 Flávio Henrique Jesus-Santos1,2
Flávio Henrique Jesus-Santos1,2 Jéssica Lobo-Silva1
Jéssica Lobo-Silva1 Pablo Ivan Pereira Ramos3
Pablo Ivan Pereira Ramos3 Albert Descoteaux4
Albert Descoteaux4 Jonilson Berlink Lima5
Jonilson Berlink Lima5 Valéria Matos Borges1,2
Valéria Matos Borges1,2 Leonardo Paiva Farias1,2*
Leonardo Paiva Farias1,2*- 1Laboratory of Inflammation and Biomarkers, Instituto Gonçalo Moniz, Fundação Oswaldo Cruz (FIOCRUZ), Salvador, Brazil
- 2Faculdade de Medicina da Bahia, Federal University of Bahia (UFBA), Salvador, Brazil
- 3Center for Data and Knowledge Integration for Health (CIDACS), Instituto Gonçalo Moniz, Fundação Oswaldo Cruz, Salvador, Brazil
- 4Institut National de la Recherche Scientifique—Centre Armand-Frappier Santé Biotechnologie, Laval, QC, Canada
- 5Center of Biological Sciences and Health, Federal University of Western of Bahia (UFOB), Barreiras, Brazil
On the surface of the Leishmania promastigote, phosphoglycans (PG) such as lipophosphoglycan (LPG), proteophosphoglycan (PPG), free phosphoglycan polymers (PGs), and acid phosphatases (sAP), are dominant and contribute to the invasion and survival of Leishmania within the host cell by modulating macrophage signaling and intracellular trafficking. Phosphoglycan synthesis depends on the Golgi GDP-mannose transporter encoded by the LPG2 gene. Aiming to investigate the role of PG-containing molecules in Leishmania infantum infection process, herein we describe the generation and characterization of L. infantum LPG2-deficient parasites. This gene was unexpectedly identified as duplicated in the L. infantum genome, which impaired gene targeting using the conventional homologous recombination approach. This limitation was circumvented by the use of CRISPR/Cas9 technology. Knockout parasites were selected by agglutination assays using CA7AE antibodies followed by a lectin (RCA 120). Five clones were isolated and molecularly characterized, all revealing the expected edited genome, as well as the complete absence of LPG and PG-containing molecule expression. Finally, the deletion of LPG2 was found to impair the outcome of infection in human neutrophils, as demonstrated by a pronounced reduction (~83%) in intracellular load compared to wild-type parasite infection. The results obtained herein reinforce the importance of LPG and other PGs as virulence factors in host-parasite interactions.
Introduction
Leishmania promastigotes are coated by a thick glycocalyx consisting of glycoconjugates crucial to parasite pathogenesis. Lipophosphoglycan (LPG), proteophosphoglycan (PPG), and glycophosphatidylinositol lipids (GIPL), as well as the GP63 metalloprotease, comprise the vast majority of these molecules. Leishmania also secrete protein-linked phosphoglycans (PG) (e.g., secreted proteophosphoglycan (sPPG) and secreted acid phosphatase (sAP) (reviewed in Guha-Niyogi et al., 2001; Franco et al., 2012; Forestier et al., 2015). In promastigotes, LPG plays an important role in parasite survival inside the sand fly vector, in addition to macrophage infection (Sacks et al., 2000; Balaraman et al., 2005; Moradin and Descoteaux, 2012). Moreover, intracellular survival and the multiplication of amastigotes in macrophages is enhanced by other PG-containing molecules (e.g., PPG and sAP), which are highly expressed on the surface of amastigotes (Gaur et al., 2009).
LPG is organized into four domains: a conserved 1-O-alkyl-2-lyso-phosphatidyl(myo)inositol membrane anchor, a conserved diphosphoheptasaccharide core structure, a polymer that consists of repeating phosphodisaccharide units (phosphoglycan or PG) and carries species-specific side chains, as well as and variable mannose-rich cap structures (reviewed in McConville et al., 1993; Descoteaux and Turco, 1999). The PPGs comprise a heterogeneous family of cell surface and secreted proteins containing Ser-Thr rich regions to which phosphodisaccharide repeating units (Manα1-PO4 residue) (PG) are covalently linked, similarly to the LPG molecule (reviewed in Ilg, 2000b; de Assis et al., 2012). This type of phosphoglycosylation is the most abundant type of protein glycosylation found in Leishmania. Investigations focused on the synthesis of LPG and PG-containing molecules (PPG, PGs, sAP) have attracted considerable interest, and several enzymes and transporters involved in this process have been identified either biochemically, genetically, or both (Ryan et al., 1993; Descoteaux et al., 1995, 1998, 2002).
An enzyme critical for the synthesis of LPG and PG-containing molecules is the Golgi GDP-mannose transporter (encoded by LPG2) (Descoteaux et al., 1995), which contains up to nine transmembrane domains and presents a TPT domain, which is found in many transporters with affinity for triose phosphate. In Leishmania, this gene is required for the addition of disaccharide-phosphate units to lipophosphoglycan and related glycoconjugates (Ma et al., 1997; Hong et al., 2000; Segawa et al., 2005).
Over the past decades, the development of Leishmania mutants deficient in LPG or other PG-containing molecules has provided researchers with powerful tools to analyze the function of these structures/molecules (McNeely et al., 1990; Descoteaux et al., 1995; Butcher et al., 1996). L. major and L. donovani Δlpg2 mutants failed to survive in the midgut of the sand fly vector and were unable to establish infection in macrophages. In an animal infection model, L. major parasites were found to exhibit persistence without causing pathological manifestations, with parasites persisting at low levels throughout the life of the infected animals (Spath et al., 2003b). However, whether LPG and PGs are necessary for parasite survival in all Leishmania species has been contested based on L. mexicana studies. Although Δlpg1 and Δlpg2 parasites of this species exhibit complement sensitivity, no decreases in infectivity were observed in vitro in macrophages or in vivo in mice (Ilg, 2000a; Ilg et al., 2001; Gaur et al., 2009). Our group recently generated an LPG-deficient mutant of L. infantum through the deletion of the putative galactofuranosyl transferase gene (LPG1) involved in the synthesis of the LPG glycan core. Phenotypically, this deletion impaired the outcome of infection in murine bone marrow-derived macrophages, likely due to the activation of the iNOS promoter in an NF-κB-dependent manner (Lazaro-Souza et al., 2018). While these parasites expressed a truncated LPG molecule lacking the PG domain, they were still able to assemble and secrete other PG-containing molecules (e.g., PPG, sAP, and other PGs) (Dermine et al., 2000; Spath et al., 2000).
In an effort to investigate the role of PG-containing molecules in the Leishmania infantum infection process, we applied gene targeting by homologous recombination and also CRISPR/Cas9 technology to generate an L. infantum mutant lacking the Golgi GDP-mannose transporter gene (Δlpg2). Our results demonstrate both the value and the caveats of using of both systems for genome editing. The generated mutant produced distinct infection outcomes in comparison to WT parasites, and can therefore be usefully applied to investigate the role of L. infantum PG-containing molecules in host-parasite interactions.
Methods
Ethics Statement
This study was approved by the Institutional Review Board of Human Ethical Research Committee of Fundação Oswaldo Cruz-Bahia, under number 100/2006.
Parasite Cultures
Leishmania infantum Ba262 (MCAN/BR/89/BA262) promastigotes were cultured in HOMEM medium supplemented with 10% inactivated Fetal Bovine Serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin and 2 mM L-glutamine in 25 cm2 flasks at 25°C until late log-phase. The number of promastigotes was determined by counting in a Neubauer chamber.
LPG2 Gene Targeting by Homologous Recombination
Initially, we attempted to obtain an L. infantum LPG-deficient mutant (Δlpg2) by homologous recombination, using a previously described strategy (Scianimanico et al., 1999) with modifications. Briefly, the gene sequences from the resistance markers Hygromycin and Neomycin were amplified by PCR using specific oligonucleotides (Supplementary Table 1) and ligated in the pUC18 vector (Sigma, USA), thusly generating the pUC18-Hyg and pUC18-Neo constructs. Next, the 5' and 3' UTR LPG2 gene regions, consisting of 838 bp and 858 bp fragments respectively, were amplified by PCR from L. infantum Ba262 (MCAN/BR/89/BA262) genomic DNA using a specific oligonucleotide design based on the 19 kb genomic contig (GenBank accession CACT01000040.1), which was predicted to encode the LPG2 gene (LinJ_34_4290) (Supplementary Table 1). The respective fragments were then digested with EcoRV/KpnI and XbaI/Hind III enzymes, and ligated in the pUC18-Hyg and pUC18-Neo constructs. The final constructs were sequenced for confirmation and designated pLPG2-Hyg and pLPG2-Neo. Log-phase WT L. infantum promastigotes were electroporated in two steps with 10 μg of purified fragments (pLPG2-Neo and pLPG2-Hyg) using 0.4 cm cuvettes in a Gene Pulser II (BIO-RAD) under electroporation conditions based on a previously described high voltage protocol (Robinson and Beverley, 2003). Briefly, two pulses were applied at 10-s intervals (25 μF, 1,500 V). Following electroporation, promastigotes were incubated for 24 h at 25°C in drug-free medium, and the neo-resistant parasites were subsequently selected in the presence of 70 μg/mL G418 for 22 days at 25°C. The resistant parasites (lpg2+/−) were isolated and submitted to a second round of electroporation using the pLPG2-Hyg fragment to disrupt the second allele of the LPG2 gene. Double knockout (KO) Δlpg2 parasites were then selected in the presence of both 70 μg/mL G418 and 50 μg/mL Hygromycin B for 22 days at 25°C. The absence of the LPG2 gene in the resulting double drug-resistant promastigotes was verified by PCR following DNA extraction using “NucleoSpin tissue” kit (Macherey Nagel).
gRNA Design and Cloning Into pLdCN
The present procedures involving the CRISPR/Cas9 system were based on pioneering work previously performed by Zhang and Matlashewski (2015) and Zhang et al. (2017) with minimal modifications. Two gRNA-targeting sequences and the respective oligonucleotide donors (oligodonors) were selected using the gRNA designer tool (http://grna.ctegd.uga.edu/) and then manually inspected (Supplementary Table 1). The complementary guide sequence oligonucleotides were first phosphorylated in T4 DNA ligase buffer with T4 polynucleotide kinase and then annealed in a thermocycler (MJ Research: PTC-200, DNA Engine) (program: 95°C for 5 min, ramp to 25°C at a rate of −0.1°C/s). The annealed guide sequence was then cloned into the gRNA and Cas9 coexpression vector (pLdCN) (Addgene plasmid #84290; http://n2t.net/addgene:84290; RRID:Addgene_84290) previously digested with BbsI.
Parasite Transfection (CRISPR/Cas9)
To obtain L. infantum parasites expressing Cas9, log-phase WT L. infantum promastigotes were electroporated with purified pLdCN (gRNA440 and gRNA516) under the same conditions described in the section above (LPG2 gene targeting by homologous recombination). After selection with G418 (70 μg/mL), four rounds of transfection were performed using 100 μM single-stranded oligodonors at 3-day intervals (Zhang et al., 2017).
LPG2 Knockout Selection and Limiting Dilution Assay
After transfection with the oligodonors, the putative Δlpg2 parasites were selected by consecutive rounds of agglutination with the CA7AE monoclonal IgM antibody (MediMabs) (1:2,000 dilution), which recognizes the Gal(β1,4)Man(α1-PO4) repeating units contained in LPG and PG-containing molecules (Descoteaux et al., 1998). Briefly, parasites were incubated for 2 h with the antibody and then centrifuged at 100 × g for 7 min to remove any agglutinated promastigotes. The supernatant was then transferred to a new tube and centrifuged at 1,300 × g for 7 min to sediment non-agglutinated parasites (theoretically Δlpg2). Subsequently, these non-agglutinated parasites were incubated in fresh culture medium containing CA7AE (1:2,000) for 3 days, followed by five rounds of the above-described agglutination steps. As a further selection step, non-agglutinated parasites were selected with 100 μg/ml of Ricin 120 (Vector Laboratories, USA) as described in King and Turco (1988). After selecting the Δlpg2 parasites, the pLdCN vector, as well as Cas9 protein expression, were eliminated by growing parasites in the absence of the G418 marker, which was achieved after 5–6 passages. Finally, a limiting dilution assay was performed for clonal isolation using 96-well plates with culture medium containing 100 μg/mL of ricin, incubated for 3 weeks at 25°C with medium supplementation when necessary. To calculate doubling times, cells were seeded at 105 cells/ml and density was measured every 24 h. The average doubling time, from day zero until reaching stationary phase, was calculated by (24/(log2(t2/t1)))/number of days evaluated (Beneke et al., 2019). Subsequently, genomic DNA was extracted from the last dilution in which parasite growth was observed.
Sequencing Analysis
The sequence corresponding to the 5'UTR-Neo/Hyg/LPG2-3'UTR genomic DNA region was amplified with specific oligonucleotides (Supplementary Table 1) by PCR. The PCR products were purified on agarose gels (Wizard® SV Gel and PCR Clean-Up System) and sequenced using the Gonçalo Moniz Institute sequencing platform.
Western Blotting
Late log-phase promastigotes at a concentration of 2 × 107 cells were centrifuged and resuspended in 100 μl of RIPA Lysis Buffer (150 mM NaCl, 0.5 M EDTA, pH 8.0, 1 M Tris, pH 8.0, 1%NP-40, 1% sodium deoxycholate, 0.1% SDS and dH2O) containing a protease inhibitor (Sigma, cat. No. P-2,714). The extract was incubated on ice and vortexed for approximately 5 min. The lysated material was separated by SDS-PAGE on a 12% polyacrylamide gel and Western blotting was performed as previously described (Lazaro-Souza et al., 2018).
T7 Endonuclease I Assay
Briefly, genomic DNA from L. infantum WT and Δlpg clones was extracted using a “NucleoSpin tissue” kit (Macherey Nagel). The sequences corresponding to the LPG2 and Δlpg2 genes were amplified by PCR with specific oligonucleotides (Supplementary Table 1) and purified from agarose gels using the Wizard® SV Gel and PCR Clean-Up System. Subsequently, 200 ng of the purified PCR product was denatured and re-annealed at a final volume of 19 μl in 1 × NEBuffer2 (NEB) using a thermocycler in accordance with the following protocol: 95°C for 5 min; ramp to 85°C at a rate of −2°C/s; ramp to 25°C at a rate of −1°C/s; hold at 4°C. The PCR products were then treated with T7 E1 enzyme (NEB) at 37°C for 15 min at a reaction volume of 20 μl. An aliquot (10 μl) of the reaction was subsequently analyzed on a 1% agarose gel.
Confocal Immunofluorescence Microscopy
Late log-phase promastigotes were adhered on Poly-L-Lysine-coated glass coverslips (BD Biosciences, San Jose, CA) by centrifugation, fixed with 4% paraformaldehyde for 20 min and simultaneously blocked and permeabilized for 20 min using a solution containing 0.1% Triton X-100, 1% BSA, 6% non-fat dry milk, 20% goat serum and 50% FBS. The distribution of LPG and other PGs containing the Gal(β1,4)Man(α1-PO4) repeating unit epitope was visualized using the CA7AE mouse monoclonal antibody (MediMabs, 1:2,000) after a 2 h incubation period, followed by an additional 30 min incubation with Alexa Fluor 488 goat anti-mouse IgM (Molecular Probes) at 1:500. Parasite nuclei were stained with DAPI (Molecular Probes) at 1:17,000. All steps were performed at room temperature. The coverslips were then mounted in Fluoromount-G (Interscience) and sealed with nail polish. Parasites were observed under a Plan APOCHROMAT 63 × oil-immersion DIC 1.4 NA objective on a Leica SP8 confocal microscope in reflective mode, using a 488 nm laser, with an LP505 filter.
Infection Assay
Human blood was obtained from volunteers at the Bahia Foundation of Hematology and Hemotherapy (HEMOBA). Human neutrophils were isolated by gradient separation in polymorphonuclear medium (PMN) according to the manufacturer's instructions (Axis-ShieldPoc AS, Oslo, Norway). Neutrophils were collected and washed three times with saline at 4°C for 10 min at 300 x g. Neutrophils were plated with RPMI-1640 medium, supplemented with 1% Nutridoma-SP, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml of streptomycin. Cells were infected with stationary WT or Δlpg2 promastigotes at an MOI of 10 for 3 h at 37°C under 5% CO2. The intracellular load of L. infantum was then evaluated in neutrophils by assessing parasite viability as previously described (Quintela-Carvalho et al., 2017). Briefly, after 3 h of incubation, infected neutrophils were centrifuged at 300 × g for 10 min at 4°C, the supernatant containing non-internalized promastigotes was discarded and replaced by 250 μl of HOMEM medium. After a final incubation at 23°C for 24 h, the number of viable proliferating extracellular promastigotes was counted in a Neubauer chamber.
Statistical Analysis
Diferences in growth curves were analyzed using Area under the Curve (AUC) analysis. Neutrophil infection assays were performed twice from at least five donors, data are presented as means and SE (standard error) of representative experiments. Wild-type and knockout groups were compared using the Student's t-test. Differences were considered statistically significant when p ≤ 0.05. All figures and statistical analyses were performed using PRISM version 5.02 software (GraphPad, San Diego, CA).
Results
LPG2 Is Duplicated in L. infantum
After selection of double drug-resistant (Neo/Hyg) parasites, the absence of the LPG2 gene was assessed by PCR. Electrophoresis results confirmed the expected occurrence of homologous recombination and the successful integration of the resistance markers (Neo and Hyg). However, surprisingly, the coding region of the LPG2 gene was still detected following amplification (Figure 1A).
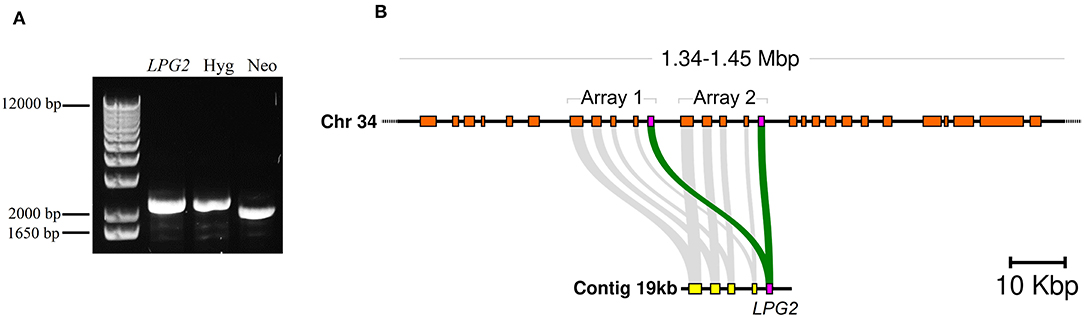
Figure 1. Evidence of LPG2 duplication in L. infantum. (A) Genomic DNA from L. infantum Hyg/NeoR strain was amplified by PCR using specific oligonucleotides (Supplementary Table 1) to verify LPG2 gene targeting by homologous recombination. (B) Comparison of the genomic contexts where LPG2 (magenta box) was originally located (in an unplaced 19 kbp contig shown at the bottom) to the recent resequencing and reassembly of the L. infantum JPCM5 genome (Gonzalez-de la Fuente et al., 2017), where the duplication of the LPG2 gene array is evidenced in chromosome 34 (top). Gray connecting segments indicate sequence conservation, and the coordinates at the top refer to the novel chromosome 34 assembly.
This effectively indicated that either homologous recombination did not occur, or that the LPG2 gene is duplicated in this species. No available evidence suggested the latter possibility, since the L. infantum JPCM5 genome assembly published at the time of the initial recombination experiments showed LPG2 as a single-copy gene located in an unplaced 19 kb contig (GenBank accession CACT01000040.1). However, a more recent resequencing of the JPCM5 genome, which employed a hybrid sequencing approach with PacBio long reads and Illumina short reads, yielded a more robust assembly containing 36 scaffolds accounting for the 36 chromosomes present in this species (Gonzalez-de la Fuente et al., 2017) (EMBL accession GCA_900500625.1). BLAST searches of the 19 kb contig as a query against this novel assembly revealed that the entire contig, including the LPG2 gene, was duplicated in a tandem array in chromosome 34 of L. infantum JPCM5 (Figure 1B and Supplementary Figure 1A). This finding highlights a limitation associated with the obtainment of knockout parasites using conventional homologous recombination procedures.
Generation of L. infantum Expressing Cas9
As a first step to perform LPG2 gene editing, we developed L. infantum parasites expressing the Cas9 enzyme. WT parasites were transfected with the pLdCN plasmid and selected in G418 culture medium to generate the strain constitutively expressing the Cas9 nuclease. The expression of the Cas9 enzyme was confirmed in G418-resistant parasites by Western blotting (Figure 2A). The growth curves of promastigotes revealed that the expression of Cas9-gRNAs influenced parasite growth. A delay in the replication capability of the Cas9-gRNA expressing parasites was observed in comparison to WT. In addition, the former reached stationary phase by day 6, with a cell density of ~5 × 107, while the latter reached this phase 2 days later, with a cell density of ~3 × 107 (Figure 2B). Sequencing data revealed no evidence of LPG2 gene editing at this initial stage (data not shown). Next, L infantum expressing Cas9 were submitted to four rounds of transfection using single-stranded oligodonors to improve LPG2 gene disruption and knockouts were subsequently selected.
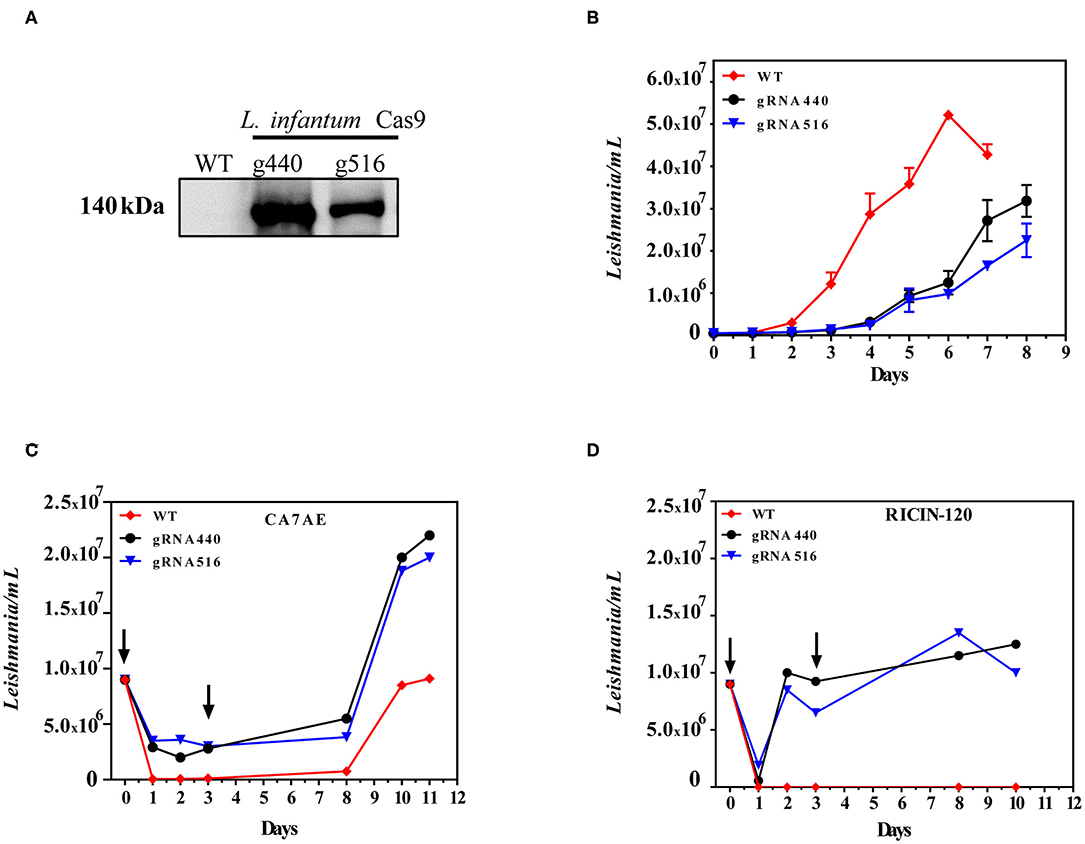
Figure 2. Generation of LPG2 knockout using CRISPR/Cas9. (A) Western blot analysis of L. infantum promastigotes expressing Cas9 (gRNA440 and gRNA516). (B) Growth curves of L. infantum wild-type (WT) and L. infantum-Cas9 (gRNA440 and gRNA516) promastigotes. (C) Agglutination assay using the CA7AE monoclonal antibody and associated growth curves. (D) Agglutination assay using Ricin 120 lectin and associated growth curves, demonstrating the selection of Δlpg2. The arrows indicate the time points where antibody (CA7AE) or lectin (Ricin-120) were added to the cultures, exemplifying the typical agglutination results observed.
Selection of L. infantum Δlpg2
Five rounds of selection using the CA7AE antibody were unexpectedly unsuccessful in completely removing WT parasites by agglutination (Figure 2C). This was circumvented through the use of Ricin 120, after which complete depletion of WT L. infantum and the selection of Δlpg2 was observed (Figure 2D).
Characterization of L. infantum Δlpg2
To examine the types of mutations created by the CRISPR-Cas9 system at the LPG2-targeting sites, genomic DNA was extracted from these non-agglutinating parasites after clonal isolation (clones denominated E3, E4, F1, G2, and G6). PCR was performed with primers designed to amplify the LPG2 gene, followed by DNA sequencing of the targeted sites (Supplementary Figure 1B and Figure 3A). All five sequenced clones revealed the expected genome editing with oligodonor insertions, and no random insertions/deletions were observed within the sequenced region (nucleotides 15–870, Supplementary Figure 1C).
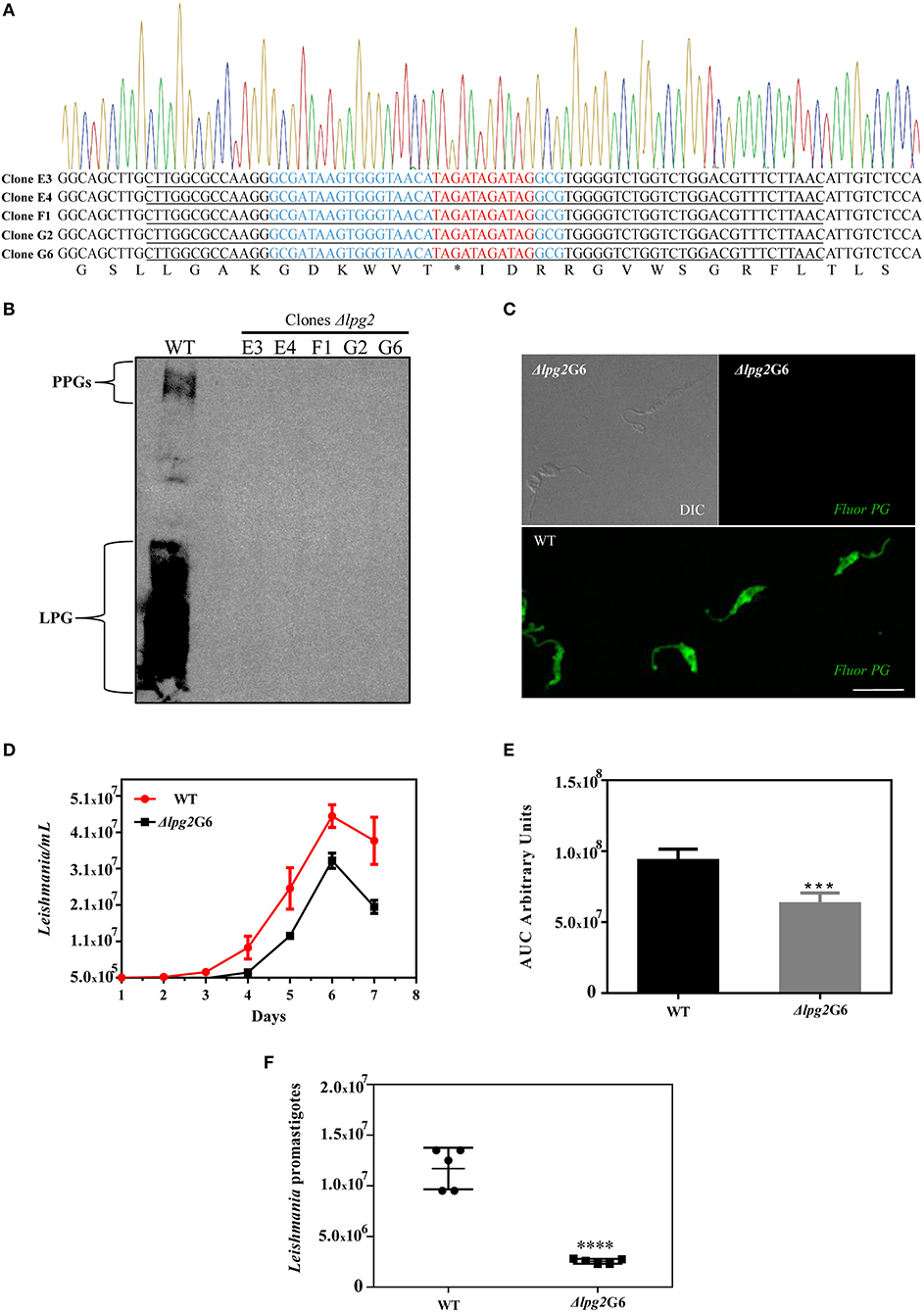
Figure 3. Molecular characterization of Δlpg2 and reduced virulence phenotype. (A) Chromatogram and translated sequence showing the region of the LPG2 gene in which the precise insertion of a stop codon (denoted by an *) occurred by homologous recombination at the cleavage site of the Cas9 enzyme (nucleotides in red). The oligodonor sequence is underlined and the gRNA440 sequence is highlighted in blue. (B) Western blot analysis of the expression of LPG and PPGs in L. infantum promastigotes WT and Δlpg2 clones. (C) Confocal immunofluorescence analysis of WT and Δlpg2 (clone G6) parasites. Late log-phase promastigotes were adhered on Poly-L-Lysine-coated glass coverslips, fixed and incubated with the CA7AE antibody to visualize LPG and other Gal(β1,4)Man(α1-PO4) repeating unit-containing PGs (green). Differential interference contrast (DIC) for Δlpg2 is shown in the upper left panel. Scale bar: 10 μm. (D) Axenic growth curves of late log-phase promastigotes of L. infantum wild-type (WT) and clone G6 Δlpg2, each point represents mean and SE. Data are representative of at least three independent assays and were collected in triplicate for each experimental condition. (E) Area under the curve (AUC) analysis of growth curves presented in (D), ***p < 0.01. (F) Reduced virulence of Δlpg2 parasites in human neutrophils. Human neutrophils were infected with L. infantum Ba262 wild-type and Δlpg2 promastigotes for 3 h. Numbers of viable promastigotes shown after 24 h, with each point on the graph representing the cells from a health donor. Statistical differences were evaluated using the Student t-test, ****p < 0.001.
The loss of LPG expression in L. infantum Δlpg2 was demonstrated in promastigotes of all five isolated clones by Western blot, and by confocal immunofluorescence microscopy for clone G6 (Figures 3B,C). To better characterize these five knockout clones, growth curves were constructed and compared to wild-type parasites (Supplementary Figure 2A). This assay revealed no apparent differences in the growth profile of the five Δlpg2 clones. However, Area Under the Curve (AUC) analysis demonstrated a significant impairment in the growth rate of knockout parasites compared to WT. Our AUC analysis compared the average growth rate of all five knockout clones to WT (Supplementary Figures 2B,C), in addition to comparing one specific clone (G6) to WT (Figures 3D,E). The doubling time was calculated at 8.1 (G6) and 23.7 (WT). Despite differences in cell densities among Δlpg2 parasites, no significant delays were observed in comparison to wild-type parasites with respect to the time needed to reach stationary phase (Figure 3D and Supplementary Figure 2A). While it appeared that knockout and WT parasites seemed to differentiate into metacyclic forms at similar frequencies, since no apparent morphological differences were observed during growth phase, this warrants further investigation (e.g., through the analysis of ultrastructural and morphometric data or stage-specific gene expression).
Finally, the T7 Endonuclease I assay confirmed the absence of any non-edited versions of LPG2 in the genomic DNA of clones (G6 and E4) as reveled by the absence of digested products following enzyme incubation (Supplementary Figure 2D). After selecting the Δlpg2 parasites, Cas9 protein expression was eliminated by removing the selection marker (G418) for the pLdCN vector, which was achieved after 5-6 passages (Supplementary Figure 2E). Together, these data indicate that the LPG2 gene was successfully edited in the Δlpg2 mutants, resulting in the generation of a parasite deficient in LPG, as well as PG-containing molecules (e.g., PPGs).
Δlpg2 Parasites Exhibit Limited Survival in Neutrophils
To evaluate differences in parasite survival between WT and genome-edited parasites in vitro, human neutrophils were experimentally infected and incubated for 3 h. Neutrophils infected with Δlpg2 parasites exhibited an ~83% reduction in intracellular parasite load as estimated by parasite viability determined at 3 h after infection (Figure 3F).
Discussion
We recently confirmed the involvement of LPG as a virulence factor in L. infantum through the use of parasites that had the LPG1 gene knocked out (Lazaro-Souza et al., 2018). However, other phosphoglycan-containing molecules (PPGs, PGs and sAPs) may also play a role in parasite survival within host cells (Spath et al., 2003a; Gaur et al., 2009). Here we developed an L. infantum species parasite knocked-out for the LPG2 gene, which did not express LPG or other PG-containing molecules (PPG, PGs, sAP).
Initially, we sought to obtain knockouts using the conventional method of homologous recombination involving resistance marker genes (Cruz et al., 1991). Despite some limitations, this method has been useful for single gene disruption and was shown to successfully knock out the LPG2 gene in L. donovani, L. Mexicana, and L. major (Scianimanico et al., 1999; Ilg et al., 2001; Spath et al., 2003b). After performing two transformation rounds and selecting for the potential Δlpg2 parasites, the presence of an integral allele of the LPG2 gene was still observed in genomic DNA, suggesting either that the homologous recombination process was inefficient, or that this gene was possibly duplicated in the L. infantum genome.
The homologous recombination gene disruption strategy was designed based on information present in an unplaced 19 kb genomic contig of L. infantum JPCM5 (GenBank accession CACT01000040.1), in which the LPG2 gene was originally mapped (LinJ_34_4290). It is important to note that although this contig was described in association with chromosome 34, it was not physically mapped to this chromosome, which raised doubts regarding the consistency of this contig. Subsequent analysis in a new assembly of the L. infantum JPCM5 genome (Gonzalez-de la Fuente et al., 2017) indicated that this 19 Kb contig (including the LPG2 gene) is duplicated in a tandem array located in chromosome 34. This new assembly, performed using PacBio and Illumina technology, was more robust, as evidenced by fewer gaps and increased identification of duplicate regions (tandem arrays). Interestingly, the duplication of the LPG2 gene in the tandem array was not mentioned by these authors in their list of duplicated genes. More recently the LPG2 duplication was spotted by a study that performed a comparative genomic and evolutionary analysis of the proteins involved in the biosynthesis of LPG in Leishmania (Azevedo et al., 2020).
The fact that the duplication of LPG2 was not initially identified highlights limitations regarding the way Leishmania genomes are traditionally assembled, which usually entails the use of low-coverage whole-genome sequencing using short reads, with subsequent assembly performed using a reference genome obtained from a previously sequenced genome that can even belong to a different Leishmania species (Peacock et al., 2007). The presence of two copies of LPG2 in L. infantum limited our ability to perform genetic manipulation via homologous recombination in this parasite species, which emphasizes the need to improve the overall quality of Leishmania genomes, i.e., completeness and contiguity, preferentially through the use of recently employed hybrid sequencing strategies (short and long reads) (Gonzalez-de la Fuente et al., 2017, 2019; Lypaczewski et al., 2018). In the end, we overcame this limitation through the use of the CRISPR/Cas9 system for genome editing.
The CRISPR/Cas9 system used here proved more flexible in producing Leishmania knockouts without the use of genetic resistance markers for selection, thereby allowing for more direct comparisons with WT parasites. In addition, the use of oligodonors containing stop codons permitted more specific editing of the target gene as opposed to random insertions/deletions (Zhang et al., 2017; Zhang and Matlashewski, 2019).
Previous studies found no evidence of Cas9 toxicity in Leishmania mexicana, since promastigote forms grew and reached stationary phase at similar rates and densities as wild-type cells (Beneke et al., 2017; Martel et al., 2017). However, others have reported some reduced phenotypic growth in the late stages of culturing (Ishemgulova et al., 2018). The reasons behind these divergent results remain unclear and could rely on the way that this expression is conducted (episomal vs. genomic integration) and also the window of time analyzed, but further study will be necessary.
As the generated L. infantum strain expressing Cas9-gRNA presented delayed growth, we speculate that this could be due to the action of Cas9-gRNA and the low efficiency of mechanisms involved in repairing double-strand breaks in Leishmania, which may lead to cell cycle arrest and even cell death, as demonstrated by (Zhang and Matlashewski, 2019). Importantly, future studies must address this issue, which remains unclear: Does the expression of Cas9 per se negatively affect Leishmania growth, or this is a result of Cas9 expression together with a Leishmania-DNA-targeting-gRNA?
For the selection and clonal isolation of the Δlpg2 parasites, we initially used the CA7AE monoclonal antibody that recognizes the Gal(β1,4)Man(α1-PO4) repeat region of the LPG molecule (Descoteaux et al., 1998). However, after five rounds of agglutination, there was reason to believe that wild-type parasites could still be present in knockout cultures, due to the fact that it was not possible to completely eliminate wild-type parasites in control cultures. These results highlight a limitation associated with the use of this antibody to select Δlpg2 parasites in an L. infantum model without the additional presence of a resistance gene marker. To overcome this limitation, we chose to employ a lectin (RCA 120), which recognizes terminal β-galactose residues, that was shown to be cytotoxic to wild-type L. donovani and L. major parasites, but did not affect parasites that do not present LPG and other PG-containing molecules (King and Turco, 1988; Cappai et al., 1994; Opat et al., 1996). After two rounds of agglutination, the use of this lectin resulted in wild-type parasite death and complete elimination. Despite the absence of resistance markers to select knockout parasites, the CRISPR/Cas9 system, coupled to oligodonors, demonstrated high efficiency in target gene editing, as evidenced by the analysis of sequencing data from five of the generated clones, which all presented the expected result at the site of Cas9 cleavage. In addition, Western blotting and confocal microscopy demonstrated the complete elimination of PG-repeating units in both LPG and other PG-containing molecules, which is consistent with the literature (Descoteaux et al., 1995; Dermine et al., 2000; Spath et al., 2003b; Lazaro-Souza et al., 2018). However, it will be important for future experimental design to quantitatively investigate system efficiency by determining how many null mutants are generated as a fraction of oligodonor-transfected parasites.
After selecting the Δlpg2 parasites, Cas9 protein expression was eliminated by removing the selection marker (G418) for the pLdCN vector, which was achieved after 5–6 passages (Supplementary Figure 2E). No differences in phenotype or growth rates were observed among the five isolated clones, which suggest that, if present, off-target effects had little impact. Although low cell density was observed when the Δlpg2 parasites reached stationary phase, the time required to reach this phase was not significantly delayed. This finding stands in contrast to delays seen in achieving stationary phase when knockout parasites are obtained by homologous recombination using resistance markers, which likely occurs due to the presence of antibiotics in culture medium (Lazaro-Souza et al., 2018). In addition, we did not observe any significant morphological or ultrastructural changes, indicating that the deletion of the LPG2 gene did not interfere with the cellular biology of L. infantum promastigotes.
In an attempt to obtain insights regarding the role of LPG and PG-containing molecules in cells present at initial stages of infection, human neutrophils were infected for 3 h with L. infantum Δlpg2, revealing a significant reduction in infection rate compared to WT. Neutrophils, essential innate immune system cells, are rapidly recruited to the site of inflammation and have been described as playing a critical role in Leishmania infection (Ribeiro-Gomes et al., 2004; Hurrell et al., 2016). In addition, LPG was shown to be important to the induction of autophagy in neutrophils using a model of L. donovani infection (Pitale et al., 2019). However, our findings in this cell type must be considered preliminary, as it will be necessary to investigate additional time points and the underling mechanisms. Future studies should also extend the scope of investigation to include macrophages.
In sum, the results presented herein confirm the unexpected duplication of the LPG2 gene in the L. infantum genome. The successful inactivation of both copies of this gene using the CRISPR/Cas9 system highlights the potential of using these KO parasites to assess signal transduction pathways elicited by PG-containing molecules from a variety of Leishmania species in different types of immune cells. In addition, the technique demonstrated herein of editing the LPG2 gene using CRISPR exemplifies the prospects of inactivating other virulence factors sequentially, which could pave the way for new vaccine development.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Human Ethical Research Committee of Fundação Oswaldo Cruz-Bahia. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
FJ-S, PR, AD, JL, VB, and LF conceived and designed the study and contributed to data analysis. FJ-S, JL, JL-S, and LF performed the experiments. FJ-S, JL, PR, AD, JL-S, VB, and LF wrote and critically revised the manuscript. All authors have read and approved the final version of this manuscript.
Funding
This work was supported by the Brazilian National Research Council (CNPq) (Grant Number: 431857/2018-0) and the Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB) (Grant Number: 04/2015) to VB. VB is a senior investigator funded by CNPq. AD holds the Canada Research Chair in Biology of intracellular parasitism. The fellowships received by FJ-S, JL-S, and part of this study was financed by the program: Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES) –Finance Code 001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Rede de Plataformas Tecnológicas (FIOCRUZ-Bahia) for the use of its DNA Sequencing Platform, Dr. Ricardo Khouri (FIOCRUZ-Bahia) for technical assistance with chemicals and reagents and Astrid Goicochea for her assistance with the neutrophil assays. The authors are also grateful to Andris K. Walter for English language revision and manuscript copyediting assistance (FIOTEC-Programa de Capacitação).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00408/full#supplementary-material
Supplementary Figure 1. Detail of LPG2 gene locus indicating gRNA targeting sites. (A) Schematic representation of the wild-type alleles of the LPG2 gene (magenta box) present in a duplicated tandem array. (B) Schematic representation showing the insertion of a stop codon in LPG2 alleles following Cas9 genome editing (white line interrupting the magenta box). (C) LPG2 sequence depicting: gRNA440 sequence in (blue), oligonucleotide donor sequence (underlined) and inserted stop codons (red). Arrows represents primers used for LPG2 sequence and Δlpg2 characterization.
Supplementary Figure 2. Characterization of Δlpg2 clones. (A) Growth curve of wild-type (WT) L. infantum promastigotes and five different Δlpg2 clones. (B) Analysis of growth curve of L. infantum wild-type (WT) promastigotes and mean growth rates of all five different Δlpg2 clones (C) Area under the curve (AUC) of growth curve presented in (B). (D) T7 Endonuclease I assay using genomic DNA from L. infantum wild-type (WT) promastigotes and two clones (E4 and G6) of L. infantum Δlpg2. A positive control containing equal amounts of WT and KO DNA demonstrates the presence of two bands following digestion. (E) Western blot analysis of L. infantum promastigotes (clone G6) demonstrating the loss of the pLdCN vector after 5–6 passages in the absence of the G418 marker.
Supplementary Table 1. Oligonucleotides sequences used to assemble the fragments pLPG2-Hyg and pLPG2-Neo for gene targeting of LPG2 by homologous recombination and CRISPR/Cas9.
References
Azevedo, L. G., de Queiroz, A. T. L., Barral, A., Santos, L. A., and Ramos, P. I. P. (2020). Proteins involved in the biosynthesis of lipophosphoglycan in Leishmania: a comparative genomic and evolutionary analysis. Parasit. Vectors 13:44. doi: 10.1186/s13071-020-3914-9
Balaraman, S., Singh, V. K., Tewary, P., and Madhubala, R. (2005). Leishmania lipophosphoglycan activates the transcription factor activating protein 1 in J774A.1 macrophages through the extracellular signal-related kinase (ERK) and p38 mitogen-activated protein kinase. Mol. Biochem. Parasitol. 139, 117–127. doi: 10.1016/j.molbiopara.2004.10.006
Beneke, T., Demay, F., Hookway, E., Ashman, N., Jeffery, H., Smith, J., et al. (2019). Genetic dissection of a Leishmania flagellar proteome demonstrates requirement for directional motility in sand fly infections. PLoS. Pathog. 15:e1007828. doi: 10.1371/journal.ppat.1007828
Beneke, T., Madden, R., Makin, L., Valli, J., Sunter, J., and Gluenz, E. (2017). A CRISPR Cas9 high-throughput genome editing toolkit for kinetoplastids. R Soc. Open Sci. 4:170095. doi: 10.1098/rsos.170095
Butcher, B. A., Turco, S. J., Hilty, B. A., Pimenta, P. F., Panunzio, M., and Sacks, D. L. (1996). Deficiency in beta1,3-galactosyltransferase of a Leishmania major lipophosphoglycan mutant adversely influences the Leishmania-sand fly interaction. J. Biol. Chem. 271, 20573–20579. doi: 10.1074/jbc.271.34.20573
Cappai, R., Morris, L., Aebischer, T., Bacic, A., Curtis, J. M., Kelleher, M., et al. (1994). Ricin-resistant mutants of Leishmania major which express modified lipophosphoglycan remain infective for mice. Parasitology 108 (Pt 4), 397–405. doi: 10.1017/S0031182000075946
Cruz, A., Coburn, C. M., and Beverley, S. M. (1991). Double targeted gene replacement for creating null mutants. Proc. Natl. Acad. Sci. U. S. A. 88, 7170–7174. doi: 10.1073/pnas.88.16.7170
de Assis, R. R., Ibraim, I. C., Nogueira, P. M., Soares, R. P., and Turco, S. J. (2012). Glycoconjugates in New World species of Leishmania: polymorphisms in lipophosphoglycan and glycoinositolphospholipids and interaction with hosts. Biochim Biophys. Acta 1820, 1354–1365. doi: 10.1016/j.bbagen.2011.11.001
Dermine, J. F., Scianimanico, S., Prive, C., Descoteaux, A., and Desjardins, M. (2000). Leishmania promastigotes require lipophosphoglycan to actively modulate the fusion properties of phagosomes at an early step of phagocytosis. Cell Microbiol. 2, 115–126. doi: 10.1046/j.1462-5822.2000.00037.x
Descoteaux, A., Avila, H. A., Zhang, K., Turco, S. J., and Beverley, S. M. (2002). Leishmania LPG3 encodes a GRP94 homolog required for phosphoglycan synthesis implicated in parasite virulence but not viability. EMBO J. 21, 4458–4469. doi: 10.1093/emboj/cdf447
Descoteaux, A., Luo, Y., Turco, S. J., and Beverley, S. M. (1995). A specialized pathway affecting virulence glycoconjugates of Leishmania. Science 269, 1869–1872. doi: 10.1126/science.7569927
Descoteaux, A., Mengeling, B. J., Beverley, S. M., and Turco, S. J. (1998). Leishmania donovani has distinct mannosylphosphoryltransferases for the initiation and elongation phases of lipophosphoglycan repeating unit biosynthesis. Mol. Biochem. Parasitol. 94, 27–40. doi: 10.1016/S0166-6851(98)00047-4
Descoteaux, A., and Turco, S. J. (1999). Glycoconjugates in Leishmania infectivity. Biochim Biophys. Acta 1455, 341–352. doi: 10.1016/S0925-4439(99)00065-4
Forestier, C. L., Gao, Q., and Boons, G. J. (2015). Leishmania lipophosphoglycan: how to establish structure-activity relationships for this highly complex and multifunctional glycoconjugate? Front. Cell. Infect. Microbiol. 4:193. doi: 10.3389/fcimb.2014.00193
Franco, L. H., Beverley, S. M., and Zamboni, D. S. (2012). Innate immune activation and subversion of Mammalian functions by leishmania lipophosphoglycan. J. Parasitol. Res. 2012:165126. doi: 10.1155/2012/165126
Gaur, U., Showalter, M., Hickerson, S., Dalvi, R., Turco, S. J., Wilson, M. E., et al. (2009). Leishmania donovani lacking the Golgi GDP-Man transporter LPG2 exhibit attenuated virulence in mammalian hosts. Exp. Parasitol. 122, 182–191. doi: 10.1016/j.exppara.2009.03.014
Gonzalez-de la Fuente, S., Camacho, E., Peiro-Pastor, R., Rastrojo, A., Carrasco-Ramiro, F., Aguado, B., et al. (2019). Complete and de novo assembly of the Leishmania braziliensis (M2904) genome. Mem. Inst. Oswaldo Cruz 114:e180438. doi: 10.1590/0074-02760180438
Gonzalez-de la Fuente, S., Peiro-Pastor, R., Rastrojo, A., Moreno, J., Carrasco-Ramiro, F., Requena, J. M., et al. (2017). Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci Rep. 7:18050. doi: 10.1038/s41598-017-18374-y
Guha-Niyogi, A., Sullivan, D. R., and Turco, S. J. (2001). Glycoconjugate structures of parasitic protozoa. Glycobiology 11(4), 45R-59R. doi: 10.1093/glycob/11.4.45R
Hong, K., Ma, D., Beverley, S. M., and Turco, S. J. (2000). The Leishmania GDP-mannose transporter is an autonomous, multi-specific, hexameric complex of LPG2 subunits. Biochemistry 39, 2013–2022. doi: 10.1021/bi992363l
Hurrell, B. P., Regli, I. B., and Tacchini-Cottier, F. (2016). Different Leishmania Species Drive Distinct Neutrophil Functions. Trends Parasitol. 32, 392–401. doi: 10.1016/j.pt.2016.02.003
Ilg, T. (2000a). Lipophosphoglycan is not required for infection of macrophages or mice by Leishmania mexicana. EMBO J 19, 1953–1962. doi: 10.1093/emboj/19.9.1953
Ilg, T. (2000b). Proteophosphoglycans of Leishmania. Parasitol. Today 16, 489–497. doi: 10.1016/S0169-4758(00)01791-9
Ilg, T., Demar, M., and Harbecke, D. (2001). Phosphoglycan repeat-deficient Leishmania mexicana parasites remain infectious to macrophages and mice. J Biol. Chem. 276, 4988–4997. doi: 10.1074/jbc.M008030200
Ishemgulova, A., Hlavacova, J., Majerova, K., Butenko, A., Lukes, J., Votypka, J., et al. (2018). CRISPR/Cas9 in Leishmania mexicana: A case study of LmxBTN1. PLoS One 13:e0192723. doi: 10.1371/journal.pone.0192723
King, D. L., and Turco, S. J. (1988). A ricin agglutinin-resistant clone of Leishmania donovani deficient in lipophosphoglycan. Mol. Biochem. Parasitol. 28, 285–293. doi: 10.1016/0166-6851(88)90013-8
Lazaro-Souza, M., Matte, C., Lima, J. B., Arango Duque, G., Quintela-Carvalho, G., de Carvalho Vivarini, A., et al. (2018). Leishmania infantum Lipophosphoglycan-Deficient Mutants: A Tool to Study Host Cell-Parasite Interplay. Front. Microbiol. 9:626. doi: 10.3389/fmicb.2018.00626
Lypaczewski, P., Hoshizaki, J., Zhang, W. W., McCall, L. I., Torcivia-Rodriguez, J., Simonyan, V., et al. (2018). A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence. Sci. Rep. 8:16549. doi: 10.1038/s41598-018-34812-x
Ma, D., Russell, D. G., Beverley, S. M., and Turco, S. J. (1997). Golgi GDP-mannose uptake requires Leishmania LPG2. A member of a eukaryotic family of putative nucleotide-sugar transporters. J. Biol. Chem. 272, 3799–3805. doi: 10.1074/jbc.272.6.3799
Martel, D., Beneke, T., Gluenz, E., Spath, G. F., and Rachidi, N. (2017). Characterisation of Casein Kinase 1.1 in Leishmania donovani Using the CRISPR Cas9 Toolkit. Biomed. Res. Int. 2017:4635605. doi: 10.1155/2017/4635605
McConville, M. J., Collidge, T. A., Ferguson, M. A., and Schneider, P. (1993). The glycoinositol phospholipids of Leishmania mexicana promastigotes. Evidence for the presence of three distinct pathways of glycolipid biosynthesis. J. Biol. Chem. 268, 15595–15604.
McNeely, T. B., Tolson, D. L., Pearson, T. W., and Turco, S. J. (1990). Characterization of Leishmania donovani variant clones using anti-lipophosphoglycan monoclonal antibodies. Glycobiology 1, 63–69. doi: 10.1093/glycob/1.1.63
Moradin, N., and Descoteaux, A. (2012). Leishmania promastigotes: building a safe niche within macrophages. Front. Cell. Infect. Microbiol. 2:121. doi: 10.3389/fcimb.2012.00121
Opat, A., Ng, K., Currie, G., Handman, E., and Bacic, A. (1996). Characterization of lipophosphoglycan from a ricin-resistant mutant of Leishmania major. Glycobiology 6, 387–397. doi: 10.1093/glycob/6.4.387
Peacock, C. S., Seeger, K., Harris, D., Murphy, L., Ruiz, J. C., Quail, M. A., et al. (2007). Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 39, 839–847. doi: 10.1038/ng2053
Pitale, D. M., Gendalur, N. S., Descoteaux, A., and Shaha, C. (2019). Leishmania donovani Induces Autophagy in Human Blood-Derived Neutrophils. J. Immunol. 202, 1163–1175. doi: 10.4049/jimmunol.1801053
Quintela-Carvalho, G., Luz, N. F., Celes, F. S., Zanette, D. L., Andrade, D., Menezes, D., et al. (2017). Heme Drives Oxidative Stress-Associated Cell Death in Human Neutrophils Infected with Leishmania infantum. Front. Immunol. 8:1620. doi: 10.3389/fimmu.2017.01620
Ribeiro-Gomes, F. L., Otero, A. C., Gomes, N. A., Moniz-De-Souza, M. C., Cysne-Finkelstein, L., Arnholdt, A. C., et al. (2004). Macrophage interactions with neutrophils regulate Leishmania major infection. J. Immunol. 172, 4454–4462. doi: 10.4049/jimmunol.172.7.4454
Robinson, K. A., and Beverley, S. M. (2003). Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Mol. Biochem. Parasitol. 128, 217–228. doi: 10.1016/S0166-6851(03)00079-3
Ryan, K. A., Garraway, L. A., Descoteaux, A., Turco, S. J., and Beverley, S. M. (1993). Isolation of virulence genes directing surface glycosyl-phosphatidylinositol synthesis by functional complementation of Leishmania. Proc. Natl. Acad. Sci. U. S. A. 90, 8609–8613. doi: 10.1073/pnas.90.18.8609
Sacks, D. L., Modi, G., Rowton, E., Spath, G., Epstein, L., Turco, S. J., et al. (2000). The role of phosphoglycans in Leishmania-sand fly interactions. Proc. Natl. Acad. Sci. U. S. A. 97, 406–411. doi: 10.1073/pnas.97.1.406
Scianimanico, S., Desrosiers, M., Dermine, J. F., Meresse, S., Descoteaux, A., and Desjardins, M. (1999). Impaired recruitment of the small GTPase rab7 correlates with the inhibition of phagosome maturation by Leishmania donovani promastigotes. Cell Microbiol. 1, 19–32. doi: 10.1046/j.1462-5822.1999.00002.x
Segawa, H., Soares, R. P., Kawakita, M., Beverley, S. M., and Turco, S. J. (2005). Reconstitution of GDP-mannose transport activity with purified Leishmania LPG2 protein in liposomes. J. Biol. Chem. 280, 2028–2035. doi: 10.1074/jbc.M404915200
Spath, G. F., Epstein, L., Leader, B., Singer, S. M., Avila, H. A., Turco, S. J., et al. (2000). Lipophosphoglycan is a virulence factor distinct from related glycoconjugates in the protozoan parasite Leishmania major. Proc. Natl. Acad. Sci. U. S. A. 97, 9258–9263. doi: 10.1073/pnas.160257897
Spath, G. F., Garraway, L. A., Turco, S. J., and Beverley, S. M. (2003a). The role(s) of lipophosphoglycan (LPG) in the establishment of Leishmania major infections in mammalian hosts. Proc. Natl. Acad. Sci. U. S. A. 100, 9536–9541. doi: 10.1073/pnas.1530604100
Spath, G. F., Lye, L. F., Segawa, H., Sacks, D. L., Turco, S. J., and Beverley, S. M. (2003b). Persistence without pathology in phosphoglycan-deficient Leishmania major. Science 301, 1241–1243. doi: 10.1126/science.1087499
Zhang, W. W., Lypaczewski, P., and Matlashewski, G. (2017). Optimized CRISPR-Cas9 Genome Editing for Leishmania and Its Use To Target a Multigene Family, Induce Chromosomal Translocation, and Study DNA Break Repair Mechanisms. mSphere 2, e00340-16 doi: 10.1128/mSphere.00340-16
Zhang, W. W., and Matlashewski, G. (2015). CRISPR-Cas9-Mediated Genome Editing in Leishmania donovani. mBio 6, e00861. doi: 10.1128/mBio.00861-15
Keywords: GDP-mannose transporter, lipophosphoglycan, Leishmania infantum, gene targeting, CRISPR/CAS9
Citation: Jesus-Santos FH, Lobo-Silva J, Ramos PIP, Descoteaux A, Lima JB, Borges VM and Farias LP (2020) LPG2 Gene Duplication in Leishmania infantum: A Case for CRISPR-Cas9 Gene Editing. Front. Cell. Infect. Microbiol. 10:408. doi: 10.3389/fcimb.2020.00408
Received: 20 December 2019; Accepted: 02 July 2020;
Published: 13 August 2020.
Edited by:
Prajwal Gurung, The University of Iowa, United StatesReviewed by:
Ciaran Michael Lee, University College Cork, IrelandEva Gluenz, University of Oxford, United Kingdom
Copyright © 2020 Jesus-Santos, Lobo-Silva, Ramos, Descoteaux, Lima, Borges and Farias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonardo Paiva Farias, bGVvbmFyZG8uZmFyaWFzQGJhaGlhLmZpb2NydXouYnI=