 Sandra Appelt1
Sandra Appelt1 Kristin Köppen2Aleksandar Radonić3Oliver Drechsel4
Kristin Köppen2Aleksandar Radonić3Oliver Drechsel4 Daniela Jacob1
Daniela Jacob1 Roland Grunow1
Roland Grunow1 Klaus Heuner2*
Klaus Heuner2*- 1Centre for Biological Threats and Special Pathogens (ZBS2), Robert Koch Institute, Berlin, Germany
- 2Working Group Cellular Interactions of Bacterial Pathogens, ZBS2, Robert Koch Institute, Berlin, Germany
- 3Methodology and Research Infrastructure Genome Sequencing (MF2), Robert Koch Institute, Berlin, Germany
- 4Bioinformatics (MF1), Robert Koch Institute, Berlin, Germany
Francisella tularensis is an intracellular pleomorphic bacterium and the causative agent of tularemia, a zoonotic disease with a wide host range. Among the F. tularensis subspecies, especially F. tularensis subsp. holarctica is of clinical relevance for European countries. The study presented herein focuses namely on genetic diversity and spatial segregation of F. tularensis subsp. holarctica in Germany, as still limited information is available. The investigation is based on the analysis of 34 F. tularensis subsp. holarctica isolates and one draft genome from an outbreak strain. The isolates were cultured from sample material being that of primarily human patients (n = 25) and free-living animals (n = 9). For six of 25 human isolates, epidemiological links between disease onset and tick bites could be established, confirming the importance of arthropod linked transmission of tularemia in Germany. The strains were assigned to three of four major F. tularensis subsp. holarctica clades: B.4, B.6, and B.12. Thereby, B.6 and B.12 clade members were predominantly found; only one human isolate was assigned to clade B.4. Also, it turned out that eight isolates which caused pneumonia in patients clustered into the B.6 clade. Altogether, eight different final subclades were assigned to clade B.6 (biovar I, erythromycin sensitive) and six to B.12 (biovar II, erythromycin resistant) in addition to one new final B.12 subclade. Moreover, for 13 human and 3 animal isolates, final subclade subdivisions were not assigned (B.12 subdivisions B.33 and B.34, and B.6 subdivision B.45) because official nomenclatures are not available yet. This gives credit to the genetic variability of F. tularensis subsp. holarctica strains in Germany. The results clearly point out that the given genetic diversity in Germany seems to be comparably high to that found in other European countries including Scandinavian regions. A spatial segregation of B.6 and B.12 strains was found and statistically confirmed, and B.12 clade members were predominantly found in eastern parts and B.6 members more in western to southern parts of Germany. The portion of B.12 clade members in northeastern parts of Germany was 78.5% and in southwestern parts 1.9%.
Introduction
Francisella tularensis is a small, intracellular, non-motile, Gram-negative pleomorphic bacterium and the causative agent of tularemia, a zoonotic disease with a wide range of hosts (mammals, birds, amphibians, fishes, and invertebrates) (Ellis et al., 2002; Maurin and Gyuranecz, 2016; Schulze et al., 2016). Two F. tularensis subspecies are of clinical relevance: F. tularensis subspecies tularensis (Jellison Type A) and F. tularensis subsp. holarctica (Jellison Type B). F. tularensis subsp. tularensis is prevalent in North America, whereas the subspecies holarctica is found all over the northern hemisphere. The subtype A2 of subspecies tularensis is described to be more virulent than F. tularensis subsp. holarctica (Jellison, 1961; Farlow et al., 2005; Vogler et al., 2009a; Molins et al., 2014; Dwibedi et al., 2016). In Germany, F. tularensis subsp. holarctica is the only Francisella subspecies which is known to cause disease in both animals and humans. Recently, one additional Francisella species (Isolate W12-1067) has been identified in Germany, yet pathogenicity needs to be evaluated (Rydzewski et al., 2014; Faber et al., 2018). However, F. tularensis subsp. holarctica originates from North America or Asia from where the bacteria spread (Vogler et al., 2009a; Karlsson et al., 2013; Dwibedi et al., 2016; Hestvik et al., 2018). Additionally, it was proposed that within the postulated spread of the pathogen from east to west, Germany might be a “melting pot,” a region where strains are mixed, reassorted, and give rise to further variants (Jusatz, 1952, 1961; Faber et al., 2018). Also, phylogenetic studies have already revealed a spread of the pathogen from Scandinavia to the southern parts of Europe (Karlsson et al., 2013; Dwibedi et al., 2016).
The minimal number of bacteria needed to cause an infection in humans depends on the route of infection. Intradermal and inhalational tularemia can already be caused by 10–25 bacteria (Saslaw et al., 1961; Jones et al., 2005). Primary infection sources for humans are free-living lagomorphs (hares and rabbits), other mammals, animal carcasses, and insects (mosquitoes and ticks) and the environment (water, dust, aerosol, and soil) (Oyston and Griffiths, 2009; Maurin and Gyuranecz, 2016). A broad host species diversity was also reported in Germany (Schulze et al., 2016), and especially hunters bear a high risk of getting infected by skinning, preparing, or consuming meat of infected hares. The high rate of seropositive animals in Germany indicated that the frequency as well as the occurrence of the pathogen in the environment and wild animals might be underestimated (Jenzora et al., 2008; Gehringer et al., 2013; Kuehn et al., 2013; Muller et al., 2013; Otto et al., 2014). There might be also a high diversity of different F. tularensis subsp. holarctica strains in northeastern parts of Germany (Antwerpen et al., 2015; Schulze et al., 2016; Faber et al., 2018). For distinguishing between F. tularensis subsp. holarctica strains which display within its subspecies little genetic variation, canonical single-nucleotide polymorphisms (canSNPs) can be used (Svensson et al., 2009a; Vogler et al., 2009b; Karlsson et al., 2013; Dwibedi et al., 2016). Based on this analysis and an erythromycin-resistant/erythromycin-sensitive phenotype and genotype, F. tularensis subsp. holarctica can be subdivided into two biovars (biovar I and biovar II) and four major clades: B.4, B.6, B.12, and B.16 (Vogler et al., 2009b; Karlsson et al., 2013). These clades can be subdivided further into subclades. The subdivision into different clades and subclades is so far not performed consistently. For instance, B.12 subclade B.75 is designated as subclade and by others as clade. However, an up-to-date typing scheme, also used in this study, was recently published (Wittwer et al., 2018).
Spatial segregation of clades predominantly found in Europe (B.6 and B.12) has already been reported (Gyuranecz et al., 2012), pointing out that B.6 is primarily found in western parts of Europe and B.12 in central to eastern parts (Koene et al., 2019). Both clades are postulated to display differences in pathogenicity in lagomorphs (Origgi et al., 2014; Origgi and Pilo, 2016; Kreizinger et al., 2017; Hestvik et al., 2018). B.6 and B.12 clade members exhibit also a different resistance to erythromycin due to a mutation in the rrl gene (Kudelina and Olsufiev, 1980; Karlsson et al., 2016). As mentioned above, B.6 clade members are sensitive to erythromycin and B.12 clade members are resistant (Kudelina and Olsufiev, 1980; Karlsson et al., 2016).
The objective of the study presented herein was to enhance our understanding about the genetic diversity of B.6 and B.12 clade members in Germany with a specific focus on human isolates. Also, the geographical distribution pattern of B.6 and B.12 clade members was investigated. To this end, the genomes of 34 F. tularensis subsp. holarctica isolated from mainly human and animal hosts were sequenced and compared by computational analysis based on phylogenetic constructions and canSNP analysis. The analysis also includes a draft genome of a F. tularensis subsp. holarctica strain which has caused an outbreak in Germany recently (Jacob et al., 2019).
Materials and Methods
Bacterial Isolates
A total of 34 F. tularensis subsp. holarctica isolates from Germany were investigated, including 25 bacterial isolates from human specimens in addition to 9 isolates from samples collected from free-living animals (wild boar, raccoon dog, fox, and hare, Table 1). All bacterial strains were isolated or received from third parties to the German National Consultant Laboratory for Tularemia in human medicine between 2007 and 2018. For the isolation of Francisella from different sample materials, species and subspecies identification routine diagnostic tools were applied (Broekhuijsen et al., 2003; Versage et al., 2003; Jacob et al., 2011).
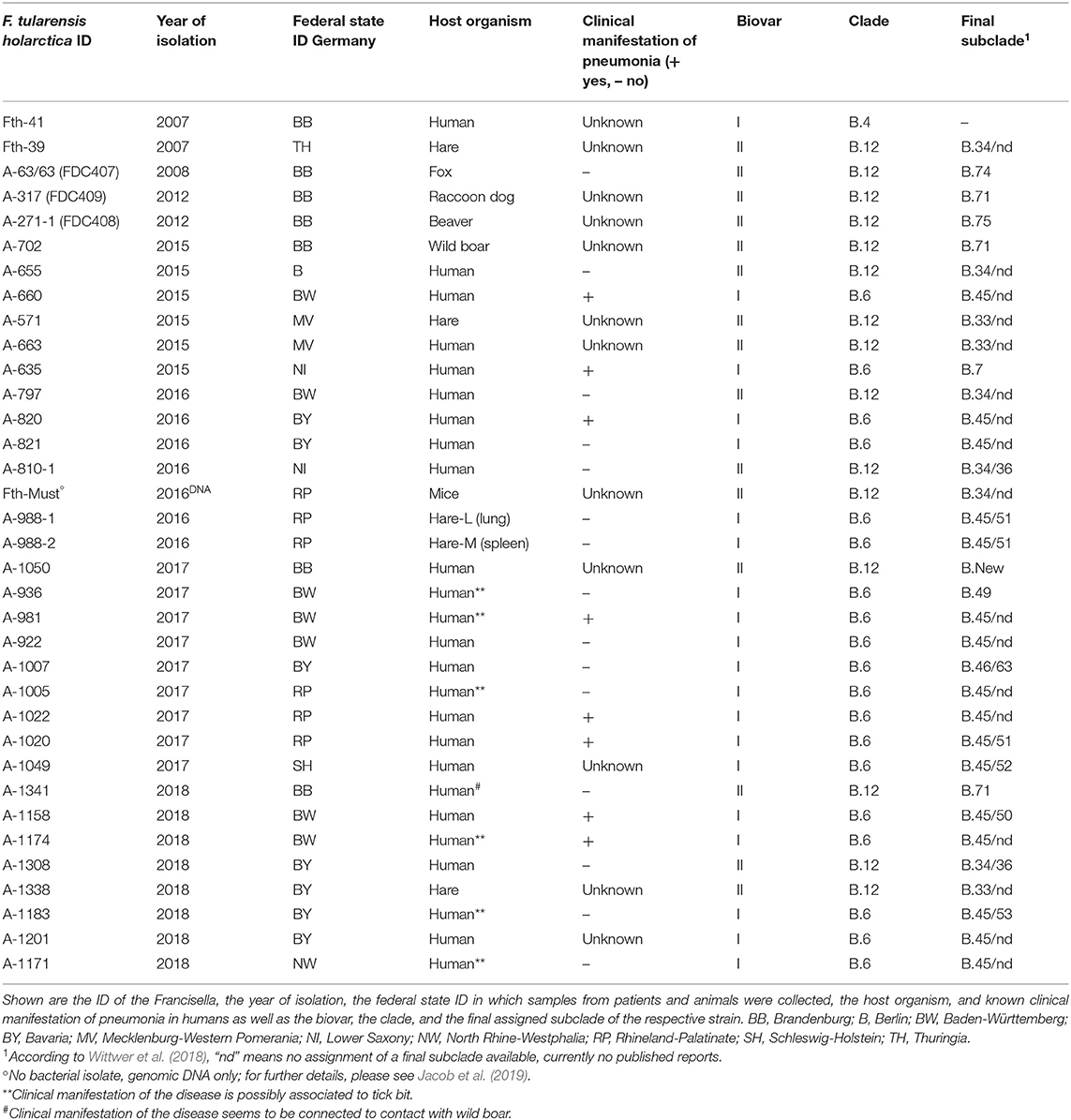
Table 1. Overview on investigated Francisella tularensis subsp. holarctica genomes from Germany.
In addition, six isolates from different European countries were included: one F. tularensis subsp. holarctica strain from Lithuania (Fth-40), three F. tularensis subsp. holarctica strains (Fth-34, Fth-35, and Fth-38), and two recently isolated from hares in Austria and two isolated from ticks in Switzerland (A-328-25 and A-328-2). The isolate Fth-40 was obtained from the Lithuanian National Public Health Surveillance Laboratory. The isolates Fth-34, Fth-35, and Fth-38 were received from Germany's Federal Institute for Risk Assessment, and the tick isolates provided by the Spiez Laboratory, Bacteriology Branch, Switzerland.
Antimicrobial Susceptibility Testing (AST) of Bacterial Isolates and In silico Analysis
To collect information about antimicrobial resistances of bacterial isolates to erythromycin, 26 of 34 isolates were tested using the disk diffusion method (n = 17) or the microdilution methods (n = 16). The disk diffusion method was performed for erythromycin using only two different agar plates: Mueller–Hinton agar plates with 5% sheep blood (Becton Dickinson GmbH, Heidelberg, Germany) and chocolate plates (Oxoid, Munich, Germany). The microdilution method was performed in compliance with the Clinical and Laboratory Standards Institute (CLSI) standards (Clinical Laboratory Standards Institute, 2011). For the interpretation of minimum inhibitory concentration (MIC) values, determined MICs are in general compared to defined clinical breakpoint standards. Yet, for F. tularensis subsp. holarctica, officially released breakpoints for erythromycin, are so far not available. Therefore, MIC values higher than 16 μg/ml were interpreted as resistant, corresponding to results obtained by phylogenetic analysis of genomes. Recommendations provided in World Health Organization (2007) guidelines on tularemia were followed.
For 7 of 34 F. tularensis subsp. holarctica isolates, the erythromycin resistance was assigned in silico according to Karlsson et al. (2016) exclusively. The erythromycin resistance of the F. tularensis subsp. holarctica isolate Fth-41 was not investigated since this is an isolate not clustering into the B.6 or B.12 clade. The outcome of the tests was compared to results obtained by phylogenetic analysis of genomes.
Recovery of Genomic DNA for Genome Sequencing From Bacterial Isolates
DNA extraction was performed out of bacterial colony material using the QIAGEN DNeasy Blood and Tissue Kit (Hilden, Germany) following the manufacturer's instructions. DNA elution was performed in 100 μl of QIAGEN Elution Buffer (Hilden, Germany).
Genome Sequencing
DNA quantification was performed with the Qubit™ 4 fluorometer (Invitrogen by Thermo Fisher Scientific) using the Qubit dsDNA HS assay kit (Life Technologies, Darmstadt, Germany). To generate the libraries, the NextEra XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) was used; the library normalization step described in the manufacturer's instructions was thereby skipped. For the estimation of the DNA fragment sizes of the libraries, the Agilent 2100 Bioanalyzer was used (Agilent Technologies, Waldbronn, Germany) utilizing the High-Sensitivity DNA Analysis Kit (Agilent Technologies, Waldbronn, Germany) and electrophoresis DNA chips.
Library pool sequencing was performed in paired-end mode on a MiSeq instrument (Illumina, San Diego, CA, USA) as previously described (Jacob et al., 2019). All genome sequences have been uploaded to the European Nucleotide Archive (ENA: www.ebi.ac.uk/ena). The BioProject ID is PRJEB33006; IDs of single data sets are provided in Supplementary Table S1.
Computational Analysis and Phylogenetic Classification
For quality trimming and adapter clipping of Illumina raw data, an in-house pipeline QCumber was used. The pipeline comprises the following tools: FastQC version 0.11 (Andrews, 2014), trimmomatic version 0.36 (Bolger et al., 2014), and KRAKEN (Wood and Salzberg, 2014). The mapping was performed with Bowtie version 2.3 using default setting parameters (Langmead, 2010). F. tularensis subsp. holarctica LVS [National Center for Biotechnology Information (NCBI) reference: NC_007880.1] was used as the reference genome for the assembly of draft genomes (Barabote et al., 2009). BAM files were uploaded into Geneious version R9.3 (Kearse et al., 2012) for further analysis. The consensus sequences of genomes were extracted and aligned using a progressive Mauve alignment for collinear genomes applying the Muscle (version 3.6; Edgar, 2004) alignment algorithm. The alignment was used for determining canSNPs (Svensson et al., 2009b; Vogler et al., 2009a; Karlsson et al., 2013) and for phylogenetic constructions. The phylogenetic tree was constructed based on entire genome sequences and in addition for comparison also on sequences of Francisella pathogenicity islands (FPIs) only. To generate the phylogenetic tree, the neighbor joining method for clustering was used, applying a bootstrap of 100 (Saitou and Nei, 1987). Reference genomes included in the phylogenetic reconstructions were F. tularensis subsp. holarctica OSU18 (NCBI reference: NC_017463.1) (Petrosino et al., 2006; Puiu and Salzberg, 2008), F. tularensis subsp. holarctica FSC162 (NCBI reference: PRJNA89145) (Karlsson et al., 2013), F. tularensis subsp. holarctica FSC200 (NCBI reference: NC_019551.1) (Svensson et al., 2012), F. tularensis subsp. holarctica LVS (NCBI reference: NC_007880.1) (Larsson et al., 2005), and FTNF002-00 (NCBI reference: NC_009749.1) (Haristoy et al., 2003; Barabote et al., 2009).
In addition, seven genomes of F. tularensis subsp. holarctica strains from different European countries (A-328-25, A-328-2, Fth-40, Fth-34; Fth-35, and Fth-38) were included. Genome sequences generated during the study have been uploaded to ENA (www.ebi.ac.uk/ena). The BioProject ID is PRJEB33006; IDs of single data sets are provided in Supplementary Table S1.
The Pearson chi-squared test with Yates's correction was applied to test if the geographical distribution of Francisella clades (clade B.12 vs. B.6) within northern and southern parts of Germany is possible (Pearson, 1900). The Yates (1934) correction was applied to prevent overestimation of statistical significance in the small dataset. To run the statistical computing, the free software R version 3.5.1 was used (Dessau and Pipper, 2008). To perform the assessment, Germany was geographically divided into a northeastern part (group 1) and a southwestern part (group 2). Group 1 comprised a total of 13 F. tularensis subsp. holarctica; of these, 11 were classified into clade B.12 and 2 were classified into clade B.6. Group 2 comprised a total of 21 F. tularensis subsp. holarctica; of these, 4 were classified into clade B.12 and 17 were classified into clade B.6.
Results
Genetic Diversity in Germany and Analysis of canSNP Analysis
The typing results could show that one out of 35 F. tularensis subsp. holarctica (Fth-41, Table 1) genomes clustered into clade B.4, next to the reference strain OSU18 (Figure 1). No other genome clustered into clade B.4. Altogether, 19 genomes (17 from humans and 2 from animals) were assigned to clade B.6 and 15 genomes to clade B.12 (seven from humans and eight from animals). Surprisingly, it turned out that all F. tularensis subsp. holarctica genomes associated to samples taken from patients with pneumonia (n = 8) clustered into clade B.6. Also, for six of 17 Francisella human isolates belonging to clade B.6, links between the onset of tularemia in patients and tick bites could be established (Table 1). No link between onset of tularemia and tick bites could be established for any human isolate which was assigned to clade B.12.
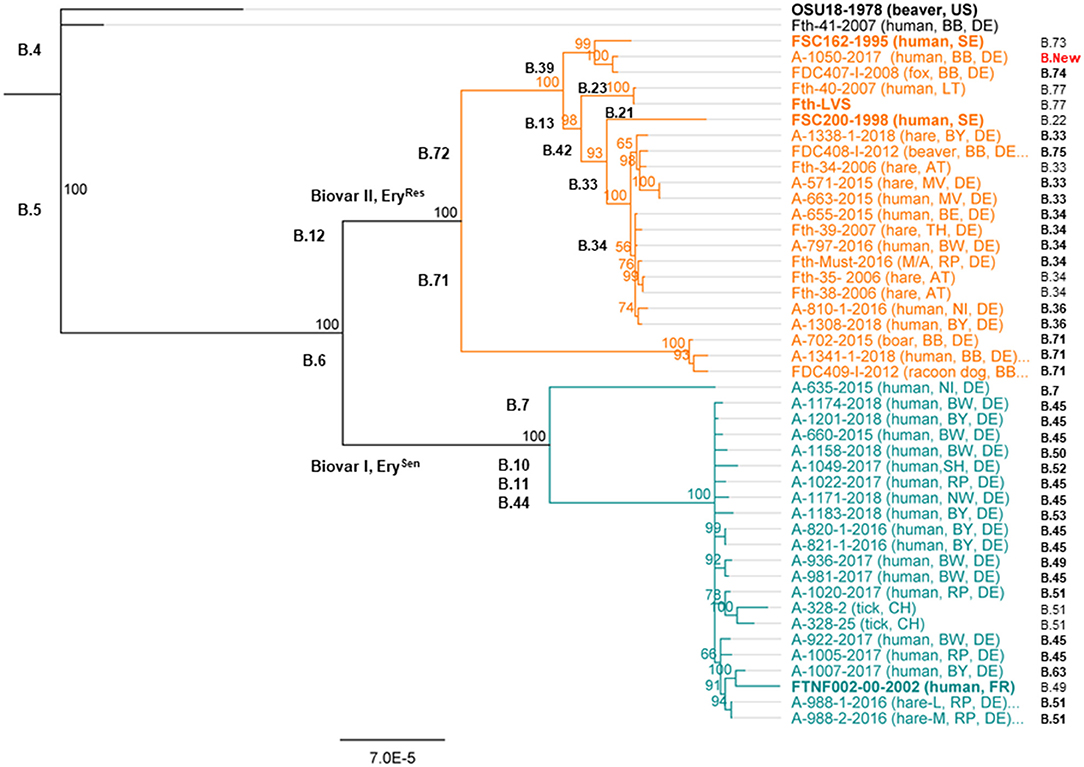
Figure 1. Phylogenetic relationship of Francisella tularensis subsp. holarctica in Germany. The phylogenetic analysis was based on a Mauve alignment for collinear genomes, and for the clustering, the neighbor joining bootstrap method (Fth OSU18 as an out-group) was chosen. Outlined for each genome are the identifier of the investigated Francisella and the year of sampling; the host organism (human or animal) and the sampling spot are indicated by the identifier of Germany's federal states. Also, the different Francisella clades are given in addition to the lowest assignable subclade (final subclade) for each genome. Also, reference genomes were included in the analysis; these genomes are highlighted in bold. These Francisella isolates come from different countries including the United States (US), France (FR), Lithuania (LT), Austria (AT), Switzerland (CH), and Sweden (SE). Germany's federal states, BB, Brandenburg; B, Berlin; BW, Baden-Württemberg; BY, Bavaria; MV, Mecklenburg-Western Pomerania; NI, Lower Saxony; NW, North Rhine-Westphalia; RP, Rhineland-Palatinate; SH, Schleswig-Holstein; TH, Thuringia.
For clade B.6, a total of eight different lowest assignable subclades (final subclades) were determined. The dominating final subclade was B.45, followed by the final subclades B. 51, B.49, and B.63. In addition, for 10 human isolates and the draft genome of the outbreak strain (Fth-Must), a final subclade subdivision of B.45 was not assigned as no official nomenclature is available yet (Table 1).
Referring to clade B.12, six final subclades were identified, predominantly Francisella belonging to subclade B.33 followed by subclade B.71. In addition, a new B.12 subclade of branch B.39 could be identified, namely, B.39-New (Figure 1). For three human isolates and three animal isolates, a final subclade of subdivisions B.33 and B.34 was not assigned as no official nomenclature is available. No correlation between clade or subclades and different hosts could be identified (Table 1).
To test the reproducibility of results, biological genome duplicates were included. The results could show that these duplicates, clustered identically: A-821 and A-820, as well as A-988-2 and A-988-1 (Figure 1). The samples A-988-2 and A-988-1 were sampled from the same hare (Table 1), one from a sample taken from the spleen and one lung sample. The isolates A-821 and A-820 were both obtained from specimens coming from the same patient, taken at different time points of the infection. Interestingly, a third cluster of two F. tularensis subsp. holarctica genomes was identified. One genome from a dead hare isolate (A-663) clustered next to one human isolate (A-571) (Figure 1). Both genomes were, in terms of SNPs, identical. Furthermore, the F. tularensis subsp. holarctica genome obtained from a hunter (A-1341) grouped next to a genome from an isolate obtained from a wild boar (A-702). The wild boar Francisella isolate was obtained in 2015, and the hunter was infected in the same region by a wild boar in 2018. One isolate [A-317 (FDC-409)] from a raccoon dog hunted in the same region [Brandenburg (BB)] was also found to cluster in B.71 (Schulze et al., 2016), together with an isolate obtained from the hunter (A-1341) (Figure 1).
Phylogenetic constructions performed herein were based on comparison of entire genomes among each other. Analysis performed on the entire genomes was also performed on selected sequences, being those of FPIs only. The analysis was performed to gather more information on minimal input of sequence needed for phylogenetic analysis allowing drawing of correct conclusions of Francisella biovars, clades, and subclades (Supplementary Figure S1). It turned out that to a certain point, comparable results were achieved by relying on selected sequences only. The Francisella grouped into the same main clades, comprising B.4, B.6, and B.12. However, the informative and discriminative value of FPIs beyond the classification into main clades and some subclades (B.71, B.72, and B.42) seems compromised, as the assignment of other subclades could not be performed properly (Supplementary Figure S1).
Erythromycin Resistances of Clades B.6 and B.12
The biovar typical erythromycin resistance could be confirmed by laboratory tests (microdilution method) and by in silico analysis for all F. tularensis subsp. holarctica strains tested. The disk diffusion methods as well as the microdilution method, yet applied without predetermined comparative clinical breakpoint values, yielded consistent results. The results obtained by phylogenetic analysis were confirmed.
Geographical Distribution of Clades B.6 and B.12 in Germany
During the investigation, differences of geographical distribution of F. tularensis subsp. holarctica clade B.6 and clade B.12 in Germany were specifically searched. A pure, perfect pattern could not be identified. However, striking the higher portion of B.12 clade members in northeastern parts of Germany, a total of 78.5% of strains of the region were assigned to clade B.12 [(group 1) 11 of 14 genomes, Figure 2], whereas in southwestern parts, the portion of B.12 clade members was 1.9% [(group 2) 4 of 21 genomes, Figure 2]. The possible geographical segregation between northeastern (group 1) and southwestern parts (group 2) of Germany is indicated by a dashed line in Figure 2. To test if the distribution of Francisella clades (B.12 vs. B.6 clades) within Germany is different, Pearson's chi-squared test with Yates's correction was applied. The x-squared was determined to 11.468 and the p-value to 0.000707, showing indeed that the distribution of Francisella clades in both groups is different. In fact, the results indicate that a geographical segregation in Germany seems to be highly likely; still further confirmation is required by testing larger sample sizes. In these lines, it could be shown that B.6 members are primarily found in southwestern parts and B.12 clade members in northeastern parts of Germany (Figure 2). F. tularensis subsp. holarctica clade B.6 was primarily found in Rhineland-Palatinate (RP), Bavaria (BY), and Baden-Württemberg (BW), whereas clade B.12 members were predominantly found in northeast Germany [Mecklenburg-Western Pomerania (MV), Brandenburg (BB), and Berlin (B)] (Figure 2). Additionally, it turned out that B.12 clade members were assigned to two additional regions [Thuringia (TH) and Lower Saxony (NI)] and B.6 members to three additional regions in Germany [North Rhine-Westphalia (NW), Schleswig-Holstein (SH), and Lower Saxony (NI)].
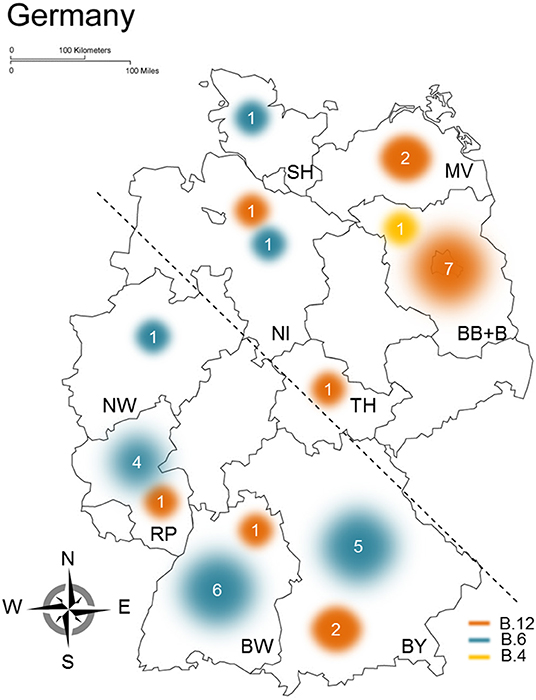
Figure 2. Geographical distribution of clades B.6 and B.12 in Germany. The results gathered from the analysis of different Francisella tularensis subsp. holarctica genomes are shown, outlined are assigned Francisella clades (yellow, B.4; orange, B.12; blue, B.6), and the sample size clustering in the respective clades is proportional to circles used for illustrating the distribution of clades in Germany. The different federal states in Germany are indicated by identifiers, BB, Brandenburg; B, Berlin; BW, Baden-Württemberg; BY, Bavaria; MV, Mecklenburg-Western Pomerania; NI, Lower Saxony; NW, North Rhine-Westphalia; RP, Rhineland-Palatinate; SH, Schleswig-Holstein; TH, Thuringia. For statistical evaluations, Germany was split into two parts (northeastern and southwestern) indicated by the dashed line.
Discussion
Tularemia is a rarely reported but reemerging infectious disease in Germany (Kaysser et al., 2008; Splettstoesser et al., 2009; Faber et al., 2018). A recent review has outlined aspects of the genetic diversity, epidemiological situation, and surveillance data of tularemia in Germany (Faber et al., 2018). The objective herein was to focus tighter on elucidating the genetic diversity of F. tularensis subsp. holarctica strains in Germany, with a main focus on human isolates classified into clades B.6 and B.12.
Thirty-five genomes were included in our analysis: 34 F. tularensis subsp. holarctica genomes of strains isolated mainly from humans and animal hosts and 1 draft genome from an outbreak (Fth-Must) (Jacob et al., 2019). Included in the panel are also two sets of biological duplicates (A-988-1 and A-998-2; A-820 and A-821) which clustered in the phylogenetic tree next to each other and were in terms of canSNPs identical. These findings indicate that no bias was introduced during the analysis. Also, one F. tularensis subsp. holarctica genome obtained from a strain isolated from a wild hare (A-663) clustered in the phylogenetic tree together with an isolate from a human patient (A-571). Both genomes were, in terms of SNPs, identical (Figure 1), and based on the patient report, it seems highly possible that there is an epidemiological link between both cases.
Of the 35 genomes, 34 clustered into the B.6 and B.12 clades and one genome was assigned to clade B.4. That only one of 35 analyzed genomes clustered into the B.4 clade was not surprising, as, in Europe, strains of clades B.6 and B.12 are dominant (Gyuranecz et al., 2012) and known to be present in Germany (Muller et al., 2013; Tomaso et al., 2017, 2018). The erythromycin susceptibility of biovar I (B.6 clade), as well as the erythromycin resistance of biovar II (B.12 clade), was confirmed by both experimental and in silico analyses. The findings are in compliance with results obtained by others showing that experimental results obtained by different means, e.g., AST using the microdilution method or disk diffusion method, can confirm in silico data based on phylogenetic reconstructions (Tomaso et al., 2017) or in silico assessment of erythromycin resistances by specifically investigating the rrl gene (Karlsson et al., 2016).
Besides, it turned out that all F. tularensis subsp. holarctica genomes associated to samples taken from patients with pneumonia clustered in this study into clade B.6. In hares, it was recently reported that pneumonia is significantly more common in B.6 than in B.12 cases (Koene et al., 2019). However, referring to human cases, different clinical manifestations are known to be caused by both clades (Johansson et al., 2014; Afset et al., 2015). To investigate a possible biovar-associated manifestation of pneumonia in humans would be of importance for public health matters, showing the need for analyzing genetic diversity and phylogeny of Francisella. In addition, the ratio of putative tick-borne tularemia in clade B.6 was surprisingly high, but this finding needs to be corroborated with more data. The ratio of almost 1:2 (8 out of 19) underlines the importance of tularemia transmitted by arthropods in Germany (Gehringer et al., 2013; Boone et al., 2015; Borde et al., 2017).
The study could clearly emphasize that a geographical segregation or clustering of F. tularensis subsp. holarctica in Germany is highly likely. Findings could show that clade B.12 members were more frequently found in northeastern parts of Germany and B.6 clade members in southwestern parts (Muller et al., 2013). A similar geographic distribution, meaning that biovar I is mainly found in Western Europe and biovar II in Northern and Eastern Europe, was already described (Kudelina, 1973; Ellis et al., 2002; Svensson et al., 2009a; Vogler et al., 2009a; Gyuranecz et al., 2012; Dwibedi et al., 2016; Karlsson et al., 2016; Origgi and Pilo, 2016; Faber et al., 2018).
Different to former analyses which were mainly based on F. tularensis subsp. holarctica isolates from brown hares, a broader host spectrum (humans and wild animals) was included next to a broader geographical scope covered (e.g., Berlin, Brandenburg) (Muller et al., 2013). But no correlation between host and subclade could be identified (Farlow et al., 2005; Pilo, 2018) as already described for F. tularensis subsp. tularensis.
Altogether, 14 different F. tularensis subsp. holarctica B.6 and B.12 final subclades were identified. For 15 isolates and the outbreak strain, the final B.12 and B.6 subclade subdivisions (subdivisions of B.33, B.34, and B.45) were not assigned because an official nomenclature is still lacking until today. Moreover, one new B.12 final subclade closely related to B.39 was identified, yet not defined. The identification of a new subclade distantly related to all other strains of subclade B.45 or B.33, and further two members of a recently identified new B.71 subcluster in Berlin-Brandenburg (Schulze et al., 2016) showed that there are still open gaps in Francisella phylogeny, still to be addressed by further analysis (Wittwer et al., 2018). In addition, these results show that the genetic diversity of F. tularensis subsp. holarctica strains in Germany seems to have been underestimated as initially thought (Gehringer et al., 2013; Muller et al., 2013; Schulze et al., 2016). There seems to be still room for discussions if a “sub-sub”-clades definition is needed for further phylogenetic analysis of F. tularensis subsp. holarctica. However, to find high genetic diversity gives credit to studies presuming that the diversity of tularemia in non-Scandinavian countries seems to be higher than initially expected (Chanturia et al., 2011; Gyuranecz et al., 2012; Gehringer et al., 2013; Muller et al., 2013; Antwerpen et al., 2015; Borde et al., 2017; Wittwer et al., 2018). The diversity seems even to be comparably high to Scandinavian countries known for being the source of the historical spread of the bacteria (Chanturia et al., 2011; Gyuranecz et al., 2012; Gehringer et al., 2013; Karlsson et al., 2013, 2016; Muller et al., 2013; Antwerpen et al., 2015; Schulze et al., 2016; Borde et al., 2017; Wittwer et al., 2018). For instance, a final B.6 subclade, namely, B.52, was reported to be found in Spain exclusively (Dwibedi et al., 2016) and now also assigned during the study for German Francisella isolates.
To conclude, the study presented herein represents a comprehensive analysis of F. tularensis subsp. holarctica strains recovered from both wild animals and human patients and is extending our current understanding about genotypic diversity of tularemia and spatial segregation in Germany.
Data Availability Statement
The datasets generated for this study can be found in the European Nucleotide Archive, BioProject ID: PRJEB33006.
Author Contributions
KH, RG, and DJ provided the expertise in the field of tularemia, and provided theoretical and practical advices. KH coordinated and supervised the present work. OD and AR performed whole-genome sequencing, quality trimming, and mapping. SA and KH analyzed the sequence data including the functional SNP analysis and phylogenetic constructions. RG, DJ, and SA were involved in the differential diagnostic performed on animal and human bacterial isolates as well as in the AST using the microdilution method on Francisella isolates. KK performed the AST using the disk diffusion method and analyzed the data. KH and SA drafted the manuscript. DJ and KK revised the manuscript critically. All authors contributed to the revision of manuscript and read and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the public health departments of the Federal States, as well as the physicians and veterinarians who kindly contributed F. tularensis samples or isolates for the study. We would like to acknowledge also the German public health department for providing information about the tularemia cases described herein. Additionally, we would like to thank Nathalie Fiedler, Silke Becker, and Petra Lochau for technical support. The German public health department had no role in the study design, data collection, interpretation, or decision to submit the work for publication.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00376/full#supplementary-material
References
Afset, J. E., Larssen, K. W., Bergh, K., Larkeryd, A., Sjodin, A., Johansson, A., et al. (2015). Phylogeographical pattern of Francisella tularensis in a nationwide outbreak of tularaemia in Norway, 2011. Euro Surveill. 20, 9–14. doi: 10.2807/1560-7917.ES2015.20.19.21125
Andrews, S. (2014). FastQC A Quality Control Tool for High Throughput Sequence Data. Version 0.11.5.2010. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Antwerpen, M. H., Prior, K., Mellmann, A., Hoppner, S., Splettstoesser, W. D., and Harmsen, D. (2015). Rapid high resolution genotyping of Francisella tularensis by whole genome sequence comparison of annotated genes (“MLST+”). PLoS ONE 10:e0123298. doi: 10.1371/journal.pone.0123298
Barabote, R. D., Xie, G., Brettin, T. S., Hinrichs, S. H., Fey, P. D., Jay, J. J., et al. (2009). Complete genome sequence of Francisella tularensis subspecies holarctica FTNF002-00. PLoS ONE 4:e7041. doi: 10.1371/journal.pone.0007041
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boone, I., Hassler, D., Nguyen, T., Splettstoesser, W. D., Wagner-Wiening, C., and Pfaff, G. (2015). Tularaemia in southwest Germany: three cases of tick-borne transmission. Ticks Tick Borne Dis. 6, 611–614. doi: 10.1016/j.ttbdis.2015.05.004
Borde, J. P., Zange, S., Antwerpen, M. H., Georgi, E., Von Buttlar, H., Kern, W. V., et al. (2017). Five cases of vector-borne Francisella tularensis holarctica infections in south-western Germany and genetic diversity. Ticks Tick Borne Dis. 8, 808–812. doi: 10.1016/j.ttbdis.2017.06.009
Broekhuijsen, M., Larsson, P., Johansson, A., Bystrom, M., Eriksson, U., Larsson, E., et al. (2003). Genome-wide DNA microarray analysis of Francisella tularensis strains demonstrates extensive genetic conservation within the species but identifies regions that are unique to the highly virulent F. tularensis subsp. tularensis. J. Clin. Microbiol. 41, 2924–2931. doi: 10.1128/JCM.41.7.2924-2931.2003
Chanturia, G., Birdsell, D. N., Kekelidze, M., Zhgenti, E., Babuadze, G., Tsertsvadze, N., et al. (2011). Phylogeography of Francisella tularensis subspecies holarctica from the country of Georgia. BMC Microbiol. 11:139. doi: 10.1186/1471-2180-11-139
Clinical and Laboratory Standards Institute (2011). CLSI addresses susceptibility testing of fastidious bacteria in new guideline. Lab. Med. 42, 374–375. doi: 10.1309/LM3ILTL0TCE3RFBT
Dessau, R. B., and Pipper, C. B. (2008). [”R"–project for statistical computing]. Ugeskr. Laeg. 170, 328–330.
Dwibedi, C., Birdsell, D., Larkeryd, A., Myrtennas, K., Ohrman, C., Nilsson, E., et al. (2016). Long-range dispersal moved Francisella tularensis into Western Europe from the East. Microb. Genom. 2:e000100. doi: 10.1099/mgen.0.000100
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ellis, J., Oyston, P. C., Green, M., and Titball, R. W. (2002). Tularemia. Clin. Microbiol. Rev. 15, 631–646. doi: 10.1128/CMR.15.4.631-646.2002
Faber, M., Heuner, K., Jacob, D., and Grunow, R. (2018). Tularemia in Germany-A re-emerging zoonosis. Front. Cell. Infect. Microbiol. 8:40. doi: 10.3389/fcimb.2018.00040
Farlow, J., Wagner, D. M., Dukerich, M., Stanley, M., Chu, M., Kubota, K., et al. (2005). Francisella tularensis in the United States. Emerg. Infect. Dis. 11, 1835–1841. doi: 10.3201/eid1112.050728
Gehringer, H., Schacht, E., Maylaender, N., Zeman, E., Kaysser, P., Oehme, R., et al. (2013). Presence of an emerging subclone of Francisella tularensis holarctica in Ixodes ricinus ticks from south-western Germany. Ticks Tick Borne Dis. 4, 93–100. doi: 10.1016/j.ttbdis.2012.09.001
Gyuranecz, M., Birdsell, D. N., Splettstoesser, W., Seibold, E., Beckstrom-Sternberg, S. M., Makrai, L., et al. (2012). Phylogeography of Francisella tularensis subsp. holarctica, Europe. Emerg. Infect. Dis. 18, 290–293. doi: 10.3201/eid1802.111305
Haristoy, X., Lozniewski, A., Tram, C., Simeon, D., Bevanger, L., and Lion, C. (2003). Francisella tularensis bacteremia. J. Clin. Microbiol. 41, 2774–2776. doi: 10.1128/JCM.41.6.2774-2776.2003
Hestvik, G., Uhlhorn, H., Mattsson, R., Westergren, E., Sodersten, F., Akerstrom, S., et al. (2018). Pathology of natural Francisella tularensis subsp. holarctica infection in two yellow-necked mice (Apodemus flavicollis). Acta Vet. Scand. 60:26. doi: 10.1186/s13028-018-0381-9
Jacob, D., Koppen, K., Radonic, A., Haldemann, B., Zanger, P., Heuner, K., et al. (2019). Molecular identification of the source of an uncommon tularaemia outbreak, Germany, autumn 2016. Euro Surveill. 24. doi: 10.2807/1560-7917.ES.2019.24.18.1800419
Jacob, D., Wahab, T., Edvinsson, B., Peterzon, A., Boskani, T., Farhadi, L., et al. (2011). Identification and subtyping of Francisella by pyrosequencing and signature matching of 16S rDNA fragments. Lett. Appl. Microbiol. 53, 592–595. doi: 10.1111/j.1472-765X.2011.03158.x
Jellison, W. L. (1961). Tularemia and animal populations: ecology and epizootiology. Wildl. Dis. 17, 1–22.
Jenzora, A., Jansen, A., Ranisch, H., Lierz, M., Wichmann, O., and Grunow, R. (2008). Seroprevalence study of Francisella tularensis among hunters in Germany. Pathogens Dis. 53, 183–189. doi: 10.1111/j.1574-695X.2008.00408.x
Johansson, A., Larkeryd, A., Widerstrom, M., Mortberg, S., Myrtannas, K., Ohrman, C., et al. (2014). An outbreak of respiratory tularemia caused by diverse clones of Francisella tularensis. Clin. Infect. Dis. 59, 1546–1553. doi: 10.1093/cid/ciu621
Jones, R. M., Nicas, M., Hubbard, A., Sylvester, M. D., and Reingold, A. (2005). The infectious dose of Francisella Tularensis (Tularemia). Appl. Biosafety 10, 227–239. doi: 10.1177/153567600501000405
Jusatz, H. J. (1952). Zweiter Bericht über das Vordringen der Tularämie nach Mittel- und Westeuropa in der Gegenwart. Zeitschrift für Hygiene und Infektionskrankheiten 134, 350–374. doi: 10.1007/BF02149615
Jusatz, H. J. (1961). Dritter Bericht über das Vordringen der Tularämie nach Mittel- und Westeuropa über den Zeitraum von 1950 bis 1960. Zeitschrift für Hygiene und Infektionskrankheiten, medizinische Mikrobiologie, Immunologie und Virologie 148, 69–93. doi: 10.1007/BF02151963
Karlsson, E., Golovliov, I., Larkeryd, A., Granberg, M., Larsson, E., Ohrman, C., et al. (2016). Clonality of erythromycin resistance in Francisella tularensis. J. Antimicrob. Chemother. 71, 2815–2823. doi: 10.1093/jac/dkw235
Karlsson, E., Svensson, K., Lindgren, P., Bystrom, M., Sjodin, A., Forsman, M., et al. (2013). The phylogeographic pattern of Francisella tularensis in Sweden indicates a Scandinavian origin of Eurosiberian tularaemia. Environ. Microbiol. 15, 634–645. doi: 10.1111/1462-2920.12052
Kaysser, P., Seibold, E., Matz-Rensing, K., Pfeffer, M., Essbauer, S., and Splettstoesser, W. D. (2008). Re-emergence of tularemia in Germany: presence of Francisella tularensis in different rodent species in endemic areas. BMC Infect. Dis: 8, 157. doi: 10.1186/1471-2334-8-157
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Koene, M., Rijks, J., Maas, M., Ruuls, R., Engelsma, M., Van Tulden, P., et al. (2019). Phylogeographic distribution of human and hare Francisella Tularensis subsp. holarctica strains in the Netherlands and its pathology in European brown hares (Lepus europaeus). Front. Cell. Infect. Microbiol. 9:11. doi: 10.3389/fcimb.2019.00011
Kreizinger, Z., Erdelyi, K., Felde, O., Fabbi, M., Sulyok, K. M., Magyar, T., et al. (2017). Comparison of virulence of Francisella tularensis ssp. holarctica genotypes B.12 and B.FTNF002-00. BMC Vet. Res. 13:46. doi: 10.1186/s12917-017-0968-9
Kudelina, R. I. (1973). [Change in the properties of the causative agent of tularemia due to erythromycin]. Zh. Mikrobiol. Epidemiol. Immunobiol. 50, 98–101.
Kudelina, R. I., and Olsufiev, N. G. (1980). Sensitivity to macrolide antibiotics and lincomycin in Francisella tularensis holarctica. J. Hyg. Epidemiol. Microbiol. Immunol. 24, 84–91.
Kuehn, A., Schulze, C., Kutzer, P., Probst, C., Hlinak, A., Ochs, A., et al. (2013). Tularaemia seroprevalence of captured and wild animals in Germany: the fox (Vulpes vulpes) as a biological indicator. Epidemiol. Infect. 141, 833–840. doi: 10.1017/S0950268812001008
Langmead, B. (2010). Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics Chapter 11:Unit 11.17. doi: 10.1002/0471250953.bi1107s32
Larsson, P., Oyston, P. C., Chain, P., Chu, M. C., Duffield, M., Fuxelius, H. H., et al. (2005). The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat. Genet. 37, 153–159. doi: 10.1038/ng1499
Maurin, M., and Gyuranecz, M. (2016). Tularaemia: clinical aspects in Europe. Lancet Infect. Dis. 16, 113–124. doi: 10.1016/S1473-3099(15)00355-2
Molins, C. R., Delorey, M. J., Yockey, B. M., Young, J. W., Belisle, J. T., Schriefer, M. E., et al. (2014). Virulence difference between the prototypic Schu S4 strain (A1a) and Francisella tularensis A1a, A1b, A2 and type B strains in a murine model of infection. BMC Infect. Dis. 14:67. doi: 10.1186/1471-2334-14-67
Muller, W., Hotzel, H., Otto, P., Karger, A., Bettin, B., Bocklisch, H., et al. (2013). German Francisella tularensis isolates from European brown hares (Lepus europaeus) reveal genetic and phenotypic diversity. BMC Microbiol. 13:61. doi: 10.1186/1471-2180-13-61
Origgi, F. C., Frey, J., and Pilo, P. (2014). Characterisation of a new group of Francisella tularensis subsp. holarctica in Switzerland with altered antimicrobial susceptibilities, 1996 to 2013. Euro Surveill. 19:20858. doi: 10.2807/1560-7917.ES2014.19.29.20858
Origgi, F. C., and Pilo, P. (2016). Francisella Tularensis clades B.FTN002-00 and B.13 are associated with distinct pathology in the European brown hare (Lepus europaeus). Vet. Pathol. 53, 1220–1232. doi: 10.1177/0300985816629718
Otto, P., Chaignat, V., Klimpel, D., Diller, R., Melzer, F., Muller, W., et al. (2014). Serological investigation of wild boars (Sus scrofa) and red foxes (Vulpes vulpes) as indicator animals for circulation of Francisella tularensis in Germany. Vector Borne Zoonotic Dis. 14, 46–51. doi: 10.1089/vbz.2013.1321
Oyston, P. C., and Griffiths, R. (2009). Francisella virulence: significant advances, ongoing challenges and unmet needs. Expert Rev. Vaccines 8, 1575–1585. doi: 10.1586/erv.09.114
Pearson, K. (1900). X. On the criterion that a given system of deviations from the probable in the case of a correlated system of variables is such that it can be reasonably supposed to have arisen from random sampling. Lond. Edinburgh Dublin Philosoph. Magaz. J. Sci. 50, 157–175. doi: 10.1080/14786440009463897
Petrosino, J. F., Xiang, Q., Karpathy, S. E., Jiang, H., Yerrapragada, S., Liu, Y., et al. (2006). Chromosome rearrangement and diversification of Francisella tularensis revealed by the type B (OSU18) genome sequence. J. Bacteriol. 188, 6977–6985. doi: 10.1128/JB.00506-06
Pilo, P. (2018). Phylogenetic lineages of Francisella tularensis in animals. Front. Cell. Infect. Microbiol. 8:258. doi: 10.3389/fcimb.2018.00258
Puiu, D., and Salzberg, S. L. (2008). Re-assembly of the genome of Francisella tularensis subsp. holarctica OSU18. PLoS ONE 3:e3427. doi: 10.1371/journal.pone.0003427
Rydzewski, K., Schulz, T., Brzuszkiewicz, E., Holland, G., Luck, C., Fleischer, J., et al. (2014). Genome sequence and phenotypic analysis of a first German Francisella sp. isolate (W12-1067) not belonging to the species Francisella tularensis. BMC Microbiol. 14:169. doi: 10.1186/1471-2180-14-169
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Saslaw, S., Eigelsbach, H. T., Wilson, H. E., Prior, J. A., and Carhart, S. (1961). Tularemia vaccine study. I. Intracutaneous challenge. Arch. Intern. Med. 107, 689–701. doi: 10.1001/archinte.1961.03620050055006
Schulze, C., Heuner, K., Myrtennas, K., Karlsson, E., Jacob, D., Kutzer, P., et al. (2016). High and novel genetic diversity of Francisella tularensis in Germany and indication of environmental persistence. Epidemiol. Infect. 144, 3025–3036. doi: 10.1017/S0950268816001175
Splettstoesser, W. D., Piechotowski, I., Buckendahl, A., Frangoulidis, D., Kaysser, P., Kratzer, W., et al. (2009). Tularemia in Germany: the tip of the iceberg? Epidemiol. Infect. 137, 736–743. doi: 10.1017/S0950268808001192
Svensson, K., Back, E., Eliasson, H., Berglund, L., Granberg, M., Karlsson, L., et al. (2009a). Landscape epidemiology of tularemia outbreaks in Sweden. Emerg. Infect. Dis. 15, 1937–1947. doi: 10.3201/eid1512.090487
Svensson, K., Granberg, M., Karlsson, L., Neubauerova, V., Forsman, M., and Johansson, A. (2009b). A real-time PCR array for hierarchical identification of Francisella isolates. PLoS ONE 4:e8360. doi: 10.1371/journal.pone.0008360
Svensson, K., Sjodin, A., Bystrom, M., Granberg, M., Brittnacher, M. J., Rohmer, L., et al. (2012). Genome sequence of Francisella tularensis subspecies holarctica strain FSC200, isolated from a child with tularemia. J. Bacteriol. 194, 6965–6966. doi: 10.1128/JB.01040-12
Tomaso, H., Hotzel, H., Otto, P., Myrtennas, K., and Forsman, M. (2017). Antibiotic susceptibility in vitro of Francisella tularensis subsp. holarctica isolates from Germany. J. Antimicrob. Chemother. 72, 2539–2543. doi: 10.1093/jac/dkx182
Tomaso, H., Otto, P., Peters, M., Suss, J., Karger, A., Schamoni, H., et al. (2018). Francisella tularensis and other bacteria in hares and ticks in North Rhine-Westphalia (Germany). Ticks Tick Borne Dis. 9, 325–329. doi: 10.1016/j.ttbdis.2017.11.007
Versage, J. L., Severin, D. D., Chu, M. C., and Petersen, J. M. (2003). Development of a multitarget real-time TaqMan PCR assay for enhanced detection of Francisella tularensis in complex specimens. J. Clin. Microbiol. 41, 5492–5499. doi: 10.1128/JCM.41.12.5492-5499.2003
Vogler, A. J., Birdsell, D., Price, L. B., Bowers, J. R., Beckstrom-Sternberg, S. M., Auerbach, R. K., et al. (2009a). Phylogeography of Francisella tularensis: global expansion of a highly fit clone. J. Bacteriol. 191, 2474–2484. doi: 10.1128/JB.01786-08
Vogler, A. J., Birdsell, D., Wagner, D. M., and Keim, P. (2009b). An optimized, multiplexed multi-locus variable-number tandem repeat analysis system for genotyping Francisella tularensis. Lett. Appl. Microbiol. 48, 140–144. doi: 10.1111/j.1472-765X.2008.02484.x
Wittwer, M., Altpeter, E., Pilo, P., Gygli, S. M., Beuret, C., Foucault, F., et al. (2018). Population Genomics of Francisella tularensis subsp. holarctica and its implication on the eco-epidemiology of tularemia in Switzerland. Front. Cell. Infect. Microbiol. 8:89. doi: 10.3389/fcimb.2018.00089
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Keywords: Francisella, tularemia, canonical SNPs, genome sequencing, genetic diversity
Citation: Appelt S, Köppen K, Radonić A, Drechsel O, Jacob D, Grunow R and Heuner K (2019) Genetic Diversity and Spatial Segregation of Francisella tularensis Subspecies holarctica in Germany. Front. Cell. Infect. Microbiol. 9:376. doi: 10.3389/fcimb.2019.00376
Received: 22 July 2019; Accepted: 17 October 2019;
Published: 06 November 2019.
Edited by:
Caspar Yvan, Centre Hospitalier Universitaire de Grenoble, FranceReviewed by:
Dawn Birdsell, Northern Arizona University, United StatesMichael L. Vasil, University of Colorado Denver, United States
Herbert Tomaso, Friedrich Loeffler Institut, Germany
Copyright © 2019 Appelt, Köppen, Radonić, Drechsel, Jacob, Grunow and Heuner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaus Heuner, aGV1bmVya0Bya2kuZGU=