 Wenhao Su
Wenhao Su Yongyu Chen1,2
Yongyu Chen1,2 Yuanmei Guo
Yuanmei Guo Weiguo Dong
Weiguo Dong- 1Department of Gastroenterology, Renmin Hospital of Wuhan University, Wuhan, China
- 2Key Laboratory of Hubei Province for Digestive System Disease, Renmin Hospital of Wuhan University, Wuhan, China
There is a growing body of evidence which suggests that intestinal microbiota, especially Fusobacterium nucleatum (F. nucleatum), are associated with intestinal immune disease such as ulcerative colitis (UC). The mechanism by which F. nucleatum promotes intestinal epithelial cell (IEC) death remained undefined. Here, we investigated the potential mechanisms about how F. nucleatum aggravates IEC death in UC. We first detected the abundance of F. nucleatum in UC tissues and analyzed its relationship with the clinical characteristics of UC. Next, we explored whether F. nucleatum promotes intestinal epithelial cell death in vitro and in vivo. Furthermore, we extracted lipopolysaccharide (LPS) of the F. nucleatum and examined whether F. nucleatum exacerbates UC via LPS. Our results indicated that F. nucleatum was abundant in UC tissues and was correlated with clinical characteristics. In addition, we demonstrated that F. nucleatum and its LPS aggravated IEC death by promoted IEC autophagy. Furthermore, autophagy inhibitors, chloroquine (CQ), 3-methyladenine (3-MA) or Atg5 silencing prevented IEC death mediated by F. nucleatum, which suggests F. nucleatum may contribute to UC by activating autophagic cell death. All our results uncover a vital role of F. nucleatum in autophagic cell death and UC, giving rise to a new sight for UC therapy by inhibiting excessive IEC autophagy and autophagic cell death.
Introduction
Ulcerative colitis (UC) is a chronic, idiopathic, relapsing immunologic disorder of the gastrointestinal tract (Park et al., 2017). UC is considered a worldwide healthcare issue, with its steady increase in prevalence (Jeong et al., 2019). In recent years, the incidence and prevalence of UC have a dramatic rise in Asia, which makes it get more attention (Hou et al., 2009). The current treatment strategy of UC is mainly to use anti-inflammatory drugs, immunosuppressive agents, steroids and biological agents to control disease activity. Thus, it is essential to deepen the understanding of the pathogenesis in UC for further treatment.
Intestinal microbes have been taken into account as an important factor in the pathogenesis of UC (Bashir et al., 2016). Fusobacterium nucleatum (F. nucleatum) is a common colonizer in the oral cavity and intestinal mucosa of human hosts. Studies have found that F. nucleatum is closely related to intestinal diseases such as inflammatory bowel disease (IBD) and colorectal cancer (CRC) by promoting intestinal inflammation and the release of inflammatory factors (Kumar et al., 2017; Weng et al., 2019). It has been shown that F. nucleatum can aggravate UC by regulating M1 microphage polarization (Liu et al., 2019). However, the mechanism of F. nucleatum in the pathogenesis of UC has not been fully elucidated.
Autophagy plays a crucial role in regulating the interaction between gut microbiota and host immunity (Kim et al., 2019). Genetic variations of autophagy genes has been identified to be the pathogenic factors of IBD (Nguyen et al., 2013). Studies has showed a mutual regulation between autophagy and inflammation. Previous studies have showed that loss of functional autophagy promote inflammasome activation and secretion of inflammatory factors such as IL-18 and IL-β, which leads to a higher susceptibility to inflammation (Larabi et al., 2020). However, some studies found that autophagy activation is accompanied with excessive intestinal inflammatory response and epithelial injury in colitis mice (Shen et al., 2018) and high levels of autophagy is a potential signal for cell death (Denton and Kumar, 2019). F. nucleatum has been found to activate IEC autophagy pathway, promote cell apoptosis and proinflammatory cytokine production in oral and gut (Dharmani et al., 2011; Yu et al., 2017; Kang et al., 2019). However, the exact role of autophagy in F. nucleatum-associated UC is still unclear.
Here we studied whether and how F. nucleatum affects IEC function in UC patients. Our results demonstrate that F. nucleatum is abundant in UC patients compared with healthy controls. Moreover, enrichment of F. nucleatum activates IEC autophagy and exacerbates IEC autophagic death in vivo and in vitro. Our data provide a potential strategy for UC treatment by inhibiting excessive autophagy flux and epithelial cell death.
Materials and Methods
Collection of Clinical Samples
All study samples were obtained from patients between 2018 and 2019 who were treated at Wuhan University People’s Hospital (Hubei, China). This study was approved by the Medical Ethics Committee of Wuhan University People’s Hospital (approval number: 2018K-C089), and written informed consent was obtained from each participant. Endoscopic biopsies were collected from inflamed intestinal mucosal from UC patients. The diagnosis of UC was based on clinical, endoscopic and histological criteria. The clinical data of each patient were collected from the hospital records. Formalin-fixed and paraffin-embedded samples were retrieved from Pathology department archives. The control group was composed of age-matched healthy patients who underwent an endoscopy which resulted in a normal endoscopy and biopsies. Clinical disease activity was assessed using the Colitis Activity Index. Fresh UC tissue specimens were collected from participants of ≥19 years of age with biopsy proven. Patients were screened and those with Crohn’s disease, infectious diarrhea, clinical conditions requiring emergency management, primary sclerosing cholangitis, pregnancy, prednisolone use within the last 2 weeks, biologicals or immunosuppressants within the past 2 years, history of fecal microbiota transplantation (FMT), antibiotic use within previous 3 months and recent malignancy were excluded. Formalin-fixed, paraffin-embedded UC intestinal tissues for FISH analysis were obtained from the pathology department archives between February 2018 and December 2019. Clinicopathologic data for each patient were obtained from the hospital records.
Bacterial Strains and Cell Lines
The human normal epithelial cell line NCM460 was maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum at 37°C, 5% CO2. F. nucleatum was incubated for 3 days in fastidious anaerobe broth (FAB) under anaerobic conditions (10% H2, 5% CO2, and 85% N2) at 37°C. Escherichia coli strain (Tiangen, China) was cultured in Luria–Bertani (LB) medium for 16 h at 37°C with shaking at 200–220 rpm. The F. nucleatum suspension was centrifuged at 2,500×g for 5 min and then resuspended in antibiotic-free DMEM before infecting normal epithelial cells.
DSS-Induced Mouse Model of Colitis
All mice experiments were performed with male mice of 6 to 8 weeks age unless otherwise stated. C57BL/6 mice were given 2 mg/ml streptomycin in drinking water for 3 days to ensure the consistency of common microflora and facilitate colonization of F. nucleatum. F. nucleatum resuspended in PBS (109 CFU/ml) or PBS alone was administered to mice by gavage every day for 2 weeks. After 2 weeks of infection, mice were administered either normal water or water containing 2.5% (wt/vol) dextran sulfate sodium (DSS) (molecular weight 36 to 50 kDa; MP Biomedicals) for 7 days to induce colitis. To examine the effect of CQ on colitis, mice received F. nucleatum gavage or PBS meanwhile injected intraperitoneally with CQ (50 mg/kg of body weight) dissolved in PBS every two days.
Measurement of Colitis Severity
Mice were observed once daily for weight, body weight, morbidity, stool consistency, and the presence of gross blood in the feces and at the anus. On day 7 after 2.5% DSS treatment, mice were sacrificed, and the colon was quickly isolated for measurement of length. The distal colon was collected and fixed in 10% buffered formalin for 24 h for histological staining (H&E) and fluorescence in situ hybridization (FISH). The histological scores were calculated by three pathology staffs. The histological scores were determined blindly as follows: Damaged area: 0: n/a, 1: ≤25%, 2: ≤50%, 3: ≤75%, 4: ≤100%. Muco-depletion of glands: 0 = none, 1 = mild, 2 and 3 = moderate, 4 = severe. Tissue damage: 0= no mucosal damage, 1 = discrete epithelial lesions, 2 = surface mucosal erosion or focal ulceration, 3 = extensive mucosal damage and extension into deeper structures of the bowel wall. Inflammatory cell infiltration: 0 = occasional inflammatory cells in the lamina propria, 1 =increased numbers of inflammatory cells in the lamina propria, 2 = confluence inflammatory cells, extending into the submucosa, 3 = transmural extension of the infiltrate. The total histopathological score was determined by summation of the scores from each category.
RNA Extraction and Real-Time PCR
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA), and 1 μg of total RNA was reverse transcribed using the PrimeScript RT Reagent Kit (Perfect Real Time; Takara, Shiga, Japan). Real-time quantitative PCR was performed in triplicates on an Applied Biosystem7900 quantitative PCR system (Applied Biosystems, Foster City, CA, USA). The Ct values obtained from different samples were compared using the 2−ΔΔCt method. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the internal reference transcripts. The primers are shown in Supplementary Table 1.
Fluorescence In Situ Hybridization
Five-micrometer-thick sections were prepared and hybridized following the manufacturer’s instructions (FOCOFISH, Guangzhou, China). The sequence of the F. nucleatum-targeted probe (FUS664; FITC-labeled) was 5’-CTT GTA GTT CCG C(C/T) TAC CTC-3’. Slides were examined using a microscope (BX53F; Olympus, Tokyo, Japan). Five random 200× magnification fields per sample were evaluated by an observer who was blinded to the experimental protocol, and the average number of bacteria per field was calculated. We defined a negative or positive of F. nucleatum as an average of < 10 or > 10 visualized FUS664 probes per field, respectively.
Electron Microscopy
NCM460 cells were treated with PBS, E.coli (DH5α) or F. nucleatum and fixed with 2.5% glutaraldehyde. Samples were post-fixed with 1% osmium tetroxide, dehydrated in ethanol and propylene oxide, and embedded in epoxy resin. Subsequently, embedded samples were cut into ultra-thin sections (50–70 nm) stained with 3% uranyl acetate and lead citrate. Finally, the sections were observed by JEM-1230 electron microscope (JEOL, Tokyo, Japan).
Knockdown of Atg5 With shRNA
siRNA targeting the human Atg5 gene (siAtg5) and nontargeting siRNAs (control siRNAs) were purchased from Genechem (Shanghai, China). Cells were cultured and transfected with siRNAs according to the supplier’s instructions. Lipid-based transfections were achieved with Lipofectamine 6000 (Beyotime, China) according to the manufacturer’s protocol. Cells were incubated with the siRNA complex for 72 h, and protein was extracted for assessing transfection efficiency by WB.
Western Blotting
Total cellular protein was isolated from cultured cells and mice colons with a protein extraction solution (Beyotime, china). Proteins were subjected to 10 and 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes at 0.25 A (1 h, 4°C) using a wet-blotting apparatus (Bio-Rad). Membranes were blocked with 5% non-fat milk for 2 h at room temperature, and then incubated overnight at 4°C with the primary antibodies diluted in PBST. Membranes were then incubated with appropriate secondary antibodies for 1 h at room temperature. The protein signals were detected by using the ChemiDocTMXRS+ system (Bio-Rad). For WB, primary antibodies against the following targets were used:LC3 (CST), P62 (CST), Atg5 (CST), Bcl-2 (Proteintech), Bax (Proteintech), and GAPDH (Bioworld).
Statistical Analyses
Statistical analysis was performed using GraphPad Prism software version 8.0 and SPSS Statistics 20.0 software. Data are expressed as the means ± SDs. Associations between F. nucleatum abundance and patient’s clinical characteristics were determined using Pearson’s Chi-square test or Fisher’s exact test. Normally distributed data were analyzed by Student’s t test. Differences among multiple groups were evaluated for significance using one-way ANOVA combined with Bonferroni’s post hoc test. Statistical significance was defined as P <0.05.
Results
F. nucleatum Is Abundant in UC Tissues and Linked to Clinical Characteristics
To examine the potential relationship between F. nucleatum and UC, we examined F. nucleatum abundance in paraffin sections of 44 UC tissues from patients and 9 normal tissues using FISH. F. nucleatum was detected in a higher percentage of UC tissues (24/44, 54.5%) than that in normal tissues (1/9, 11.1%; P <0.01; Figure 1). We then analyzed the relationship between F. nucleatum abundance and different clinical characteristics as shown in Table 1. The amount of F. nucleatum was positively correlated with the clinical course and clinical activity (P <0.05). Thus, these data suggest that the abundance F. nucleatum is possibly associated with clinical characteristics of UC.
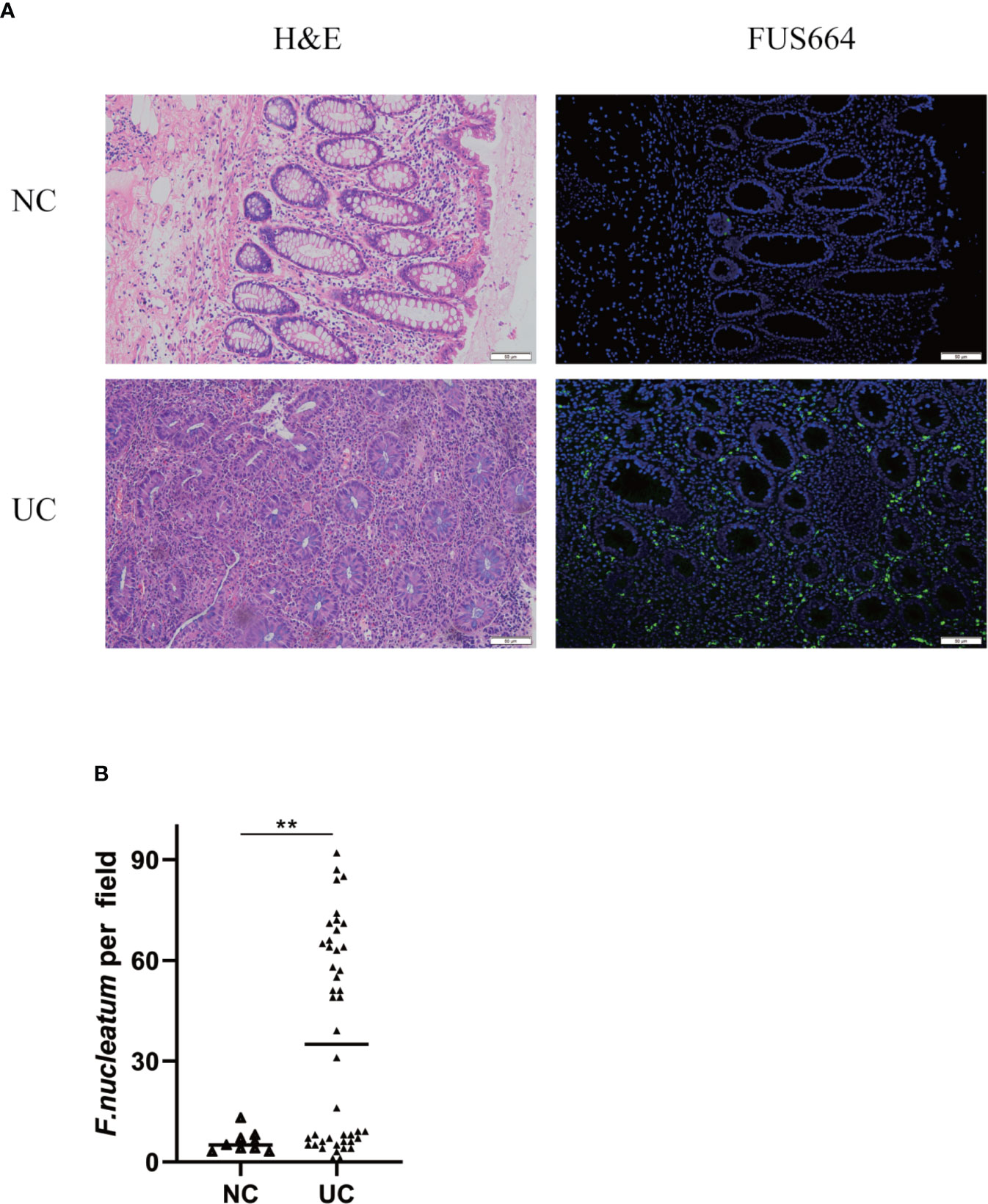
Figure 1 F. nucleatum is associated with UC. (A) Representative FISH images to assess the amount of F. nucleatum in UC (n = 44) and healthy control tissues (n = 9). FUS664 (green) is a FITC-conjugated F. nucleatum-specific oligonucleotide probe. Magnification, 200×. (B) The number of F. nucleatum was quantified in panel (**P < 0.01).
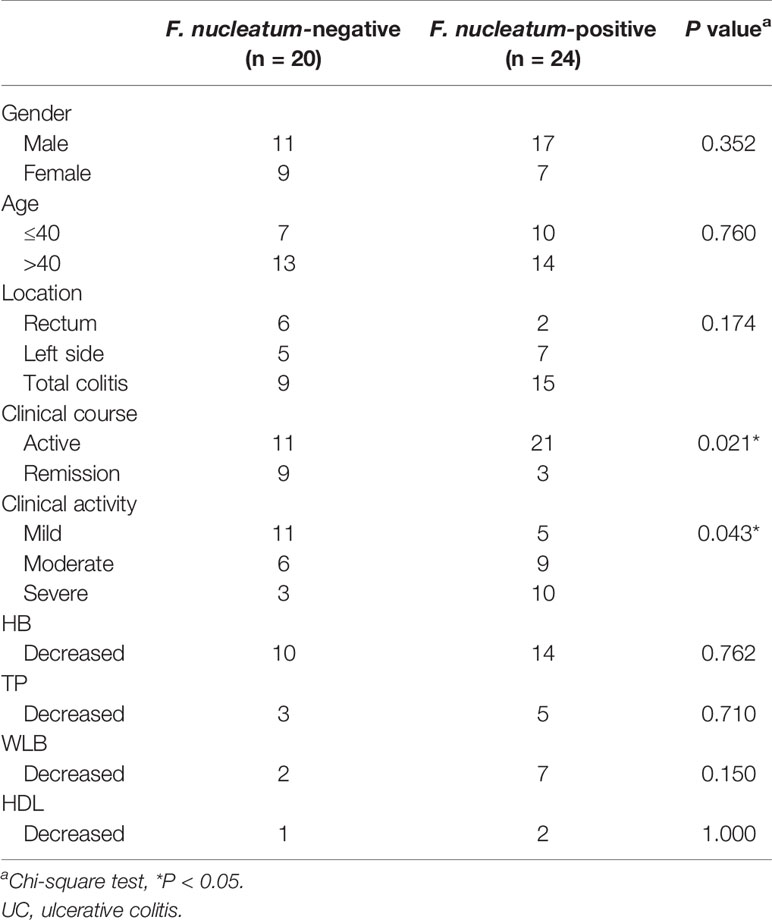
Table 1 Clinical features of F. nucleatum-negative vs. F. nucleatum-positive UC.
F. nucleatum Promotes IEC Autophagy In Vitro
We hypothesized that F. nucleatum infection may aggravate intestinal inflammation by activating autophagy in UC. To test this hypothesis, we co-cultured NCM460 cells with F. nucleatum, E. coli (DH5α) or PBS (Control). Real-time PCR showed that F. nucleatum elevated autophagy-related gene ATG5 and ATG12 mRNA levels (Figures 2A, B). In line with this, Western blot results showed that F. nucleatum increased the expression of Atg5 and LC3II, and decreased the expression of P62 in a time-dependent manner (Figure 2C). These effects were not found in the NCM460 cells co-cultured with E. coli. These data suggest that F. nucleatum may drive autophagy activation in intestinal cells. Furthermore, Transmission electron microscopy confirmed the increase in the formation of autophagic vesicles in the F. nucleatum co-cultured NCM460 cells (Figures 2D, E). Collectively, our data indicate that F. nucleatum activates the autophagy pathway in IEC cells.
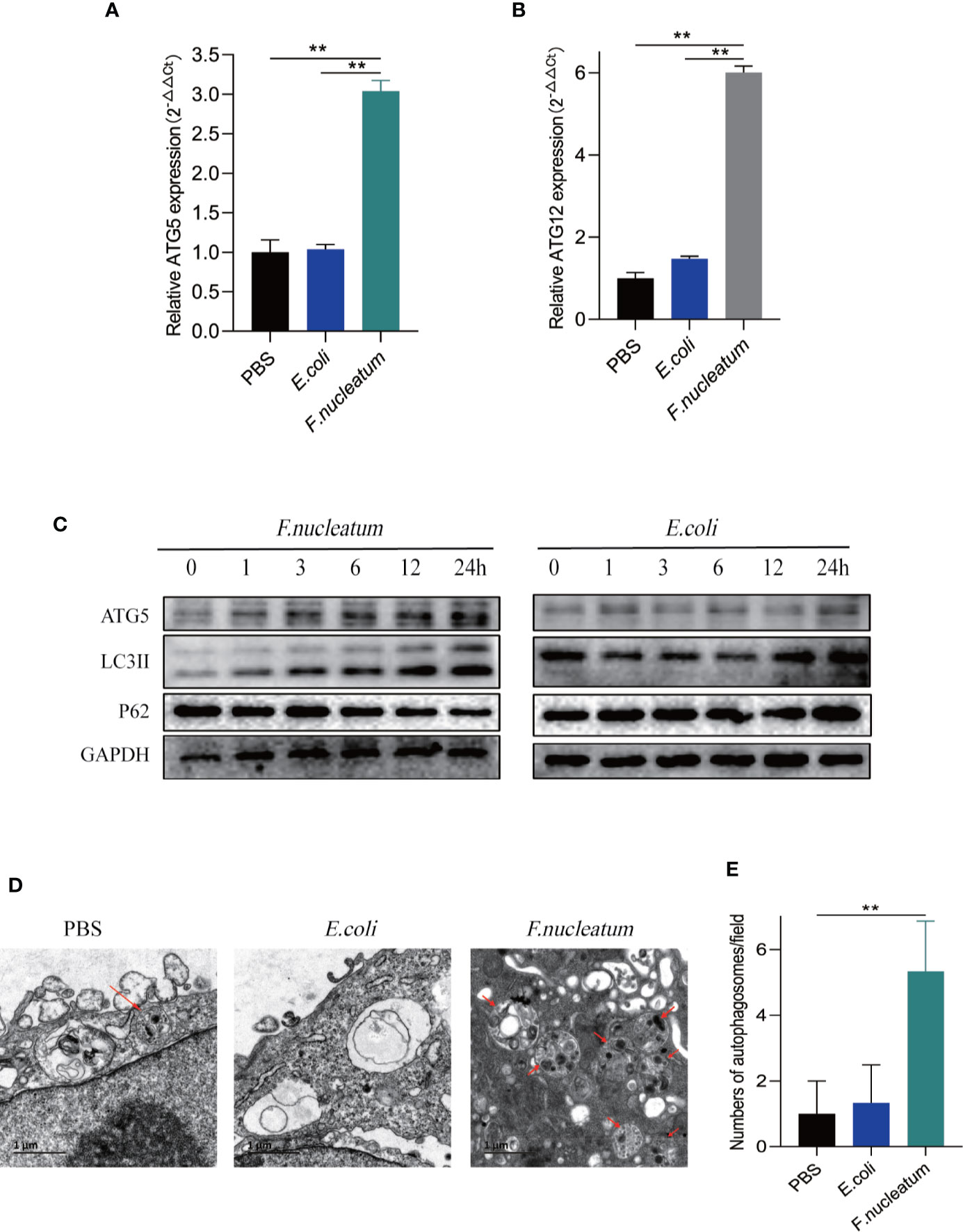
Figure 2 F. nucleatum promotes IEC autophagy pathway in vitro. (A, B) The mRNA expression of Atg5 and Atg12 was measured in NCM460 cells cocultured with PBS, F. nucleatum or E. coli for 24 h (*P < 0.05, **P < 0.01, and ***P < 0.001; unpaired Student’s t-test; the error bars indicate the SDs). (C) Western blot analysis was performed to measure the protein levels of Atg5, LC3 and P62 in NCM460 cells cocultured with E. coli or F. nucleatum. (D) Representative electron micrographs of autophagosomes (red arrows) in NCM460 cells infected with PBS, E. coli or F. nucleatum. Bar scale, 1 um. (E) Quantification of cells containing autophagosomes (from D) (**P < 0.01; unpaired Student’s t-test).
F. nucleatum Promotes IEC Autophagy Activation In Vivo
To further understand the role of F. nucleatum in the development of UC, we established a DSS-induced experimental colitis model with F. nucleatum infection. We observed that mice co-treated with DSS and F. nucleatum showed more severe gut damage, including rapid weight loss (Figure 3A) and a higher disease activity index (DAI) (Figure 3B). Aggravation of the disease is accompanied by high abundance of F. nucleatum and F. nucleatum enhanced epithelial damage, including mucosal erosion, crypt loss, lymphocyte infiltration compared with E. coli (DH5α) (Figure 3C). The colon length of F. nucleatum + DSS group was significantly shorter than other groups (Figure 3D). In line with these results, a higher histological score (HS) was observed in F. nucleatum + DSS group than group treated with DSS, F. nucleatum or E. coli + DSS (Figure 3E). These data suggest that F. nucleatum, contrary to E. coli (DH5α), may exacerbate the histological features of DSS-induced colitis. Western blotting results showed higher levels of ATG5 and LC3II and a lower level of P62 in colitis tissues from F. nucleatum + DSS treated mice compared with that in the other groups (F. nucleatum group, DSS group and E. coli + DSS group) (Figure 3F), indicating that F. nucleatum specifically aggravates DSS-induced colitis through autophagy pathway. These results indicate that F. nucleatum may contribute to the development of UC via the autophagy pathway in vivo.
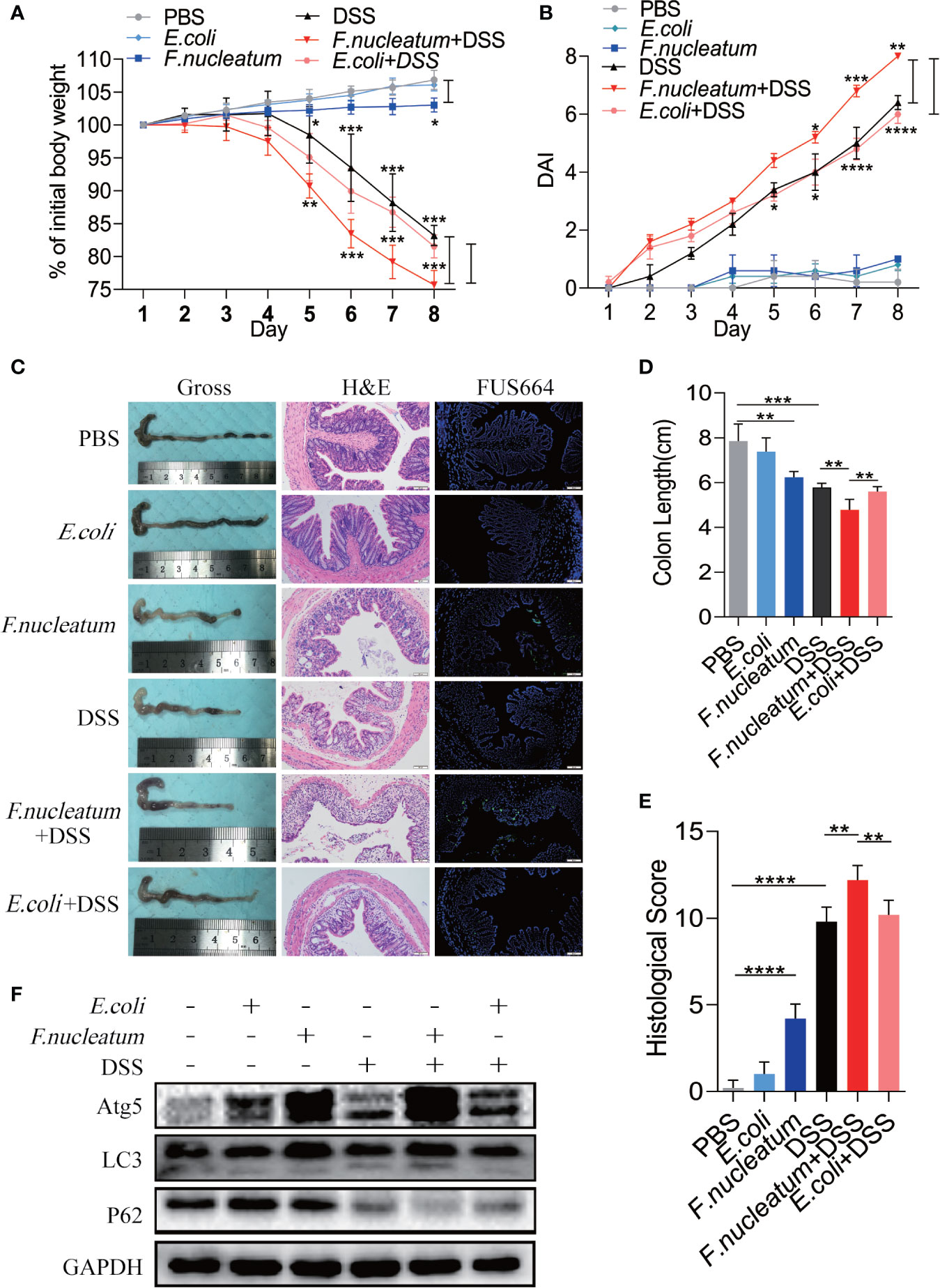
Figure 3 F. nucleatum promotes IEC Autophagy activation in vivo (A, B) Mice (n = 6 per group) were administered F. nucleatum, E. coli or PBS for 2 weeks and treated with 2.5% DSS for another 7 days. Colitis induction was evaluated by body weight loss (A) expressed as a percentage of the initial body weight and by the disease activity index (DAI) (B) (*P < 0.05, **P < 0.01, and ***P <0.001; one-way ANOVA combined with Bonferroni’s post hoc test. Error bars indicate SD). (C–E) Representative colon morphology and length of the mice described in (A) are shown in (C) and are quantified in (D)). Representative images of histological analyses are shown in (C) and are quantified in (E) (*P < 0.05, **P < 0.01, and ***P < 0.001; ****P < 0.0001; nonparametric Mann–Whitney test. Error bars indicate SD; 200 × magnification). (F) Western blot analysis was performed to measure the protein levels of Atg5, LC3 and P62 in the mouse tissues described in (A).
F. nucleatum Leads to IEC Death In Vitro
Study found F. nucleatum could promote cell death in human gingival fibroblasts lymphocytes (Kaplan et al., 2010; Kang et al., 2019). To detect whether F. nucleatum promote cell death in IEC, we coculture NCM460 cells with F. nucleatum. RT-PCR and western bolt results showed that antiapoptotic protein Bcl-2 decreased in mRNA and protein levels, and proapoptotic protein Bax increased, indicating an increased level of apoptosis (Figures 4A, B, C). In addition, autophagy inhibitor of CQ and 3-MA or silencing Atg5 by siRNA knockdown reversed the level of F. nucleatum-induced apoptosis (Figures 4C, D), suggesting that F. nucleatum may promote apoptosis by activating autophagy.
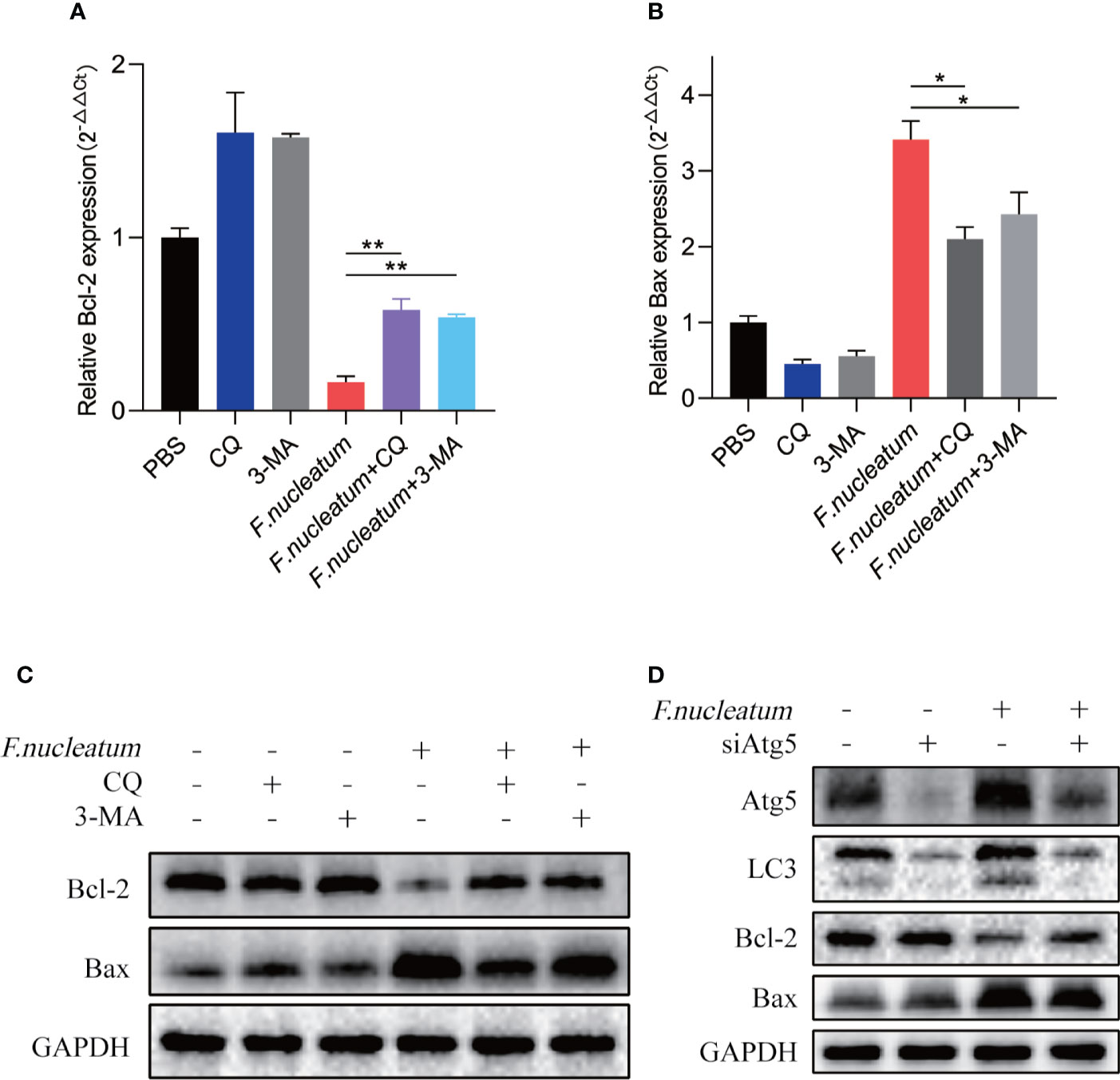
Figure 4 F. nucleatum promotes IEC death in vitro. (A, B) The mRNA expression of Bcl-2 and Bax was measured in NCM460 cells cocultured with PBS, CQ, 3-MA and F. nucleatum (*P < 0.05 and **P < 0.01; unpaired Student’s t-test; the error bars indicate the SDs). (C) Western blot analysis was performed to measure the protein levels of Bcl-2 and Bax in NCM460 cells cocultured with PBS, CQ, 3-MA and F. nucleatum. (D) Western blot analysis was performed to measure the protein levels of Atg5, LC3, Bcl-2 and Bax with NCM460 cells transfected with Atg5-targeting siRNA and cocultured with either PBS or F. nucleatum for 24 h.
Chloroquine Deadens Excessive IEC Death in Colitis Mouse
We found F. nucleatum promoted autophagy pathway in IECs in vivo and in vitro, therefore we want to know whether F. nucleatum will contribute to autophagic cell death to exacerbate the colitis. In our following experiments, CQ was used to inhibit lysosomal acidification, thus preventing autophagy by preventing the fusion and degradation of autophagosomes. We found that colitis mice treated with CQ showed a slower decline in body weight (Figure 5A), a lower DAI (Figure 5B), a longer colon length (Figures 5C, D), a lower HS (Figure 5E). HE results showed that CQ treatment attenuated inflammatory cell infiltration into mucosa and extensive damage of epithelium along with crypt destruction in F. nucleatum + DSS induced mice (Figure 5C). As a signaling pathway that regulates cell apoptosis and survival, increased level of Bax and decreased level of Bcl-2 has been studied as biomarkers of cell death. Western blot results showed that the use of CQ reversed the increased level of Bax and reduced level of Bcl-2 in vivo (Figure 5F). These results indicate that F. nucleatum may aggravate the development of IBD by promoting IEC autophagic death.
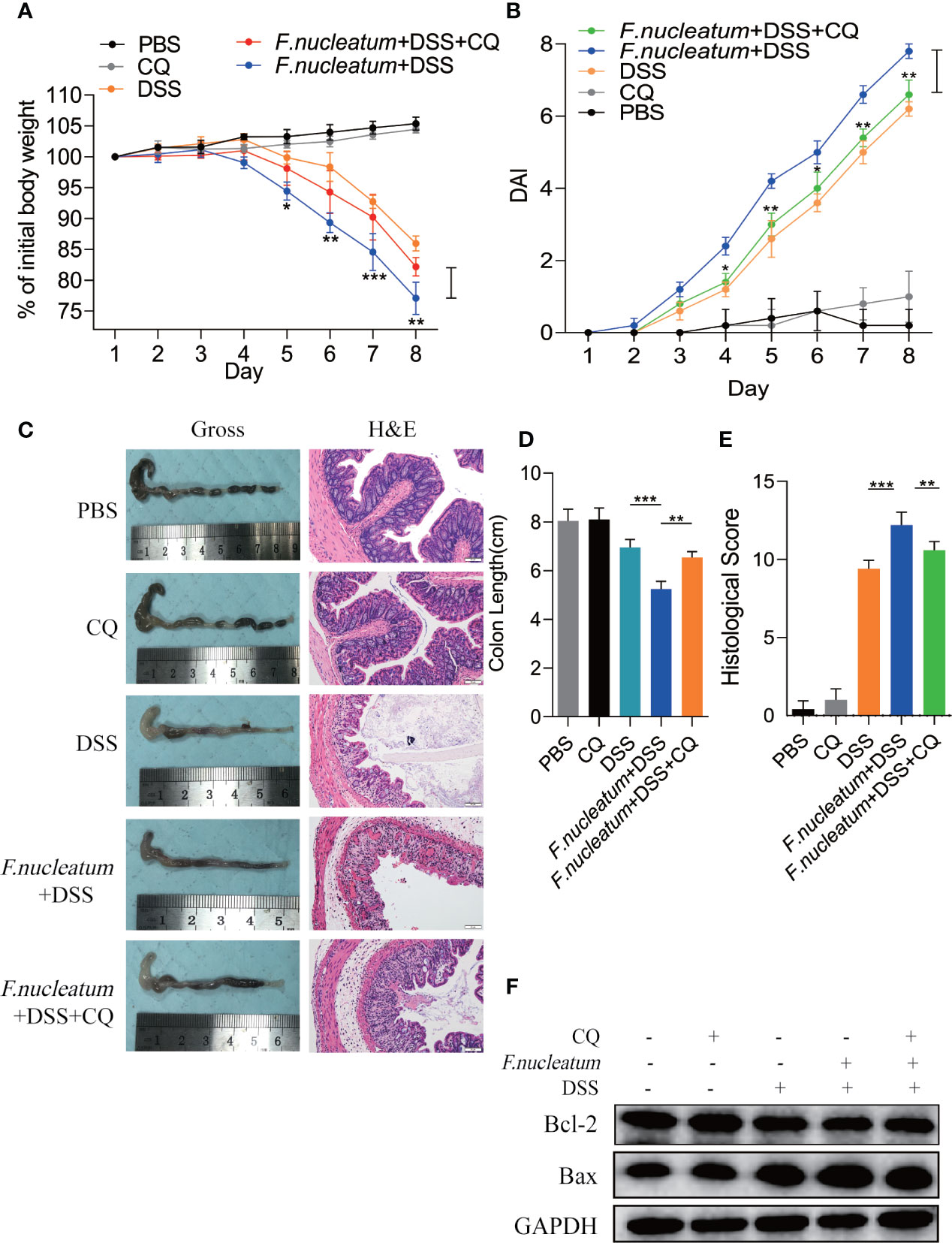
Figure 5 Inhibition of autophagy by CQ attenuates excessive cell death in the DSS-induced colitis mouse model in vivo and in vitro (A, B) For two weeks, Mice (n = 6 per group) were administered F. nucleatum, PBS and CQ was injected intraperitoneally every two days and then treated with 2.5% DSS for another 7 days. Colitis induction was evaluated by body weight loss (A) expressed as a percentage of the initial body weight and by the disease activity index (DAI) (B) (*P < 0.05, **P < 0.01, and ***P < 0.001; one-way ANOVA combined with Bonferroni’s post hoc test. Error bars indicate SD). (C–E) Representative colon morphology and length of the mice described in (A) are shown in (C) and are quantified in (D). Representative images of histological analyses are shown in (C) and are quantified in (E) (*P < 0.05, **P < 0.01, and ***P < 0.001; one-way ANOVA combined with Bonferroni’s post hoc test. Error bars indicate SD; 200 × magnification). (F) Western blot analysis was performed to measure the protein levels of Bcl-2 and Bax in the mouse tissues described in (A).
LPS of F. nucleatum Promotes Autophagic Cell Death of IEC
To further understand how F. nucleatum affect the autophagy pathway, we extracted LPS from cultured F. nucleatum and treated NCM460 cells. Western blot results showed that LPS of F. nucleatum activated autophagy pathway in a time-dependent manner and it promotes the autophagic cell death of IEC (Figure 6A). Real-time PCR results showed that LPS also elevated ATG5 mRNA level and contributed to apoptosis level (Figures 6B–D). These findings indicate that F. nucleatum may contribute to autophagic cell death of IEC through its LPS.
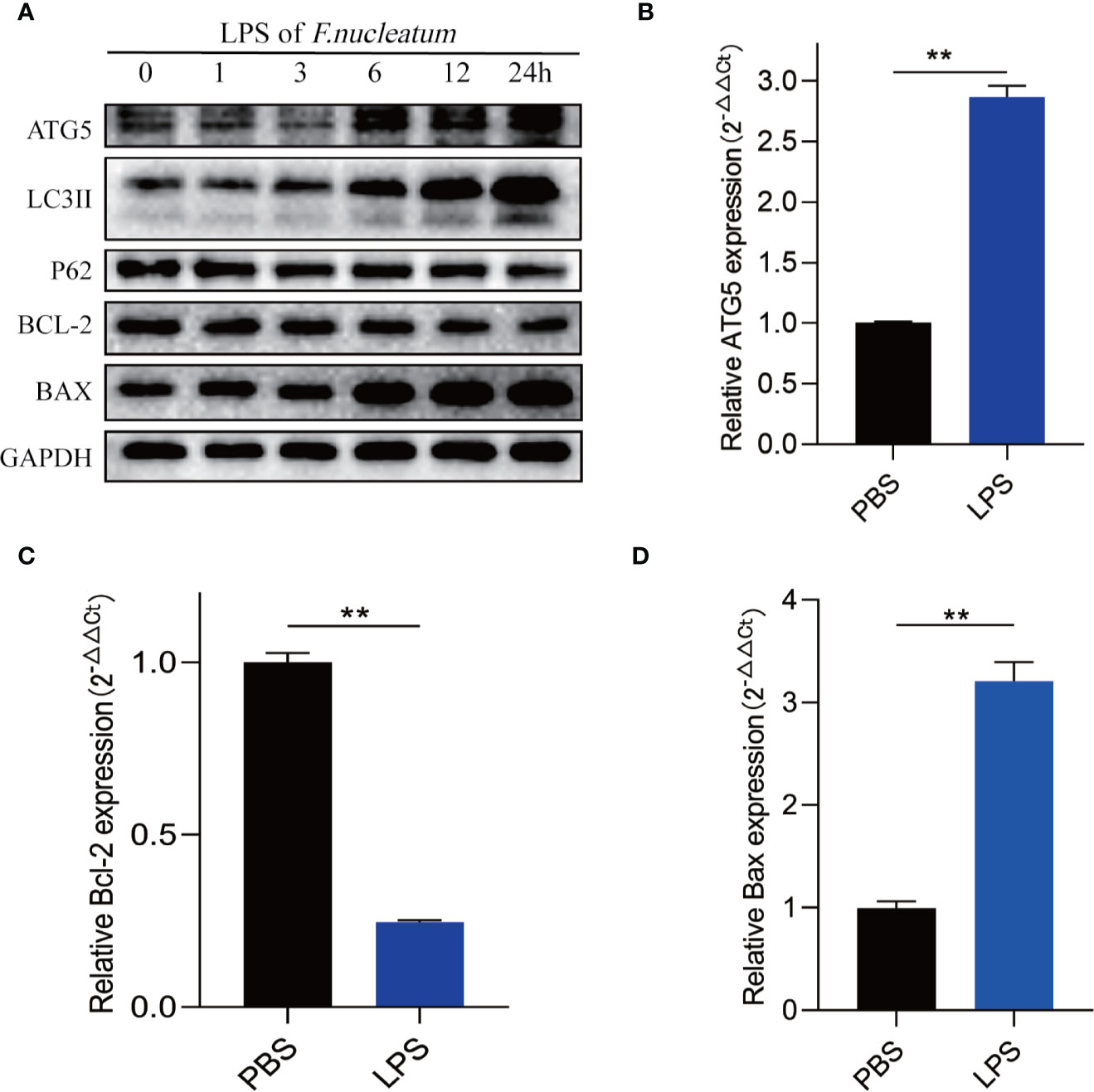
Figure 6 LPS of F. nucleatum promote autophagic cell death in UC. (A) Western blots showing that levels of ATG5, LC3II, Bax increase and expression of P62 and Bcl-2 decrease when NCM460 cells are challenged with LPS extracted from F. nucleatum over increasing time periods. (B–D) The mRNA expression of Atg5, Bcl-2 and Bax in NCM460 cells cocultured with LPS of F. nucleatum and PBS for 24 h (**P < 0.01; unpaired Student’s t-test; the error bars indicate the SDs).
Discussion
Intestinal microbiota has been thought one of the major contributor in the pathogenesis of UC (Weingarden and Vaughn, 2017). Recent studies have found that F. nucleatum, a commensal within the oral cavity, is an emergent opportunistic pathogen of the gastrointestinal tract, having been abundantly found in patients suffering from UC (Nishida et al., 2018; Dinakaran et al., 2019). Here, we confirmed that F. nucleatum is an opportunistic risk factor of UC, which is closely related to the clinical characteristics of UC. In addition, we used DSS-induced colitis models to assess the effect of F. nucleatum in colitis. Through clinical specimen analysis and animal model verification, we have demonstrated that F. nucleatum is enriched in colorectal tissues of UC patients and exacerbates the severity of UC.
F. nucleatum have been confirmed to contribute to the CRC through promoting autophagy pathway and TNF-α expression in IEC (Yu et al., 2017). Infection with F. nucleatum was also found in UC patients (Zhou et al., 2016). However, the mechanisms of how F. nucleatum exacerbates colitis remains unclear. We examined the autophagy-associated protein (LC3 and p62) expression of IEC and colon tissues in vivo and in vitro. We found the presence of F. nucleatum was accompanied with elevated autophagy levels in IEC and colon tissues of DSS mice.
Next, we studied the mechanism by which F. nucleatum lead to IEC death. Elevated levels of apoptosis rate have been observed in the intestinal epithelium of patients with UC (Souza et al., 2004). Autophagy activation may be beneficial to cell survival by eliminating excess and damaged organelles, but excessive autophagy activation may cause autophagic cell death (Liu and Levine, 2015). We cocultured E. coli and F. nucleatum with intestinal epithelial cell lines, and found with F. nucleatum infection, the levels of autophagy and apoptosis were increased contrary to E. coli, but the use of CQ and 3-MA reduced the F. nucleatum-induce elevated apoptosis level of IEC and improved the integrity of mucosa. Therefore, we concluded that F. nucleatum may induce autophagic death of IEC to aggravate UC, and the effect can be mitigated by autophagy inhibitor CQ and 3-MA. One study found that calpain-mediated Atg5 cleavage provokes apoptosis cell death, which showed Atg5 could lead to cell death in an autophagy-independent manner (Yousefi et al., 2006). However, a previous study found that ATG5 can promote autophagy for cell death (Arakawa et al., 2017). Study in normal fibroblast cell lines also indicated that accumulation of autophagosomes and upregulation of ATG5 lead to cell death, which are attenuated by autophagy inhibition (Byun et al., 2009). Thus we examine the level of ATG5 in RNA and protein level, we found that when autophagic cell death occurs in IEC, the level of ATG5 increases, and silencing Atg5 could reduce the cell death and autophagy, confirming that ATG5 could induce IEC apoptosis in an autophagy-dependent manner.
As inhibitor of autophagy, CQ and 3-MA was widely-used in autophagy-related researches (Mauthe et al., 2018; Yang et al., 2019). Our study found that CQ and 3-MA inhibit F. nucleatum-induced autophagic cell death, CQ treatment attenuates the levels of the extent of inflammation, range of inflammation and crypt damage in F. nucleatum-treatment mice. In recent years, the role of autophagy regulators in IBD has been extensively studied. Therefore, our study finds that inhibiting autophagy by CQ and 3-MA may be used in the treatment of UC patient with high F. nucleatum, and this conclusion needs to be confirmed in clinical studies.
In addition, we investigated which component of F. nucleatum plays a role in mediating the autophagy of IEC. As the major component of the Gram-negative bacteria outer membrane, LPS is commonly used to establish intestinal injury models and induce inflammation with its antigenicity and cytotoxicity (Zhang et al., 2018; Xiong et al., 2019). Study found that LPS recognized by TLR4 to initiate a signaling cascade, of which NF-κB signaling pathway plays an vital role in inducing the inflammation and organ injury (Sun et al., 2015). LPS has been shown to induce intestinal epithelial cell autophagy, reduce intestinal cell proliferation, increase intestinal cell apoptosis, and ultimately lead to intestinal damage (Xie et al., 2019). Study have reported that F. nucleatum-related lipopolysaccharide (LPS) can induce macrophages to secrete TNF-α and promote inflammation (Grenier and Grignon, 2006), which both play an important role in pathogenesis of UC. Therefore, we chose to extract LPS to verify whether F. nucleatum exerts autophagy induction through its LPS. We demonstrated that LPS extracted from F. nucleatum can lead to autophagic death of IEC. However, we did not explore whether other components such as FadA play a role in autophagy activation.
From a clinical perspective, as the abundance of F. nucleatum is related with the severity of UC, treatment for autophagy may be a new target for UC patients with a high abundance of F. nucleatum. In addition, some conventional drugs used to treat UC have been shown to have autophagy regulating effect (Hooper et al., 2017), thus whether UC patients with F. nucleatum infection benefit from autophagy regulation of conventional treatment remains to be further elucidated.
Overall, our results demonstrate that F. nucleatum contribute to the development UC by inducing autophagic cell death of IEC. Moreover, the clinical study indicates that F. nucleatum is a risk factor for a high degree of disease activity in UC patients. Our research provides new evidence demonstrating the pathogenicity of F. nucleatum in UC and offer new sight to the treatment of intestinal microbe and autophagy regulation for the treatment of UC.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Renmin Hospital of Wuhan University, China. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Institutional Review Board of Renmin Hospital of Wuhan University, China.
Author Contributions
Study conception and design: WD, WS, YoC. Acquisition of clinical data: WS, YC, PC. Data analysis and interpretation and statistical analysis: WS, YoC, PC. Animal experiments: WS, YoC, PC, YC. Manuscript drafting: WS, YoC, WD. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81870392, No. 81372551, and No. 81572426) and the Guiding Foundation of Renmin Hospital of Wuhan University (No. RMYD2018Z01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.594806/full#supplementary-material
Supplementary Figure 1 | F. nucleatum promotes IEC autophagy in vitro and in vivo. (A–F) The protein expression of Atg5, LC3 and P62 in NCM460 cells cocultured with F. nucleatum and E. coli were quantified. (G–I) The protein expression of Atg5, LC3 and P62 in mouse tissues were quantified. Data are expressed as mean ± SD for three independent experiments. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01 and ***P < 0.001. (J) Western blotting was performed to measure the expression of Atg5 in NCM460 cells transfected with Atg5-targeting siRNA (siAtg5) and nontargeting siRNAs (NC).
Supplementary Figure 2 | F. nucleatum and LPS promotes IEC autophagic cell death in vitro and in vivo. (A, B) The protein expression of Bcl-2 and Bax in NCM460 cells cocultured with PBS, CQ, 3-MA with or without F. nucleatum were quantified. (C–F) The protein expression of Atg5, LC3, Bcl-2 and Bax in NCM460 cells cocultured with PBS, F. nucleatum or siAtg5 were quantified. (G, H) The protein expression of Bcl-2 and Bax in colon tissues from mice were quantified. (I–M) The protein expression of Atg5, LC3, P62, Bcl-2 and Bax in NCM460 cells cocultured with LPS of F. nucleatum were quantified. Data are expressed as mean ± SD for three independent experiments. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Figure 3 | Low power field image corresponding to HE and FISH images of UC in Figure 1.
Supplementary Table 1 | Clinical characteristics of F. nucleatum-negative vs. F. nucleatum-positive UC.
References
Arakawa S., Tsujioka M., Yoshida T., Tajima-Sakurai H., Nishida Y., Matsuoka Y., et al. (2017). Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 24 (9), 1598–1608. doi: 10.1038/cdd.2017.84
Bashir A., Miskeen A. Y., Hazari Y. M., Asrafuzzaman S., Fazili K. M. (2016). Fusobacterium nucleatum, inflammation, and immunity: the fire within human gut. Tumour Biol. 37 (3), 2805–2810. doi: 10.1007/s13277-015-4724-0
Byun J. Y., Yoon C. H., An S., Park I. C., Kang C. M., Kim M. J., et al. (2009). The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis. Carcinogenesis 30 (11), 1880–1888. doi: 10.1093/carcin/bgp235
Denton D., Kumar S. (2019). Autophagy-dependent cell death. Cell Death Differ 26 (4), 605–616. doi: 10.1038/s41418-018-0252-y
Dharmani P., Strauss J., Ambrose C., Allen-Vercoe E., Chadee K. (2011). Fusobacterium nucleatum infection of colonic cells stimulates MUC2 mucin and tumor necrosis factor alpha. Infect. Immun. 79 (7), 2597–2607. doi: 10.1128/IAI.05118-11
Dinakaran V., Mandape S. N., Shuba K., Pratap S., Sakhare S. S., Tabatabai M. A., et al. (2019). Identification of Specific Oral and Gut Pathogens in Full Thickness Colon of Colitis Patients: Implications for Colon Motility. Front. Microbiol. (9), 3220. doi: 10.3389/fmicb.2018.03220
Grenier D., Grignon L. (2006). Response of Human Macrophage-Like Cells to Stimulation by Fusobacterium Nucleatum Ssp. Nucleatum Lipopolysaccharide. Oral. Microbiol Immunol. 21 (3), 190–196. doi: 10.1111/j.1399-302X.2006.00278.x
Hooper K. M., Barlow P. G., Stevens C., Henderson P. (2017). Inflammatory Bowel Disease Drugs: A Focus on Autophagy. J. Crohns Colitis 11 (1), 118–127. doi: 10.1093/ecco-jcc/jjw127
Hou J. K., El-Serag H., Thirumurthi S. (2009). Distribution and manifestations of inflammatory bowel disease in Asians, Hispanics, and African Americans: a systematic review. Am. J. Gastroenterol. 104 (8), 2100–2109. doi: 10.1038/ajg.2009.190
Jeong D. Y., Kim S., Son M. J., Son C. Y., Kim J. Y., Kronbichler A., et al. (2019). Induction and maintenance treatment of inflammatory bowel disease: A comprehensive review. Autoimmun Rev. 18 (5), 439–454. doi: 10.1016/j.autrev.2019.03.002
Kang W., Jia Z., Tang D., Zhang Z., Gao H., He K., et al. (2019). Fusobacterium nucleatum Facilitates Apoptosis, ROS Generation, and Inflammatory Cytokine Production by Activating AKT/MAPK and NF-kappaB Signaling Pathways in Human Gingival Fibroblasts. Oxid. Med. Cell Longev 2019, 1681972. doi: 10.1155/2019/1681972
Kaplan C. W., Ma X., Paranjpe A., Jewett A., Lux R., Kinder-Haake S., et al. (2010). Fusobacterium nucleatum Outer Membrane Proteins Fap2 and RadD Induce Cell Death in Human Lymphocytes. Infect Immun. 78 (11), 4773–4778. doi: 10.1128/iai.00567-10
Kim S., Eun H. S., Jo E. K. (2019). Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 8 (1):77. doi: 10.3390/cells8010077
Kumar A., Saranathan R., Prashanth K., Tiwary B. K., Krishna R. (2017). Inhibition of the MurA enzyme in Fusobacterium nucleatum by potential inhibitors identified through computational and in vitro approaches. Mol. Biosyst. 13 (5), 939–954. doi: 10.1039/c7mb00074j
Larabi A., Barnich N., Nguyen H. T. T. (2020). New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 16 (1), 38–51. doi: 10.1080/15548627.2019.1635384
Liu Y., Levine B. (2015). Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 22 (3), 367–376. doi: 10.1038/cdd.2014.143
Liu L., Liang L., Liang H., Wang M., Lu B., Xue M., et al. (2019). Fusobacterium nucleatum Aggravates the Progression of Colitis by Regulating M1 Macrophage Polarization via AKT2 Pathway. Front. Immunol. 10, 1324. doi: 10.3389/fimmu.2019.01324
Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K. J., et al. (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14 (8), 1435–1455. doi: 10.1080/15548627.2018.1474314
Nguyen H. T., Lapaquette P., Bringer M. A., Darfeuille-Michaud A. (2013). Autophagy and Crohn’s disease. J. Innate Immun. 5 (5), 434–443. doi: 10.1159/000345129
Nishida A., Inoue R., Inatomi O., Bamba S., Naito Y., Andoh A. (2018). Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 11 (1), 1–10. doi: 10.1007/s12328-017-0813-5
Park J. H., Peyrin-Biroulet L., Eisenhut M., Shin J. I. (2017). IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun Rev. 16 (4), 416–426. doi: 10.1016/j.autrev.2017.02.013
Shen T., Li S., Cai L. D., Liu J. L., Wang C. Y., Gan W. J., et al. (2018). Erbin exerts a protective effect against inflammatory bowel disease by suppressing autophagic cell death. Oncotarget 9 (15), 12035–12049. doi: 10.18632/oncotarget.23925
Souza H. S. P., Tortori C. J. A., Castelo-Branco M. T. L., Carvalho A. T. P., Margallo V. S., Delgado C. F. S., et al. (2004). Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease: evidence of altered expression of FasL and perforin cytotoxic pathways. Int. J. Colorectal Dis. 20 (3), 277–286. doi: 10.1007/s00384-004-0639-8
Sun X. J., Li X. Q., Wang X. L., Tan W. F., Wang J. K. (2015). Sevoflurane Inhibits Nuclear factor-κB Activation in Lipopolysaccharide-Induced Acute Inflammatory Lung Injury via Toll-Like Receptor 4 Signaling. PloS One 10 (4), e0122752. doi: 10.1371/journal.pone.0122752
Weingarden A. R., Vaughn B. P. (2017). Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 8 (3), 238–252. doi: 10.1080/19490976.2017.1290757
Weng Y. J., Gan H. Y., Li X., Huang Y., Li Z. C., Deng H. M., et al. (2019). Correlation of diet, microbiota and metabolite networks in inflammatory bowel disease. J. Dig Dis. 20 (9), 447–459. doi: 10.1111/1751-2980.12795
Xie M. Y., Hou L. J., Sun J. J., Zeng B., Xi Q. Y., Luo J. Y., et al. (2019). Porcine Milk Exosome MiRNAs Attenuate LPS-Induced Apoptosis through Inhibiting TLR4/NF-κB and p53 Pathways in Intestinal Epithelial Cells. J. Agric. Food Chem. 67 (34), 9477–9491. doi: 10.1021/acs.jafc.9b02925
Xiong W., Ma H., Zhang Z., Jin M., Wang J., Xu Y., et al. (2019). The protective effect of icariin and phosphorylated icariin against LPS-induced intestinal epithelial cells injury. BioMed. Pharmacother. 118:109246. doi: 10.1016/j.biopha.2019.109246
Yang H. L., Lin R. W., Karuppaiya P., Mathew D. C., Way T. D., Lin H. C., et al. (2019). Induction of autophagic cell death in human ovarian carcinoma cells by Antrodia salmonea through increased reactive oxygen species generation. J. Cell Physiol. 234 (7), 10747–10760. doi: 10.1002/jcp.27749
Yousefi S., Perozzo R., Schmid I., Ziemiecki A., Schaffner T., Scapozza L., et al. (2006). Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8 (10), 1124–1132. doi: 10.1038/ncb1482
Yu T., Guo F., Yu Y., Sun T., Ma D., Han J., et al. (2017). Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 170 (3), 548–563.e516. doi: 10.1016/j.cell.2017.07.008
Zhang X. Y., Chen X., Zhang H. F., Guan S., Wen S. H., Huang W. Q., et al. (2018). Propofol Does Not Reduce Pyroptosis of Enterocytes and Intestinal Epithelial Injury After Lipopolysaccharide Challenge. Dig Dis. Sci. 63 (1), 81–91. doi: 10.1007/s10620-017-4801-x
Keywords: Fusobacterium nucleatum, ulcerative colitis, autophagy, cell death, chloroquine
Citation: Su W, Chen Y, Cao P, Chen Y, Guo Y, Wang S and Dong W (2020) Fusobacterium nucleatum Promotes the Development of Ulcerative Colitis by Inducing the Autophagic Cell Death of Intestinal Epithelial. Front. Cell. Infect. Microbiol. 10:594806. doi: 10.3389/fcimb.2020.594806
Received: 25 August 2020; Accepted: 30 October 2020;
Published: 27 November 2020.
Edited by:
Yongqun Oliver He, University of Michigan, United StatesReviewed by:
Mario Mauthe, University Medical Center Groningen, NetherlandsBeatriz Leon Ruiz, University of Alabama at Birmingham, United States
Copyright © 2020 Su, Chen, Cao, Chen, Guo, Wang and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiguo Dong, ZG9uZ3dlaWd1b0B3aHUuZWR1LmNu