Abstract
Few genetic tools were available to work with Trypanosoma cruzi until the recent introduction of the CRISPR/Cas9 technique for gene knockout, gene knock-in, gene complementation, and endogenous gene tagging. Riboswitches are naturally occurring self-cleaving RNAs (ribozymes) that can be ligand-activated. Results from our laboratory recently demonstrated the usefulness of the glmS ribozyme from Bacillus subtilis, which has been shown to control reporter gene expression in response to exogenous glucosamine, for gene silencing in Trypanosoma brucei. In this work we used the CRISPR/Cas9 system for endogenously tagging T. cruzi glycoprotein 72 (TcGP72) and vacuolar proton pyrophosphatase (TcVP1) with the active (glmS) or inactive (M9) ribozyme. Gene tagging was confirmed by PCR and protein downregulation was verified by western blot analyses. Further phenotypic characterization was performed by immunofluorescence analysis and quantification of growth in vitro. Our results indicate that the method was successful in silencing the expression of both genes without the need of glucosamine in the medium, suggesting that T. cruzi produces enough levels of endogenous glucosamine 6-phosphate to stimulate the glmS ribozyme activity under normal growth conditions. This method could be useful to obtain knockdowns of essential genes in T. cruzi and to validate potential drug targets in this parasite.
Introduction
Infection by Trypanosoma cruzi is the main cause of congestive heart failure in Latin America (Rassi et al., 2000). The disease affects 8–10 million people in the Americas. The FDA approval of a test for these parasites in donated blood (Kessler et al., 2013) emphasizes the relevance of this parasite to human health in the United States, where physicians are largely unaware of the cardiac symptoms of chronic T. cruzi infection. Treatment of Chagas disease is limited to drugs with relatively high toxicity and partial efficacy (Urbina and Docampo, 2003). The study of metabolic pathways in these parasites that could be important for their viability but that do not affect their host could result in the finding of specific inhibitors to control the parasites without altering the hosts. A drawback for these studies in T. cruzi has been the lack of genetic tools, such as inducible downregulation, which are essential for the demonstration of the essentiality of these metabolic pathways and for the validation of new drug targets.
Few genetic tools were available to work with T. cruzi (Docampo, 2011; Burle-Caldas Gde et al., 2015) until the recent introduction of the CRISPR/Cas9 technique for gene knockout (Peng et al., 2014; Lander et al., 2015; Costa et al., 2018; Romagnoli et al., 2018; Takagi et al., 2019) endogenous gene tagging (Lander et al., 2016b, 2017; Soares Medeiros et al., 2017; Costa et al., 2018), gene complementation (Chiurillo et al., 2017), and gene knock-in (Chiurillo et al., 2019). These studies have been recently reviewed in Lander and Chiurillo (2019).
Control of gene expression can be achieved at the transcriptional, translational or posttranslational levels (Ganesan et al., 2016). In the case of the related trypanosomatid Trypanosoma brucei, the fastest method for the generation of conditional mutants is the use of RNA interference (Alibu et al., 2005). This pathway, however, is absent in T. cruzi (Darocha et al., 2004). Integration of a tetracycline-regulated extra copy of the gene of interest to allow the knockout of the endogenous alleles in a cell line stably expressing a tet repressor and the T7 RNA polymerase (inducible knockout) has also been successfully employed in T. brucei (Clayton, 1999). Efforts to develop a similar method for T. cruzi have mostly failed until now (Darocha et al., 2004; Burle-Caldas Gde et al., 2015). In contrast to the control of a reporter gene expression over a range of four orders of magnitude in response to tetracycline in T. brucei, relatively high expression levels of the gene was detected in T. cruzi in the absence of tetracycline and little increase was detected after tetracycline addition [reviewed by (Burle-Caldas Gde et al., 2015)]. Inducible systems using destabilization domains of dihydrofolate reductase (DDD), or the rapamycin binding protein (ddFKBP), were only used to either create suicidal T. cruzi strains (Ma et al., 2015), or did not mediate the efficient knockdown of the genes (Burle-Caldas Gde et al., 2015). Inducible expression of dimerizable CRE recombinase (DiCRE system) was also tried in T. cruzi but has been only used for removal of exogenous selectable markers from the parasite's genome with limited success (Kangussu-Marcolino et al., 2014).
We recently reported the use of an alternative method for downregulation of gene expression in T. brucei, mediated at the mRNA level, using riboswitches (Cruz-Bustos et al., 2018b), which are naturally occurring self-cleaving RNAs (ribozymes) that can be modified to respond to ligands (Winkler et al., 2004). We used the glmS gene from Bacillus subtilis, which can control reporter gene expression in response to exogenous glucosamine in other eukaryotes, such as Saccharomyces cerevisiae (Watson and Fedor, 2011) and Plasmodium falciparum (Prommana et al., 2013). The glmS gene encodes the enzyme glutamine-fructose 6-phosphate amidotransferase that uses fructose 6-phosphate and glutamine to generate glucosamine 6-phosphate (GlcN6P). A conserved element in the 5′-unstranslated region of this gene acts, when transcribed into RNA, as a self-cleaving riboswitch stimulated by glucosamine 6-phosphate (GlcN6P) (Winkler et al., 2004). When this conserved element is inserted in the 5′-UTR or the 3′-UTR of a gene of interest the self-cleaving RNA motif will silence it when in the presence of GlcN6P produced within the cells. Addition of glucosamine to the culture medium stimulates this activity through the endogenous generation of GlcN6P. A mutant glmS gene whose RNA has no self-cleaving activity (M9) can be used as negative control (Winkler et al., 2004). Tagging only one allele of the gene of interest (GOI) in T. brucei with glmS was sufficient to down-regulate gene expression at the mRNA level, and in some cases, produce phenotypic changes (Cruz-Bustos et al., 2018b). Since this technique requires the endogenous tagging of the genes that are targeted for down-regulation, our recent development of C-terminal endogenous tagging of genes in T. cruzi using CRISPR/Cas9 (Lander et al., 2016b, 2017) made this approach feasible.
In this work, we report the use of the glmS ribozyme for silencing the expression of endogenous genes without the need to add glucosamine to the medium, suggesting that T. cruzi produces enough levels of endogenous GlcN6P to stimulate the glmS ribozyme activity under normal growth conditions. This method could be useful to validate potential drug targets in this parasite.
Results
Effect of Glucosamine on Growth of Epimastigotes
We first tested whether glucosamine has any effect on parasite growth. Figure S1 shows the effects of 0–30 mM glucosamine added to a modified SDM-79 medium. This medium, which has been used to grow T. cruzi (Hasne et al., 2010), has a known concentration of glucosamine (1 mM) that could be varied to stimulate the riboswitch. We found that 10 mM was the maximum concentration that did not affect growth during the evaluated period and used that concentration in subsequent experiments.
Downregulation of the Expression of T. cruzi Glycoprotein 72 (TcGP72)
We first used the CRISPR/Cas9 system (Lander et al., 2016b, 2017) for endogenously tagging T. cruzi glycoprotein 72 (TcGP72) with the active (glmS) or inactive (M9) ribozyme following the strategy shown in Figure 1. TcGP72 is a glycoprotein responsible for adhesion of the flagellum to the cell body and is not essential for the parasite survival (Cooper et al., 1993). We co-transfected a specific GP72-3'end-sgRNA/Cas9/pTREX-n construct with a specific DNA donor molecule amplified from the pMOTag-M9/glmS-4H vectors (Cruz-Bustos et al., 2018b), as described under Experimental Procedures and grew the cells initially in a modified SDM-79 medium. However, when glucosamine addition was shown to be unnecessary to downregulate TcGP72 expression further experiments were done in regular LIT medium. We obtained G418/hygromycin resistant cells after 3 weeks under selective pressure. Transfectants were analyzed by PCR, using gDNA isolated from them, and a specific primer set to distinguish between the wild type and the tagged cell lines (Figure 1). After 21 days in culture it was possible to confirm the presence of the tagged gene (band of 779 bp) in TcGP72-3xHA-glmS (glmS) and TcGP72-3xHA-M9 (M9) transfectants, but not in wild type (WT) parasites (Figure 2A). Insertion of glmS and M9 constructs at the 3′UTR of GP72 gene was confirmed by sequencing (Sequences S1, S2, respectively). qRT-PCR showed complete downregulation of TcGP72 expression (Figure 2B). Western blot analysis using monoclonal antibody WIC 29.26 showed disappearance of the 72-KDa band after 45 days in culture compared to WT and TcGP72-3xHA-M9-transfectants (Figure 2C). As reported before (Cooper et al., 1993), this antibody recognizes the glycan epitope in additional proteins. Figure 2D shows the presence of parasites with flagellar detachment and greatly reduced labeling with monoclonal antibody WIC 29.26. More than 90% of the cells showed this phenotype.
Figure 1

Schematic representation of the strategy used in Trypanosoma cruzi. (A) (a) pMOTag-4H-M9/glmS vector map. HR1 Fw, and HR2 Rv, ultramers indicate oligonucleotides used to amplify the DNA donor. (b) A doubled strand break was produced in the gDNA by Cas9 targeted by the sgRNA downstream of the STOP codon of the gene of interest (GOI), both expressed from 3′end-sgRNA/Cas9/pTREX-n plasmid. Homologous directed repair was induced co-transfecting epimastigotes with the DNA donor cassette, containing homologous regions to the GOI 3′ end (dark blue) and to the GOI 3′UTR (light blue). (c) Integration of 3xHA, M9/glmS and antibiotic resistance genes at 3′end of GOI by homologous recombination. (B) Diagram representing the positions of the primers (arrows) used to verify the integration of the donor DNA at the 3′ end of TcGP72 ORF.
Figure 2

Integration of glmS/M9 ribozyme sequences into the TcGP72 gene. (A) PCR analysis using gDNA isolated from WT and TcGP72-M9/glmS (M9, glmS) cell lines. A DNA fragment of 779 bp was amplified in 3xHA-glmS/M9-tagged epimastigotes, whereas the band is absent in WT cells. (B) qRT-PCR shows down-regulation of TcGP72. (C) Western-blot analysis of TcGP72-M9/glmS cell lines. Antibody WIC 29.26 labels a band of 72 kDa in TcGP72-3xHA-M9- but not in TcGP72-3xHA-glmS-tranfected cells. Anti-tubulin antibody (Tub) was used as loading control. Antibodies are indicated on the right side of the blots, and molecular weights (kDa) are on the left side. (D) Immunofluorescence microscopy of TcGP72-3xHA-M9- and TcGP72-3xHA-glmS-transfected epimastigotes. There is co-localization in the flagellum of anti-HA and WIC 29.26 antibodies in TcGP72-3xHA-M9-transfected epimastigotes (merge) while the flagellum is detached and has little labeling with anti-HA and reduced labeling with WIC 29.26 in TcGP72-3xHA-glmS-expressing cells. DIC, differential interference contrast microscopy. Scale bars = 5 μm.
To exclude the possibility of off target effects, we investigated whether an exogenous TcGP72 gene could complement the TcGP72 knock down epimastigotes. We found that the exogenous gene (with changes in the PAM sequence to prevent disruption by CRISPR/Cas9) bearing a Ty epitope was targeted to the epimastigote flagellum, as shown in Figure 3A. Western blot analysis showed recovery of the labeling with antibody WIC 29.96 in complemented epimastigotes as compared to TcGP72-3xHA-glmS transfectants (Figure 3B). In conclusion, CRISPR/Cas9-mediated endogenous C-terminal tagging of T. cruzi GP72 with glmS was successful in silencing the gene without the need to add glucosamine to the medium. These results suggest that T. cruzi produces high levels of endogenous GlcN6P and that these levels are sufficient to stimulate the glmS ribozyme activity under normal growth conditions.
Figure 3

Complementation of TcGP72-3xHA-glmS. (A) Immunofluorescence microscopy of TcGP72-3xHA-glmS epimastigotes complemented with an exogenous copy of TcGP72 gene. Labeling of the flagellum was detected with anti-Ty and WIC 29.26 antibodies, while no flagellar detachment was observed. Bars = 5 μm. (B) Western-blot analysis of total protein extracts of TcGP72-3xHA-M9 (M9), TcGP72-3xHA-glmS (glmS), TcGP72-3xHA-glmS complemented with TcGP72-Ty (lane C) and wild type (WT) epimastigotes, using WIC 29.26 antibodies. Anti-α-tubulin antibodies (Tub) were used as a loading control.
Downregulation of the Expression of T. cruzi Vacuolar Proton Pyrophosphatase (TcVP1)
The T. cruzi vacuolar proton pyrophosphatase (TcVP1) is an electrogenic proton pump mainly localized to acidocalcisomes (Scott et al., 1998; Lander et al., 2016b), where it maintains their acidity. In T. brucei, RNAi experiments have shown that the enzyme is essential for normal growth of procyclic and bloodstream forms in vitro (Lemercier et al., 2002). We followed the same procedure used to downregulate the expression of TcGP72. Chimeric sgRNA was cloned into Cas9/pTREX-n vector and co-transfected with DNA donor containing glmS ribozyme sequence or its M9 inactive version into T. cruzi epimastigotes. Donor DNAs were amplified from pMOTag-glmS-4H and pMOTag-M9-4H vectors, respectively (Cruz-Bustos et al., 2018b). Transfectant parasites were obtained after 3 weeks of selection with G418 and hygromycin. We then obtained clonal populations from both TcVP1-3xHA-M9 and TcVP1-3xHA-glmS tagged cell lines by serial dilutions. We confirmed TcVP1 tagging in mixed and clonal populations by PCR (Figure 4A). We chose TcVP1-3xHA-M9, clone F9, and TcVP1-3xHA-glmS, clone E5 for further experiments. TcVP1 downregulation in glmS-tagged but not in M9-tagged epimastigotes was confirmed by western blot analysis using monoclonal antibodies anti-TcVP1 in the absence of 10 mM glucosamine (day 0) or at days 1, 2, and 3 after addition of glucosamine to the medium (Figure 4B). TcVP1 expression was also evaluated at the RNA level by quantitative RT-PCR under the same conditions (Figure 4C). Downregulation of the gene in TcVP1-glmS epimastigotes in the absence of glucosamine (day 0) or at days 1, 2, and 3 post-induction (dpi) was confirmed, although basal levels of TcVP1 expression were detected at all time points in TcVP1-glmS cells, relative to TcVP1-M9 uninduced epimastigotes (day 0). No significant differences were observed in TcVP1-M9 parasites at days 1, 2, and 3 post induction relative to day 0 (uninduced) (Figure 4C). We also evaluated the growth in vitro of TcVP1-M9- and TcVP1-glmS-tagged epimastigotes (Figures 4D,E). Growth of TcVP1-glmS-expressing epimastigotes in LIT medium was significantly affected as compared with the growth of TcVP1-M9-tagged cells but the presence of 10 mM glucosamine did not change the growth rate of these mutants (Figure 4D). The growth of these cell lines was then monitored for a longer period (10 days) in LIT medium, including a cell line expressing Cas9 and a scrambled sgRNA as control (Scrambled) (Figure 4E). Again, the results show a significant lower growth of TcVP1-glmS-expressing epimastigotes as compared with those expressing TcVP1-M9 or a scrambled sgRNA, thus confirming the results observed with downregulation of TcGP72.
Figure 4
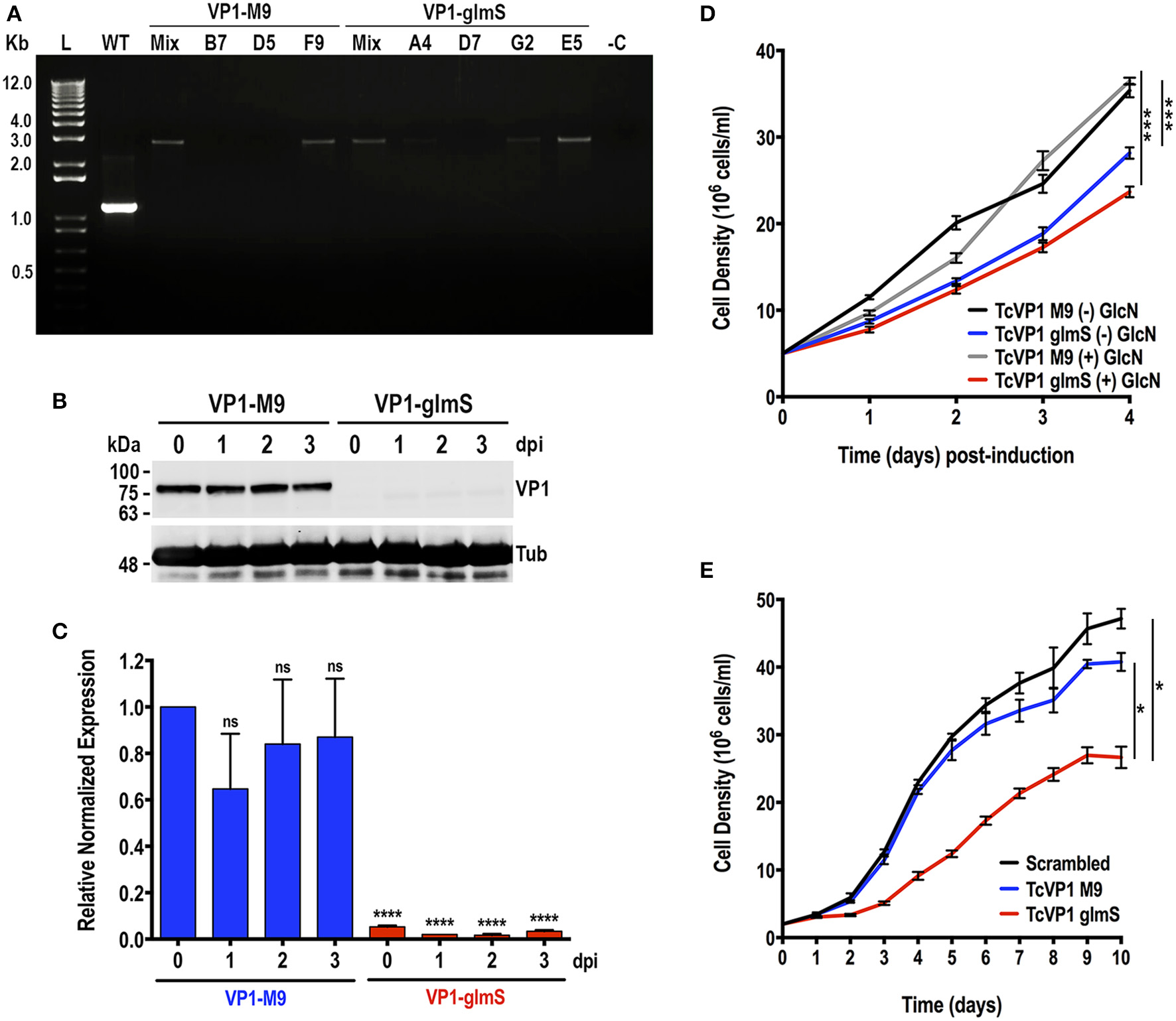
Integration of glmS/M9 ribozyme sequences into the TcVP1 gene. (A) PCR analysis using gDNA isolated from WT and TcVP1-M9/glmS mixed (Mix) and clonal populations (TcVP1-M9, clones B7, D5, and F9; TcVP1-glmS, clones D7, G2, and E5). The primer set used (Table S1, primers 11 and 12) generates a band of 1,144 bp in wild type parasites (WT) and a band of 2,758 bp in TcVP1-tagged (M9 or glmS) epimastigotes. Transfectant mixed and clonal populations were analyzed from both tagged versions of TcVP1, and in both cases the band of 2,758 bp was detected in the originally transfected cell lines (Mix) and in some of the clones. Clones F9 (TcVP1-M9) and E5 (TcVP1-glmS) were chosen for further phenotypic analysis. (B) Western-blot analysis of TcVP1-M9 and TcVP1-glmS cell lines. Anti TcVP1 monoclonal antibody labels a band of 85 kDa in TcVP1-3xHA-M9- but not in TcVP1-3xHA-glmS- epimastigotes. Expression of TcVP1 was monitored during 3 days after addition of 10 mM glucosamine to the medium (days post induction, dpi). Anti-tubulin antibody (Tub) was used as loading control. Antibodies are indicated on the right side of the blots, and molecular weights (kDa) are on the left side. (C) qRT-PCR analysis of TcVP1-M9 and TcVP1-glmS cell lines from 0 to 3 days post induction (dpi). (D) Growth in vitro of TcVP1-M9 and TcVP1-glmS epimastigotes cultured with [(+) GlcN] or without [(–) GlcN] 10 mM glucosamine. (E) Growth in vitro of scrambled, TcVP1-M9 and TcVP1-glmS epimastigotes cultured in LIT medium until reaching the stationary phase, without addition of glucosamine. In (C), qRT-PCR data analysis was performed using one-way ANOVA with multiple comparisons (n = 3; ****p < 0.0001; ns, no significant). In (D,E), one-way ANOVA with multiple comparisons was applied to growth rates calculated from each growth curve (n = 3; *p <0.05; ***p <0.001).
We induced the differentiation of TcVP1-glmS and TcVP1-M9 epimastigotes to metacyclic forms and infected Vero cells as described in Experimental procedures. Once enough culture-derived trypomastigotes were obtained we used them to test their ability to infect Vero cells and replicate intracellularly as amastigotes. Figure 5 shows that the ability of TcVP1-glmS trypomastigotes to infect tissue-culture cells, but not the amastigote replication, was significantly impaired, as compared with that of TcVP1-M9 parasites. The presence of added glucosamine had no effect in either invasion or replication, thus confirming the results with epimastigotes in both TcGP72-glmS and TcVP1-glmS cells. Downregulation of TcVP1 in the TcVP1-glmS but not in the TcVP1-M9 trypomastigotes used for the invasion assays was confirmed by western blot analysis (Figure 5C).
Figure 5

Invasion and intracellular replication of TcVP1-M9 and TcVP1-glmS cell lines. (A)TcVP1-M9 and TcVP1-glmS trypomastigote infection of Vero cells. There was a significant difference in the percentage of infected Vero cells but not in the number of intracellular amastigotes per infected host cell observed 48 h post infection (B). Values are mean ± s.d.; n = 3; **p < 0.01; ns, not significant (Two-way ANOVA with multiple comparisons test). (C) Western-blot analysis of TcVP1-M9 and TcVP1-glmS trypomastigotes. Anti TcVP1 antibody labels an expected band of 85 kDa in TcVP1-3xHA-M9- but not in TcVP1-3xHA-glmS- epimastigotes. Tissue culture-derived trypomastigotes collected at day 6 post-infection of Vero cells were induced with 10 mM glucosamine (GlcN) overnight at 4°C. After induction, trypomastigotes were used for infection of Vero cells and 1 × 107 trypomastigotes were reserved for protein extraction. Proteins were analyzed by western blot using TcVP1 mAb (1:2,000). Anti-tubulin antibody (Tub) was used as loading control. Antibodies are indicated on the right side of the blots, and molecular weights (kDa) are on the left side.
Discussion
CRISPR/Cas9-mediated endogenous C-terminal tagging of TcGP72 and TcVP1 with glmS, but not with M9, was successful in knocking down the gene expression without the addition of glucosamine to the medium. These results suggest that T. cruzi produces high levels of endogenous glucosamine 6-phosphate and that these levels are sufficient to stimulate the glmS ribozyme activity under normal growth conditions. In this regard, it has been found that T. cruzi (TcCLB.506507.10), as well as Leishmania major (LmjF32.3260), possesses an N-acetyl glucosamine 6-phosphate deacetylase (NAGD) that generates glucosamine 6-phosphate while T. brucei lacks this enzyme (Naderer et al., 2010). It is tempting to speculate that this enzyme could be involved in the production of high endogenous levels of glucosamine 6-phoshate in T. cruzi. However, it is important to indicate that the method, as it has been developed until now, could be useful to obtain knockdowns of essential genes in T. cruzi. If the gene is essential, and depending on the ribozyme used, the cells will die (glmS transfectants) or survive (M9 transfectants). This method could then be used to validate the essentiality of potential targets. In addition, as the glmS ribozyme acts at the mRNA level, it is possible to detect basal levels of expression in the transfectants, thus allowing the selection of parasites expressing glmS-tagged essential genes, as shown in Figure 4C for TcVP1 gene. C-terminal endogenous tagging also facilitates localization studies of the genes of interest.
One advantage of using our previously reported CRISPR/Cas9 methodology (Lander et al., 2016b, 2017) for tagging genes with glmS ribozyme, is that the constitutive expression of Cas9 and the sgRNA in the presence of a DNA donor conferring antibiotic resistance, allows tagging both alleles of the gene using a single resistance marker, as previously shown (Lander et al., 2015, 2018; Chiurillo et al., 2017, 2019; Cruz-Bustos et al., 2018a; Bertolini et al., 2019). In this way, the downregulation efficiency is significantly improved because only one transfection is necessary to tag and inactivate both alleles of the gene.
It has been reported that in S. cerevisiae (Meaux and Van Hoof, 2006) and in P. falciparum (Prommana et al., 2013) the ribozyme-cleaved mRNA 5′ fragment separated from its polyA tail could be degraded by the 3′exosome. This pathway has also been reported to be active in trypanosomatids (Estevez et al., 2001). When the glmS ribozyme is in the 5′UTR, mRNA cleavage would separate the mRNA from its 5′cap structure and the de-capped mRNA would be degraded by the 5′-3′-exonucleases, which are also present in trypanosomatids (Li et al., 2006).
Interestingly, our studies revealed that, in contrast to what occurs with TbVP1 in T. brucei (Lemercier et al., 2002), TcVP1 is not essential for the viability of epimastigotes or the infective stages of T. cruzi, although is important for normal growth of epimastigotes in rich medium, and for trypomastigote invasion of host cells. TcVP1 is mostly localized to acidocalcisomes where it contributes to the acidification of the organelle together with a vacuolar proton ATPase (Docampo et al., 1995; Scott et al., 1998).
In conclusion, the CRISPR/Cas9/riboswitch method developed in this work will enable the downregulation of gene expression in T. cruzi, and potentially in Leishmania spp. in which endogenous gene tagging using CRISPR/Cas9 has been achieved (Lander et al., 2016a,b).
Experimental Procedures
Culture Methods
Trypanosoma cruzi Y strain epimastigotes were cultured in liver infusion tryptose (LIT) medium (Bone and Steinert, 1956) containing 10% heat-inactivated fetal bovine serum at 28°C. The endogenously tagged cell line was maintained in medium containing 250 μg/ml G418 and 150 μg/ml of hygromycin. For initial experiments to test the effect of glucosamine, epimastigotes were also cultured in medium SDM-79, without the addition of D-glucosamine and supplemented with hemin (7.5 μg/mL), 200 μM putrescine, and 10% heat-inactivated fetal bovine serum (Hasne et al., 2010; Jimenez and Docampo, 2015). Cell growth was determined by counting cells in a Neubauer chamber.
Chemicals and Reagents
The pMOTag4H vector was a gift from Dr. Thomas Seebeck (University of Bern, Bern, Switzerland). Monoclonal antibody WIC 29.26 was a gift from Dr. George A.M. Cross (Rockefeller University). Wild type and M9 mutated Bacillus subtilis glmS ribozymes were gifts from Dr. Vasant Muralidharan (University of Georgia). Monoclonal antibody BB2 against the S. cerevisiae Ty1 virus-like particle was a gift from Dr. R. Drew Etheridge (University of Georgia). Monoclonal antibody against T. cruzi vacuolar H+-pyrophosphatase (TcVP1) was described before (Seufferheld et al., 2004). GoTaq DNA polymerase and T4 DNA ligase were from Promega. Antarctic phosphatase, restriction enzymes, and Q5 high fidelity DNA polymerase were from New England Biolabs (Ipswich, MA). Fluoromount-G was from SouthernBiotech (Birmingham, AL). Pierce BCA protein assay, Hygromycin B, BenchMark prestained protein ladder, BenchMark protein ladder, MagicMark™ XP Western Protein Standard, Alexa-conjugated secondary antibodies, and HRP-conjugated secondary antibodies were from Thermo Fisher Scientific. Anti-HA high affinity rat monoclonal antibody (clone 3F10) was purchased from Roche Applied Science. IRDye-conjugated secondary antibodies were from LI-COR Biosciences (Lincoln, NE). Benzonase nuclease was from Novagen (EMD Milli- pore, Billerica, MA). Nitrocellulose membranes were from Bio-Rad. The primers were purchased from Integrated DNA Technologies. TRI® reagent, anti-tubulin monoclonal antibody, G418, mammalian cell protease inhibitor mixture (P8340), other protease inhibitors, and all other reagents of analytical grade were from Sigma-Aldrich.
Molecular Constructs
The pMOTag4H vector designed for endogenous C-terminal tagging of T. brucei (Oberholzer et al., 2006) was used to construct the template plasmids for DNA donor amplification. This vector contains a 3xHA tag and the hygromycin resistance marker. Wild-type and M9 mutated Bacillus subtilis glmS ribozyme sequences (Winkler et al., 2004) were used to generate the pMOTag-glmS-4H and pMOTag-M9-4H vectors (Addgene plasmids #106378 and #106379) (Cruz-Bustos et al., 2018b). For CRISPR/Cas9-mediated endogenous C-terminal tagging in T. cruzi we used the Cas9/pTREX-n vector (Addgene plasmid #68708) (Lander et al., 2015) to clone a specific single guide RNA (sgRNA) sequence targeting the 3′end of TcGP72 (TcCLB.50956120) and TcVP1 (TcCLB. 510773.20) genes. The sgRNAs were amplified by PCR (Table S1, primers 1–3), using pUC_sgRNA plasmid as template (Addgene plasmid #68710) (Lander et al., 2015). We co-transfected the GP72-3′end-sgRNA/Cas9/pTREX-n or VP1-3′end-sgRNA/Cas9/pTREX-n construct with the specific DNA donor template for tagging the 3′ end of each gene, right upstream the stop codon, with a 3xHA tag sequence and the glmS/M9 ribozyme amplified from pMOTag-glmS-4H and pMOTag-M9-4H vectors, respectively, as previously described (Lander et al., 2016b; Cruz-Bustos et al., 2018b) (Table S1, primers 4–7). sgRNA correct orientation in Cas9/pTREX-n vector was determined by PCR (Table S1, primers 1, 2, and 8) and sequencing using primer 8. Endogenous tagging of TcVP1 and TcGP72 was confirmed by PCR (Table S1, primers 9–12). A cell line expressing Cas9 and a scrambled sgRNA (Lander et al., 2015) was used a control for growth experiments in vitro.
Complementation of TcGP72-KO Cells
To revert the phenotype exhibited by TcGP72-g3xHA-glmS epimastigotes we used an exogenous TcGP72 gene to complement the mutants. Following a PCR strategy, we eliminated the PAM sequence (TGG–TGT) specific for the TcGP72-sgRNA used to obtain the TcGP72-g3xHA-glmS cells, therefore avoiding constitutively expressed Cas9 to target the inserted sequence (Table S1, primers 13 and 14). The PCR product was cloned into pTREX-p vector (Chiurillo et al., 2017), which confers resistance to puromycin, by XbaI and XhoI restriction sites. We also included a C-terminal Ty1 tag in the reverse primer 14 (Table S1) in order to detect the overexpressed protein using anti-Ty1 antibody.
Cell Transfection
Trypanosoma cruzi epimastigotes were grown to a density of 1–2 × 107 cells/ml, washed once with cold PBS, pH 7.4, and resuspended in ice-cold Cytomix (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4, 2 mM EDTA, 5 mM MgCl2, pH 7.6) at a density of 108 cells/ml in electroporation buffer. Transfections were carried out in a 4-mm cuvette with 25 μg of plasmid DNA and 25 μg of DNA donor, using the Bio-Rad Gene Pulser Xcell electroporator set at 1.5 kV and 25 μF with three pulses, allowing at least 1 min for cells to recover in ice between pulses, and then incubated at room temperature for 15 min. Parasites were recovered in 5 ml of LIT supplemented with 20% fetal bovine serum at 28°C and after 24 h in culture, geneticin (G418), and hygromycin B were added to a final concentration of 250 and 150 μg/ml, respectively.
Western Blot Analyses
Electrophoresed proteins were transferred to nitrocellulose membranes using a Bio-Rad transblot apparatus for 1 h at 100 V at 4°C. Following transfer, the membrane blots were blocked with 5% non-fat dry milk in PBS containing 0.1% Tween-20 (PBS-T) overnight at 4°C. Blots were probed with primary antibody (WIC 29.26 monoclonal antibody (1:1,000), anti-Ty1 monoclonal antibody (1:1,000), anti TcVP1 monoclonal antibody (1:2,000) or anti-tubulin monoclonal antibody [1:40,000)] for 1 h, at RT. After washing three times with PBS-T, the blots were incubated with goat anti-rabbit antibody (1:20,000) or goat anti-mouse antibody (1:20,000). The membranes were washed three times with PBS-T, and western blot images were processed and analyzed using the Odyssey infrared system software (LICOR Biosciences) or a ChemiDoc™ Imaging System (Bio-Rad).
Immunofluorescence Analyses
Epimastigotes in log phase were washed once with PBS at room temperature and fixed with 4% paraformaldehyde in PBS for 30 min at room temperature. The cells were allowed to adhere to poly-L-lysine-coated coverslips and then permeabilized for 3 min with 0.3% Triton X-100. Permeabilized cells were blocked with PBS containing 3% BSA, 1% fish gelatin, 50 mM NH4Cl, and 5% goat serum 1 h at room temperature. Then cells were incubated with the primary antibody (1:100 rat anti-HA tag, 1:500 anti-Ty1 monoclonal antibody and 1:100 mouse WIC 29.26 diluted in PBS (pH 7.4) for 1 h at room temperature. The cells were washed three times with in PBS (pH 7.4) and then incubated for 1 h at room temperature in the dark with Alexa Fluor 488-conjugated goat anti-mouse and Alexa Fluor 546-conjugated goat anti-rat (1:1,000). Following incubation with the secondary antibody, the cells were washed five times in PBS and once in water and then mounted on slides. DAPI (5 μg/ml) was included in the Fluoromount-G mounting medium to stain DNA. Controls were performed as described above using M9-tagged epimastigotes. Specimens were imaged using the Delta Vision Elite deconvolution microscope (Applied Precision).
Metacyclogenesis
Trypanosoma cruzi epimastigotes were in vitro differentiated into infective metacyclic trypomastigotes by aging in LIT medium for 10 days at 28°C. Cultures were started at 5 × 106 cells/mL and after 10 days, 1.5 mL aged culture was centrifuged at 1,000 × g for 7 min and resuspended in 5 mL RPMI supplemented with 20% FBS fresh. The complement in fresh FBS kills epimastigotes, whereas metacyclic trypomastigotes survive. Cells were examined under the microscope to confirm the presence of about 10% metacyclic trypomastigotes, and used immediately to infect Vero cells as described below.
Host Cell Invasion and Intracellular Replication Assays
Gamma-irradiated (2,000 radiation-absorbed doses) Vero cells (4.5 × 105 cells) were plated onto sterile coverslips in a 12-well plate and incubated overnight at 35°C, 7% CO2, in RPMI medium plus 10% fresh FBS. Tissue culture-derived trypomastigotes were incubated at 4°C overnight to allow amastigotes to settle from swimming trypomastigotes. Trypomastigotes from the supernatants of these collections were counted and used to infect the coverslips at a 10:1 ratio of parasites to host cells. At 4 h post-infection, coverslips were washed extensively with Hank's balanced salt solution, followed by PBS, pH 7.4, to remove any extracellular parasites. Coverslips were fixed immediately in 4% paraformaldehyde in PBS, pH 7.4, at 4°C for 30 min. Coverslips were washed once with PBS and mounted onto glass slides in Fluoromount-G containing 15 μg/ml DAPI, which stains host and parasite DNA. Coverslips were viewed on an Olympus BX60 microscope to quantify the number of host cells that contained intracellular parasites and the number of intracellular parasites per cell in randomly selected fields. Three hundred host cells were counted per sample in three independent experiments. To quantify amastigote replication, the following modifications were used: host cells were infected at a ratio of 10 parasites to one host cell, and coverslips were allowed to incubate for 48 h post-infection at 35°C, 7% CO2, prior to fixation and DAPI staining. Coverslips were mounted onto glass slides and analyzed by fluorescence microscopy. Amastigotes in infected cells were counted using a 100 × objective.
Quantitative Real-Time PCR
Total RNA was isolated from trypanosomes using the TRI® reagent (Sigma) by following the manufacturer's instructions. The total RNA was treated with DNase I to remove genomic DNA contamination. cDNA synthesis was accomplished using the iScript cDNA synthesis kit (Bio-Rad) with 100 ng of total RNA used per reaction. Real-time PCR was done using a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad) and set up in hard-shell/clear 96-well PCR plates, in a final volume of 10 μl per reaction. The primers for gene amplification are listed in Table S1 (primers 15–24). Reaction mixtures contained 2 μl of sample DNA (100 ng/μl), 5 μl of a master mix iQ™ SYBR® Green Supermix (Bio-Rad) and 4 μl of nuclease-free water with primers at a final concentration of 300 nM. Activation of polymerase was performed at 95°C for 2 min. PCR cycling conditions included 39 cycles of denaturation at 95°C for 10 s, and annealing and extension at 60°C for 30 s (GP72 gene) or 55 °C for 45 s (TcVP1 gene). SYBR Green fluorescent emission was measured at the end of the elongation step. Subsequently, a melting curve program was applied with a continuous fluorescent measurement starting at 65°C and ending at 95°C (ramping rate of 0.1°C/s). In order to normalize the expression of the genes, we used primers for P0 and L3 housekeeping genes (Table S1, primers 15–18 used for GP72 normalization) and α-Tubulin (Table S1, primers 21 and 22 used for TcVP1 normalization) from T. cruzi. Relative quantification normalized to reference genes was performed according to the ΔCT method and all the assays were performed at least three times.
Statistical Analysis
All values are expressed as means ± s.d. Significant differences between treatments were compared using the tests indicated in the figure legends. Differences were considered statistically significant at P < 0.05, and n refers to the number of independent biological experiments performed. All statistical analyses were conducted using GraphPad Prism 5 (GraphPad Software, San Diego, CA).
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author contributions
NL, TC-B, and RD designed the experiments and analyzed the data. NL and TC-B conducted the experiments. RD wrote the majority of the manuscript with specific sections contributed by NL and TC-B. RD supervised the work and contributed to the analysis of the experiments.
Funding
Funding for his work was provided by the U.S. National Institutes of Health (Grant AI140421 to RD).
Acknowledgments
We thank Thomas Seebeck for pMOTag4H plasmid, George A. M. Cross for monoclonal antibody WIC 29.26, Vasant Muralidharan for plasmids containing glmS and M9 ribozymes and useful discussions, Drew Etheridge for BB2 monoclonal antibody, and Muthugapatti Kandasamy and the Biomedical Microscopy Core of the University of Georgia for the use of microscopes.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00068/full#supplementary-material
Figure S1Effect of different glucosamine concentrations on growth of epimastigotes in SDM-79 medium. Values are means ± s.d. of n = 3.
Table S1Primers used in this work.
Sequence S1GP72-glmS locus sequenced with primer 9 (Table S1).
Sequence S2GP72-M9 locus sequenced with primer 9 (Table S1).
References
1
AlibuV. P.StormL.HaileS.ClaytonC.HornD. (2005). A doubly inducible system for RNA interference and rapid RNAi plasmid construction in Trypanosoma brucei. Mol. Biochem. Parasitol.139, 75–82. 10.1016/j.molbiopara.2004.10.002
2
BertoliniM. S.ChiurilloM. A.LanderN.VercesiA. E.DocampoR. (2019). MICU1 and MICU2 play an essential role in mitochondrial Ca2+ uptake, growth, and infectivity of the human pathogen Trypanosoma cruzi. mBio10:e00348–e00319. 10.1128/mBio.00348-19
3
BoneG. J.SteinertM. (1956). Isotopes incorporated in the nucleic acids of Trypanosoma mega. Nature178, 308–309. 10.1038/178308a0
4
Burle-Caldas GdeA.Grazielle-SilvaV.LaibidaL. A.DarochaW. D.TeixeiraS. M. (2015). Expanding the tool box for genetic manipulation of Trypanosoma cruzi. Mol. Biochem. Parasitol.203, 25–33. 10.1016/j.molbiopara.2015.10.004
5
ChiurilloM. A.LanderN.BertoliniM. S.StoreyM.VercesiA. E.DocampoR. (2017). Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio8, e00574–e00517. 10.1128/mBio.00574-17
6
ChiurilloM. A.LanderN.BertoliniM. S.VercesiA. E.DocampoR. (2019). Functional analysis and importance for host cell infection of the Ca2+-conducting subunits of the mitochondrial calcium uniporter of Trypanosoma cruzi. Mol. Biol. Cell30, 1676–1690. 10.1091/mbc.E19-03-0152
7
ClaytonC. E. (1999). Genetic manipulation of kinetoplastida. Parasitol. Today15, 372–378. 10.1016/S0169-4758(99)01498-2
8
CooperR.De JesusA. R.CrossG. A. (1993). Deletion of an immunodominant Trypanosoma cruzi surface glycoprotein disrupts flagellum-cell adhesion. J. Cell Biol.122, 149–156. 10.1083/jcb.122.1.149
9
CostaF. C.FranciscoA. F.JayawardhanaS.CalderanoS. G.LewisM. D.OlmoF.et al. (2018). Expanding the toolbox for Trypanosoma cruzi: a parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Negl. Trop. Dis.12:e0006388. 10.1371/journal.pntd.0006388
10
Cruz-BustosT.PotapenkoE.StoreyM.DocampoR. (2018a). An intracellular ammonium transporter is necessary for replication, differentiation, and resistance to starvation and osmotic stress in Trypanosoma cruzi. mSphere3, e00377–e00317. 10.1128/mSphere.00377-17
11
Cruz-BustosT.RamakrishnanS.CordeiroC. D.AhmedM. A.DocampoR. (2018b). A riboswitch-based inducible gene expression system for Trypanosoma brucei. J. Eukaryot. Microbiol.65, 412–421. 10.1111/jeu.12493
12
DarochaW. D.OtsuK.TeixeiraS. M.DonelsonJ. E. (2004). Tests of cytoplasmic RNA interference (RNAi) and construction of a tetracycline-inducible T7 promoter system in Trypanosoma cruzi. Mol. Biochem. Parasitol133, 175–186. 10.1016/j.molbiopara.2003.10.005
13
DocampoR. (2011). Molecular parasitology in the 21st century. Essays Biochem.51, 1–13. 10.1042/bse0510001
14
DocampoR.ScottD. A.VercesiA. E.MorenoS. N. (1995). Intracellular Ca2+ storage in acidocalcisomes of Trypanosoma cruzi. Biochem. J. 310 (Pt 3), 1005–1012. 10.1042/bj3101005
15
EstevezA. M.KempfT.ClaytonC. (2001). The exosome of Trypanosoma brucei. EMBO J.20, 3831–3839. 10.1093/emboj/20.14.3831
16
GanesanS. M.FallaA.GoldflessS. J.NasamuA. S.NilesJ. C. (2016). Synthetic RNA-protein modules integrated with native translation mechanisms to control gene expression in malaria parasites. Nat. Commun.7:10727. 10.1038/ncomms10727
17
HasneM. P.CoppensI.SoysaR.UllmanB. (2010). A high-affinity putrescine-cadaverine transporter from Trypanosoma cruzi. Mol. Microbiol.76, 78–91. 10.1111/j.1365-2958.2010.07081.x
18
JimenezV.DocampoR. (2015). TcPho91 is a contractile vacuole phosphate sodium symporter that regulates phosphate and polyphosphate metabolism in Trypanosoma cruzi. Mol. Microbiol.97, 911–925. 10.1111/mmi.13075
19
Kangussu-MarcolinoM. M.CunhaA. P.AvilaA. R.HermanJ. P.DarochaW. D. (2014). Conditional removal of selectable markers in Trypanosoma cruzi using a site-specific recombination tool: proof of concept. Mol. Biochem. Parasitol198, 71–74. 10.1016/j.molbiopara.2015.01.001
20
KesslerD. A.ShiP. A.AvecillaS. T.ShazB. H. (2013). Results of lookback for chagas disease since the inception of donor screening at New York blood center. Transfusion53, 1083–1087. 10.1111/j.1537-2995.2012.03856.x
21
LanderN.ChiurilloM. A. (2019). State-of-the-art CRISPR/Cas9 technology for genome editing in trypanosomatids. J. Eukaryot. Microbiol. 66, 981–991. 10.1111/jeu.12747
22
LanderN.ChiurilloM. A.BertoliniM. S.StoreyM.VercesiA. E.DocampoR. (2018). Calcium-sensitive pyruvate dehydrogenase phosphatase is required for energy metabolism, growth, differentiation, and infectivity of Trypanosoma cruzi. J. Biol. Chem.293, 17402–17417. 10.1074/jbc.RA118.004498
23
LanderN.ChiurilloM. A.DocampoR. (2016a). Genome editing by CRISPR/Cas9: a game change in the genetic manipulation of protists. J. Eukaryot. Microbiol.63, 679–690. 10.1111/jeu.12338
24
LanderN.ChiurilloM. A.StoreyM.VercesiA. E.DocampoR. (2016b). CRISPR/Cas9-mediated endogenous C-terminal tagging of Trypanosoma cruzi genes reveals the acidocalcisome localization of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem.291, 25505–25515. 10.1074/jbc.M116.749655
25
LanderN.ChiurilloM. A.VercesiA. E.DocampoR. (2017). Endogenous C-terminal tagging by CRISPR/Cas9 in Trypanosoma cruzi. Bio. Protoc.7, 10:21769/BioProtoc 2299. 10.21769/BioProtoc.2299
26
LanderN.LiZ. H.NiyogiS.DocampoR. (2015). CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio6, e01012–e01015. 10.1128/mBio.01012-15
27
LemercierG.DutoyaS.LuoS.RuizF. A.RodriguesC. O.BaltzT.et al. (2002). A vacuolar-type H+-pyrophosphatase governs maintenance of functional acidocalcisomes and growth of the insect and mammalian forms of Trypanosoma brucei. J. Biol. Chem.277, 37369–37376. 10.1074/jbc.M204744200
28
LiC. H.IrmerH.Gudjonsdottir-PlanckD.FreeseS.SalmH.HaileS.et al. (2006). Roles of a Trypanosoma brucei 5'->3' exoribonuclease homolog in mRNA degradation. RNA12, 2171–2186. 10.1261/rna.291506
29
MaY.WeissL. M.HuangH. (2015). Inducible suicide vector systems for Trypanosoma cruzi. Microbes. Infect.17, 440–450. 10.1016/j.micinf.2015.04.003
30
MeauxS.Van HoofA. (2006). Yeast transcripts cleaved by an internal ribozyme provide new insight into the role of the cap and poly(A) tail in translation and mRNA decay. RNA12, 1323–1337. 10.1261/rna.46306
31
NadererT.HengJ.McconvilleM. J. (2010). Evidence that intracellular stages of Leishmania major utilize amino sugars as a major carbon source. PLoS Pathog.6:e1001245. 10.1371/journal.ppat.1001245
32
OberholzerM.MorandS.KunzS.SeebeckT. (2006). A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol. Biochem. Parasitol.145, 117–120. 10.1016/j.molbiopara.2005.09.002
33
PengD.KurupS. P.YaoP. Y.MinningT. A.TarletonR. L. (2014). CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio6, e02097–e02014. 10.1128/mBio.02097-14
34
PrommanaP.UthaipibullC.WongsombatC.KamchonwongpaisanS.YuthavongY.KnuepferE.et al. (2013). Inducible knockdown of Plasmodium gene expression using the glmS ribozyme. PLoS ONE8:e73783. 10.1371/journal.pone.0073783
35
RassiA.Jr.RassiA.LittleW. C. (2000). Chagas' heart disease. Clin. Cardiol.23, 883–889. 10.1002/clc.4960231205
36
RomagnoliB. A.PicchiG. F. A.HiraiwaP. M.BorgesB. S.AlvesL. R.et al. (2018). Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta. Trop.178, 190–195. 10.1016/j.actatropica.2017.11.013
37
ScottD. A.De SouzaW.BenchimolM.ZhongL.LuH. G.MorenoS. N.et al. (1998). Presence of a plant-like proton-pumping pyrophosphatase in acidocalcisomes of Trypanosoma cruzi. J. Biol. Chem.273, 22151–22158. 10.1074/jbc.273.34.22151
38
SeufferheldM.LeaC. R.VieiraM.OldfieldE.DocampoR. (2004). The H+-pyrophosphatase of Rhodospirillum rubrum is predominantly located in polyphosphate-rich acidocalcisomes. J. Biol. Chem.279, 51193–51202. 10.1074/jbc.M406099200
39
Soares MedeirosL. C.SouthL.PengD.BustamanteJ. M.WangW.BunkofskeM.et al. (2017). Rapid, selection-free, high-efficiency genome editing in protozoan parasites using CRISPR-Cas9 ribonucleoproteins. mBio8, e01788–e01717. 10.1128/mBio.01788-17
40
TakagiY.AkutsuY.DoiM.FurukawaK. (2019). Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLoS Negl. Trop. Dis. 13:e0007088. 10.1371/journal.pntd.0007088
41
UrbinaJ. A.DocampoR. (2003). Specific chemotherapy of chagas disease: controversies and advances. Trends Parasitol19, 495–501. 10.1016/j.pt.2003.09.001
42
WatsonP. Y.FedorM. J. (2011). The glmS riboswitch integrates signals from activating and inhibitory metabolites in vivo. Nat. Struct. Mol. Biol.18, 359–363. 10.1038/nsmb.1989
43
WinklerW. C.NahviA.RothA.CollinsJ. A.BreakerR. R. (2004). Control of gene expression by a natural metabolite-responsive ribozyme. Nature428, 281–286. 10.1038/nature02362
Summary
Keywords
acidocalcisome, CRISPR/Cas9, glmS, GP72, riboswitch, Trypanosoma cruzi, vacuolar proton pyrophosphatase
Citation
Lander N, Cruz-Bustos T and Docampo R (2020) A CRISPR/Cas9-riboswitch-Based Method for Downregulation of Gene Expression in Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 10:68. doi: 10.3389/fcimb.2020.00068
Received
25 November 2019
Accepted
11 February 2020
Published
27 February 2020
Volume
10 - 2020
Edited by
Julius Lukes, Institute of Parasitology (CAS), Czechia
Reviewed by
Eva Gluenz, University of Oxford, United Kingdom; Drahomíra Faktorová, Institute of Parasitology (CAS), Czechia
Updates
Copyright
© 2020 Lander, Cruz-Bustos and Docampo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto Docampo rdocampo@uga.edu
†Present address: Teresa Cruz-Bustos, Institute of Parasitology, University of Veterinary Medicine, Vienna, Austria
This article was submitted to Parasite and Host, a section of the journal Frontiers in Cellular and Infection Microbiology
‡These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.