 Qingtian Guan1
Qingtian Guan1 Roy Ummels2
Roy Ummels2 Fathia Ben-Rached1Yara Alzahid1Mohammad S. Amini1
Fathia Ben-Rached1Yara Alzahid1Mohammad S. Amini1 Sabir A. Adroub1Jakko van Ingen3
Sabir A. Adroub1Jakko van Ingen3 Wilbert Bitter2
Wilbert Bitter2 Abdallah M. Abdallah4,1*†
Abdallah M. Abdallah4,1*† Arnab Pain1,5*†
Arnab Pain1,5*†- 1Pathogen Genomics Laboratory, BESE Division, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 2Department of Medical Microbiology and Infection Control, Amsterdam University Medical Centers, Amsterdam, Netherlands
- 3Department of Medical Microbiology, Radboud UMC Center for Infectious Diseases, Radboud University Medical Center, Nijmegen, Netherlands
- 4Department of Basic Medical Sciences, College of Medicine, QU Health, Qatar University, Doha, Qatar
- 5Center for Zoonosis Control, Global Institution for Collaborative Research and Education (GI-CoRE), Hokkaido University, Sapporo, Japan
Mycobacterium kansasii is an important opportunistic pathogen of humans and has a close phylogenetic relationship with Mycobacterium tuberculosis. Seven subtypes (I–VII) have been identified using molecular biology approaches, of which subtype I is the most frequent causative agent of human disease. To investigate the genotypes and pathogenic components of M. kansasii, we sequenced and compared the complete base-perfect genomes of different M. kansasii subtypes. Our findings support the proposition that M. kansasii “subtypes” I-VI, whose assemblies are currently available, should be considered as different species. Furthermore, we identified the exclusive presence of the espACD operon in M. kansasii subtype I, and we confirmed its role in the pathogenicity of M. kansasii in a cell infection model. The espACD operon is exclusively present in mycobacterial species that induce phagosomal rupture in host phagocytes and is known to be a major determinant of ESX1-mediated virulence in pathogenic mycobacteria. Comparative transcriptome analysis of the M. kansasii I-V strains identified genes potentially associated with virulence. Using a comparative genomics approach, we designed primers for PCR genotyping of M. kansasii subtypes I-V and tested their efficacy using clinically relevant strains of M. kansasii.
Introduction
Nontuberculous mycobacteria (NTM) are increasingly being recognized as important opportunistic pathogens of humans. Reports have shown that more than 50 species of mycobacteria are associated with human diseases (Griffith et al., 2007), and Mycobacterium kansasii is the one of the most common causes of NTM disease in South America, South Africa and Europe (Hoefsloot et al., 2013).
M. kansasii was named after it was isolated from two patients suspected of having tuberculosis in Kansas, USA; it was previously known as the “yellow bacillus” (Pollak, 1953). This raised attention because it is difficult to treat patients co-infected with human immunodeficiency virus (HIV) (Levine and Chaisson, 1991; Corbett et al., 1999). In addition to systemic infection, M. kansasii also causes lung, cervical lymph node, and skin infections (Breathnach et al., 1995). Seven major subtypes have been described based on PCR-restriction fragment length polymorphism (RFLP) of hsp65 and internal transcribed spacer (ITS) (Taillard et al., 2003), while subtype I remains the most commonly isolated from clinical environments (Taillard et al., 2003; Borówka et al., 2017; Machado et al., 2018). A recent study proposed that the “subspecies” of M. kansasii should be considered as new species (Tagini et al., 2019).
Previous phylogenetic studies have suggested that Mycobacterium marinum (Stinear et al., 2008), Mycobacterium lacus (Tortoli et al., 2017), Mycobacterium decipiens (Brown-Elliott et al., 2018), Mycobacterium shinjukuense (Saito et al., 2011), Mycobacterium. riyadhense (Fedrizzi et al., 2017; Guan et al., 2019; Sapriel and Brosch, 2019) along with M. kansasii are closely related to the free-living ancestor of the Mycobacterium tuberculosis complex (MTBC). Phylogenetically, M. kansasii is closely related to M. tuberculosis (Wang et al., 2015), and the clinical manifestation of M. kansasii infections also shows significant overlap with the clinical profile of M. tuberculosis infections. M. kansasii has been suggested to represent the environmental ancestor of M. tuberculosis and can serve as a potential model organism to study evolutionary aspects of the switch from an opportunistic environmental pathogen to a professional host-restricted pathogen (Wang et al., 2015).
The pathogenesis of mycobacteria depends on the secretion of key virulence factors by the ESX-1 secretion system, which is absent in the vaccine strain Mycobacterium bovis BCG (Mahairas et al., 1996; Pym et al., 2002; Hsu et al., 2003). Substrates of this secretion system are responsible for phagosomal escape of pathogenic mycobacteria in macrophages and thereby for successful completion of the intracellular infection cycle (van der Wel et al., 2007). The espACD operon (Rv3616c~Rv3614c, espA, espC, and espD), a group of non-RD1 loci (Fortune et al., 2005; MacGurn et al., 2005), is essential for ESX-1 function, and its presence in the NTM pathogen M. kansasii subtype I is of particular interest in this study.
To explore the complexity of M. kansasii subtypes and understand why M. kansasii subtype I is most commonly associated with human diseases, we generated high-quality genomes of all five available subtypes of M. kansasii using a combination of Illumina and Pacific Biosciences sequencing technologies. Furthermore, we undertook a comprehensive comparative genomics and transcriptomics approach to identify components of potential M. kansasii subtype I virulence factors, as well as establish the role of the espACD operon in the pathogenicity of M. kansasii. By utilizing genome sequences, we also designed PCR-based genotyping primers for distinguishing M. kansasii subtypes.
Methods and Materials
Ethics Statement
The research protocol was approved by the Institutional Biosafety and Bioethics Committee of King Abdullah University of Science and Technology (Jeddah, Saudi Arabia; #18IBEC23). We confirm that all adult subjects provided informed consent, and a parent of the child participant provided informed consent on his behalf. Written informed consent was given.
Bacterial Strains and Cell Culture Conditions
We collected five environmental M. kansasii strains, designated KAUST-I to KAUST-V, that were originally isolated from either water or soil samples from five different European countries. Sixteen gDNA samples isolated from clinically relevant strains were also included in the study. These strains were collected from Radboud UMC Center of Infectious Diseases in the Netherlands. The isolates we collected were subtyped using the hsp65 gene as discussed before (Telenti et al., 1993) and subtype I-V were found, which are mostly isolated in many clinical cases (Taillard et al., 2003; Houben et al., 2012; Borówka et al., 2017; Machado et al., 2018) and no subtype VI and VII was detected.
M. kansasii subtypes I-V (strains KAUST-I to KAUST-V) were grown in Middlebrook 7H9 (Saitoh et al., 2000) broth with 0.5% glycerol, 0.05% polysorbate 80 and OADC (0.85% sodium chloride, 5% bovine albumin, 2% dextrose, 0.003% catalase) at 37°C. The human monocytic cell line THP-1 (ATCC® TIB-202™) was grown in Roswell Park Memorial Institute medium (RPMI)-1640 supplemented with 10% fetal bovine serum, 100 μg/mL penicillin and 100 μg/mL streptomycin at 37°C with 5% CO2.
DNA Isolation, Library Preparation, Assembly, and Genome Annotation
M. kansasii subtypes I-V DNA were isolated using a phenol-chloroform extraction protocol (Belisle and Sonnenberg, 1998). The extracted genomic DNA molecules were then sequenced using PacBio RSII sequencer with a 10 kb SMRT library. In parallel, genomic DNAs from M. kansasii subtypes I-V were sheared into ~500 bp fragments using CovarisTM. Paired-end, Illumina TruSeq PCR-FreeTM libraries were generated following the manufacturer's instruction, and the libraries were sequenced on a HiSeq2000 platform. The genomes were assembled into contigs using Canu (Koren et al., 2017) and corrected using Pilon (Walker et al., 2014) with PCR-free Illumina reads, and the complete assemblies were annotated by Prokka (Seemann, 2014).
Genotyping and Comparative Genomics of M. kansasii
To gain a better understanding of the M. kansasii pan-genome and develop a quick and accurate genotyping protocol, we downloaded forty M. kansasii genome sequences available on the NCBI genome database before Oct. 15th, 2018. We compared the phylogeny of these strains based on the hsp65 gene and ITS using the RaxML maximum-likelihood method (Stamatakis, 2014) with 100,000 replicates. Given that the standard method for reconstructing phylogenies of closely related microbes is the core-genome single-nucleotide polymorphism sites (SNPs) typing method, we also called the SNPs from the downloaded strain assemblies using Parsnp from the Harvest suite (Treangen et al., 2014) and generated the phylogenomic relationship map of M. kansasii. The phylogenetic tree was generated based on 135,969 core SNPs from each of the assemblies using the RaxML maximum-likelihood method (Stamatakis, 2014) with 100,000 replicates. In addition, the average nucleotide identity (ANI) between every two assemblies was calculated by OrthoANI (Lee et al., 2016). To determine the paralogous gene groups of the M. kansasii subspecies, the predicted protein sequences of M. kansasii subtypes I–V and M. tuberculosis were analyzed using OrthoMCL (Li et al., 2003) with a 50% identity cut-off and an inflation parameter of 1.5. The ortholog groups were visualized as a Venn diagram with the R VennDiagram package (Chen and Boutros, 2011). Mauve (Darling et al., 2004) was used to align the genomes of the subtypes to reveal the structure of the backbones of the chromosomes. The whole-genome comparison against subtype I was analyzed and visualized with BLAST Ring Image Generator (BRIG) (Alikhan et al., 2011). The uniqueness of singletons from each subtype and unique ortholog groups was further examined by BlastN (Version 2.2.26) against each other with 60% identity and an E-value of 0.00001 as the cutoff. The functional annotation of these genes was analyzed by EggNOG (Huerta-Cepas et al., 2016).
Investigation of the Role of the espACD Operon in M. kansasii Pathogenicity
During our comparative genome analysis of the M. kansasii subtypes and the list of genes that are uniquely present in the subtype I, we have observed the presence of an espACD operon, which is known to have an important role in ESX-1 secretion (Ates and Brosch, 2017). To investigate the functional role of the espACD operon in M. kansasii subtype I, we constructed the pSMT3-espACD-GFP plasmid, and it was transformed into M. kansasii subtype II using the electroporation method (Goude et al., 2015) to investigate the function of the espACD operon.
The espACD operon of M. kansasii subtype I was amplified with the primers EspACD-HindIII (TTTTAAGCTTCGGGACTTGCGCTTAGTCTG) and EspACD- AflII (TTTTCTTAAGGTGGCCGCCCGTTTATGTAG). The DNA fragments were digested with HindIII and AflII and cloned into an AflII-restriction site containing a variant of pSMT3 digested with the same enzymes. The resulting plasmid, pSMT3-espACD-GFP, is a shuttle plasmid that is difficult to incorporate directly into M. kansasii. Therefore, we introduced the OriT region of pRAW (Ummels et al., 2014) and introduced the plasmid into Mycobacterium marinum. Subsequently, we introduced the plasmid pSMT3-espACD-GFP into M. kansasii through conjugation.
Cultures of the different subtypes of M. kansasii (wild-type subtype I, wild-type subtype II, subtype II transferred with pSMT3-espACD-GFP, and subtype II transferred with pSMT3-GFP) with an OD600nm = 0.8–1.0 were prepared. Simultaneously, THP-1 cells (Tsuchiya et al., 1980) were counted using a haemocytometer and diluted to seed half a million cells per well in 24-well plates in the presence of 25 ng/mL phorbol myristate acetate (PMA) to allow the cells to differentiate into macrophages and adhere overnight at 37°C with 5% CO2. After their differentiation, the complete medium was replaced with RPMI+10% FCS without antibiotics and incubated for 3 h. The macrophages were incubated in triplicate with M. kansasii KAUST-I, KAUST-II, KAUST-II-pSMT3-espACD-GFP, and KAUST-II-pSMT3-GFP, with a multiplicity of infection (MOI) = 5, or incubated with only medium. After 2 h of infection, the macrophages were washed with PBS and incubated with fresh medium without antibiotics. The cells were then treated with 1 mL of 1% Triton X-100 for 10 min at 37°C and collected at different time points after infection (0, 24, 48, and 72 h). Then, the obtained lysates were diluted to 1:10, 1:100, and 1:1,000, and 100 μL of each dilution was subsequently spread on Middlebrook 7H10 agar plates. The survival rate was evaluated as the percentage of colony forming units (CFUs) at different time points, taking the number of CFUs at time point “0” as the reference.
Development of Genotyping Primers for M. kansasii Subtypes
The unique genes within each singleton pool were further investigated. We used BlastN against the non-redundant (nr) nucleotide database for the unique genes and refined the list of selected unique genes after comparison against the other species in the nr database. The unique genes were also confirmed by the binary alignment map (BAM) files generated by cross-mapping of Illumina reads to each subtype. The primer sets that target the unique genes are presented in Table 1. Primer sets that target the 16S rRNA V2~V4 regions were used as internal controls. GoTaqTM Green Master Mix was used for the amplification, and the cycling conditions for these primers consisted of preheating at 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 62°C for 30 s and extension at 72°C for 30 s. The final extension was performed at 72°C for 10 min after 30 cycles.
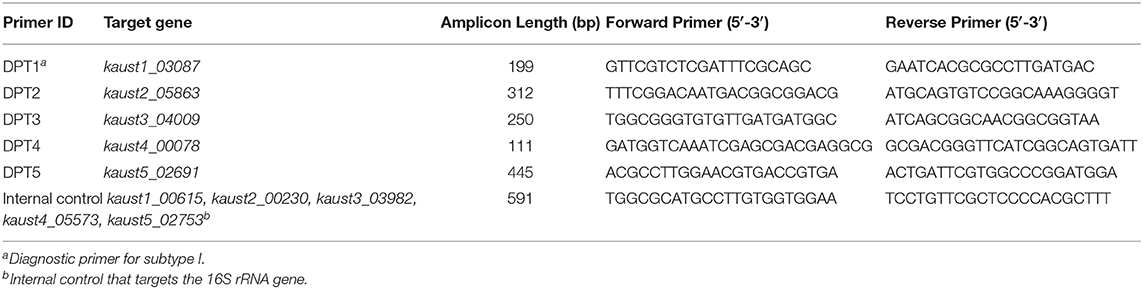
Table 1. Primer sets designed from selected sequences of M. kansasii subtypes I-V.
Transcriptome Analysis of M. kansasii Subspecies
Total RNA was extracted from 40 ml of exponential growth phase culture bacterial culture using the TRIzol protocol (Rio et al., 2010). Briefly, the bacterial cultures were centrifuged at 1,500 rcf for 15 min, suspended in 1 mL of TRIzol and incubated for 5 min. Then, 500 μL of zirconia beads (BiospecTM) were added and treated six times by beating at maximum speed for 30 s. Then, the mixture was centrifuged, and the upper layer was incubated with 200 μL of chloroform. After centrifugation at 4°C at maximum speed for 20 min, an equal volume of isopropanol was added to the aqueous layer. The mixture was centrifuged at 4°C at full speed, and the supernatant was discarded. Then, 1.5 mL of 70% cold ethanol was added and centrifuged for 10 min. The ethanol was discarded, and the RNA was air-dried. The RNA was suspended in 30 μl of RNase-free water and incubated at 60°C until all of the RNA was dissolved. The RNA was then stored at −80°C before library preparation. For library preparation, DNA was removed using TurboTM DNase, and rRNA was removed using the InvitrogenTM Ribominus Kit. Strand-specific Illumina RNAseq libraries were prepared using the TruSeq kit following the manufacturer's manual, and the libraries were sequenced on a HiSeq2000 platform (IlluminaTM). The RNAseq reads obtained were first trimmed using the Trimomatic program (Bolger et al., 2014) to remove adapters and low-quality reads (cutoff: Q30). The “clean reads” were further mapped to the annotated genomes of M. kansasii subtypes I-V. To compare the differences between M. kansasii subtype I and the four other subtypes, the transcriptomes of the 3,761 one-to-one orthologs obtained from OrthoMCL were compared. The RNAseq reads were mapped to each genome with HISAT2 (Pertea et al., 2016), and the reads mapped to each gene were counted by HTSeq (Anders et al., 2015) with union mode. DESeq2 (Anders and Huber, 2013) was used to call the differentially expressed genes. Genes with a padj value < 0.01 and an absolute Log2 fold change value greater than two were selected and further analyzed. The same method was applied to determine the differentially expressed genes (DEGs) in the other four M. kansasii subtypes.
Results
Comparative Genomics of M. kansasii Subtypes I–V
To study the variation in the M. kansasii subtypes, we obtained isolates belonging to each subtype and used a combination of Illumina and SMRT (PacBio) sequencing methods to assemble genomes into single base-perfect contigs. All of the bacterial chromosomes of M. kansasii subtypes I-V were assembled into a single contig each, and the genome of M. kansasii subtype III included a large new 301,558 bp circular plasmid pMKIII01 (Supplementary Figure 1) that has not been described before. The sequencing depths of each of the assemblies (subtype I–V) from PacBio were 251X, 122X, 98X, 148X, and 158X, respectively. The comparison of these genome assemblies with M. tuberculosis H37Rv, M. marinum M and the type strain M. kansasii 12478 are shown in Table 2. The comparison of our assemblies with previously published genomes from the same strains is described in Supplementary Table 1. The backbone structure of subtypes I–V is shown in Supplementary Figure 2, and the BlastN comparison of subtypes II–V to subtype I is shown in Figure 1.
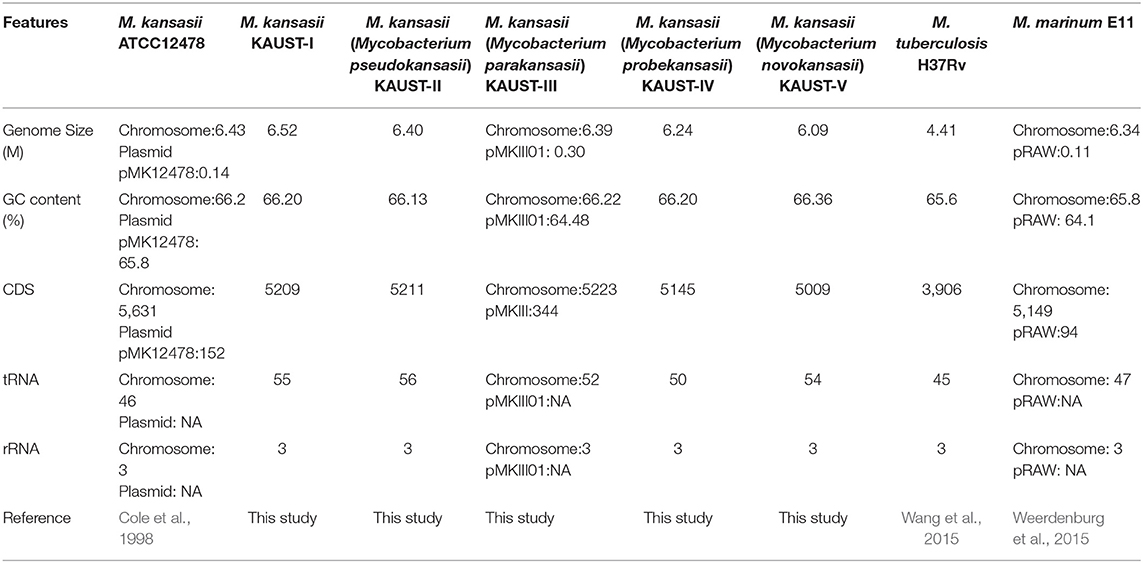
Table 2. Comparison of general features/gene families of M. kansasii subtypes I-V with those of other Mycobacteria.
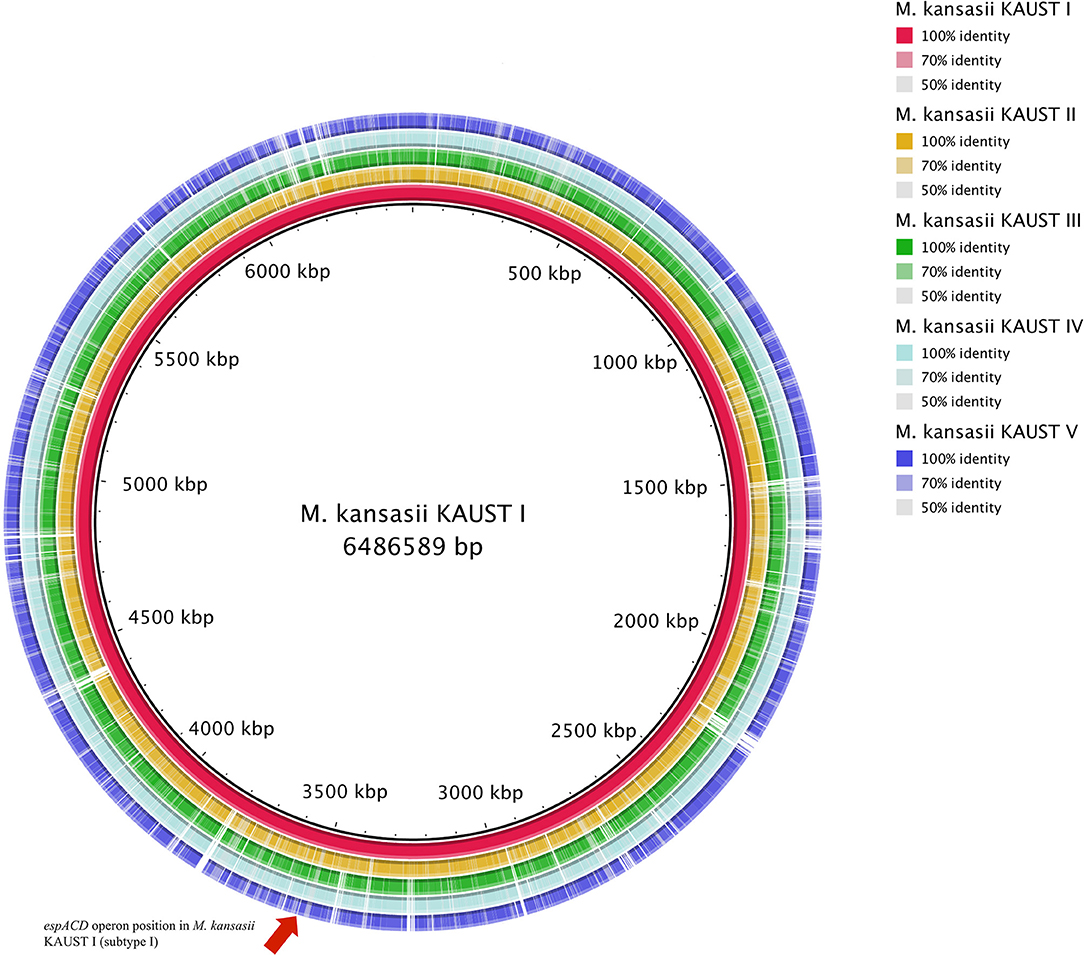
Figure 1. Circular map representation of the M. kansasii subtype II–subtype V genome showing BlastN similarities to M. kansasii subtype I. Each ring of the circle corresponds to a specific complete genome as referred to in the figure keys on the right. The espACD operon position in M. kansasii KAUST I is indicated by an arrow.
The ortholog groups cluster across the genomes of the different M. kansasii subtypes, and the functional annotation categories of the paralogues/singletons are presented in Figure 2A. The number of unique singletons of each subtype that did not appear in the Venn diagram from M. kansasii subtypes I-V was 555, 628, 608, 756, and 642, respectively, most of which were proteins with unknown functions. This means that all subtypes have a similar number of singletons, indicating that they have similar genetic headroom and evolutionary pressures. The comparison of M. kansasii subtypes and M. tuberculosis H37Rv is shown in Figure 2B. A detailed analysis of the genes shared between M. kansasii subtype I and M. tuberculosis H37Rv is presented in Supplementary Table 2.
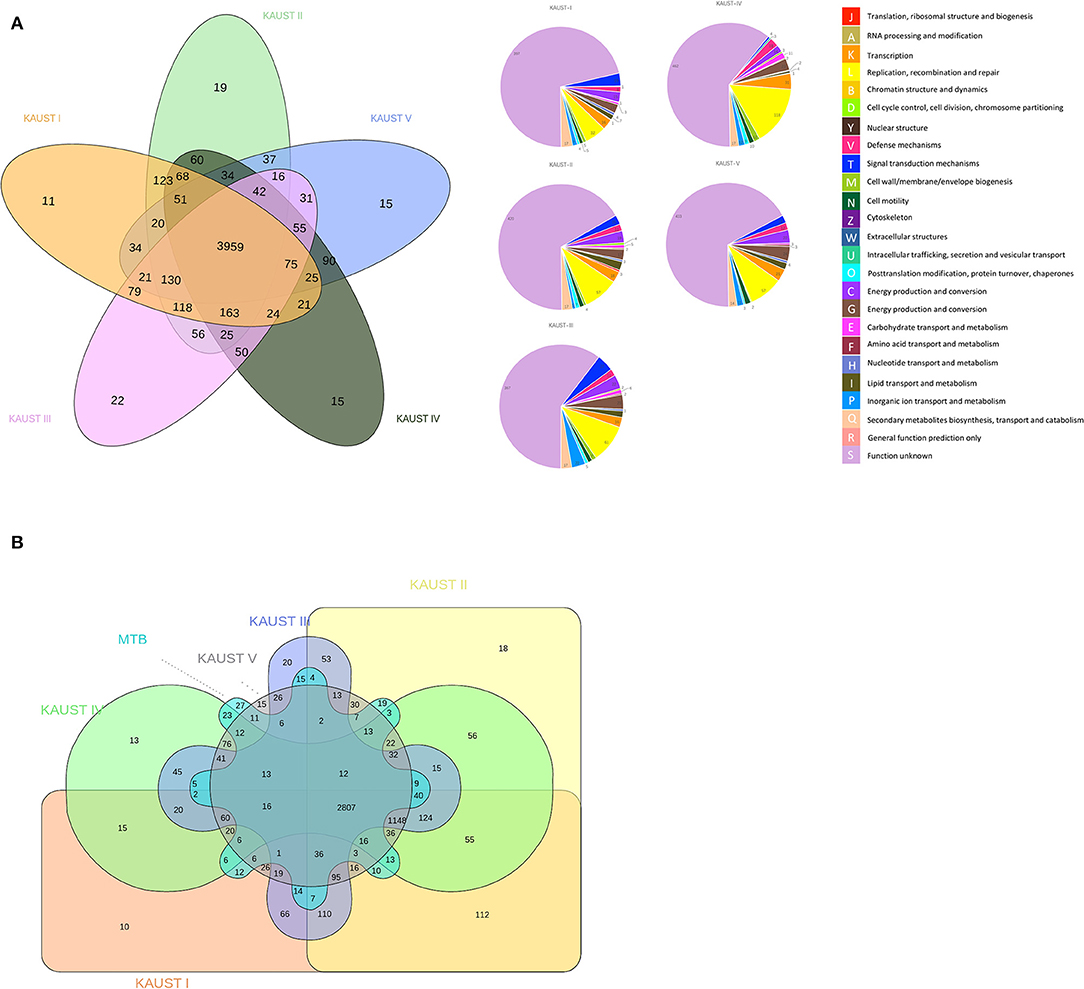
Figure 2. Comparative genomics study of the chromosomes of M. kansasii subtypes I to V. Venn Diagram of the ortholog groups and paralogue groups shared by (A) the five M. kansasii subtypes and (B) the five subtypes with M. tuberculosis H37Rv. The number represents the ortholog groups or paralogue groups present in the particular types.
One of the striking findings in the comparative genomic analysis was that one copy of the espACD operon (kaust1_3087~kaust1_3085) is present exclusively in subtype I (Figure 1, Supplementary Figure 3). We examined whether these espACD loci are involved in the virulence of M. kansasii.
Genotyping of M. kansasii
Various molecular methods for genotyping M. kansasii have been developed for comparison and strain identification (Zhang et al., 2004). To study the subtypes in detail, we analyzed the available 45 sequenced M. kansasii genome datasets. The analysis revealed that the phylogenetic tree that was constructed based on the ITS sequences grouped the M. kansasii subtypes incorrectly in some cases (Figure 3) due to limited information provided in the sequences (Supplementary Figure 4). In silico genotyping of the subtypes of the 45 M. kansasii strains based on whole-genome core SNPs and ANI revealed that there are six major subtypes of M. kansasii that have been sequenced in full length so far (Figure 3, Supplementary Table 3). The ANI value between the subtypes of M. kansasii is high, which supports the idea that M. kansasii “subtypes” I-VI should be considered as different species; e.g., the ANI value between subtype VI and subtype III is 89.35–89.48, and the value between subtype II and subtype VI is 92.86–92.95. Therefore, we conclude that M. kansasii subtypes I-VI should be assigned as different species. This finding is in line with a recently published result describing the ANI values between M. kansasii subtypes (Tagini et al., 2019) in which the authors proposed that M. kansasii subtypes III, V and VI should be considered as independent species rather than subspecies.
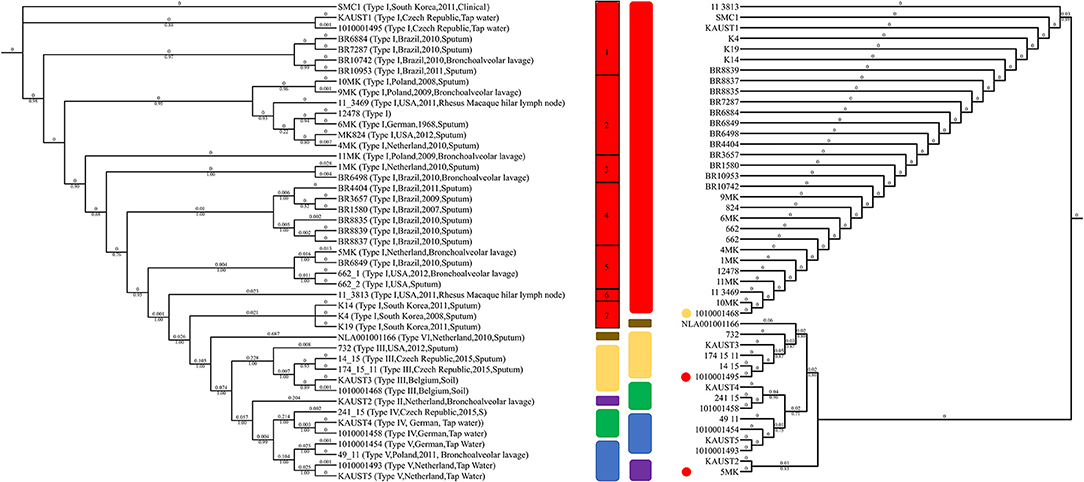
Figure 3. Genotyping of M. kansasii using ITS and SNPs from the complete genome assemblies. The maximum-likelihood tree based on the internal transcribed spacer (ITS) sequences of 45 M. kansasii strains is shown on the right. The maximum-likelihood phylogenetic tree based on 135,969 SNPs from the core genome of M. kansasii subtypes is shown on the left. Bootstrap support values are indicated on the branch as a percentage of 100,000 replicates. The branch length is ignored for illustration purposes and displayed on each branch. The color code for each genotype: red: M. kansasii subtype I; purple: M. kansasii subtype II; yellow: M. kansasii subtype III; green: M. kansasii subtype IV; blue: M. kansasii subtype V; brown: M. kansasii subtype VI. The strains within M. kansasii subtype I are separated with black rectangles in the figure. The circles indicate the difference between the ITS and SNP genotyping method, and colors represent the genotypes suggested by ANI clustering.
Comparative Transcriptome Analysis
To reveal the differences in expression profiles of M. kansasii subtypes, we performed a global transcriptome analysis across M. kansasii subtypes I-V grown in vitro, focusing on the changes in gene expression profiles during the exponential growth of the bacilli in Middlebrook 7H9 medium. To investigate this, RNA was extracted from three independent exponential phase cultures of M. kansasii subtypes I-V. Data quality was assessed using Euclidean distance matrices and demonstrated high levels of reproducibility between the biological replicates (Supplementary Figure 5). After filtering, a total of 38 genes were uniquely downregulated and 10 genes were upregulated in M. kansasii subtype I in comparison with M. kansasii subtypes II-V within the one-to-one ortholog groups (Figure 4A) (Supplementary Table 4). Analysis of differential expression in M. kansasii subtype I identified changes in genes involved in a variety of cellular processes, mostly genes associated with metabolic, respiratory, regulatory and cell wall-associated processes (Figure 4B).
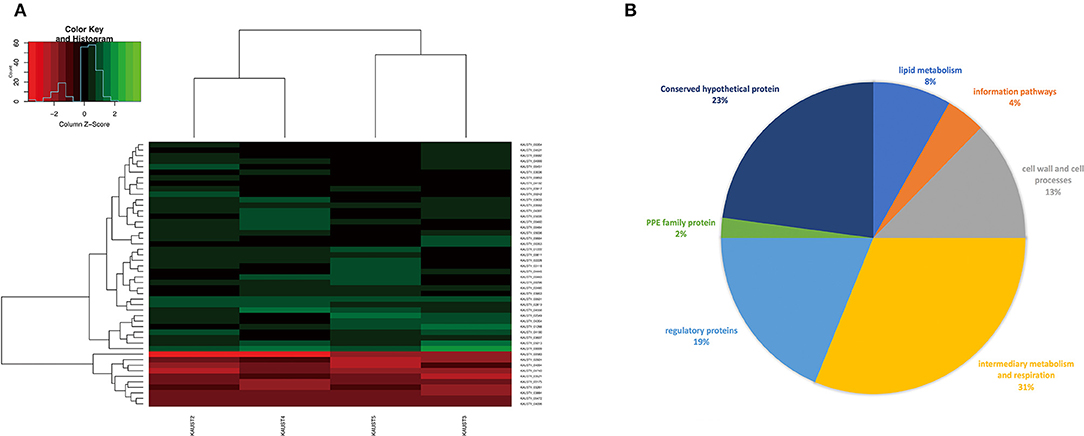
Figure 4. (A) Gene expression profiles (log2 fold change) of the differentially expressed genes of M. kansasii subtypes II-V in comparison to M. kansasii subtype I. The red color represents upregulated genes, and the green color represents downregulated genes compared with the control strain. padj < 0.01 and Log2FC > 2 were used as the cutoff for the selection of the genes. (B) Pie-chart showing a subdivision of all differentially expressed genes based on functional categories.
We noted that a substantial number of genes that are likely essential and required for mycobacterial growth (Sassetti et al., 2003; Weerdenburg et al., 2015) were differentially regulated in M. kansasii subtype I, including genes involved in cell wall-associated processes (mpt63) and lipid metabolism (accD4, mbtC) (Supplementary Table 4). In addition, many of these differentially regulated genes encode proteins of unknown function and hence need to be further characterized (Figure 4A). However, we did not notice any well-known virulence genes in the list.
To determine the functional classifications and pathways of the DEGs associated with each subtype, all upregulated DEGs from each subtype were analyzed with EggNOG Mapper (Huerta-Cepas et al., 2016) (Supplementary Table 5). The different KEGG pathways associated with the DEGs were as follows: three pathways for M. kansasii subtype I, twenty-one pathways for M. kansasii subtype II, nine pathways for M. kansasii subtype IV and 14 pathways for M. kansasii subtype V. Notably, metabolic pathways were significantly dominant in all subtypes.
Complementation of the espACD Operon Recovers the Pathogenicity of the Non-pathogenic M. kansasii Subtype II
M. kansasii subtype I contains five ESX systems, and the overall arrangement of the five ESX systems is similar to that in M. tuberculosis H37Rv (Supplementary Figure 6). This includes the ESX-1 region, which is important for virulence in M. tuberculosis (Conrad et al., 2017; Tiwari et al., 2019). Previous research has shown that the ESX-1 substrate EsxA is secreted by both M. kansasii subtype I and subtype V (Houben et al., 2012). Surprisingly, the same study also showed using electron microscopy that only M. kansasii subtype I is able to escape from the phagolysosome (Houben et al., 2012).
To test whether this espACD locus could affect the virulence of M. kansasii, we cloned the espACD gene cluster of subtype I on a shuttle plasmid and introduced this plasmid into subtype II, which does not have the espACD operon. We first confirmed that there are no in vitro growth differences among the tested strains (Supplementary Figure 7). Subsequently, differentiated THP-1 monocytes were infected with these strains. After 3 days, we observed a significant decrease in the survival rate of wild-type M. kansasii subtype II and the same bacterial strain harboring the control plasmid pSMT3-GFP (Figure 5). On the other hand, wild-type subtype I and the subtype II strain transfected with pSMT3-espACD-GFP had significantly increased CFU numbers. We concluded that the presence of the espACD operon was able to increase the virulence of M. kansasii subtype II (Figure 5).
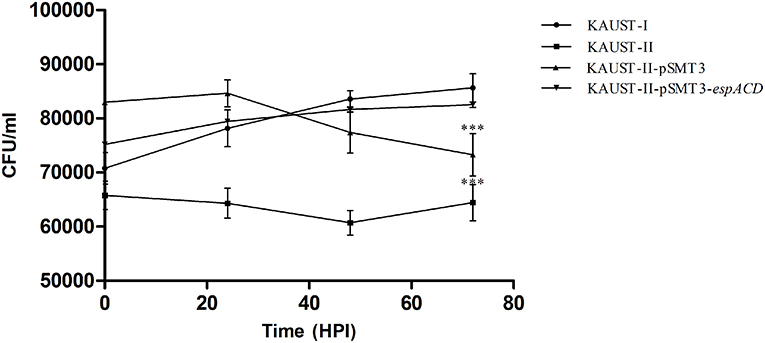
Figure 5. Complementation of the espACD operon in M. kansasii subtype II with that from M. kansasii subtype I is crucial for its virulence. The functional complementation experiment revealed that the espACD operon plays a significant role in M. kansasii subtype II pathogenicity, at least in THP-1 cells. Intracellular CFU count showing the bacterial load in THP-1 macrophages infected with KAUST-I (•), KAUST-II (•), KAUST-II-pSMT3 (▴) and KAUST-II-pSMT3-espACD (▾) at a MOI of 5. The infected macrophages were lysed at 0, 24, 48, and 72 h time-points post-infection and three dilutions of the released mycobacterial cells were plated on 7H10 agar plates. CFU were counted and recorded after 15 days of plating. Experiments were performed with three replicates, and Student's t-test for significance was calculated with the level of significance shown (***highly significant difference, p < 0.01).
M. kansasii Subspecies Genotyping
Many genotyping methods have been applied for the subtyping of M. kansasii. Several of those methods either failed to distinguish all of the subtypes, such as AccuProbeTM (Tortoli et al., 1996) and INNO LiPATM (Suffys et al., 2001), or involved sequencing (Park et al., 2000) or restriction enzyme digestion (Bakuła et al., 2013) steps. To simplify the genotyping method for these different subtypes, especially the more virulent subtype I, we developed a genotyping method that can rapidly and accurately distinguish M. kansasii subtypes I-V by PCR (Figures 6A–E). The primer sets accurately identify M. kansasii subtypes and can serve as part of an accurate and rapid diagnostic protocol in a clinical setting (Figure 6F) to overcome misdiagnosis or extensive lab work using the hsp65 and ITS regions (Figure 3).
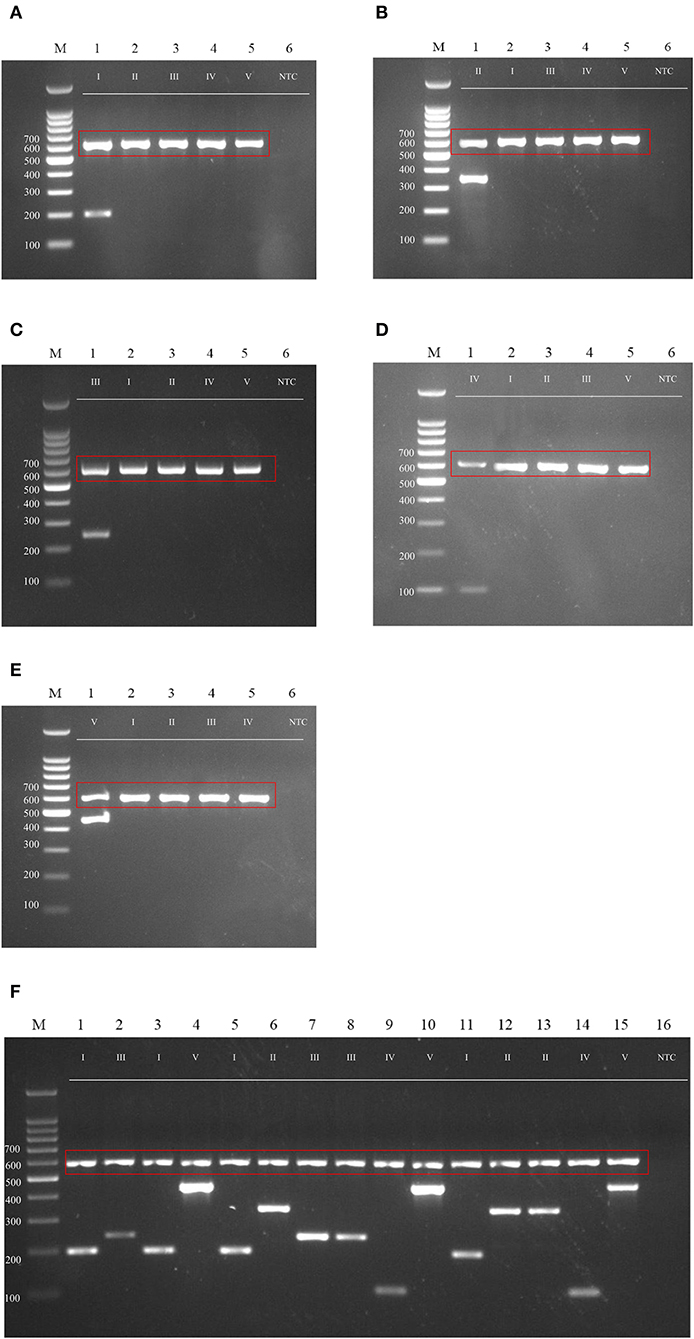
Figure 6. Agarose gel (2%) electrophoresis of diagnostic PCR tests for M. kansasii subtypes I-V. Lane M: DNA markers. The gDNA used for testing is labeled on the top, and the size of the ladder is labeled on the left side of each gel picture. The genotyping results for M. kansasii subtypes I-V are listed in (A–E). The clinical samples tested with the cocktail of primers DP1-5 is shown in (F). The 16S primer control is shown in the red box. NTC, Non-template control.
Discussion
We obtained high-quality genomes of the five most frequently observed types of M. kansasii by applying a combination of short reads and long reads from the Illumina and PacBio platforms, respectively. This allowed us to undertake a comprehensive genome comparison at single base-pair resolution and define the key hallmarks of the five subtypes of sequenced M. kansasii strains.
As expected, owing to the environmental niche of this bacteria, M. kansasii subtypes I-V share a large number of orthologs that are not present in M. tuberculosis H37Rv (1148). Toxin/antitoxin (T/A) systems were previously only reported in M. tuberculosis while recent studies shows the expansion of the T/A systems from NTMs to MTBC (Guan et al., 2019; Sapriel and Brosch, 2019). The comparison of the M. kansasii subtype I and M. tuberculosis H37Rv genomes reveal that they share 12 genes that are not present in other subspecies (Figure 2B, Supplementary Table 2). Two copies of the toxin and antitoxin proteins VapB12 and VapC4, which are known to be involved in the adaptation of M. tuberculosis (Sala et al., 2014), two mobile genetic elements genes within the RD3 of M. tuberculosis H37Rv (rv1584c, rv1585c) and one copy of a regulatory protein (rv1129c) that has been shown to be required for M. tuberculosis growth on cholesterol (Griffin et al., 2011) are uniquely present in M. kansasii subtype I and M. tuberculosis H37Rv. We also found two PPE family genes (rv1801, rv1802) that are present in subtype I, which was reported to be essential for endothelial-cell invasion and/or intracellular survival (Talaat et al., 2004; Jain et al., 2006). M. kansasii phylogenetically and clinically resembles M. tuberculosis (Wang et al., 2015), and the preservation of these genes during the reduction of the Mycobacterium tuberculosis complex (MTBC) genomes indicates that they may be important for virulence and pathogenicity.
pMKIII01 is a novel plasmid with a lower GC content (64.48%) than the M. kansasii subtype III chromosome, whose G+C content is 66.22%. This may suggest that this plasmid was not originally from M. kansasii subtype III and has been transformed into the bacterial cells during the evolutionary processes. The new plasmid pMKIII01 (Supplementary Figure 1) harbors a locus encoding a putative Type IV secretion system and a putative Type VII secretion system. This plasmid resembles the conjugative mycobacterial plasmids that have been discovered previously, such as the type-plasmid pRAW in M. marinum (Ummels et al., 2014), pMAH135 (Uchiya et al., 2015), and pMA100 (da Silva Rabello et al., 2012) of Mycobacterium avium, pMyong1 from Mycobacterium yongonense (Kim et al., 2013), pMK12478 (Veyrier et al., 2009) from M. kansasii 12478 and several plasmids from Mycobacterium chimera (van Ingen et al., 2017). The presence of a pRAW-like plasmid in M. kansasii subtype III confirms that these plasmids are widespread in the Mycobacterium genus, which seems to be logical, as they were shown to be conjugative (Ummels et al., 2014). The variation in the sizes of pRAW-like plasmids is significant, with a value of 301,558 bp; the pMKIII01 plasmid is 2.5 times the size of pRAW. Thus far, there is no evidence that pRAW-like plasmids are directly involved in virulence, but they have been instrumental in the evolution and duplication of the ESX systems (Dumas et al., 2016; Newton-Foot et al., 2016).
The commonly used method for diagnosis, which is based on the variation of hsp65 and ITS sequences, either requires restriction enzyme digestion steps or cannot distinguish the M. kansasii subtypes accurately (Figure 3, Supplementary Figure 4). While the chromosomes of all five subtypes (I-V) are syntenic and contain primarily shared orthologs (Figure 1, Supplementary Figure 2) with a limited number of uniquely shared patterns of genes across all of them, the phylogenomic analyses (Figure 3) and the ANI figures revealed unusually high levels of nucleotide diversity amongst the M. kansasii complex (Supplementary Table 3). The complexity of M. kansasii is greater than we previously thought, and M. kansasii is too diverse to be considered as a single “species” (Figure 3) given that approximately >95–96% ANI values are considered to set the species boundary (Richter and Rosselló-Móra, 2009). This proposition is also consistent with the differences in virulence characteristics amongst the subtypes.
Hence, we propose to consider M. kansasii “subspecies” as six different species: Mycobacterium pseudokansasii (previously classified as Mycobacterium kansasii subtype II); Mycobacterium parakansasii (previously classified as Mycobacterium kansasii subtype III); Mycobacterium probekansasii (previously classified as Mycobacterium kansasii subtype IV); Mycobacterium novokansasii (previously classified as Mycobacterium kansasii subtype V); and Mycobacterium eurokansasii (previously classified as Mycobacterium kansasii subtype VI).
In line with this, the function of M. kansasii subtype I DEGs is diverse and includes aminopeptidase, ADP-ribose pyrophosphatase and phenyloxazoline synthase (Figure 4A, Supplementary Table 4), suggesting the diverse metabolic capacities of M. kansasii subtypes I and II–V.
There are now approximately one dozen ESX-1 substrates identified (Champion et al., 2014; Phan et al., 2018; Sala et al., 2018; Abdallah et al., 2019) in M. tuberculosis, and most of the genes coding for these ESX-1 substrates are located within the ESX-1 locus. Although ESX-1-secreted substrates are essential for virulence, their separate roles are not well defined. This is because the secretion of some of these substrates, such as EspA and EsxA (ESAT-6), is mutually dependent (Fortune et al., 2005; Houben et al., 2012). The genes located within the espACD operon (rv3614~rv3616c) are outside the ESX-1 loci. They are homologous to rv3864~rv3867, located within the ESX-1 locus and required for ESX-1 secretion for virulence (MacGurn et al., 2005). While no evidences that suggest there are any connections of these two groups of genes. Comparative genome analyses suggest that the espACD serves as a pathogenetic island (Simeone et al., 2012; Majlessi et al., 2015) which might be introduced independently by horizontal transfer from different species (Ates and Brosch, 2017). EspC associates with EspA in the cytoplasm and membrane, then polymerizes during secretion from M. tuberculosisis. EspC forms a filamentous structure in the cell envelope of M. tuberculosis hence affects ESX-1 secretion (Lou et al., 2017). We have confirmed that the espACD operon plays a crucial role in the pathogenicity of M. kansasii. Functional complementation of the espACD operon in wild-type M. kansasii subtype II was able to recover the persistence phenotype (Figure 5). It is probable that this increased persistence is induced by complementation of the ESX secretion system, which might be essential for bacteria survival within macrophages. Not surprisingly, M. kansasii subtype II-pSMT3-espACD-GFP was still less virulent than the wild-type M. kansasii subtype I because the espACD operon is under the control of EspR (Raghavan et al., 2008; Kumar et al., 2016) and PhoPR (Frigui et al., 2008), which are missing in the plasmid.
In conclusion, our study reveals that M. kansasii subtype I has several unique features in comparison to the established subtypes II-V, consistent with its pathogenicity characteristics. We also provide evidence that the espACD operon plays an important role in acquiring virulence, at least in M. kansasii subtype II. Our results support the notion that there are six subtypes of M. kansasii, and the high levels of nucleotide variation amongst the subtypes prompt us to propose that we should consider M. kansasii subtypes I–VI as different species. Finally, we developed a simple approach to distinguish M. kansasii subtypes I-V that can be applied in clinical settings.
Data Availability Statement
The M. kansasii dataset is available at the European Nucleotide Archive (ENA) under study accession No. PRJEB32175.
Ethics Statement
The studies involving human participants were reviewed and approved by The research protocol was approved by the Institutional Biosafety and Bioethics Committee of King Abdullah University of Science and Technology (Jeddah, Saudi Arabia; #18IBEC23). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AP and AA conceived the study and obtained the funding and supervised the work. AP, QG, and AA designed the experiments. RU generated the strains for the complementation experiment. QG and FB-R performed the complementation experiment. YA, MA, and SA generated the Illumina data and helped in the analysis. QG generated the PacBio data, performed all the data analysis, and prepared the initial draft of the manuscript, followed by edits from AP, WB, AA, and JI. All authors have commented on various sections of the manuscript, which were finally curated and incorporated into the final version by QG, AP, and AA.
Funding
Work in AP's laboratory was supported by the KAUST faculty baseline fund (BAS/1/1020-01- 01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank Biological Core Lab of King Abdullah University of Science and Technology for sequencing the strains on the Illumina and PacBio platforms.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00122/full#supplementary-material
References
Abdallah, A. M., Weerdenburg, E. M., Guan, Q., Ummels, R., Borggreve, S., Adroub, S. A., et al. (2019). Integrated transcriptomic and proteomic analysis of pathogenic mycobacteria and their esx-1 mutants reveal secretion-dependent regulation of ESX-1 substrates and WhiB6 as a transcriptional regulator. PLoS ONE 14:e0211003. doi: 10.1371/journal.pone.0211003
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Anders, S., and Huber, W. (2013). Differential Expression of RNA-Seq Data at the Gene Level – The DESeq Package. Bioconductor Package Vignette, Heidelberg, Germany.
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Ates, L. S., and Brosch, R. (2017). Discovery of the type VII ESX-1 secretion needle? Mol. Microbiol. 103, 7–12. doi: 10.1111/mmi.13579
Bakuła, Z., Safianowska, A., Nowacka-Mazurek, M., Bielecki, J., and Jagielski, T. (2013). Short communication: subtyping of Mycobacterium kansasii by PCR-restriction enzyme analysis of the hsp65 gene. Biomed Res. Int. 2013:178725. doi: 10.1155/2013/178725
Belisle, J. T., and Sonnenberg, M. G. (1998). Isolation of genomic DNA from mycobacteria. Methods Mol. Biol. 101, 31–44. doi: 10.1385/0-89603-471-2:31
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Borówka, P., Lach, J., Bakuła, Z., Ingen, J., Safianowska, A., Brzostek, A., et al. (2017). Draft genome sequences of Mycobacterium kansasii clinical strains. Genome Announc. 5, 1–3. doi: 10.1128/genomeA.00406-17
Breathnach, A., Levell, N., Munro, C., Natarajan, S., and Pedler, S. (1995). Cutaneous Mycobacterium kansasii infection: case report and review. Clin. Infect. Dis. 20, 812–817. doi: 10.1093/clinids/20.4.812
Brown-Elliott, B. A., Simmer, P. J., Trovato, A., Hyle, E. P., Droz, S., Buckwalter, S. P., et al. (2018). Mycobacterium decipiens sp. nov., a new species closely related to the Mycobacterium tuberculosis complex. Int. J. Syst. Evol. Microbiol. 68, 3557–3562. doi: 10.1099/ijsem.0.003031
Champion, M. M., Williams, E. A., Pinapati, R. S., and Champion, P. A. D. (2014). Correlation of phenotypic profiles using targeted proteomics identifies mycobacterial esx-1 substrates. J. Proteome Res. 13, 5151–5164. doi: 10.1021/pr500484w
Chen, H., and Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Conrad, W. H., Osman, M. M., Shanahan, J. K., Chu, F., Takaki, K. K., Cameron, J., et al. (2017). Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc. Natl. Acad. Sci. U.S.A. 114, 1371–1376. doi: 10.1073/pnas.1620133114
Corbett, E. L., Churchyard, G. J., Hay, M., Herselman, P., Clayton, T., Williams, B., et al. (1999). The impact of HIV infection on Mycobacterium kansasii disease in South African gold miners. Am. J. Respir. Crit. Care Med. 160, 10–14. doi: 10.1164/ajrccm.160.1.9808052
da Silva Rabello, M. C., Matsumoto, C. K., de Almeida, L. G. P., Menendez, M. C., de Oliveira, R. S., Silva, R. M., et al. (2012). First description of natural and experimental conjugation between mycobacteria mediated by a linear plasmid. PLoS ONE 7:e29884. doi: 10.1371/journal.pone.0029884
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dumas, E., Boritsch, E. C., Vandenbogaert, M., De La Vega, R. C. R., Thiberge, J. M., Caro, V., et al. (2016). Mycobacterial pan-genome analysis suggests important role of plasmids in the radiation of type VII secretion systems. Genome Biol. Evol. 8, 387–402. doi: 10.1093/gbe/evw001
Fedrizzi, T., Meehan, C. J., Grottola, A., Giacobazzi, E., Fregni Serpini, G., Tagliazucchi, S., et al. (2017). Genomic characterization of nontuberculous mycobacteria. Sci. Rep. 7:45258. doi: 10.1038/srep45258
Fortune, S. M., Jaeger, A., Sarracino, D. A., Chase, M. R., Sassetti, C. M., Sherman, D. R., et al. (2005). Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. U.S.A. 102, 10676–10681. doi: 10.1073/pnas.0504922102
Frigui, W., Bottai, D., Majlessi, L., Monot, M., Josselin, E., Brodin, P., et al. (2008). Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog. 4:e33. doi: 10.1371/journal.ppat.0040033
Goude, R., Roberts, D. M., and Parish, T. (2015). Electroporation of mycobacteria. Methods Mol. Biol. 1285, 117–130. doi: 10.1007/978-1-4939-2450-9_7
Griffin, J. E., Gawronski, J. D., DeJesus, M. A., Ioerger, T. R., Akerley, B. J., and Sassetti, C. M. (2011). High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7:e1002251. doi: 10.1371/journal.ppat.1002251
Griffith, D. E., Aksamit, T., Brown-Elliott, B. A., Catanzaro, A., Daley, C., Gordin, F., et al. (2007). An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 175, 367–416. doi: 10.1164/rccm.200604-571ST
Guan, Q., Garbati, M., Mfarrej, S., AlMutairi, T., Smyth, A., Singh, A., et al. (2019). Insights into ancestry and adaptive evolution of the Mycobacterium tuberculosis complex from analysis of the emerging pathogen Mycobacterium riyadhense. bioRxiv. doi: 10.1101/728923
Hoefsloot, W., van Ingen, J., Andrejak, C., Angeby, K., Bauriaud, R., Bemer, P., et al. (2013). The geographic diversity of nontuberculous mycobacteria isolated from pulmonary samples: an NTM-NET collaborative study. Eur. Respir. J. 42, 1604–1613. doi: 10.1183/09031936.00149212
Houben, D., Demangel, C., van Ingen, J., Perez, J., Baldeón, L., Abdallah, A. M., et al. (2012). ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell. Microbiol. 14, 1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x
Hsu, T., Hingley-Wilson, S. M., Chen, B., Chen, M., Dai, A. Z., Morin, P. M., et al. (2003). The primary mechanism of attenuation of bacillus Calmette-Guérin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U.S.A. 100, 12420–12425. doi: 10.1073/pnas.1635213100
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). EGGNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Jain, S. K., Paul-Satyaseela, M., Lamichhane, G., Kim, K. S., and Bishai, W. R. (2006). Mycobacterium tuberculosis invasion and traversal across an in vitro human blood-brain barrier as a pathogenic mechanism for central nervous system tuberculosis. J. Infect. Dis. 193, 1287–1295. doi: 10.1086/502631
Kim, B.-J., Kim, B.-R., Lee, S.-Y., Seok, S.-H., Kook, Y.-H., and Kim, B.-J. (2013). Whole-genome sequence of a novel species, Mycobacterium yongonense DSM 45126 T. Genome Announc. 1, 604–613. doi: 10.1128/genomeA.00604-13
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, V. A., Goyal, R., Bansal, R., Singh, N., Sevalkar, R. R., Kumar, A., et al. (2016). Espr-dependent ESAT-6 protein secretion of Mycobacterium tuberculosis requires the presence of virulence regulator phoP. J. Biol. Chem. 291, 19018–19030. doi: 10.1074/jbc.M116.746289.
Lee, I., Kim, Y. O., Park, S. C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Levine, B., and Chaisson, R. E. (1991). Mycobacterium kansasii: a cause of treatable pulmonary disease associated with advanced human immunodeficiency virus (HIV) infection. Ann. Intern. Med. 114, 861–868. doi: 10.7326/0003-4819-114-10-861
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lou, Y., Rybniker, J., Sala, C., and Cole, S. T. (2017). EspC forms a filamentous structure in the cell envelope of Mycobacterium tuberculosis and impacts ESX-1 secretion. Mol. Microbiol. 103, 36–38. doi: 10.1111/mmi.13575
MacGurn, J. A., Raghavan, S., Stanley, S. A., and Cox, J. S. (2005). A non-RD1 gene cluster is required for snm secretion in Mycobacterium tuberculosis. Mol. Microbiol. 57, 1653–1663. doi: 10.1111/j.1365-2958.2005.04800.x
Machado, E., Vasconcellos, S. E. G., Cerdeira, C., Gomes, L. L., Junqueira, R., Carvalho, L. D., et al. (2018). Whole genome sequence of Mycobacterium kansasii isolates of the genotype 1 from Brazilian patients with pulmonary disease demonstrates considerable heterogeneity. Mem. Inst. Oswaldo Cruz 113:e180085. doi: 10.1590/0074-02760180085
Mahairas, G. G., Sabo, P. J., Hickey, M. J., Singh, D. C., and Stover, C. K. (1996). Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J. Bacteriol. 178, 1274–1282. doi: 10.1128/jb.178.5.1274-1282.1996
Majlessi, L., Prados-Rosales, R., Casadevall, A., and Brosch, R. (2015). Release of mycobacterial antigens. Immunol. Rev. 264, 25−45. doi: 10.1111/imr.12251
Newton-Foot, M., Warren, R. M., Sampson, S. L., van Helden, P. D., and Gey Van Pittius, N. C. (2016). The plasmid-mediated evolution of the mycobacterial ESX (Type VII) secretion systems. BMC Evol. Biol. 16:62. doi: 10.1186/s12862-016-0631-2
Park, H., Jang, H., Kim, C., Chung, B., Chang, C. L., Park, S. K., et al. (2000). Detection and identification of mycobacteria by amplification of the internal transcribed spacer regions with genus- and species-specific PCR primers. J. Clin. Microbiol. 38, 4080–4085. doi: 10.1128/JCM.38.11.4080-4085.2000
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Phan, T. H., van Leeuwen, L. M., Kuijl, C., Ummels, R., van Stempvoort, G., Rubio-Canalejas, A., et al. (2018). EspH is a hypervirulence factor for Mycobacterium marinum and essential for the secretion of the ESX-1 substrates EspE and EspF. PLoS Pathog. 14:e1007247. doi: 10.1371/journal.ppat.1007247
Pollak, M. (1953). Human infection with atypical acid-fast organisms report of two cases with pathologic findings. Am J Clin Pathol. 23, 363–374. doi: 10.1093/ajcp/23.4.363
Pym, A. S., Brodin, P., Brosch, R., Huerre, M., and Cole, S. T. (2002). Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 46, 709–717. doi: 10.1046/j.1365-2958.2002.03237.x
Raghavan, S., Manzanillo, P., Chan, K., Dovey, C., and Cox, J. S. (2008). Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 454, 717–721. doi: 10.1038/nature07219
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rio, D. C., Ares, M., Hannon, G. J., and Nilsen, T. W. (2010). Purification of RNA using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. 5:pdb.prot5439. doi: 10.1101/pdb.prot5439
Saito, H., Iwamoto, T., Ohkusu, K., Otsuka, Y., Akiyama, Y., Sato, S., et al. (2011). Mycobacterium shinjukuense sp. nov., a slowly growing, non-chromogenic species isolated from human clinical specimens. Int. J. Syst. Evol. Microbiol. 61, 1927–1932. doi: 10.1099/ijs.0.025478-0
Saitoh, H., Yamane, N., Miyagi, C., and Nakasone, I. (2000). [Comparative evaluation of two different formulae of Middlebrook 7H9 broth in a fully automated mycobacteria culture system, MB/BacT; the effect of Tween 80]. Rinsho Biseibutshu Jinsoku Shindan Kenkyukai Shi 11, 79–85.
Sala, A., Bordes, P., and Genevaux, P. (2014). Multiple toxin-antitoxin systems in Mycobacterium tuberculosis. Toxins 6, 1002–1020. doi: 10.3390/toxins6031002
Sala, C., Odermatt, N. T., Soler-Arnedo, P., Gülen, M. F., von Schultz, S., Benjak, A., et al. (2018). EspL is essential for virulence and stabilizes EspE, EspF and EspH levels in Mycobacterium tuberculosis. PLoS Pathog. 14:e1007491. doi: 10.1371/journal.ppat.1007491
Sapriel, G., and Brosch, R. (2019). Shared pathogenomic patterns characterize a new phylotype, revealing transition toward host-adaptation long before speciation of Mycobacterium tuberculosis. Genome Biol. Evol. 11, 2420–2438. doi: 10.1093/gbe/evz162
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Simeone, R., Bobard, A., Lippmann, J., Bitter, W., Majlessi, L., Brosch, R., et al. (2012). Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 8:e1002507. doi: 10.1371/journal.ppat.1002507
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stinear, T. P., Seemann, T., Harrison, P. F., Jenkin, G. A., Davies, J. K., Johnson, P. D. R., et al. (2008). Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 18, 729–741. doi: 10.1101/gr.075069.107
Suffys, P. N., Da Silva Rocha, A., De Oliveira, M., Dias Campos, C. E., Werneck Barreto, A. M., Portaels, F., et al. (2001). Rapid identification of Mycobacteria to the species level using INNO-LiPA Mycobacteria, a reverse hybridization assay. J. Clin. Microbiol. 39, 4477–4482. doi: 10.1128/JCM.39.12.4477-4482.2001
Tagini, F., Aeby, S., Bertelli, C., Droz, S., Casanova, C., Prod'hom, G., et al. (2019). Phylogenomics reveal that Mycobacterium kansasii subtypes are species-level lineages. Description of Mycobacterium pseudokansasii sp. nov., Mycobacterium innocens sp. nov. and Mycobacterium attenuatum sp. nov. Int. J. Syst. Evol. Microbiol. 69, 1696–1704. doi: 10.1099/ijsem.0.003378
Taillard, C., Greub, G., Weber, R., Gaby, E., Bodmer, T., Zimmerli, S., et al. (2003). Clinical Implications of Mycobacterium kansasii species heterogeneity : Swiss National Survey. J. Clin. Microbiol. 41, 1240–1244. doi: 10.1128/jcm.41.3.1240-1244.2003
Talaat, A. M., Lyons, R., Howard, S. T., and Johnston, S. A. (2004). The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc. Natl. Acad. Sci. U.S.A. 101, 4602–4607. doi: 10.1073/pnas.0306023101
Telenti, A., Marchesi, F., Balz, M., Bally, F., Bottger, E. C., and Bodmer, T. (1993). Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 31, 175–178. doi: 10.1128/JCM.31.2.175-178.1993
Tiwari, S., Casey, R., Goulding, C. W., Hingley-Wilson, S., and Jacobs, W. R. Jr. (2019). Infect and inject: how Mycobacterium tuberculosis exploits its major virulence-associated type VII secretion system, ESX-1. Microbiol. Spectr. 7. doi: 10.1128/9781683670261.ch8
Tortoli, E., Fedrizzi, T., Meehan, C. J., Trovato, A., Grottola, A., Giacobazzi, E., et al. (2017). The new phylogeny of the genus Mycobacterium: the old and the news. Infect. Genet. Evol. 56, 19–25. doi: 10.1016/j.meegid.2017.10.013
Tortoli, E., Simonetti, M. T., and Lavinia, F. (1996). Evaluation of reformulated chemiluminescent DNA probe (AccuProbe) for culture identification of Mycobacterium kansasii. J. Clin. Microbiol. 34, 2838–2840. doi: 10.1128/JCM.34.11.2838-2840.1996
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Tsuchiya, S., Yamabe, M., Yamaguchi, Y., Kobayashi, Y., Konno, T., and Tada, K. (1980). Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 26, 171–176. doi: 10.1002/ijc.2910260208
Uchiya, K. I., Takahashi, H., Nakagawa, T., Yagi, T., Moriyama, M., Inagaki, T., et al. (2015). Characterization of a novel plasmid, pMAH135, from Mycobacterium avium subsp. hominissuis. PLoS ONE. 10:e0117797. doi: 10.1371/journal.pone.0117797
Ummels, R., Abdallah, A. M., Kuiper, V., Aâjoud, A., Sparrius, M., Naeem, R., et al. (2014). Identification of a novel conjugative plasmid in mycobacteria that requires both type IV and type VII secretion. MBio 5:e01744-14. doi: 10.1128/mBio.01744-14
van der Wel, N., Hava, D., Houben, D., Fluitsma, D., van Zon, M., Pierson, J., et al. (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298. doi: 10.1016/j.cell.2007.05.059
van Ingen, J., Kohl, T. A., Kranzer, K., Hasse, B., Keller, P. M., Katarzyna Szafranska, A., et al. (2017). Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect. Dis. 17, 1033–1041. doi: 10.1016/S1473-3099(17)30324-9
Veyrier, F., Pletzer, D., Turenne, C., and Behr, M. A. (2009). Phylogenetic detection of horizontal gene transfer during the step-wise genesis of Mycobacterium tuberculosis. BMC Evol. Biol. 9:196. doi: 10.1186/1471-2148-9-196
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, J., McIntosh, F., Radomski, N., Dewar, K., Simeone, R., Enninga, J., et al. (2015). Insights on the emergence of Mycobacterium tuberculosis from the analysis of Mycobacterium kansasii. Genome Biol. Evol. 7, 856–870. doi: 10.1093/gbe/evv035
Weerdenburg, E. M., Abdallah, A. M., Rangkuti, F., Abd El Ghany, M., Otto, T. D., Adroub, S., et al. (2015). Genome-wide transposon mutagenesis indicates that Mycobacterium marinum customizes its virulence mechanisms for survival and replication in different hosts. Infect. Immun. 83, 1778–1788. doi: 10.1128/IAI.03050-14
Keywords: non-tuberculous mycobacteria, comparative genomics, M. kansasii subtypes, espACD operon, virulence factor
Citation: Guan Q, Ummels R, Ben-Rached F, Alzahid Y, Amini MS, Adroub SA, van Ingen J, Bitter W, Abdallah AM and Pain A (2020) Comparative Genomic and Transcriptomic Analyses of Mycobacterium kansasii Subtypes Provide New Insights Into Their Pathogenicity and Taxonomy. Front. Cell. Infect. Microbiol. 10:122. doi: 10.3389/fcimb.2020.00122
Received: 20 November 2019; Accepted: 04 March 2020;
Published: 24 March 2020.
Edited by:
Baolei Jia, Chung-Ang University, South KoreaReviewed by:
Brosch Roland, Université Louis-Pasteur, FranceLaurent Kremer, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Seyed Ehtesham Hasnain, Jamia Hamdard University, India
Copyright © 2020 Guan, Ummels, Ben-Rached, Alzahid, Amini, Adroub, van Ingen, Bitter, Abdallah and Pain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdallah M. Abdallah, YWJkYWxsYWgubXVzYUBxdS5lZHUucWE=; Arnab Pain, YXJuYWIucGFpbkBrYXVzdC5lZHUuc2E=
†These authors have contributed equally to this work