lindemann, M. J., et al. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206, 299–311. doi: 10.1084/jem.20081463
de Vos, W. M. (2015). Microbial biofilms and the human intestinal microbiome. NPJ Biofilms Microbiomes 1:15005. doi: 10.1038/npjbiofilms.2015.5
Dominguez-Andres, J., Feo-Lucas, L., Minguito de la Escalera, M., Gonzalez, L., Lopez-Bravo, M., and Ardavin, C. (2017). Inflammatory Ly6Chigh monocytes protect against candidiasis through IL-15-driven NK cell/neutrophil activation. Immunity 46, 1059–1072 e1054. doi: 10.1016/j.immuni.2017.05.009
Ells, R., Kock, J. L., Albertyn, J., Kemp, G., and Pohl, C. H. (2011). Effect of inhibitors of arachidonic acid metabolism on prostaglandin E(2) production by Candida albicans and Candida dubliniensis biofilms. Med. Microbiol. Immunol. 200, 23–28. doi: 10.1007/s00430-010-0169-7
Erb-Downward, J. R., Noggle, R. M., Williamson, P. R., and Huffnagle, G. B. (2008). The role of laccase in prostaglandin production by Cryptococcus neoformans. Mol. Microbiol. 68, 1428–1437. doi: 10.1111/j.1365-2958.2008.06245.x
Erb-Downward, J. R., and Noverr, M. C. (2007). Characterization of prostaglandin E2 production by Candida albicans. Infect. Immun. 75, 3498–3505. doi: 10.1128/IAI.00232-07
Fan, D., Coughlin, L. A., Neubauer, M. M., Kim, J., Kim, M. S., Zhan, X., et al. (2015). Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 21, 808–814. doi: 10.1038/nm.3871
Fidel, P. L. Jr. (2005). Immunity in vaginal candidiasis. Curr. Opin. Infect. Dis. 18, 107–111. doi: 10.1097/01.qco.0000160897.74492.a3
Gagliardi, M. C., Teloni, R., Mariotti, S., Bromuro, C., Chiani, P., Romagnoli, G., et al. (2010). Endogenous PGE2 promotes the induction of human Th17 responses by fungal ss-glucan. J. Leukoc. Biol. 88, 947–954. doi: 10.1189/jlb.0310139
Ganguly, S., and Mitchell, A. P. (2011). Mucosal biofilms of Candida albicans. Curr. Opin. Microbiol. 14, 380–385. doi: 10.1016/j.mib.2011.06.001
Grozer, Z., Toth, A., Toth, R., Kecskemeti, A., Vagvolgyi, C., Nosanchuk, J. D., et al. (2015). Candida parapsilosis produces prostaglandins from exogenous arachidonic acid and OLE2 is not required for their synthesis. Virulence 6, 85–92. doi: 10.4161/21505594.2014.988097
Herve, M., Angeli, V., Pinzar, E., Wintjens, R., Faveeuw, C., Narumiya, S., et al. (2003). Pivotal roles of the parasite PGD2 synthase and of the host D prostanoid receptor 1 in schistosome immune evasion. Eur. J. Immunol. 33, 2764–2772. doi: 10.1002/eji.200324143
Hoffmann, E., Kotsias, F., Visentin, G., Bruhns, P., Savina, A., and Amigorena, S. (2012). Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 109, 14556–14561. doi: 10.1073/pnas.1203912109
Kalinski, P. (2012). Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28. doi: 10.4049/jimmunol.1101029
Kalo-Klein, A., and Witkin, S. S. (1990). Prostaglandin E2 enhances and gamma interferon inhibits germ tube formation in Candida albicans. Infect. Immun. 58, 260–262.
Kashem, S. W., Igyarto, B. Z., Gerami-Nejad, M., Kumamoto, Y., Mohammed, J. A., Jarrett, E., et al. (2015). Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity 42, 356–366. doi: 10.1016/j.immuni.2015.01.008
Kim, Y. G., Udayanga, K. G., Totsuka, N., Weinberg, J. B., Nunez, G., and Shibuya, A. (2014). Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell Host Microbe 15, 95–102. doi: 10.1016/j.chom.2013.12.010
Koh, A. Y., Kohler, J. R., Coggshall, K. T., Van Rooijen, N., and Pier, G. B. (2008). Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog. 4:e35. doi: 10.1371/journal.ppat.0040035
Krause, J., Geginat, G., and Tammer, I. (2015). Prostaglandin E2 from Candida albicans stimulates the growth of Staphylococcus aureus in mixed biofilms. PLoS ONE 10:e0135404. doi: 10.1371/journal.pone.0135404
Kundu, G., and Noverr, M. C. (2011). Exposure to host or fungal PGE(2) abrogates protection following immunization with Candida-pulsed dendritic cells. Med. Mycol. 49, 380–394. doi: 10.3109/13693786.2010.532514
Lagree, K., and Mitchell, A. P. (2017). Fungal biofilms: inside out. Microbiol. Spectr. 5:FUNK-0024-2016. doi: 10.1128/microbiolspec.FUNK-0024-2016
Leonardi, I., Li, X., Semon, A., Li, D., Doron, I., Putzel, G., et al. (2018). CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236. doi: 10.1126/science.aao1503
Ma, H., Wan, S., and Xia, C. Q. (2016). Immunosuppressive CD11b+Ly6Chi monocytes in pristane-induced lupus mouse model. J. Leukoc. Biol. 99, 1121–1129. doi: 10.1189/jlb.3A0415-158R
MacCallum, D. M. (2010). “Candida infections and modelling disease,” in Pathogenic Yeasts, eds R. Ashbee and E. M. Bignell (Berlin: Springer-Verlag, 41–67.
Mamouei, Z., Zeng, G., Wang, Y. M., and Wang, Y. (2017). Candida albicans possess a highly versatile and dynamic high-affinity iron transport system important for its commensal-pathogenic lifestyle. Mol. Microbiol. 106, 986–998. doi: 10.1111/mmi.13864
Martin, S. A., Brash, A. R., and Murphy, R. C. (2016). The discovery and early structural studies of arachidonic acid. J. Lipid Res. 57, 1126–1132. doi: 10.1194/jlr.R068072
Medeiros, A., Peres-Buzalaf, C., Fortino Verdan, F., and Serezani, C. H. (2012). Prostaglandin E2 and the suppression of phagocyte innate immune responses in different organs. Mediators Inflamm. 2012:327568. doi: 10.1155/2012/327568
Mishra, N. N., Ali, S., and Shukla, P. K. (2014). Arachidonic acid affects biofilm formation and PGE2 level in Candida albicans and non-albicans species in presence of subinhibitory concentration of fluconazole and terbinafine. Braz. J. Infect. Dis. 18, 287–293. doi: 10.1016/j.bjid.2013.09.006
Nguyen, L. N., Trofa, D., and Nosanchuk, J. D. (2009). Fatty acid synthase impacts the pathobiology of Candida parapsilosis in vitro and during mammalian infection. PLoS ONE 4:e8421. doi: 10.1371/journal.pone.0008421
Noble, S. M., French, S., Kohn, L. A., Chen, V., and Johnson, A. D. (2010). Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet. 42, 590–598. doi: 10.1038/ng.605
Noble, S. M., and Johnson, A. D. (2005). Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryotic Cell 4, 298–309. doi: 10.1128/EC.4.2.298-309.2005
Noverr, M. C., Phare, S. M., Toews, G. B., Coffey, M. J., and Huffnagle, G. B. (2001). Pathogenic yeasts Cryptococcus neoformans and Candida albicans produce immunomodulatory prostaglandins. Infect. Immun. 69, 2957–2963. doi: 10.1128/IAI.69.5.2957-2963.2001
Noverr, M. C., Toews, G. B., and Huffnagle, G. B. (2002). Production of prostaglandins and leukotrienes by pathogenic fungi. Infect. Immun. 70, 400–402. doi: 10.1128/IAI.70.1.400-402.2002
Nucci, M., and Anaissie, E. (2001). Revisiting the source of candidemia: skin or gut? Clin. Infect. Dis. 33, 1959–1967. doi: 10.1086/323759
Odds, F. C. (1987). Candida infections: an overview. Crit. Rev. Microbiol. 15, 1–5. doi: 10.3109/10408418709104444
Odds, F. C., Davidson, A. D., Jacobsen, M. D., Tavanti, A., Whyte, J. A., Kibbler, C. C., et al. (2006). Candida albicans strain maintenance, replacement, and microvariation demonstrated by multilocus sequence typing. J. Clin. Microbiol. 44, 3647–3658. doi: 10.1128/JCM.00934-06
Pande, K., Chen, C., and Noble, S. M. (2013). Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat. Genet. 45, 1088–1091. doi: 10.1038/ng.2710
Perez, J. C., Kumamoto, C. A., and Johnson, A. D. (2013). Candida albicans commensalism and pathogenicity are intertwined traits directed by a tightly knit transcriptional regulatory circuit. PLoS Biol. 11:e1001510. doi: 10.1371/journal.pbio.1001510
Pierce, J. V., and Kumamoto, C. A. (2012). Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. MBio 3:e00117-12. doi: 10.1128/mBio.00117-12
Pradhan, A., Avelar, G. M., Bain, J. M., Childers, D. S., Larcombe, D. E., Netea, M. G., et al. (2018). Hypoxia promotes immune evasion by triggering β-glucan masking on the Candida albicans cell surface via mitochondrial and cAMP-protein kinase A signaling. MBio 9:e01318-18. doi: 10.1128/mBio.01318-18
Rosenbach, A., Dignard, D., Pierce, J. V., Whiteway, M., and Kumamoto, C. A. (2010). Adaptations of Candida albicans for growth in the mammalian intestinal tract. Eukaryotic Cell 9, 1075–1086. doi: 10.1128/EC.00034-10
Russell, C., and Lay, K. M. (1973). Natural history of Candida species and yeasts in the oral cavities of infants. Arch. Oral Biol. 18, 957–962. doi: 10.1016/0003-9969(73)90176-3
Sasse, C., and Morschhauser, J. (2012). Gene deletion in Candida albicans wild-type strains using the SAT1-flipping strategy. Methods Mol. Biol. 845, 3–17. doi: 10.1007/978-1-61779-539-8_1
Scott, C. L., Bain, C. C., Wright, P. B., Sichien, D., Kotarsky, K., Persson, E. K., et al. (2015). CCR2(+)CD103(-) intestinal dendritic cells develop from DC-committed precursors and induce interleukin-17 production by T cells. Mucosal Immunol. 8, 327–339. doi: 10.1038/mi.2014.70
Sem, X., Le, G. T., Tan, A. S., Tso, G., Yurieva, M., Liao, W. W., et al. (2016). β-glucan exposure on the fungal cell wall tightly correlates with competitive fitness of Candida species in the mouse gastrointestinal tract. Front. Cell. Infect. Microbiol. 6:186. doi: 10.3389/fcimb.2016.00186
Serezani, C. H., Kane, S., Medeiros, A. I., Cornett, A. M., Kim, S. H., Marques, M. M., et al. (2012). PTEN directly activates the actin depolymerization factor cofilin-1 during PGE2-mediated inhibition of phagocytosis of fungi. Sci. Signal 5:ra12. doi: 10.1126/scisignal.2002448
Shiraki, Y., Ishibashi, Y., Hiruma, M., Nishikawa, A., and Ikeda, S. (2008). Candida albicans abrogates the expression of interferon-gamma-inducible protein-10 in human keratinocytes. FEMS Immunol. Med. Microbiol. 54, 122–128. doi: 10.1111/j.1574-695X.2008.00457.x
Smeekens, S. P., van de Veerdonk, F. L., van der Meer, J. W., Kullberg, B. J., Joosten, L. A., and Netea, M. G. (2010). The Candida Th17 response is dependent on mannan- and beta-glucan-induced prostaglandin E2. Int. Immunol. 22, 889–895. doi: 10.1093/intimm/dxq442
Solis, N. V., and Filler, S. G. (2012). Mouse model of oropharyngeal candidiasis. Nat. Protoc. 7, 637–642. doi: 10.1038/nprot.2012.011
Sparber, F., Dolowschiak, T., Mertens, S., Lauener, L., Clausen, B. E., Joller, N., et al. (2018). Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS Pathog. 14:e1007069. doi: 10.1371/journal.ppat.1007069
Suram, S., Silveira, L. J., Mahaffey, S., Brown, G. D., Bonventre, J. V., Williams, D. L., et al. (2013). Cytosolic phospholipase A(2)alpha and eicosanoids regulate expression of genes in macrophages involved in host defense and inflammation. PLoS ONE 8:e69002. doi: 10.1371/journal.pone.0069002
Szkudlinski, J. (2000). Occurrence of prostaglandins and other eicosanoids in parasites and their role in host-parasite interaction. Wiad. Parazytol. 46, 439–446.
Tan, T. G., Lim, Y. S., Tan, A. S., Leong, R., and Pavelka, N. (2018). Fungal symbionts produce prostaglandin E2 to promote their intestinal colonization. bioRxiv [preprint]. doi: 10.1101/477117
Tsitsigiannis, D. I., Bok, J. W., Andes, D., Nielsen, K. F., Frisvad, J. C., and Keller, N. P. (2005). Aspergillus cyclooxygenase-like enzymes are associated with prostaglandin production and virulence. Infect. Immun. 73, 4548–4559. doi: 10.1128/IAI.73.8.4548-4559.2005
Valdez, P. A., Vithayathil, P. J., Janelsins, B. M., Shaffer, A. L., Williamson, P. R., and Datta, S. K. (2012). Prostaglandin E2 suppresses antifungal immunity by inhibiting interferon regulatory factor 4 function and interleukin-17 expression in T cells. Immunity 36, 668–679. doi: 10.1016/j.immuni.2012.02.013
Weisser, S. B., van Rooijen, N., and Sly, L. M. (2012). Depletion and reconstitution of macrophages in mice. J. Vis. Exp. e4105. doi: 10.3791/4105
White, S. J., Rosenbach, A., Lephart, P., Nguyen, D., Benjamin, A., Tzipori, S., et al. (2007). Self-regulation of Candida albicans population size during GI colonization. PLoS Pathog. 3:e184. doi: 10.1371/journal.ppat.0030184
Yano, J., and Fidel, P. L. Jr. (2011). Protocols for vaginal inoculation and sample collection in the experimental mouse model of Candida vaginitis. J. Vis. Exp. 8:3382. doi: 10.3791/3382
Yao, C., Sakata, D., Esaki, Y., Li, Y., Matsuoka, T., Kuroiwa, K., et al. (2009). Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 15, 633–640. doi: 10.1038/nm.1968
Yun, B., Lee, H., Jayaraja, S., Suram, S., Murphy, R. C., and Leslie, C. C. (2016). Prostaglandins from cytosolic phospholipase A2alpha/cyclooxygenase-1 pathway and mitogen-activated protein kinases regulate gene expression in Candida albicans-infected macrophages. J. Biol. Chem. 291, 7070–7086. doi: 10.1074/jbc.M116.714873
Keywords: prostaglandin E2, Candida albicans, symbiont, host-microbe interactions, phagocyte, virulence, arachidonic acid
Citation: Tan TG, Lim YS, Tan A, Leong R and Pavelka N (2019) Fungal Symbionts Produce Prostaglandin E2 to Promote Their Intestinal Colonization. Front. Cell. Infect. Microbiol. 9:359. doi: 10.3389/fcimb.2019.00359
Received: 24 August 2019; Accepted: 03 October 2019;
Published: 18 October 2019.
Edited by:
Robert T. Wheeler, University of Maine, United StatesReviewed by:
Ana Traven, Monash University, AustraliaCarolina Henritta Pohl, University of the Free State, South Africa
Copyright © 2019 Tan, Lim, Tan, Leong and Pavelka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norman Pavelka, bm9ybWFucGF2ZWxrYUBnbWFpbC5jb20=; Tze Guan Tan, dGFuX3R6ZV9ndWFuQGltbXVub2wuYS1zdGFyLmVkdS5zZw==
†deceased