 Sara Becker1
Sara Becker1 Annette Fink1†
Annette Fink1† Jürgen Podlech1
Jürgen Podlech1 Irina Giese2
Irina Giese2 Julia K. Schmiedeke1Thomas Bukur2
Julia K. Schmiedeke1Thomas Bukur2 Matthias J. Reddehase1
Matthias J. Reddehase1 Niels A. Lemmermann1*
Niels A. Lemmermann1*- 1Institute for Virology, Research Center for Immunotherapy (FZI), University Medical Center of the Johannes Gutenberg-University of Mainz, Mainz, Germany
- 2TRON - Translational Oncology, Medical Center of the Johannes Gutenberg-University Mainz gGmbH, Mainz, Germany
Murine cytomegalovirus (mCMV) codes for MHC class-I trafficking modulators m04/gp34, m06/gp48, and m152/gp40. By interacting with the MHC class-Iα chain, these proteins disconnect peptide-loaded MHC class-I (pMHC-I) complexes from the constitutive vesicular flow to the cell surface. Based on the assumption that all three inhibit antigen presentation, and thus the recognition of infected cells by CD8 T cells, they were referred to as “immunoevasins.” Improved antigen presentation mediated by m04 in the presence of m152 after infection with deletion mutant mCMV-Δm06W, compared to mCMV-Δm04m06 expressing only m152, led us to propose renaming these molecules “viral regulators of antigen presentation” (vRAP) to account for both negative and positive functions. In accordance with a positive function, m04-pMHC-I complexes were found to be displayed on the cell surface, where they are primarily known as ligands for Ly49 family natural killer (NK) cell receptors. Besides the established role of m04 in NK cell silencing or activation, an anti-immunoevasive function by activation of CD8 T cells is conceivable, because the binding site of m04 to MHC class-Iα appears not to mask the peptide binding site for T-cell receptor recognition. However, functional evidence was based on mCMV-Δm06W, a virus of recently doubted authenticity. Here we show that mCMV-Δm06W actually represents a mixture of an authentic m06 deletion mutant and a mutant with an accidental additional deletion of a genome region encompassing also gene m152. Reanalysis of previously published experiments for the authentic mutant in the mixture confirms the previously concluded positive vRAP function of m04.
Introduction
Human cytomegalovirus (hCMV) is the prototype member of the ß-subfamily of the herpesviruses [for an overview, see Davison et al. (2013)]. It is a clinically relevant pathogen leading to birth defects upon congenital infection, and it causes severe organ diseases in immunocompromised recipients of solid organ transplantation (SOT) and of hematopoietic cell transplantation (HCT) [for overviews, see Ho (2008), Boppana and Britt (2013), Emery et al. (2013), and Seo and Boeckh (2013)]. Despite expression of “immunoevasins” that limit the presentation of antigenic peptides to T cells [for reviews, see Wiertz et al. (1997), Hengel et al. (1998), Reddehase (2002), Doom and Hill (2008), Powers et al. (2008), Hansen and Bouvier (2009), and Berry et al. (2019)] unlimited viral spread and cytopathogenic tissue infection is prevented in the immunocompetent host, accompanied by the establishment of viral latency in certain cell types (Elder and Sinclair, 2019; Reddehase and Lemmermann, 2019). Reactivation to recurrent productive infection can occur when immune surveillance wanes due to immunocompromising conditions such as hematoablative leukemia therapy and immunosuppressive graft-vs.-host disease (GvHD) prophylaxis in HCT or immunosuppressive prophylaxis against graft rejection in SOT.
As experimental approaches and studies with recombinant viruses carrying targeted mutations for addressing mechanistic questions are not feasible in clinical investigations, the mouse model based on infection with murine cytomegalovirus (mCMV) has been developed as a versatile preclinical model. It has already provided “proof of principle” for basic aspects of viral pathogenesis and immune control, including cytoimmunotherapy with antiviral CD8 T cells in HCT recipients (Krmpotic et al., 2003; Reddehase, 2016; Reddehase and Lemmermann, 2018; Renzaho et al., 2020). Although the host species-specific CMVs differ in many genes involved in host adaptation, co-speciation of hosts and their respective CMVs has led to biological convergence in fundamental principles of virus-host interaction.
Three “immune evasion” proteins encoded by mCMV have been reported to bind to MHC class-I (MHC-I) molecules and to thereby disconnect them from the constitutive vesicular flow of trafficking to the cell surface in the MHC-I pathway of antigen processing and presentation to CD8 T cells (Lemmermann et al., 2012). These include the m02-m16 gene family members m04/gp34 (Kleijnen et al., 1997) and m06/gp48 (Reusch et al., 1999), as well as the m145 gene family member m152/gp40 (Ziegler et al., 1997, 2000; Fink et al., 2013). As far as analyzed, molecules of the m02-m16 gene family share a β-sandwich immunoglobulin variable (Ig-V)-like fold (Berry et al., 2014; Sgourakis et al., 2014, 2015), whereas members of the m145 gene family mimic the structure of MHC-I molecules, thus representing MHC-I-like virally encoded (MHC-Iv) glycoproteins (Wang et al., 2012). As shown by Fink et al. (2013), glycosylation of m152 is not required for m152 function, so that binding of the p36 isoform in the ER to nascent peptide-loaded MHC-I (pMHC-I) complexes and to the MHC-I-like ligand RAE-1 of the natural killer (NK) cell receptor NKG2D is sufficient for the dual role of m152 in inhibiting the activation of CD8 T cells and NK cells, respectively (Krmpotic et al., 2002). As concluded by implication from studies with immune evasion gene deletion mutants of mCMV (Wagner et al., 2002), evasion of antiviral CD8 T cells leads to enhanced and prolonged viral replication in recipients of experimental HCT with the consequence of an elevated latent viral genome load and increased risk of reactivation (Böhm et al., 2009). Immune evasion proteins reduce the efficacy of cytoimmunotherapy of mCMV infection by adoptive transfer of antiviral CD8 T cells (Krmpotic et al., 1999, 2002; Holtappels et al., 2004). More recently, mouse models of allogeneic HCT with immunogenetic donor-recipient mismatch in MHC-I or in minor histocompatibility loci revealed a decisive impact of viral immune evasion proteins on viral spread and lethal organ failure due to extensive histopathology (Gezinir et al., 2020; Holtappels et al., 2020). Thus, immune evasion is predictably of significant clinical relevance in HCT patients.
Regarding the molecular mechanisms, m152 mediates the retention of pMHC-I complexes in ER-Golgi intermediate/cis-Golgi compartments (Ziegler et al., 1997, 2000; Janßen et al., 2016). More recently, m152 has been identified to target the type I interferon response by binding to STING (Stempel et al., 2019). The closely related glycoproteins m04 and m06 compete for pMHC-I cargo by forming complexes and connect it to cellular adapter proteins (AP) of cargo sorting pathways via motifs in their cytosolic tails. Specifically, m06 contains a functional di-leucine motif that links the m06-pMHC-I complexes to AP-1A and AP-3A, eventually resulting in lysosomal disposal (Reusch et al., 2002). Recent work has shown that inactivation of the sorting motif by mutation does not prevent cell surface downmodulation of MHC-I molecules but inhibits the transition of m06-MHC-I complexes from early endosomes to late endosomes (Fink et al., 2019). In contrast, m04 contains a tyrosine-based AP-2 binding motif and escorts pMHC-I complexes to the cell surface (Kleijnen et al., 1997; Kavanagh et al., 2001; Lu et al., 2006; Fink et al., 2015) in association with the recently discovered viral protein MATp1. MATp1 turned out to be essential for m04-mediated MHC-I cell surface rescue resulting in silencing of NK cells via ligation of inhibitory Ly49 family NK cell receptors to overcome missing-self activation (Železnjak et al., 2019). Inactivation of the endocytic AP-2 motif was found to stabilize the complex at the cell surface, resulting in an enhanced NK cell silencing (Fink et al., 2015). The question remained if rescue of cell surface expression of pMHC-I complexes by m04 or m04-MATp1 also rescues recognition of infected cells by antiviral CD8 T cells. Although the m04 binding site to MHC-I has not been precisely mapped yet, structural data suggest that the peptide-binding platform is not masked (Berry et al., 2014), so that the TCR of CD8 T cells should still be able to recognize presented antigenic peptide.
When immune evasion proteins are expressed separately in viral mutants mCMV-Δm06m152 (selectively expressing m04), mCMV-Δm04m152 (selectively expressing m06), and mCMV-Δm04m06 (selectively expressing m152), inhibition of cell surface presentation of pMHC-I complexes ranked in the order of m04 << m06 < m152, with actually no notable inhibition by m04 [(Wagner et al., 2002; Holtappels et al., 2006); discussed in Lemmermann et al. (2012)]. This finding gave a first hint to raise doubt as to an “immune evasion” function of m04 with respect to CD8 T cells. Moreover, previous work revealed that m04 expressed in deletion mutant mCMV-Δm06W (Wagner et al., 2002) relieves the inhibition mediated by co-expressed m152 as compared to mCMV-Δm04m06 expressing only m152 (Wagner et al., 2002; Holtappels et al., 2006). This was first shown on the basis of MHC-I cell surface levels detected cytofluorometrically (Wagner et al., 2002) and later extended functionally to target cell recognition (Holtappels et al., 2006; Pinto et al., 2006) and to in vivo protection by adoptively transferred antiviral CD8 T cells (Holtappels et al., 2006). This positive function of m04 prompted us to suggest the acronym “viral regulator of antigen presentation” (vRAP) to cover both positive and negative modulation of antigen presentation.
All conclusions based on virus mutant mCMV-Δm06W, expected to express normal levels of m152, needed to be drawn into question, because the authenticity of this virus was doubted based on the observation of an unexplained reduction of m152 protein expression that included all its glycosylation isoforms and that could not be reproduced with independently generated new mutants mCMV-Δm06L1 and mCMV-Δm06L2 (Fink et al., 2012, 2013; Lemmermann et al., 2012). As reduced expression of m152 relieves immune evasion, this alone might explain enhanced antigen presentation by cells infected with virus mutant mCMV-Δm06W as compared to mutant mCMV-Δm04m06 expressing normal levels of m152. Misleadingly, genetic authenticity of mCMV-Δm06W was suggested by qualitative detection of gene m152 in liver tissue infected with mCMV-Δm06W and analyzed by in situ hybridization (Holtappels et al., 2006). Accordingly, all attempts to explain the reduced expression by mutations in the m152 gene coding region, the 3′ as well as 5′ untranslated regions, or the promoter region failed (unpublished own data).
As the problem not just casts doubt on our own previous conclusion on a positive vRAP function of m04 (Holtappels et al., 2006) but also might affect work of other groups who published data based on mCMV-Δm06W (LoPiccolo et al., 2003; Pinto et al., 2006, 2007; Babic et al., 2010), we decided to compare the m152 expression-deficient mutant mCMV-Δm06W and the m152 expression-sufficient mutant mCMV-Δm06L by next-generation sequencing (NGS) of the full-length viral genomes purified from infectious virions. We identified two genomic variants within the pool of Δm06W genomes: a small proportion of genomes with the correct, selective deletion of gene m06 and a majority with an additional large deletion encompassing open reading frame (ORF) m152 and spanning ORFs m145-m158. As implied by the growth curve of mutant mCMV-Δm06W (Wagner et al., 2002), the large deletion does not lead to attenuated growth in cell culture and therefore is maintained in purified virus stocks. Mapping of the large deletion allowed us to design specific probes for in situ hybridization distinguishing between the correct deletion mutant mCMV-Δm06 and the wrong mutant with the additional off-target site deletion. Reanalysis of stored tissue specimens from key experiments of the original work (Holtappels et al., 2006) confirmed a positive vRAP function of m04 in liver cells infected with the minority fraction of the correct m06 gene deletion mutant present in the mixed pool.
Materials and Methods
Cells and Viruses
High titer virus stocks of mCMVSmith sequence-derived mCMV-WT.BAC (MW97.01, Wagner et al., 1999), mCMV-Δm06W (Wagner et al., 2002), and mCMV-Δm06L (Fink et al., 2012), were prepared from infected murine embryonic fibroblasts (MEF) according to standard procedures (Podlech et al., 2002; Lemmermann et al., 2010).
Purification of Viral DNA From High Titer Virus Stocks
Viral DNA corresponding to 1 × 107 plaque forming units (PFU) was purified from a high titer virus stock by using Roche High Pure Viral Nucleic Acid Kit (Roche) following the manufacturer's instruction. Viral DNA was eluted in 50 μL of elution buffer and stored at 4°C until use for DNA library preparation or for PCR.
NEBNext Ultra Library Preparation
In total, eight DNA libraries were prepared using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB). As the first step, 100 ng of viral DNA was sheared to an average size of around 250 bp by ultrasonication. Fragments were checked via capillary gel electrophoresis, end-repaired, A-tailed, and Illumina-specific barcoded adapters were ligated. These adapters also serve as annealing regions for the amplification primers during a PCR. The libraries were checked for quantity and quality using Bioanalyzer2100 (Agilent) and Qubit 3.0 Fluorometer (Thermo Fisher Scientific). Subsequently, the libraries were normalized to 10 nM and pooled to be equimolar before sequencing.
Next-Generation Sequencing on Illumina MiSeq
Sequencing templates were immobilized on a flow cell surface designed to present the DNA in a manner that facilitates access to enzymes while ensuring high stability of surface-bound template and low non-specific binding of fluorescent-labeled nucleotides. Solid-phase amplification creates up to 1,000 identical copies of each single template molecule in close proximity. Sequencing by synthesis (SBS) technology uses four fluorescent-labeled nucleotides to sequence the tens of millions of clusters on the flow cell surface in parallel. During each sequencing cycle, a single labeled deoxynucleotide triphosphate (dNTP) is added to the nucleic acid chain. The nucleotide label serves as a terminator for the extension. So, after each dNTP incorporation, the fluorescent dye is imaged to identify the base and is then enzymatically cleaved to allow incorporation of the next nucleotide. Since all four reversible terminator-bound dNTPs (A, C, T, G) are present as single separate molecules, natural competition minimizes incorporation bias. Base calls are made directly from signal intensity measurements during each cycle. Here, libraries have been sequenced in four batches on an Illumina MiSeq Nano Flow cell (2 × 150 bp, paired-end) with an average output of 1 million clusters passing filter. Each batch contained a pool of two equimolar pooled libraries.
Analysis of Sequencing Data
Sequencing yielded between 1.40 and 1.91 million paired-end reads (mean: 1.59 million read pairs). Reads were trimmed of adapter sequences, filtered for read length (minimum of 15 bases by default), and overlapping read fragments were error corrected using Fastp (version 0.19.4; Chen et al., 2018). On average, 1.58 million read pairs were left for alignment (minimum: 1.39 million, maximum: 1.89 million). The reads were aligned using the Smith-Waterman alignment method implemented in Novoalign (version 3.09.01; http://www.novocraft.com/products/novoalign/) against the mCMV reference (RefSeq: NC_004065.1, INSDC: U68299.1). Alignments resulted in a mean coverage between 199.1 and 441.6 per sample on the reference. Single nucleotide variations (SNVs) and small insertions and deletions (INDELs) were identified using Fisher's Exact Test implemented in VarScan 2 (http://varscan.sourceforge.net; Koboldt et al., 2012). SNVs and INDELs were reported with a P-value below 0.01 for somatic calls. We inspected larger variations from the reference genome visually using the Integrative Genomics Viewer (version 2.4.13) (Robinson et al., 2011).
Validation of Deletion in the m145 Region
To validate a possible deletion within the m145 region, PCR was performed using the HotStarTaq DNA Polymerase (Qiagen) and oligonucleotides (m145_flank_for: CACGACAGACATACAGAG, m145_flank_rev: GCAGACTCTGAGGACCGG) with the following profile: 95°C 15 min; (95°C 15 s; 50°C 60 s; 72°C 90 s) × 35; 72°C 10 min. For the amplification of the potential 13 kbp-long inconsistent region, the LongRange PCR Kit (Qiagen) was used and PCR was performed with oligonucleotides (large_del_flk_for: GGTGAGGGGATTATGTCCTG, large_del_flk_rev: TGGTGGTGCCCTATCCTTAC) under the following conditions: 93°C 3 min; (93°C 15 s; 50°C 30 s; 68°C 13 min) × 10; (93°C 15 s; 50°C 30 s; 68°C 13 min with +20 s elongation for each cycle) × 28. The PCR products were separated on TAE agarose gel and bands of interest were cut out. DNA was purified with the QIAquick Gel Extraction Kit (Qiagen) and the PCR product was sequenced by Eurofins GATC services (Freiburg) using Sanger-Seq1_for: AGGCACGTAGCGAGGATGTC, Sanger-Seq2_for: GATGACGTACTCTCCCTG, Sanger-Seq3_for: GCGGACGACCTCGTTGAG, Sanger-Seq4_for: CGTTAACCGGGCTGCATCC.
Detection and Distinction of mCMV Variants by in situ DNA-DNA Hybridization
Sequence-specific two-color DNA-DNA in situ hybridization (2C-ISH) was used to detect and quantitate infected cells in liver tissue sections. For distinguishing cells infected with different virus variants in a mixture, variant-specific ISH probes were generated and labeled by PCR with digoxigenin-11–dUTP or fluorescein-12-dUTP, followed by peroxidase-conjugated anti-digoxigenin antibody or alkaline phosphatase-conjugated anti-fluorescein antibody for black (DAB-nickel) and red (Fuchsin) color staining, respectively (Lemmermann et al., 2010). Primers for probe synthesis were as follows: (mCMV m152-P), m152-P_for: AGTTGATGTAGACCAGGCGATAC, m152-P_rev: GCTATCACCTACTTGCTCCTCTCG. (mCMV M55/gB-P), M55-P_for: AAGCTTGCACGTCGTAGGTAAATTGC, M55-P_rev: CAGGATCCTCGTCTCTCGAGCTGGTACG. (mCMV BAC-P), BAC-P_for: CACTGTTCCACTTGTATCG, BAC-P_rev: CATGCAGCTCCCGGAGACG.
Detection and Distinction of mCMV Variants by Immunohistochemistry
As a more sensitive alternative to 2C-ISH, two-color immunohistochemistry (2C-IHC) was used to detect and quantitate infected cells in liver tissue sections. For distinguishing cells infected with different virus variants in a mixture, antibodies specific for differentially expressed viral proteins were labeled either with biotinylated second antibody and ABC-peroxidase for black (DAB-nickel) staining or with alkaline phosphatase conjugated second antibody for red (Fuchsin) staining, essentially as described (Lemmermann et al., 2010). Monoclonal rat antibody (clone M3D10) was used as the primary antibody in the red staining of m152 (Ziegler et al., 2000).
Adoptive Transfer of Antiviral CD8 T Cells
Stored tissue specimens from previously published adoptive cell transfer experiments (Holtappels et al., 2006) using CD8 T cells of cytolytic T-lymphocyte (CTL) lines specific for antigenic peptides derived from mCMV protein M45, namely peptide 507-VGPALGRGL-515 presented by the MHC-I molecule Dd (epitope M45-Dd) and peptide 985-HGIRNASFI-993 presented by Db (epitope M45-Db) (Gold et al., 2002; Holtappels et al., 2009) were reanalyzed by 2C-ISH and 2C-IHC, respectively.
Results
NGS Reveals a Large Deletion in BAC-Derived mCMV-Δm06W Genomes
To investigate the reason for the unexpectedly diminished m152 expression in cells infected with mCMV-Δm06W (Fink et al., 2012, 2013; Lemmermann et al., 2012) we tested the genetic authenticity of this mutant in comparison to mCMV-WT.BAC and the independently generated mutant mCMV-Δm06L (Fink et al., 2012) by full-length genome next-generation sequencing (NGS) of purified virion DNA. First, we verified the deletion of gene m06 in mCMV-Δm06W (nts. 5,396–6,236; GeneBank U68299.1; Rawlinson et al. (1996) and mCMV-Δm06L (nts. 5,301–6,334) (Figure 1A). Full-length genome comparison of mCMV-Δm06L and mCMV-WT.BAC revealed no unexpected differences between both genomes. In contrast, the sequencing revealed a large deletion in mCMV-Δm06W (Figure 1B) spanning ≈13 kbp (nts. 204,704–217,934) encompassing 14 ORFs from m145 to m158, many of which code for MHC-Iv glycoproteins that have been associated with modulation of host innate and/or adaptive immunity.
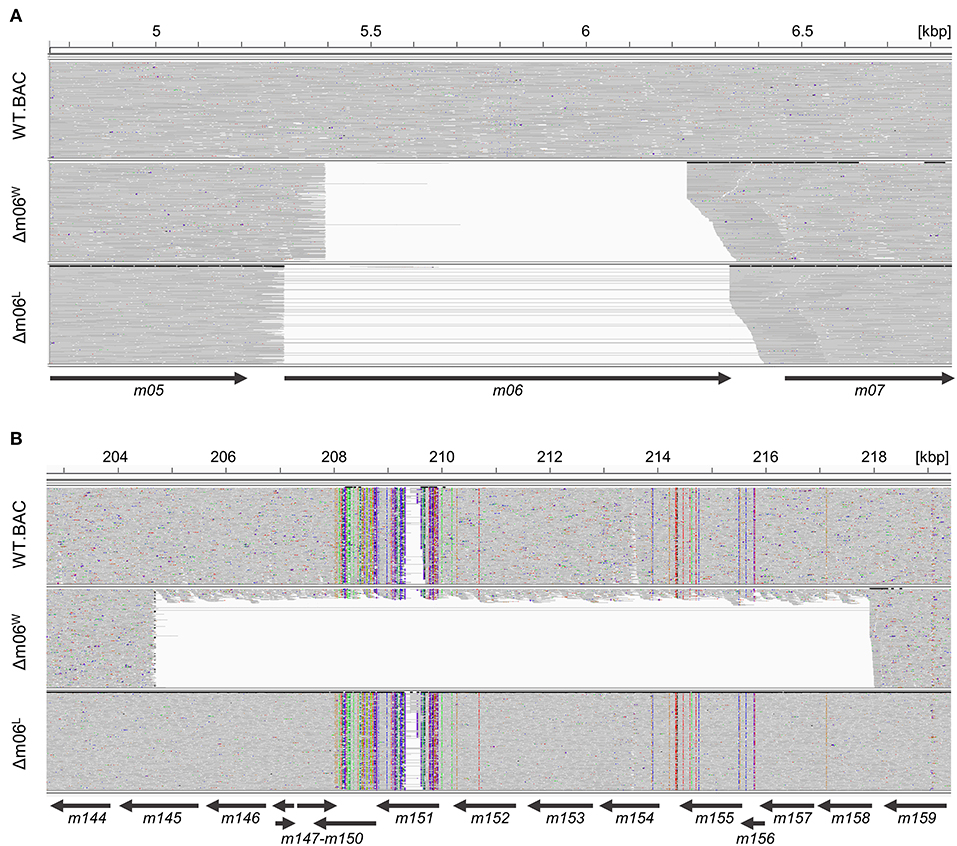
Figure 1. Comparison of Illumina-sequenced viral genomes. Viral DNA was purified from stocks of viruses mCMV-WT.BAC, mCMV-Δm06W, and mCMV-Δm06L, sequenced on Illumina MiSeq, and aligned to the mCMV WT reference sequence (RefSeq: NC_004065.1, INSDC: U68299.1). The Integrative Genomics Viewer was used for visualization. Gray areas indicate matches to the reference sequence, whereas colored spots indicate SNVs or INDELs. Regions with no successful alignment to the reference sequence are left white. Arrows represent the positions of the indicated ORFs. (A) Comparison of the viral genomes at 4.8–6.8 kbp. (B) Comparison of the viral genomes at 203–219 kbp.
Besides m152, the object of our investigation here, more prominent examples affected by the deletion include m145 and m155 that downmodulate NKG2D ligands MULT-1 (Krmpotic et al., 2005) and H60 (Hasan et al., 2005), respectively, the activatory ligand m157 of the Ly49H NK cell subset that confers genetic resistance to mCMV by controlling early virus replication in C57BL/6 (haplotype H-2b) mice (Arase et al., 2002; Scalzo et al., 2003; Voigt et al., 2003; Bubić et al., 2004; Fodil-Cornu et al., 2008), m154 that reduces the cell-surface expression of the SLAM family member CD48 (Zarama et al., 2014) and mediates broad-spectrum immune evasion by perturbing the adaptor protein-1 compartment (Strazic Geljic et al., 2020), and m147.5 that downmodulates the co-stimulatory molecule CD86/B7-2 on antigen-presenting cells (Loewendorf et al., 2004). So, obviously, a phenotype of mutant virus mCMV-Δm06W cannot be attributed with any certainty to the deletion of m06.
Notably, however, a small proportion of the annotated reads aligned within the deleted m145 region, indicating that the mCMV-Δm06W virus stock harbors a mixture of at least two different sets of viral genomes. The ratio of the two genomes can be roughly estimated from the mean coverage within the deletion (15.14 reads per base) to the mean coverage of the entire sequence except the deletion (209.6 reads per base). This estimate indicates that ≈10% of the genomes represent a correct m06 deletion mutant.
To confirm and more precisely map the deletion of the m145-m158 region, we used the primer pair m145_flank_for and m145_flank_rev for amplifying ORF m145 (Figure 2A). With the chosen assay sensitivity, m145 could be amplified from purified mCMV-WT.BAC and mCMV-Δm06L genomes, but not from mCMV-Δm06W genomes (Figure 2B). The absence of m145 amplification in mCMV-Δm06W indicated a deletion in the m145-m158 region. To further test if the missing 13 kbp represent a full deletion or an undesired recombination of viral or cellular DNA fragments, we tried to amplify a fragment spanning the potential deletion site with the primer pair large_del_flk_for and large_del_flk_rev. In case of a full deletion, a ≈1 kbp fragment was expected. However, we were not able to amplify such a fragment. Therefore, we used an optimized protocol for amplification of fragments up to 15 kbp, and this resulted in three fragments with sizes of ~2.8, 4.3, and 13 kbp (Figure 2C). The 2.8 kbp fragment was identified by sequencing as a fragment of the m145-m146 region, and the 13 kbp fragment was identified as the full-length m145-m158 region. Both fragments were derived from the small proportion of viral genomes in the virus stock. The 4.3 kbp PCR product, however, was identified as an irregular, disjointed 4,295 bp fragment of the mCMV genome containing parts of ORFs M57 and M58 (nts. 91,485–91,944) and a non-coding intergenic sequence (nts. 94,429–95,707). The following 1,429 nts proved to be a truncated BAC vector sequence of the parental mCMV-WT.BAC plasmid C3X (Wagner et al., 1999) (Figure 2D).
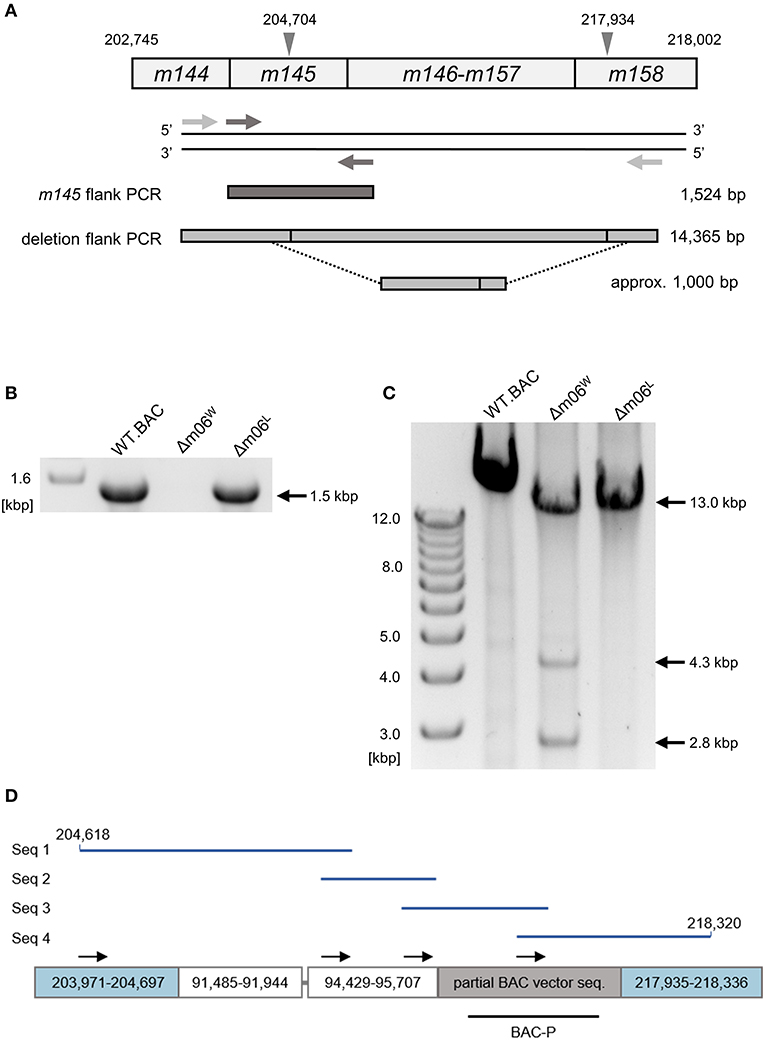
Figure 2. Mapping of the deletion in mCMV-Δm06W. (A) Map of the primer design. Gray arrowheads demarcate the suspected deletion in the m145 gene family, numbers indicate nucleotide positions in the mCMV genome. Light gray arrows represent primers flanking the site of deletion, dark gray arrows represent primers flanking the m145 gene. Expected PCR product sizes are shown on the right side. (B) PCR products of the m145-flank PCR on an agarose gel. (C) PCR products of the deletion-flank PCR on an agarose gel. Purified DNA from mCMV-WT.BAC, mCMV-Δm06W, and mCMV-Δm06L virus stocks was used as template in both (B,C). (D) Sanger sequencing of the 4.3 kbp-sized deletion-flank PCR product in mCMV-Δm06W. The PCR product was gel-purified and used for Sanger sequencing. In total, four primers were used. Black arrows indicate their map positions. Sequences (Seq 1–4, blue lines) were aligned with the mCMV genome and the matching regions are displayed by the boxes. The gray linker between the second and third box represents a short (11 bp) unmatched region. The lower black bar represents the ISH probe used to identify the “large deletion” mutant mCMV-Δm06m145-158 (probe BAC-P). Numbers indicate nucleotide positions in the mCMV genome.
Collectively, these data thus indicate that during the generation of mCMV-Δm06W virions an unwanted recombination event must have taken place at some step. This resulted in a deletion of gene region m145-m158 in the majority of the viral genomes. Apparently, the accidental “large deletion” mutant mCMV-Δm06m145-158 was not lost from the mixture during virus propagation, which is in accordance with the growth curve shown in the original report (Wagner et al., 2002) and explained by the fact that the array of deleted genes is known to be non-essential for virus growth in cell culture.
Lack of in vivo Selection Against the “Large Deletion” Mutant in Immunocompromised Mice
Based on these new data we have now learned in retrospect that our in vivo experiments performed previously with mCMV-Δm06W (Holtappels et al., 2006) were unknowingly performed as co-infection experiments with a mixture of correct mutant mCMV-Δm06 (≈10%) expressing m04 and m152, and a mutant mCMV-Δm06m145-158 with accidental co-deletion affecting m152 (≈90%), thus functionally resembling a Δm06m152 mutant. From previous work we have great experience in analyzing intended co-infections with virus variants by 2C-ISH performed with specific DNA probes to distinguish the variants in host tissues (Grzimek et al., 1999; Holtappels et al., 2004; Cicin-Sain et al., 2005). As a bottom-line message from these studies, spread- and growth-competent variants rarely co-infect tissue cells but establish separate, clonal foci of infection (Grzimek et al., 1999; Holtappels et al., 2004), whereas a requirement for functional complementation selects for co-infected cells (Cicin-Sain et al., 2005).
By using hybridization probes specific for the correct Δm06 mutant carrying gene m152 (probe m152-P, red intranuclear staining) and for mutant mCMV-Δm06m145-158 that contains BAC vector sequence (probe BAC-P, black intranuclear staining), we reanalyzed stored liver tissue from a previously published experiment (Holtappels et al., 2006), in which immunocompromised, total-body γ-irradiated BALB/c (haplotype H-2d) mice had been infected with mCMV-Δm06W. Infected liver cells, detected on day 12 after infection, mostly represent hepatocytes, but also non-parenchymal liver cells such as liver sinusoidal endothelial cells (LSEC) and liver macrophages, known as Kupffer cells (Sacher et al., 2008; Lemmermann et al., 2015). Probes m152-P and BAC-P distinguished between liver cells infected with the correct mutant mCMV-Δm06 (clean red staining) and those infected with mutant mCMV-Δm06m145-158 (clean black staining) (Figure 3A). Notably, cells infected with either virus remained spatially separate in clonal foci of infection, and dually-infected liver cells were not found. Cells infected with mCMV-Δm06m145-158 prevailed in numbers. Lack of red staining with m152-P in cells stained black with BAC-P makes absence of gene m152 in the “large deletion” mutant visible. As an alternative detection strategy, probe M55/gB-P was used to detect all infected cells by red staining, and probe BAC-P was again used to identify cells infected with mCMV-Δm06m145-158 by black staining (Figure 3B). In this case, cells infected with the correct Δm06 mutant can again be identified by clean red staining, whereas cells infected with mCMV-Δm06m145-158 show up by a speckled black-red staining. Again, foci of infection were found to be spatially separate, and cells infected with mCMV-Δm06m145-158 prevailed in numbers. Note that the deletion of genes involved in innate immune control has no phenotype in mice immunodepleted by γ-irradiation. Combined, the data show independent growth of both mutants in liver tissue of immunocompromised mice and do not indicate an in vivo growth attenuation of mCMV-Δm06m145-158 in the absence of innate and adaptive immune control.
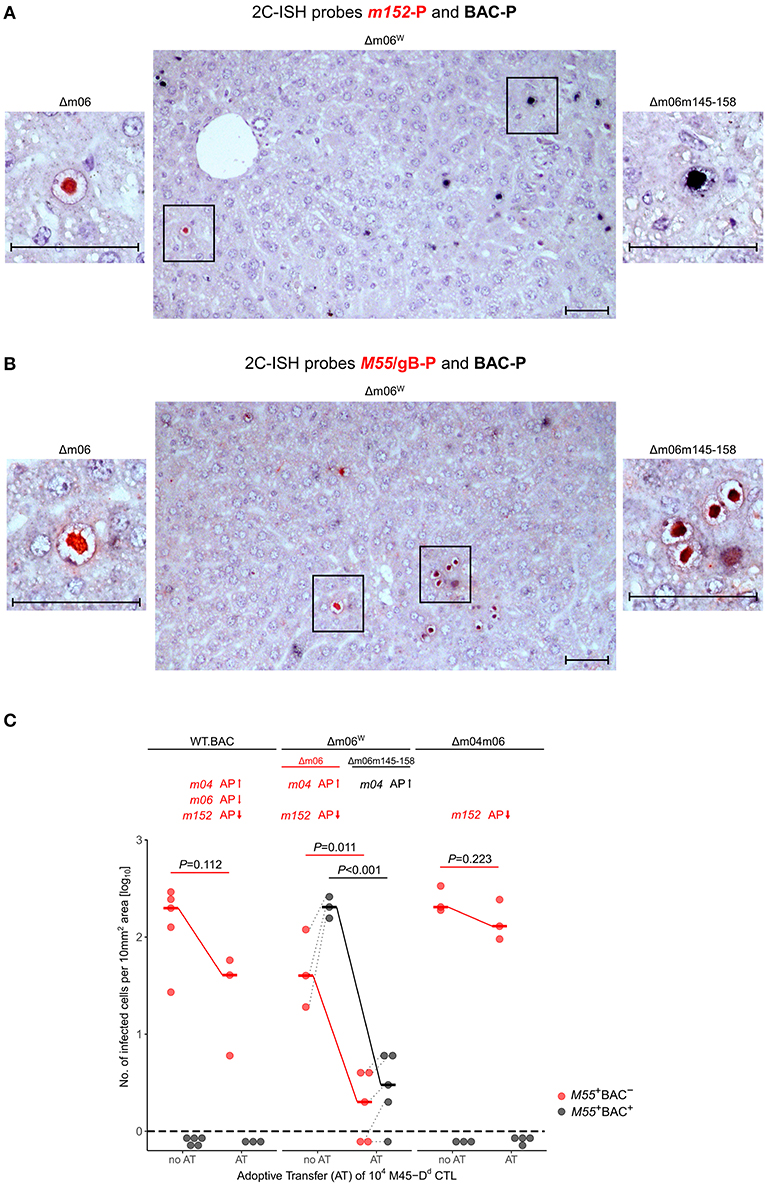
Figure 3. 2C-ISH analysis of liver infection. Reanalysis of stored liver specimens from a previously performed experiment [lung virus titers shown in Holtappels et al. (2006), Figure 8B]. BALB/c mice were immunocompromised by γ-irradiation (6.5 Gy) and infected at one footpad. Liver tissue sections were taken on day 12 after infection (A,B) Virus spread in liver tissue. Infection was performed with 105 PFU of mCMV-Δm06W, now identified to represent a mixture of correct virus mCMV-Δm06 and a “large deletion” mutant mCMV-Δm06m145-158 that includes deletion of the m152 gene. (A) 2C-ISH performed with probe m152-P (red staining) specific for mCMV-Δm06 and probe BAC-P (black staining) specific for mCMV-Δm06m145-158. (Center panel) overview image showing foci of infection for both viruses in the mixture representing mCMV-Δm06W. (Left panel) red-stained mCMV-Δm06-infected cell resolved to greater detail. (Right panel) black-stained mCMV-Δm06m145-158-infected cell resolved to greater detail. (B) 2C-ISH performed with probe M55/gB (red staining) specific for both viruses and probe BAC-P (black staining) specific for mCMV-Δm06m145-158. (Center panel) overview image showing foci of infection for both viruses in the mixture representing mCMV-Δm06W. (Left panel) red-stained mCMV-Δm06-infected cell resolved to greater detail. (Right panel) red-black speckled cells infected with mCMV-Δm06m145-158, resolved to greater detail. Frames in the overview images indicate tissue section areas resolved to greater detail in the left and right images. Note the stained CMV-typical inclusion bodies in the nuclei of infected hepatocytes. Counterstaining was performed with hematoxylin. Bar markers: 50 μm. (C) vRAP expression-dependent control of liver tissue infection with the indicated viruses on day 12 after adoptive transfer of 104 antiviral CD8 T cells specific for the viral epitope M45-Dd. 2C-ISH was performed with probes M55/gB-P (red symbols) and BAC-P (black symbols). (no AT) no adoptive transfer. (AT) adoptive transfer. vRAPs actually expressed by the viruses as well as their proposed impact on antigen presentation (AP, arrows up or down) are indicated. Data represent counts of infected liver cells in representative 10 mm2 tissue section areas. The dashed line indicates the detection limit of the assay, which is one infected cell per selected counting area. Symbols represent mice tested individually. Median values are marked and connected for the groups “no AT” and “AT” to highlight the strength of antiviral control. For statistical analysis, data were log-transformed and P-values were calculated by using the two-sided unpaired t-test with Welch's correction of unequal variances. P < 0.05 indicates statistical significance of the difference between “no AT” and “AT” groups. Linked data are connected by dotted lines, which reveals a correlation between the numbers of cells infected with the correct mutant mCMV-Δm06 and the “large deletion” mutant mCMV-Δm06m145-158 after infection with mCMV-Δm06W.
Verification of m04 as a Positive vRAP by Transfer of CTL Specific for Epitope M45-Dd
As the viruses differ in the expression of vRAPs that regulate antigen presentation to CD8 T cells, a selection pressure was introduced into the immunocompromised BALB/c mice by adoptive transfer (AT) of 104 antiviral CD8 T cells of a CTL line specific for the antigenic peptide M45507-VGPALGRGL-515 that is presented by the MHC-I molecule Dd, briefly epitope M45-Dd (Holtappels et al., 2006). Stored tissue specimens from this published experiment were reanalyzed to either confirm or disprove the previous conclusions. The reanalysis was performed by 2C-ISH of liver tissue of mice infected with viruses mCMV-WT.BAC, mCMV-Δm06W, and mCMV-Δm04m06 with hybridization probes M55/gB-P (red staining) for detection of all infected cells and BAC-P (black staining) selectively for detection of cells infected with mutant virus mCMV-Δm06m145-158, which is only present in the virus mixture of mCMV-Δm06W (Figure 3C). Confirming previous data (Holtappels et al., 2006), control of mCMV-WT.BAC expressing all three vRAPs and of mCMV-Δm04m06 expressing only the strongest inhibitory vRAP m152 was poor and did not reach statistical significance. Not unexpectedly, mCMV-Δm06m145-158 within mCMV-Δm06W was controlled most efficiently and with high statistical significance, because it lacks both inhibitory vRAPs m06 as well as m152, so that antigen presentation to the transferred CD8 T cells is optimal. For conclusions on a predicted function of m04 as a positive vRAP counteracting the negative vRAP m152, the analysis must be restricted to cells infected with the minority fraction of correct mCMV-Δm06 in the virus mixture of mCMV-Δm06W. Importantly, control of correct mutant mCMV-Δm06 that expresses m04 along with m152 was significantly more efficient than was control of mCMV-Δm04m06 expressing m152 only.
Confirmation of m04 as a Positive vRAP by Transfer of CTL Specific for Epitope M45-Db
The question remained if a positive vRAP function of m04 with respect to virus control by CD8 T cells may be an exception that applies only to antigen presentation by the MHC-I molecule Dd, a question raised because MATp1-dependent cell surface expression was found to be most prominent for m04-Dd and m04-Kb complexes and less for other MHC-I alleles as far as analyzed (Železnjak et al., 2019). We have previously intensely studied the antigenic peptide M45985-HGIRNASFI-993 that is presented by the MHC-I molecule Db, briefly epitope M45-Db (Gold et al., 2002), because it is so far the only known epitope so poorly presented in the presence of negative vRAPs that specific CTL completely fail to protect immunocompromised C57BL/6 mice upon adoptive cell transfer, unless vRAP m152 is deleted (Holtappels et al., 2004). A comparison between M45-Dd and M45-Db is particularly appealing, because both peptides are processed from the same protein, which is a cell death inhibitor (Brune et al., 2001; Daley-Bauer et al., 2017), so that differences cannot be attributed to properties of the protein. As we have shown previously, poor protection by M45-Db CTL in C57BL/6 mice compared to M45-Dd CTL in BALB/c mice relates to a very low processing efficacy yielding only few peptide molecules per infected cell, so that negative vRAPs can easily prevent cell surface presentation and thus protection (Holtappels et al., 2009).
With this rationale, we also reanalyzed the previously published control of viruses mCMV-WT.BAC, mCMV-Δm06W, and mCMV-Δm04m06 in immunocompromised C57BL/6 mice by adoptive transfer of CTL specific for epitope M45-Db (Holtappels et al., 2006). As virus levels are generally lower in tissues of C57BL/6 compared to BALB/c mice, we used the more sensitive 2C-IHC for measuring differential protein expression to distinguish between liver cells infected with the correct mutant mCMV-Δm06 that expresses m152 (black staining of intranuclear IE1 protein and red staining of cytoplasmic m152 protein) and the “large deletion” mutant mCMV-Δm06m145-158 that lacks m152 (only black staining of IE1) in mice infected with the virus mixture mCMV-Δm06W (Figure 4). In accordance with our previous work (Holtappels et al., 2004, 2006, 2009), viruses expressing the negative vRAP m152 were not controlled by M45-Db CTL, whereas again m04 relieved immune evasion by m152 expressed in liver cells infected with the correct mutant mCMV-Δm06.
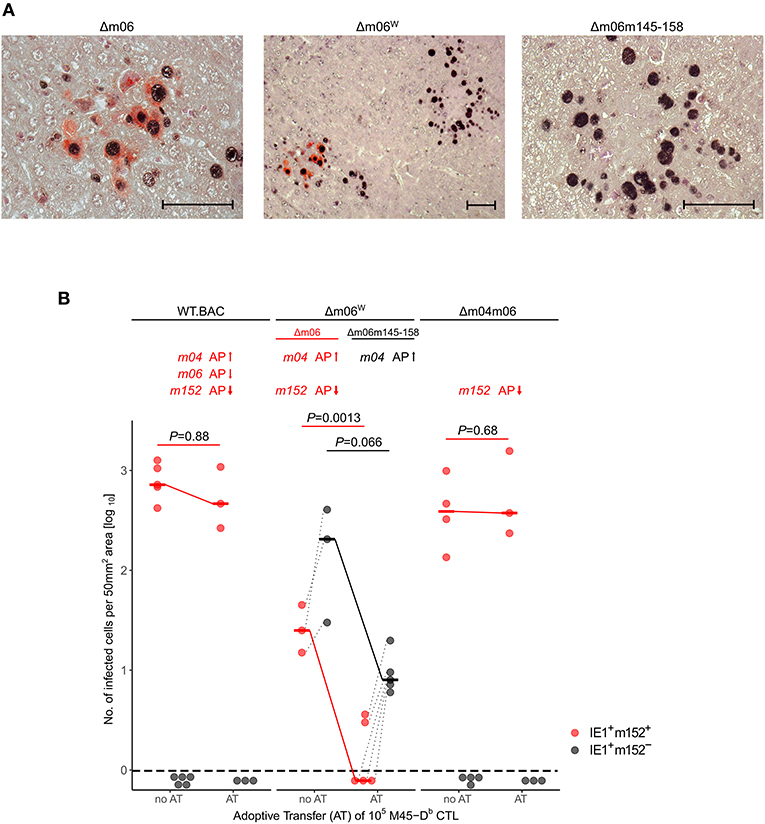
Figure 4. 2C-IHC analysis of liver infection. Reanalysis of stored liver specimens from a previously performed experiment [lung virus titers shown in Holtappels et al. (2006), Figure 8A]. C57BL/6 mice were immunocompromised by γ-irradiation (7.5 Gy) and infected at one footpad. Liver tissue sections were taken on day 12 after infection. (A) Virus spread in liver tissue. Infection was performed with 105 PFU of mCMV-Δm06W, now identified to represent a mixture of correct virus mCMV-Δm06 and a “large deletion” mutant mCMV-Δm06m145-158 that includes deletion of the m152 gene. 2C-IHC was performed to detect cytoplasmic m152 protein (red staining) specific for mCMV-Δm06 and intranuclear IE1 protein (black staining) expressed by both viruses. (Center panel) overview image showing foci of infection for both viruses in the mixture representing mCMV-Δm06W. (Left panel) detail image of cells infected with mCMV-Δm06 identified by red cytoplasmic staining of m152. (Right panel) Detail image of cells infected with mCMV-Δm06m145-158 characterized by absence of red cytoplasmic staining. Counterstaining was performed with hematoxylin. Bar markers: 50 μm. (B) vRAP expression-dependent control of liver tissue infection with the indicated viruses on day 12 after adoptive transfer of 105 antiviral CD8 T cells specific for the viral epitope M45-Db. 2C-IHC was performed to identify infected cells expressing m152 (red symbols) or lacking m152 (black symbols). (no AT) no adoptive transfer. (AT) adoptive transfer. Symbols represent mice tested individually. Data represent counts of infected liver cells in representative 50 mm2 tissue section areas. For further explanation, see the legend to Figure 3.
Discussion
Focussing first on the technical aspect of our work, we report the worrying observation of a previously unrecognized authenticity failure in a virus that has been widely distributed in the community of CMV immunologists and has led to a number of well-cited publications now to be called into question. One may wonder why an unintended deletion of ≈13 kbp spanning 14 ORFs in mCMV-Δm06W has escaped the standard quality controls employed in the early days of BAC mutagenesis of CMVs. According to the original report on the panel of combinatorial immune evasion gene deletion mutants of mCMV (Wagner et al., 2002), all recombinant BAC plasmids were controlled for correct deletions and for genome integrity by restriction pattern analysis, which reliably did not reveal a ≈13 kbp deletion in recombinant BAC plasmid pΔm06 that was used to reconstitute virus mCMV-Δm06W. In addition, correct patterns of combinatorial deletions in the panel of reconstituted viruses were tested by Western blot analysis of immune evasion protein expression (Wagner et al., 2002) and the expected patterns of immune evasion gene deletions were later also confirmed by ISH with specific DNA probes in liver tissue sections of mice infected with the respective viruses (Holtappels et al., 2006). Seen in retrospect, it was a mistake to not have analyzed the reconstituted virion genomic DNA, and that Western blot analysis and ISH were performed only qualitatively. With the knowledge of today, the seemingly correct Western blot and ISH data obtained for mCMV-Δm06W reflected the minority fraction of correct Δm06 mutant present in the now identified virus mixture. Given the structural integrity of pΔm06 (Wagner et al., 2002), the large deletion must have occurred by a recombination event during virus reconstitution and subsequent rounds of propagation in cell culture performed to excise the BAC vector sequence from the BAC-cloned viral genome (Wagner et al., 1999). Suspiciously, it was just pΔm06 that was constructed by using a shuttle plasmid for recombination, whereas all other plasmids in the combinatorial deletion panel were constructed by PCR-based en-passant mutagenesis (Wagner et al., 2002). It remains speculative if the difference in the construction of pΔm06 relates in any way to the later recombination event. To our knowledge, plaque purification was performed for mCMV-Δm06W, though even a high clone probability still bears a risk of bi-clonality based on the Poisson distribution statistics that applies to limiting dilution approaches (Lefkovits and Waldmann, 1979). We have in the meantime cloned a new combinatorial panel of immune evasion gene deletion mutants of mCMV and found a phenotype discrepancy to the “Wagner panel” only for the mutants mCMV-Δm06W and mCMV-Δm06L. If the case reported here was a rare accident or if there is reason to doubt all mutants published in the early days of CMV mutagenesis, remains unanswered and speculative. Although the previously published conclusion was not falsified, and thus requires no corrigendum, this example teaches us the lesson to always verify the authenticity of viral mutants also beyond the site of the targeted mutation. Clearly, in modern times, NGS of full-length viral genomes is the method of choice. Even NGS, however, would have misled us if performed only at the stage of recombinant BAC plasmids. Thus, the most important lesson, in our view, is to always control genetic authenticity of the full-length virion DNA.
Identification of mCMV-Δm06W as a virus mixture, combined with the fact that we routinely store paraffin-imbedded organs of previously performed animal experiments in a tissue bank, gave us the chance to reanalyze the previous experiments performed by Holtappels et al. (2006) with focus on the correct mutant mCMV-Δm06 identified in liver tissue by 2C-ISH and 2C-IHC. These reanalyses confirmed the previous conclusion of m04 acting as a positive vRAP in antiviral protection, verified for two quite different pMHC-I complexes in two MHC (H-2) haplotypes, namely two M45-derived peptides presented by Dd and Db, respectively.
The question may come up if this function applies to all pMHC-I complexes made up by different antigenic peptides and different peptide-presenting MHC-I alleles. In the original work by Kleijnen et al. (1997) m04/gp34 was described to form “a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface.” The authors found an allelic difference in that complexes could be immunoprecipitated from the cell surface of mCMVSmith-infected cells with antibodies directed against Kb, Db, Ld, and Dd, with Kd representing an exception confirming the rule. Accordingly, a more recent structural analysis revealed binding of m04 to a broad range of MHC-I alleles (Berry et al., 2014). Berry and colleagues also considered the high variability of m04 in strains of mCMV (Corbett et al., 2007) and confirmed binding of m04Smith, m04G4, and m04W8211 to Ld, Dk, and Dd. A further layer of complexity is brought in by the recent finding of allelic differences in MATp1-dependent cell surface expression of m04-MHC-I complexes (Železnjak et al., 2019). Actually, there is nothing unusual about allelic differences, since they reflect the evolutionary selection pressure that has led to MHC polymorphism.
The decisive question remained, if MHC-I still presents antigenic peptide in the complex with m04, and, if so, if interactions of pMHC-I with m04 and MATp1 interfere with recognition by the TCR of CD8 T cells. As to the first question, Kleijnen et al. (1997) have already shown TAP-dependence of cell surface expression of m04-MHC-I complexes, so that the MHC-I molecules in the complexes are peptide-loaded. Notably, the effect of MATp1 on cell surface expression was particularly strong for m04-Dd complexes, so that our data for M45-Dd imply that neither m04 nor MATp1 interaction interfere with recognition by CD8 T cells. Finally, even if exceptions exist depending on MHC-I allele and, possibly, virus strain variance in the m04 sequence, m04 positively regulates antiviral protection because virus control by CD8 T cells is secured redundantly by more than one antigenic peptide presented by any of the MHC-I molecules of a given haplotype [reviewed in Ebert et al. (2012)].
Our data also shed new light on the function of the negative vRAP m06. As the adoptive transfer experiments show, the positive effect of m04 on protection in the presence of just m152 is reduced by m06 co-expressed with m04 and m152 in mCMV-WT.BAC. Thus, while m06 when expressed alone in deletion mutant mCMV-Δm04m152 has only a moderate inhibitory effect on antigen presentation (reviewed in Lemmermann et al., 2012), its main function might be to compete with m04 for MHC-I cargo and thereby reduce the positive effect of m04. Notably, it appears that this competition does not completely abolish the positive effect of m04, as it is indicated by a somewhat better protection against mCMV-WT.BAC compared to mCMV-Δm04m06 expressing only the main negative vRAP m152 (Figures 3, 4). The balanced competitive effects of m04 and m06 also explain the previously poorly understood finding that deletion of just m152 is highly efficient in relieving virus evasion of antiviral CD8 T cells (Holtappels et al., 2004, 2009).
In conclusion, our data give new insights into the interplay between the three vRAPs in virus control by CD8 T cells.
Data Availability Statement
The datasets generated for this article can be found in European Nucleotide Archive (ENA) using the project number PRJEB38039 (http://www.ebi.ac.uk/ena/data/view/PRJEB38039).
Ethics Statement
The animal study was reviewed and approved by the ethics committee of the Landesuntersuchungsamt Rheinland-Pfalz, permission number 177-07/021-28.
Author Contributions
MR and NL were responsible for conception and design of the study, analysis, and interpretation of the data. SB, AF, JP, JS, IG, and TB conducted the work and analyzed the data. MR wrote the first draft of the manuscript. SB, TB, and NL wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft, CRC1292, individual projects TP11 (SB, JS, NL, and MR) and TP Z01 (IG and TB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Valesca Bukur (CRC1292, project Z01) and PD Dr. Claudine Graf (CRC1292, individual project TP10) for helpful scientific discussion and support.
References
Arase, H., Mocarski, E. S., Campbell, A. E., Hill, A. B., and Lanier, L. L. (2002). Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326. doi: 10.1126/science.1070884
Babic, M., Pyzik, M., Zafirova, B., Mitrovic, M., Butorac, V., Lanier, L. L., et al. (2010). Cytomegalovirus immunoevasin reveals the physiological role of “missing self” recognition in natural killer cell dependent virus control in vivo. J. Exp. Med. 207, 2663–2673. doi: 10.1084/jem.20100921
Berry, R., Vivian, J. P., Deuss, F. A., Balaji, G. R., Saunders, P. M., Lin, J., et al. (2014). The structure of the cytomegalovirus-encoded m04 glycoprotein, a prototypical member of the m02 family of immunoevasins. J. Biol. Chem. 289, 23753–23763. doi: 10.1074/jbc.M114.584128
Berry, R., Watson, G. M., Jonjic, S., Degli-Esposti, M. A., and Rossjohn, J. (2019). Modulation of innate and adaptive immunity by cytomegaloviruses. Nat Rev. Immunol. 20, 113–127. doi: 10.1038/s41577-019-0225-5
Böhm, V., Seckert, C. K., Simon, C. O., Thomas, D., Renzaho, A., Gendig, D., et al. (2009). Immune evasion proteins enhance cytomegalovirus latency in the lungs. J. Virol. 83, 10293–10298. doi: 10.1128/JVI.01143-09
Boppana, S. B., and Britt, W. J. (2013). “Synopsis of clinical aspects of human cytomegalovirus disease,” in Cytomegaloviruses: From Molecular Pathogenesis to Intervention, Vol. 2, ed M. J. Reddehase (Norfolk: Caister Academic Press), 1–25.
Brune, W., Ménard, C., Heesemann, J., and Koszinowski, U. H. (2001). A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291, 303–305. doi: 10.1126/science.291.5502.303
Bubić, I., Wagner, M., Krmpotić, A., Saulig, T., Kim, S., Yokoyama, W. M., et al. (2004). Gain of virulence caused by loss of a gene in murine cytomegalovirus. J. Virol. 78, 7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cicin-Sain, L., Podlech, J., Messerle, M., Reddehase, M. J., and Koszinowski, U. H. (2005). Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J. Virol. 79, 9492–9502. doi: 10.1128/JVI.79.15.9492-9502.2005
Corbett, A. J., Forbes, C. A., Moro, D., and Scalzo, A. A. (2007). Extensive sequence variation exists among isolates of murine cytomegalovirus within members of the m02 family of genes. J. Gen. Virol. 88, 758–769. doi: 10.1099/vir.0.82623-0
Daley-Bauer, L. P., Roback, L., Crosby, L. N., McCormick, A. L., Feng, Y., Kaiser, W. J., et al. (2017). Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc. Natl. Acad. Sci. U.S.A. 114, E2786–E2795. doi: 10.1073/pnas.1616829114
Davison, A. J., Holton, M., Dolan, A., Dargan, D. J., Gatherer, D., and Hayward, G. S. (2013). “Comparative genomics of primate cytomegaloviruses,” in Cytomegaloviruses: From Molecular Pathogenesis to Intervention, Vol. 1, ed M. J. Reddehase (Norfolk: Caister Academic Press), 1–22.
Doom, C. M., and Hill, A. B. (2008). MHC class I immune evasion in MCMV infection. Med. Microbiol. Immunol. 197, 191–204. doi: 10.1007/s00430-008-0089-y
Ebert, S., Podlech, J., Gillert-Marien, D., Gergely, K. M., Büttner, J. K., Fink, A., et al. (2012). Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med. Microbiol. Immunol. 201, 527–539. doi: 10.1007/s00430-012-0258-x
Elder, E., and Sinclair, J. (2019). HCMV latency: what regulates the regulators? Med. Microbiol. Immunol. 208, 431–438. doi: 10.1007/s00430-019-00581-1
Emery, V. C., Milne, R. S., and Griffiths, P. D. (2013). “Clinical cytomegalovirus research: liver and kidney transplantation,” in Cytomegaloviruses: From Molecular Pathogenesis to Intervention, Vol. 2, ed M. J. Reddehase (Norfolk: Caister Academic Press), 301–311.
Fink, A., Blaum, F., Babic Cac, M., Ebert, S., Lemmermann, N. A., and Reddehase, M. J. (2015). An endocytic YXXΦ (YRRF) cargo sorting motif in the cytoplasmic tail of murine cytomegalovirus AP2 'adapter adapter' protein m04/gp34 antagonizes virus evasion of natural killer cells. Med. Microbiol. Immunol. 204, 383–394. doi: 10.1007/s00430-015-0414-1
Fink, A., Lemmermann, N. A., Gillert-Marien, D., Thomas, D., Freitag, K., Böhm, V., et al. (2012). Antigen presentation under the influence of 'immune evasion' proteins and its modulation by interferon-gamma: implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 201, 513–525. doi: 10.1007/s00430-012-0256-z
Fink, A., Mikuličić, S., Blaum, F., Reddehase, M. J., Florin, L., and Lemmermann, N. A. (2019). Function of the cargo sorting dileucine motif in a cytomegalovirus immune evasion protein. Med. Microbiol. Immunol. 208, 531–542. doi: 10.1007/s00430-019-00604-x
Fink, A., Renzaho, A., Reddehase, M. J., and Lemmermann, N. A. (2013). The p36 isoform of murine cytomegalovirus m152 protein suffices for mediating innate and adaptive immune evasion. Viruses 5, 3171–3191. doi: 10.3390/v5123171
Fodil-Cornu, N., Lee, S. H., Belanger, S., Makrigiannis, A. P., Biron, C. A., Buller, R. M., et al. (2008). Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J. Immunol. 181, 6394–6405. doi: 10.4049/jimmunol.181.9.6394
Gezinir, E., Podlech, J., Gergely, K., Becker, S., Reddehase, M. J., and Lemmermann, N. A. (2020). Enhanced viral antigen presentation by deletion of viral immune evasion genes prevents lethal cytomegalovirus disease in minor histocompatibility antigen-mismatched hematopoietic cell transplantation. Front. Cell. Infect. Microbiol. 10:337. doi: 10.3389/fcimb.2020.00279
Gold, M. C., Munks, M. W., Wagner, M., Koszinowski, U. H., Hill, A. B., and Fling, S. P. (2002). The murine cytomegalovirus immunomodulatory gene m152 prevents recognition of infected cells by M45-specific CTL but does not alter the immunodominance of the M45-specific CD8 T cell response in vivo. J. Immunol. 169, 359–365. doi: 10.4049/jimmunol.169.1.359
Grzimek, N. K., Podlech, J., Steffens, H. P., Holtappels, R., Schmalz, S., and Reddehase, M. J. (1999). In vivo replication of recombinant murine cytomegalovirus driven by the paralogous major immediate-early promoter-enhancer of human cytomegalovirus. J. Virol. 73, 5043–5055. doi: 10.1128/JVI.73.6.5043-5055.1999
Hansen, T. H., and Bouvier, M. (2009). MHC class I antigen presentation: learning from viral evasion strategies. Nat. Rev. Immunol. 9, 503–513. doi: 10.1038/nri2575
Hasan, M., Krmpotic, A., Ruzsics, Z., Bubic, I., Lenac, T., Halenius, A., et al. (2005). Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 79, 2920–2930. doi: 10.1128/JVI.79.5.2920-2930.2005
Hengel, H., Brune, W., and Koszinowski, U. H. (1998). Immune evasion by cytomegalovirus–survival strategies of a highly adapted opportunist. Trends Microbiol. 6, 190–197. doi: 10.1016/S0966-842X(98)01255-4
Ho, M. (2008). The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 197, 65–73. doi: 10.1007/s00430-007-0066-x
Holtappels, R., Gillert-Marien, D., Thomas, D., Podlech, J., Deegen, P., Herter, S., et al. (2006). Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 80, 7613–7624. doi: 10.1128/JVI.00723-06
Holtappels, R., Podlech, J., Pahl-Seibert, M. F., Jülch, M., Thomas, D., Simon, C. O., et al. (2004). Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J. Exp. Med. 199, 131–136. doi: 10.1084/jem.20031582
Holtappels, R., Schader, S. I., Oettel, O., Podlech, J., Seckert, C. K., Reddehase, M. J., et al. (2020). Insufficient antigen presentation due to viral immune evasion explains lethal cytomegalovirus organ disease after allogeneic hematopoietic cell transplantation. Front. Cell. Infect. Microbiol. 10:157. doi: 10.3389/fcimb.2020.00157
Holtappels, R., Thomas, D., and Reddehase, M. J. (2009). The efficacy of antigen processing is critical for protection against cytomegalovirus disease in the presence of viral immune evasion proteins. J. Virol. 83, 9611–9615. doi: 10.1128/JVI.00936-09
Janßen, L., Ramnarayan, V. R., Aboelmagd, M., Iliopoulou, M., Hein, Z., Majoul, I., et al. (2016). The murine cytomegalovirus immunoevasin gp40 binds MHC class I molecules to retain them in the early secretory pathway. J. Cell. Sci. 129, 219–227. doi: 10.1242/jcs.175620
Kavanagh, D. G., Koszinowski, U. H., and Hill, A. B. (2001). The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-golgi compartment. J. Immunol. 167, 3894–3902. doi: 10.4049/jimmunol.167.7.3894
Kleijnen, M. F., Huppa, J. B., Lucin, P., Mukherjee, S., Farrell, H., Campbell, A. E., et al. (1997). A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16, 685–694. doi: 10.1093/emboj/16.4.685
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Krmpotic, A., Bubic, I., Polic, B., Lucin, P., and Jonjic, S. (2003). Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 5, 1263–1277. doi: 10.1016/j.micinf.2003.09.007
Krmpotic, A., Busch, D. H., Bubic, I., Gebhardt, F., Hengel, H., Hasan, M., et al. (2002). MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 3, 529–535. doi: 10.1038/ni799
Krmpotic, A., Hasan, M., Loewendorf, A., Saulig, T., Halenius, A., Lenac, T., et al. (2005). NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 201, 211–220. doi: 10.1084/jem.20041617
Krmpotic, A., Messerle, M., Crnkovic-Mertens, I., Polic, B., Jonjic, S., and Koszinowski, U. H. (1999). The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 190, 1285–1296. doi: 10.1084/jem.190.9.1285
Lefkovits, I., and Waldmann, H. (1979). “Clone size estimation,” in Limiting Dilution Analysis of Cells in Immune Systems (Cambridge: Cambridge University Press), 83–92.
Lemmermann, N. A., Fink, A., Podlech, J., Ebert, S., Wilhelmi, V., Böhm, V., et al. (2012). Murine cytomegalovirus immune evasion proteins operative in the MHC class I pathway of antigen processing and presentation: state of knowledge, revisions, and questions. Med. Microbiol. Immunol. 201, 497–512. doi: 10.1007/s00430-012-0257-y
Lemmermann, N. A., Krmpotic, A., Podlech, J., Brizic, I., Prager, A., Adler, H., et al. (2015). Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog. 11:e1004640. doi: 10.1371/journal.ppat.1004640
Lemmermann, N. A., Podlech, J., Seckert, C. K., Kropp, K. A., Grzimek, N. K., Reddehase, M. J., et al. (2010). “CD8 T cell immunotherapy of cytomegalovirus disease in the murine model,” in Methods in Microbiology. Immunology of Infection, eds D. Kabelitz and S. H. E. Kaufmann (London: Academic Press), 369–420. doi: 10.1016/S0580-9517(10)37016-4
Loewendorf, A., Krüger, C., Borst, E. M., Wagner, M., Just, U., and Messerle, M. (2004). Identification of a mouse cytomegalovirus gene selectively targeting CD86 expression on antigen-presenting cells. J. Virol. 78, 13062–13071. doi: 10.1128/JVI.78.23.13062-13071.2004
LoPiccolo, D. M., Gold, M. C., Kavanagh, D. G., Wagner, M., Koszinowski, U. H., and Hill, A. B. (2003). Effective inhibition of K(b)- and D(b)-restricted antigen presentation in primary macrophages by murine cytomegalovirus. J. Virol. 77, 301–308. doi: 10.1128/JVI.77.1.301-308.2003
Lu, X., Kavanagh, D. G., and Hill, A. B. (2006). Cellular and molecular requirements for association of the murine cytomegalovirus protein m4/gp34 with major histocompatibility complex class I molecules. J. Virol. 80, 6048–6055. doi: 10.1128/JVI.00534-06
Pinto, A. K., Jamieson, A. M., Raulet, D. H., and Hill, A. B. (2007). The role of NKG2D signaling in inhibition of cytotoxic T-lymphocyte lysis by the murine cytomegalovirus immunoevasin m152/gp40. J. Virol. 81, 12564–12571. doi: 10.1128/JVI.01328-07
Pinto, A. K., Munks, M. W., Koszinowski, U. H., and Hill, A. B. (2006). Coordinated function of murine cytomegalovirus genes completely inhibits CTL lysis. J. Immunol. 177, 3225–3234. doi: 10.4049/jimmunol.177.5.3225
Podlech, J., Holtappels, R., Grzimek, N. K., and Reddehase, M. J. (2002). “Animal models: murine cytomegalovirus,” in Methods in Microbiology. Immunology of Infection, eds S. H. E. Kaufmann and D. Kabelitz (London: Academic Press), 493–525. doi: 10.1016/S0580-9517(02)32103-2
Powers, C., DeFilippis, V., Malouli, D., and Fruh, K. (2008). Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 325, 333–359. doi: 10.1007/978-3-540-77349-8_19
Rawlinson, W. D., Farrell, H. E., and Barrell, B. G. (1996). Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 70, 8833–8849. doi: 10.1128/JVI.70.12.8833-8849.1996
Reddehase, M. J. (2002). Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2, 831–844. doi: 10.1038/nri932
Reddehase, M. J. (2016). Mutual interference between cytomegalovirus and reconstitution of protective immunity after hematopoietic cell transplantation. Front. Immunol. 7:294. doi: 10.3389/fimmu.2016.00294
Reddehase, M. J., and Lemmermann, N. A. (2018). Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: predictions for medical translation that survived the “test of time.” Viruses 10:693. doi: 10.3390/v10120693
Reddehase, M. J., and Lemmermann, N. A. (2019). Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. 208, 391–403. doi: 10.1007/s00430-019-00592-y
Renzaho, A., Podlech, J., Kühnapfel, B., Blaum, F., Reddehase, M. J., and Lemmermann, N. A. (2020). Cytomegalovirus-associated inhibition of hematopoiesis is preventable by cytoimmunotherapy with antiviral CD8 T cells. Front. Cell. Infect. Microbiol. 10:138. doi: 10.3389/fcimb.2020.00138
Reusch, U., Bernhard, O., Koszinowski, U., and Schu, P. (2002). AP-1A and AP-3A lysosomal sorting functions. Traffic 3, 752–761. doi: 10.1034/j.1600-0854.2002.31007.x
Reusch, U., Muranyi, W., Lucin, P., Burgert, H. G., Hengel, H., and Koszinowski, U. H. (1999). A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18, 1081–1091. doi: 10.1093/emboj/18.4.1081
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Sacher, T., Podlech, J., Mohr, C. A., Jordan, S., Ruzsics, Z., Reddehase, M. J., et al. (2008). The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3, 263–272. doi: 10.1016/j.chom.2008.02.014
Scalzo, A. A., Wheat, R., Dubbelde, C., Stone, L., Clark, P., Du, Y., et al. (2003). Molecular genetic characterization of the distal NKC recombination hotspot and putative murine CMV resistance control locus. Immunogenetics 55, 370–378. doi: 10.1007/s00251-003-0591-8
Seo, S., and Boeckh, M. (2013). “Clinical cytomegalovirus research: hematopoietic cell transplantation,” in Cytomegaloviruses: From Molecular Pathogenesis to Intervention, Vol. 2, ed M. J. Reddehase (Norfolk: Caister Academic Press), 337–353.
Sgourakis, N. G., May, N. A., Boyd, L. F., Ying, J., Bax, A., and Margulies, D. H. (2015). A novel MHC-I surface targeted for binding by the MCMV m06 immunoevasin revealed by solution NMR. J. Biol. Chem. 290, 28857–28868. doi: 10.1074/jbc.M115.689661
Sgourakis, N. G., Natarajan, K., Ying, J., Vogeli, B., Boyd, L. F., Margulies, D. H., et al. (2014). The structure of mouse cytomegalovirus m04 protein obtained from sparse NMR data reveals a conserved fold of the m02-m06 viral immune modulator family. Structure 22, 1263–1273. doi: 10.1016/j.str.2014.05.018
Stempel, M., Chan, B., Juranić Lisnić V., Krmpotić A., Hartung, J., Paludan, S. R., et al. (2019). The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-κB signaling and STING's dual role during MCMV infection. EMBO J. 38:e100983. doi: 10.15252/embj.2018100983
Strazic Geljic, I., Kucan Brlic, P., Angulo, G., Brizic, I., Lisnic, B., Jenus, T., et al. (2020). Cytomegalovirus protein m154 perturbs the adaptor protein-1 compartment mediating broad-spectrum immune evasion. eLife 9:e50803. doi: 10.7554/eLife.50803
Voigt, V., Forbes, C. A., Tonkin, J. N., Degli-Esposti, M. A., Smith, H. R., Yokoyama, W. M., et al. (2003). Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 13483–13488. doi: 10.1073/pnas.2233572100
Wagner, M., Gutermann, A., Podlech, J., Reddehase, M. J., and Koszinowski, U. H. (2002). Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 196, 805–816. doi: 10.1084/jem.20020811
Wagner, M., Jonjic, S., Koszinowski, U. H., and Messerle, M. (1999). Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73, 7056–7060. doi: 10.1128/JVI.73.8.7056-7060.1999
Wang, R., Natarajan, K., Revilleza, M. J., Boyd, L. F., Zhi, L., Zhao, H., et al. (2012). Structural basis of mouse cytomegalovirus m152/gp40 interaction with RAE1γ reveals a paradigm for MHC/MHC interaction in immune evasion. Proc. Natl. Acad. Sci. U.S.A. 109, 3578–3587. doi: 10.1073/pnas.1214088109
Wiertz, E., Hill, A., Tortorella, D., and Ploegh, H. (1997). Cytomegaloviruses use multiple mechanisms to elude the host immune response. Immunol. Lett. 57, 213–216. doi: 10.1016/S0165-2478(97)00073-4
Zarama, A., Pérez-Carmona, N., Farré, D., Tomic, A., Borst, E. M., Messerle, M., et al. (2014). Cytomegalovirus m154 hinders CD48 cell-surface expression and promotes viral escape from host natural killer cell control. PLoS Pathog. 10:e1004000. doi: 10.1371/journal.ppat.1004000
Železnjak, J., Lisnic, V. J., Popovic, B., Lisnic, B., Babic, M., Halenius, A., et al. (2019). The complex of MCMV proteins and MHC class I evades NK cell control and drives the evolution of virus-specific activating Ly49 receptors. J. Exp. Med. 216, 1809–1827. doi: 10.1084/jem.20182213
Ziegler, H., Muranyi, W., Burgert, H. G., Kremmer, E., and Koszinowski, U. H. (2000). The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 19, 870–881. doi: 10.1093/emboj/19.5.870
Keywords: adoptive cell transfer, antigen presentation, BAC mutagenesis, CD8 T cells, immune evasion, immunoevasin, next-generation sequencing (NGS), recombinant virus
Citation: Becker S, Fink A, Podlech J, Giese I, Schmiedeke JK, Bukur T, Reddehase MJ and Lemmermann NA (2020) Positive Role of the MHC Class-I Antigen Presentation Regulator m04/gp34 of Murine Cytomegalovirus in Antiviral Protection by CD8 T Cells. Front. Cell. Infect. Microbiol. 10:454. doi: 10.3389/fcimb.2020.00454
Received: 24 April 2020; Accepted: 23 July 2020;
Published: 26 August 2020.
Edited by:
Michael Nevels, University of St Andrews, United KingdomReviewed by:
David H. Margulies, National Institute of Allergy and Infectious Diseases (NIH), United StatesAnne Halenius, University of Freiburg, Germany
Copyright © 2020 Becker, Fink, Podlech, Giese, Schmiedeke, Bukur, Reddehase and Lemmermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niels A. Lemmermann, bGVtbWVybWFubkB1bmktbWFpbnouZGU=
†Present address: Annette Fink, Transplantation Immunology, Institute of Immunology, Heidelberg University Hospital, Heidelberg, Germany