 Moagi Shaku
Moagi Shaku Christopher Ealand
Christopher Ealand Bavesh D. Kana
Bavesh D. Kana- National Health Laboratory Service, Department of Science and Technology/National Research Foundation Centre of Excellence for Biomedical TB Research, Faculty of Health Sciences, School of Pathology, University of the Witwatersrand, Johannesburg, South Africa
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), remains the leading cause of death from an infectious bacterium and is responsible for 1.8 million deaths annually. The emergence of drug resistance, together with the need for a shorter more effective regimen, has prompted the drive to identify novel therapeutics with the bacterial cell surface emerging as a tractable area for drug development. Mtb assembles a unique, waxy, and complex cell envelope comprised of the mycolyl-arabinogalactan-peptidoglycan complex and an outer capsule like layer, which are collectively essential for growth and pathogenicity while serving as an inherent barrier against antibiotics. A detailed understanding of the biosynthetic pathways required to assemble the polymers that comprise the cell surface will enable the identification of novel drug targets as these structures provide a diversity of biochemical reactions that can be targeted. Herein, we provide an overview of recently described mycobacterial cell wall targeting compounds, novel drug combinations and their modes of action. We anticipate that this summary will enable prioritization of the best pathways to target and triage of the most promising molecules to progress for clinical assessment.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), has infected approximately a third of the global population and is currently one of the leading causes of death from an infectious disease (WHO, 2019a). The emergence and global spread of drug resistant TB (DR-TB) has hampered effective control of the disease, creating an urgent need to identify novel drug targets or to re-purpose existing antibiotics (Gautam et al., 2011; Conradie et al., 2020). Considering this, cutting-edge molecular tools such as TnSeq, CRISPRi, and high-throughput whole-cell phenotypic screening with large compound libraries coupled with whole-genome sequencing are currently being used to rapidly identify drug targets and to discover novel anti-mycobacterial drugs (Grzelak et al., 2019; Rock, 2019; De Wet et al., 2020). Recently, several promising novel compounds and targets have been identified, particularly in the biosynthetic pathways of the mycobacterial cell wall (CW) (Dulberger et al., 2019; Maitra et al., 2019). Herein, we specifically review recent developments identifying new targets and inhibitory molecules ranging from those that specifically inhibit the activity of a particular enzyme in CW biosynthesis to those that may indirectly enhance the activity of certain compounds by functionally weakening the cell wall (Jeon et al., 2017; Maitra et al., 2019). Several biochemical inhibitors of the targets discussed are not approved drugs but despite this, further characterization enable a clearer understanding of essential pathways which aid in drug discovery. We focus our review on recently discovered chemical matter with proven inhibitory properties against mycobacterial CW biosynthesis enzymes and highlight some that have entered the TB drug development pipeline. We do not extensively review the biosynthesis of all CW components nor we do we provide a historical narrative of current CW targeting drugs. For this and related information, we direct the reader to several prior reviews for further information (Bhat et al., 2017; Abrahams and Besra, 2018; Maitra et al., 2019; Vilchèze, 2020). For peptidoglycan (PG), we highlight the compounds targeting the periplasmic component of polymer biosynthesis/crosslinking specifically, the MraY/MurX translocase and amidation modifications together with drugs that can potentiate the activity of β-lactams. We also summarize WhiB4-expression and its association with augmentin sensitivity. In addition, we discuss targets associated with regulation of PG biosynthesis such as the serine/threonine protein kinases and also discuss FtsZ inhibitors. For arabinogalactan (AG), we discuss inhibitors of RmlC and GlmU inhibitors as the inhibition of DprE1 by BTZ043 is widely discussed in literature.
The Mycobacterial Cell Wall
Mycobacterial species possess a CW with biochemically diverse components, including primarily three distinct layers, namely: PG, AG, and mycolic acids (MAs) which are surrounded by a capsule (Abrahams and Besra, 2018). The capsule is comprised of proteins, polysaccharides and lipids [phosphatidyl-myo-inositol mannosides, diacyl trehaloses, phthiocerol dimycocerosates (PDIMs), and phosphatidylethanolamine] (Abrahams and Besra, 2018). In addition to these components, there are several solvent-extractable lipids including non-covalently linked glycophospholipids and inert waxes, which are well known to serve as a permeability barrier against antibiotics and play critical role in pathogenesis and survival of Mtb. The diverse pathways for biosynthesis of CW precursors, and subsequent processes required for transport and polymerization have been exploited for development of anti-mycobacterial drugs (Vilchèze, 2020). A select set of recently identified CW targeting compounds and those currently in clinical trials are summarized in Figure 1 and Table 1.
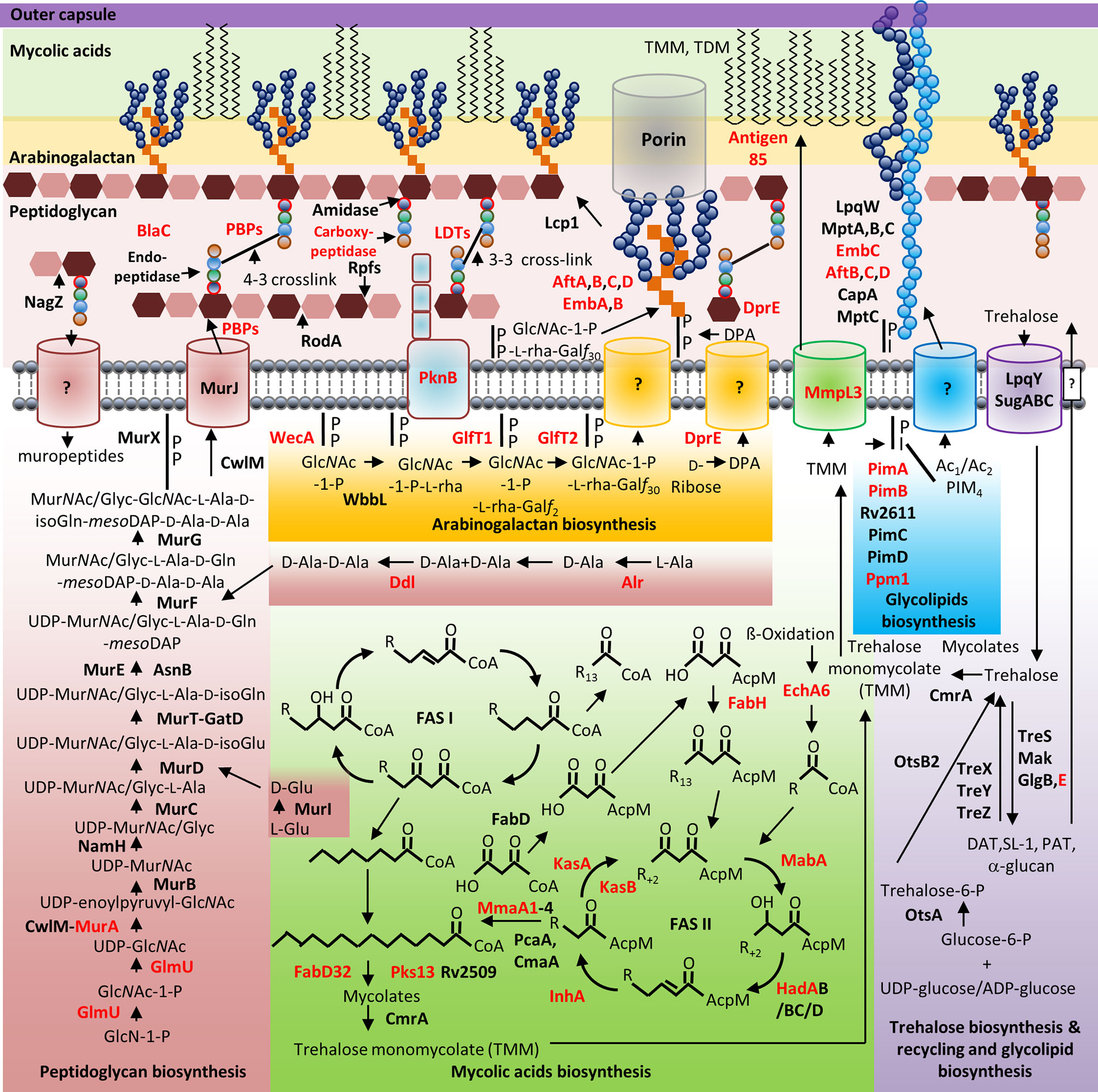
Figure 1 Mycobacterial cell wall and its validated and potential drug targets. Shown are the cytoplasmic and periplasmic biosynthetic pathways for the different polymers in the mycobacterial cell wall (peptidoglycan, arabinogalactan, mycolic acids, and glycolipids). Cytoplasmic and periplasmic enzymes already validated as drug targets and potential drug targets are shown in red text. Membrane channels involved in PG recycling, GlcNAc-1-P-L-rha-Galf30-, DPA-, Ac1/Ac2PIM4-, and surface glycolipid translocation remain to be identified (depicted by “?”).
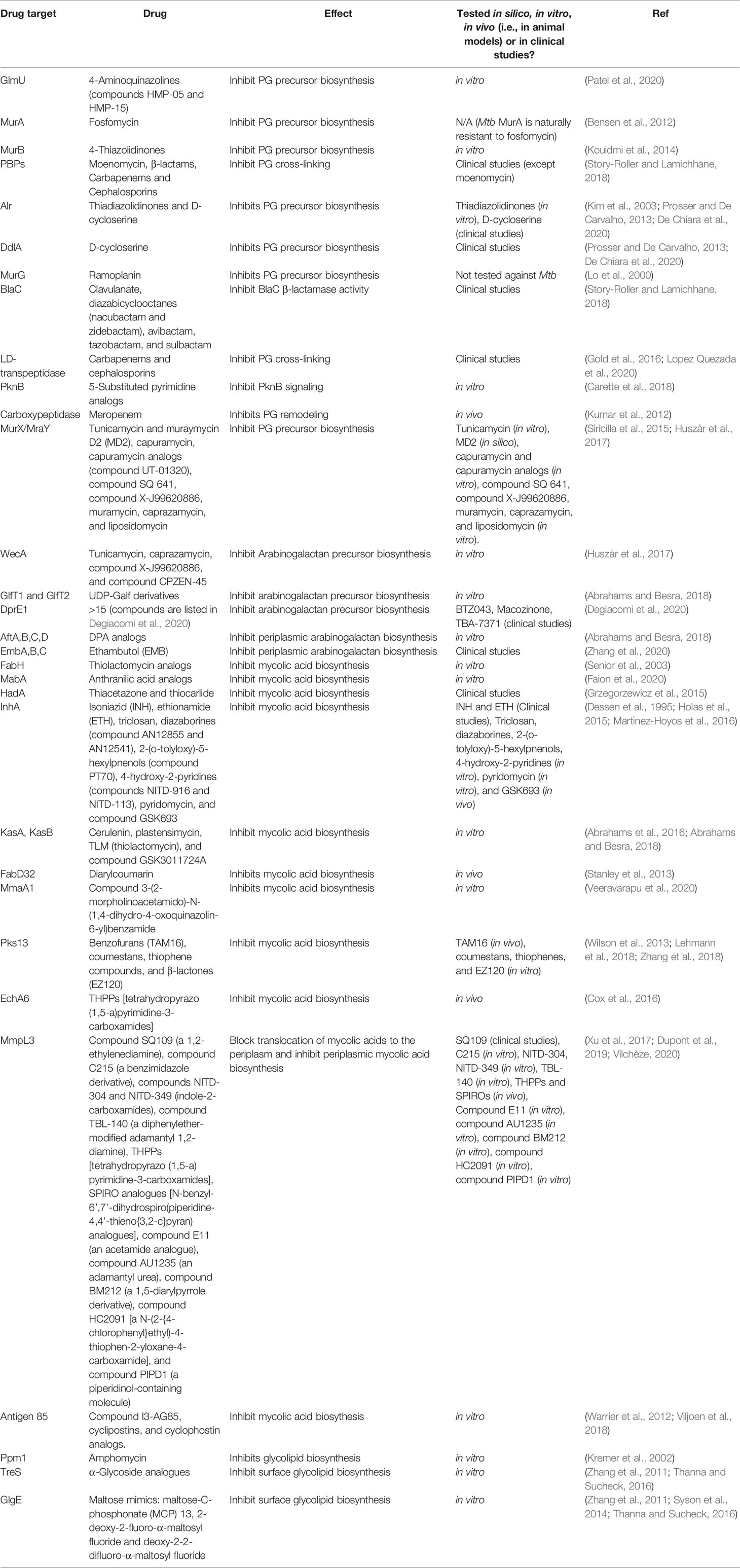
Table 1 Drugs targeting mycobacterial cell wall biosynthesis pathways.
Peptidoglycan Biosynthesis and Potential Targets
PG precursor (lipid II) biosynthesis includes major cytoplasmic enzymes (MurA–F) that produce UDP-MurNAc-penta-peptide or UDP-MurNGlyc-penta-peptide. In Gram-negative bacteria, fosfomycin targets MurA (Bensen et al., 2012) however, as the mycobacterial MurA homologue lacks a critical active site cysteine residue required for fosfomycin binding (De Smet et al., 1999). Fosfomycin mimics able to bind the Mtb MurA active site remain to be designed and tested. Interestingly, unlike in other bacteria, where MurA activity is regulated by the binding of UDP-MurNAc, mycobacterial MurA activity is regulated by interaction with phosphorylated CwlM, a catalytically inactive PG amidase (Boutte et al., 2016). Several FDA-approved drugs and known MurB inhibitors, such as 4-thiazolidinones, were screened against the Mtb MurB homologue using docking simulations and target- or inhibitor-based approaches. As there are significant similarities in the structures of MurC and MurD/MurE and MurF, it should be possible to design inhibitors that can inhibit the Mur enzymes simultaneously (Kouidmi et al., 2014). Still within the cytoplasm, alanine racemase (Alr) catalyzes the conversion of L-alanine to D-alanine which is required to synthesize the peptide component of Lipid II (De Chiara et al., 2020). There are currently several commercial antibiotics targeting Alr (see Table 1), including D-cycloserine (Kim et al., 2003). D-Cycloserine has been used to treat pulmonary and extra-pulmonary TB, including MDR-TB, but this is hampered by severe toxic side-effects (Azam et al., 2016). Recently, it was shown that Alr activity remains detectable in Mtb exposed to clinically relevant D-cycloserine concentrations (Prosser and De Carvalho, 2013; De Chiara et al., 2020). This is due to reversible binding via D-cycloserine-adduct hydrolysis thus enabling dissociation and structural rearrangement within the active enzyme to regain activity. This mechanistic insight now provides a route for discovery of improved Alr inhibitors (De Chiara et al., 2020). D-cycloserine also inhibits D-Ala:D-Ala ligase (DdlA), another essential enzyme in PG biosynthesis (Prosser and De Carvalho, 2013) and improvements on the activity of this compound may yield increased activity to DdlA also.
The next step involves the translocase MraY/MurX which links the UDP-MurNAc/Glyc-L-Ala-D-Glu-meso-DAP-D-Ala-D-Ala to a decaprenyl phosphate (C50-P) to form lipid I. This enzyme has also emerged as an attractive target as it is essential in Mtb (Hering et al., 2018). While the natural nucleoside inhibitors of MraY, tunicamycin, and muraymycin D2 (MD2), have been available, promising efforts to design new inhibitors are emerging (Tanino et al., 2011; Chen et al., 2016; Chung et al., 2016; Mashalidis and Lee, 2020).
MurG facilitates the transfer of GlcNAc from UDP-GlcNAc to MurNAc or MurNGlyc of Lipid I to generate Lipid II (Laddomada et al., 2019). MurG function is inhibited by ramoplanin, a lipoglycodepsipeptide which binds lipid I (Lo et al., 2000). Following this, the MurT-GatD complex and AsnB amidate the α-carboxyl group of D-glutamate and the D-carboxyl group of meso-DAP to form amidated Lipid II, respectively (Münch et al., 2012; Levefaudes et al., 2015). These amidation modifications are essential for PG cross-linking (Pidgeon et al., 2019) and as such, the MurT-GatD complex and AsnB remain high priority targets for development of PG targeting antibiotics.
Translocation of Lipid II into the periplasm is facilitated by MurJ, which has emerged as a possible target of newly discovered antibiotics including humimycins in gram-positive bacteria. Humimycins are potent β-lactam potentiators and display broad spectrum activity (Chu et al., 2018). However, MurJ in Mtb remains to be fully characterized and further work in this regard will guide the design of Mtb MurJ inhibitors. Ramoplanin, teixobactin, malacidin, and nisin bind periplasmic Lipid II while the glycopeptides vancomycin and teicoplanin bind the D-Ala-D-Ala terminus of lipid II preventing lipid II polymerization (Pazos and Peters, 2019). Once lipid II is translocated into the periplasm, the transglycosylase activities of PBPs and SEDS proteins facilitate the linking of the disaccharide component of Lipid II to the existing PG glycan chains (Kieser and Rubin, 2014). The transpeptidase activities of high molecular weight PBPs facilitate the formation of 4-3 cross-links between meso-DAP and D-Ala of adjacent penta-peptide chains. In mycobacteria, remodeling of 4-3 to 3-3 cross-links occurs via the co-ordinated actions of PG endopeptidases and LD-transpeptidases. The 3-3 cross-link allows for a localized increase in tensile strength of the cell wall at sub-cellular regions where there is an insufficient level of 4-3 cross linking necessary to maintain a wildtype morphology (Baranowski et al., 2018).
The moenomycin class of antibiotics inhibit transglycosylase activity of PBPs while β-lactam antibiotics inhibit the transpeptidase activity of PBPs (Ostash et al., 2010; Story-Roller and Lamichhane, 2018). Despite the successful treatment of many bacterial infections, conventional β-lactams are generally ineffective against mycobacterial species due to a chromosomally encoded β-lactamase—BlaC, which rapidly hydrolyzes the β-lactam ring (Mishra et al., 2017; Tooke et al., 2019). Consistent with this, the carbapenem class of β-lactam antibiotics are poor substrates for BlaC and when combined with a β-lactamase inhibitor, prove to be very effective in killing mycobacteria (Hugonnet et al., 2009). As a result of the early bactericidal activity (EBA) of intravenously administered meropenem plus clavulanic acid combined with oral amoxicillin (Mero/Clv/Amx) for treatment of DR-TB, the World Health Organization has recently endorsed the use of this regimen as an additional drug combination for DR-TB (WHO, 2019b). Furthermore, it was shown that carbapenems and novel cephalosporins inhibit M. abscessus growth (Kumar et al., 2017) and also kill non-replicating Mtb (Gold et al., 2016). However, the use of other carbapenems like Ertapenem in combination with Amx/Clv did not display significant EBA when compared with Mero/Clv/Amx (De Jager et al., 2020). Therefore, the search for orally bioavailable carbapenems continues.
The 2-aminoimidazoles (2-AIs) have recently been shown to potentiate the activity of β-lactams by decreasing Mtb protein secretion and also by increasing the CW permeability (Jeon et al., 2017). This was due to inhibition of the electron transport chain which resulted in impairment of protein secretion systems and MA biosynthesis (Jeon et al., 2019). Further investigations to identify novel β-lactamase inhibitors and the impact of novel β-lactam:β-lactamase inhibitor combinations for the treatment of DR-TB have therefore become an active research area (Story-Roller and Lamichhane, 2018).
Due to the potential of including β-lactamase inhibitors in the clinical setting, the mechanistic aspects of how Mtb responds to β-lactam:β-lactamase combinations, e.g., augmentin (Amx/Clv) was recently elucidated (Mishra et al., 2017). WhiB4, a cytoplasmic redox sensor, appears to co-ordinate the activity of BlaC in a redox-dependent manner. Disruption of WhiB4 increased tolerance to augmentin whereas overexpression potentiated augmentin activity against Mtb. Therefore, compounds that can induce increased expression of WhiB4 could enhance the bactericidal activity of augmentin against Mtb, this approach should be actively investigated.
Regulation of Peptidoglycan Biosynthesis and Potential Targets
Mtb expresses 11 serine/threonine protein kinases designated PknA to L, which regulate various metabolic pathways via protein phosphorylation. PknB and PknG are essential for intracellular survival of Mtb (Bellinzoni et al., 2019). PknB regulates PG biosynthesis by localizing at sites of new synthesis and activating/inhibiting PG-associated enzymes (Mir et al., 2011; Turapov et al., 2018; Kaur et al., 2019). Several kinase inhibitors that target PknA and PknB have been developed and are bactericidal for Mtb (Carette et al., 2018).
FtsZ, a tubulin homolog, is essential for bacterial cell division and remains an attractive target for novel antibiotics (Mathew et al., 2016). Dihydroquinolines have also been shown to possess inhibitory activity against mycobacterial FtsZ (Carro, 2019). Loss of LamA, another mycobacterial cytoskeletal factor, influence polar growth dynamics (a phenotype associated with heterogeneity in susceptibility to antibiotics), indirectly enhancing susceptibility to rifampicin (RIF) and cell wall targeting drugs (Rego et al., 2017). Similarly, loss of FtsX, a regulator of cell division, enhanced susceptibility to RIF (Mavrici et al., 2014). A recent whole-cell screening approach identified compound APYS1, an aminopyrimidine-sulfonamide, with potent activity indirectly inhibiting Mtb Wag31/DivIVA, an actin-like protein required for cell wall biosynthesis and cell elongation (Singh et al., 2017).
Arabinogalactan Biosynthesis and Potential Targets
AG biosynthesis is initiated in the cytoplasm by WecA, resulting in a “linker” region that serves as the site of attachment to PG (Alderwick et al., 2015; Huszár et al., 2017). WecA is a target of tunicamycins and caprazamycin derivatives (Huszár et al., 2017). The next steps yield units that are polymerized in the periplasm by AftA-D and EmbA-B (targets of ethambutol), see Figure 1. The AG complex is then ligated to PG by Lcp1, which has also recently emerged as an attractive target in the Mtb CW (Hett et al., 2008; Harrison et al., 2016). The crystal structure of a LCP homologue in Staphylococcus aureus has been solved and this provides insight into structure-guided design of inhibitors of this enzyme family (Li et al., 2020). For further insight, the reader is directed to a recent review that comprehensively describes the biosynthesis of galactan in Mtb, with a focus on drug discovery (Konyariková et al., 2020).
Another promising target, DprE1, acts as a flavoenzyme which uses the cofactor FAD (flavin adenine dinucleotide) to oxidize DPR (decaprenyl-phospho-ribose) to a keto-intermediate. This is then reduced to decaprenyl-phospho-arabinose (DPA—a substrate for AG biosynthesis) by DprE2 using NADH (Brecik et al., 2015). DprE1 localizes to the CW, negating the need of some DprE1 targeting drugs to enter into the cytoplasm (Brecik et al., 2015). DprE1 was first discovered as the target for benzothiazinones (BTZs), since then more than 15 DprE1 inhibitory compounds some of which have entered clinical trials have been identified (Degiacomi et al., 2020). The most promising BTZ compound to date, BTZ043, displayed a MIC of 1 ng/ml (0.23 nM) and is currently progressing through clinical trials. Macozinone (MCZ, also known as PBTZ169) is a BTZ043 derivative which has been medicinally optimized and has entered clinical trials. TBA-7371 is another DprE1 inhibitor with high potency and has also entered clinical trials (Degiacomi et al., 2020).
Mycolic Acid Biosynthesis and Potential Drug Targets
Targeting MA biosynthetic enzymes holds significant potential for developing new anti-tubercular drugs as MAs influence permeability and sensitivity to hydrophobic antibiotics (Jankute et al., 2015). Enzymes in the FAS-I and FAS-II pathways synthesize and facilitate the correct folding of short and long-chain fatty acids, respectively. Several enzymes in the FAS-I and FAS-II pathways are essential for viability and have been targeted, see Figure 1 and Table 1 (Sassetti et al., 2003; Baran et al., 2020). InhA is the target of isoniazid (INH) and structural analogues such as ethionamide (ETH) activated by the catalase-peroxidase KatG (Dessen et al., 1995). Novel InhA inhibitors (indicated in Table 1), unlike INH and ETH, do not require prior activation and have potential for treatment of DR-TB while some are also bactericidal against non-replicating Mtb (Holas et al., 2015; Martínez-Hoyos et al., 2016; Flint et al., 2020). KasA and KasB are targets of cerulenin, plastensimycin, thiolactomycin, and indazole sulfonamides (Abrahams et al., 2016; Abrahams and Besra, 2018). Tetrahydropyrazo (1,5-a)pyrimidine-3-carboxamides (THPPs) have recently been shown to target EchA6, a catalytically inactive enoyl-CoA hydratase required for shuttling fatty acyl-CoA esters into FAS-II for MA biosynthesis (Cox et al., 2016). MmaA1 is a target of the compound 3-(2-morpholinoacetamido)-N-(1,4-dihydro-4-oxoquinazolin-6-yl)benzamide (Veeravarapu et al., 2020). Benzofurans, coumestans, thiophenes, β-lactones target Pks13 (Wilson et al., 2013). Diarylcoumarins inhibit FabD32 and possess high bactericidal activity against Mtb (Stanley et al., 2013).
The mature MAs are translocated to the CW via the membrane embedded transporter—MmpL3 (Grzegorzewicz et al., 2012; Xu et al., 2017). Before translocation, the MAs are attached to trehalose by mycolyl-transferases to form trehalose monomycolates (TMMs) (Jankute et al., 2015). TMMs are attached to AG by the mycolyltransferase Ag85 complex. TMMs may also be converted to trehalose dimycolates (TDMs) by the Ag85 complex before being attached to AG (Jankute et al., 2015). Ag85C is a target of the compound I3-AG85 causing inhibition of incorporation of MAs into the mycobacterial CW (Warrier et al., 2012). Cyclipostins and cyclophostin analogs have been shown to inhibit Ag85 enzymes (Viljoen et al., 2018). MmpL3 is currently one of the most promising anti-TB targets (Vilchèze, 2020). Several diverse compounds (see Table 1) display inhibitory activity against MmpL3 either by direct binding or disruption of membrane potential (Xu et al., 2017; Dupont et al., 2019; Vilchèze, 2020; Yang et al., 2020).
The Variance of Cell Wall Targets in Clinical Strains
Mtb clinical isolates display differences in the composition of the CW that should be noted for drug discovery approaches (Moopanar and Mvubu, 2020). Specific differences in abundance of pthiotriol dimycerosate and PDIM, have been noted in clinical isolates of the East Asian/Beijing, Indo-Oceanic, and Euro-American lineages (Krishnan et al., 2011). These lipids form a hydrophobic barrier to antibiotics and therefore the differences in lipid profiles or differences in the abundance of enzymes associated them can be correlated with differential susceptibility to antibiotics. For example, the proteomic profile of an INH resistant clinical strain of the Euro-American lineage (T-family), and an INH resistant lab strain were analyzed by LC-MS/MS and this revealed an altered abundance of FAS-II pathway enzymes required for MA biosynthesis. This altered abundance of FAS-II enzymes was compensated for through alternative enzymes including FabG4, HtdX, FadD13, and members of the mymA operon, which has been shown to play a role in MA biosynthesis (Singh et al., 2005; Mehaffy et al., 2018). Although the change in protein abundance only affected the abundance of the two more abundant MAs in the clinical INH resistant strain, these lineage specific differences in the abundance of CW associated biosynthetic enzymes should be considered during development of new anti-mycobacterial drugs. A later study conducted by Seepe et al. in two drug susceptible and resistant East-Asian (Beijing) clinical isolates exposed to INH further elucidated the differences in MA composition amongst clinical isolates and found differential expression of FAS-II pathway enzymes highlighting the importance of considering the basis of lineage specific differences for CW targeted TB drug development (Seepe, 2011).
Despite the promise of using β-lactam:β-lactamase combinations, it should be noted that susceptibility to this combination treatment is variable among clinical isolates (Gonzalo and Drobniewski, 2013; Forsman et al., 2015; Zhang et al., 2016). A recent study assessing 89 clinical isolates from South Africa demonstrated that approximately half of the clinical isolates studied were hyper-susceptible to Amx/Clv as compared to the reference strains (Cohen et al., 2016). Whole genome sequencing of Amx/Clv hypersusceptible LAM4 isolates identified polymorphisms in the genes aftD, PE-PGRS genes, pks12, and ubiA associated with CW biosynthesis. Hence, screens for new PG targeting compounds should include different lineages of Mtb, together with representatives of drug resistant strains.
TnSeq has recently been used to conduct fitness profiling of clinical isolates, identifying notable differences in the requirement of various genes for viability and antibiotic susceptibility (Carey et al., 2018). Several CW biosynthesis genes including murI, pbpA, ldtB, otsA, pimE, pssA, papA3, and CW associated lipoprotein biosynthesis genes (lppL and lppX) were found to be differentially required for fitness in various clinical isolates (Carey et al., 2018). Moreover, TnSeq was also used recently to identify genetic variants that influence drug efficacy in vivo. Several Mtb mutants displayed altered susceptibility to TB drugs in the murine model of TB disease, many of which were associated with CW pathways described herein. The findings suggest that genetic variants that may be associated with drug resistance in clinical Mtb isolates do not alter in vitro drug susceptibility but can still influence drug efficacy in vivo (Bellerose et al., 2020). This suggested that the genetic backgrounds of different clinical strains can impact the applicability of new anti-mycobacterial drugs in the clinical setting.
Future Directions and Concluding Remarks
A significant amount of work has been done in the identification of potential drug targets in the CW and their inhibitory compounds, however, only a few lead compounds have entered clinical trials (https://www.newtbdrugs.org/pipeline/clinical). Improved strategies to test and fast-track the development of these compounds including the use of TnSeq and CRISPRi in clinical strains to study target vulnerability is of importance given the high prevalence of DR-TB. When considering these recent developments, the mycobacterial cell surface continues to hold promise for the development of shorter, more effect TB treatments.
Author Contributions
MS and CE wrote the article. MS prepared the figure. BK provided the concept, critical review, and edited the final draft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the South African National Research Foundation (to BDK and CE), the South African Medical Research Council (to BK, CE, and MS) and the Centre for Aids Prevention Research in South Africa (CAPRISA, to BK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abrahams K. A., Besra G. S. (2018). Mycobacterial cell wall biosynthesis: a multifaceted antibiotic target. Parasitology 145, 116–133. doi: 10.1017/S0031182016002377
Abrahams K. A., Chung C.-W., Ghidelli-Disse S., Rullas J., Rebollo-López M. J., Gurcha S. S., et al. (2016). Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 7, 1–13. doi: 10.1038/ncomms12581
Alderwick L. J., Harrison J., Lloyd G. S., Birch H. L. (2015). The mycobacterial cell wall—peptidoglycan and arabinogalactan. Cold Spring Harbor Perspect. Med. 5 (8), a021113. doi: 10.1101/cshperspect.a021113
Azam M. A., Jayaram U. J. J. O. E. I., Chemistry M. (2016). Inhibitors of alanine racemase enzyme: a review J. Enzyme Inhib. Med. Chem. 31, 517–526. doi: 10.3109/14756366.2015.1050010
Baran M., Grimes K. D., Sibbald P. A., Fu P., Boshoff H. I., Wilson D. J., et al. (2020). Development of small-molecule inhibitors of fatty acyl-AMP and fatty acyl-CoA ligases in. Mycobacterium Tuberculosis 201, 112408. doi: 10.1016/j.ejmech.2020.112408
Baranowski C., Welsh M. A., Sham L.-T., Eskandarian H. A., Lim H. C., Kieser K. J., et al. (2018). Maturing Mycobacterium smegmatis peptidoglycan requires non-canonical crosslinks to maintain shape. Elife 7, e37516. doi: 10.7554/eLife.37516
Bellerose M. M., Proulx M. K., Smith C. M., Baker R. E., Ioerger T. R., Sassetti C. M. (2020). Distinct Bacterial Pathways Influence the Efficacy of Antibiotics against Mycobacterium tuberculosis. Msystems 5, e00396–00320. doi: 10.1128/mSystems.00396-20
Bellinzoni M., Wehenkel A. M., Durán R., Alzari P. M. (2019). Novel mechanistic insights into physiological signaling pathways mediated by mycobacterial Ser/Thr protein kinases. Genes Immun. 20, 383–393. doi: 10.1038/s41435-019-0069-9
Bensen D., Rodriguez S., Nix J., Cunningham M., Tari L. (2012). Structure of MurA (UDP-N-acetylglucosamine enolpyruvyl transferase) from Vibrio fischeri in complex with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Acta Crystallographica Section F.: Struct. Biol. Crystallization Commun. 68, 382–385. doi: 10.1107/S1744309112006720
Bhat Z. S., Rather M. A., Maqbool M., Lah H. U., Yousuf S. K., Ahmad Z. (2017). Cell wall: a versatile fountain of drug targets in Mycobacterium tuberculosis. Biomed. Pharmacother. 95, 1520–1534. doi: 10.1016/j.biopha.2017.09.036
Boutte C. C., Baer C. E., Papavinasasundaram K., Liu W., Chase M. R., Meniche X., et al. (2016). A cytoplasmic peptidoglycan amidase homologue controls mycobacterial cell wall synthesis. Elife 5, e14590. doi: 10.7554/eLife.14590
Brecik M., Centárová I., Mukherjee R., Kolly G. S., HuszáR S., Bobovská A., et al (2015). DprE1 is a vulnerable tuberculosis drug target due to its cell wall localization. .ACS Chem. Biol. 10, 1631–1636. doi: 10.1021/acschembio.5b00237
Carette X., Platig J., Young D. C., Helmel M., Young A. T., Wang Z., et al. (2018). Multisystem analysis of Mycobacterium tuberculosis reveals kinase-dependent remodeling of the pathogen-environment interface. MBio 9 (2), e02333–02317. doi: 10.1128/mBio.02333-17
Carey A. F., Rock J. M., Krieger I. V., Chase M. R., Fernandez-Suarez M., Gagneux S., et al. (2018). TnSeq of Mycobacterium tuberculosis clinical isolates reveals strain-specific antibiotic liabilities. PloS Pathoges 14, e1006939. doi: 10.1371/journal.ppat.1006939
Carro L. (2019). Recent progress in the development of small-molecule FtsZ inhibitors as chemical tools for the development of novel antibiotics. Antibiotics 8:217. doi: 10.3390/antibiotics8040217
Chen K.-T., Chen P.-T., Lin C.-K., Huang L.-Y., Hu C.-M., Chang Y.-F., et al. (2016). Structural investigation of Park’s nucleotide on bacterial translocase MraY: Discovery of unexpected MraY inhibitors. Sci. Rep. 6, 1–11. doi: 10.1038/srep31579
Chu J., Vila-Farres X., Inoyama D., Gallardo-Macias R., Jaskowski M., Satish S., et al. (2018). Human microbiome inspired antibiotics with improved β-lactam synergy against MDR Staphylococcus aureus. ACS Infect. Dis. 4, 33–38. doi: 10.1021/acsinfecdis.7b00056
Chung B. C., Mashalidis E. H., Tanino T., Kim M., Matsuda A., Hong J., et al. (2016). Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 533, 557–560. doi: 10.1038/nature17636
Cohen K. A., El-Hay T., Wyres K. L., Weissbrod O., Munsamy V., Yanover C., et al. (2016). Paradoxical hypersusceptibility of drug-resistant Mycobacterium tuberculosis to β-lactam antibiotics. EBioMedicine 9, 170–179. doi: 10.1016/j.ebiom.2016.05.041
Conradie F., Diacon A. H., Ngubane N., Howell P., Everitt D., Crook A. M., et al. (2020). Bedaquiline, pretomanid and linezolid for treatment of extensively drug resistant, intolerant or non-responsive multidrug resistant pulmonary tuberculosis. New Engl. J. Med. 382, 893. doi: 10.1056/NEJMoa1901814
Cox J. A., Abrahams K. A., Alemparte C., Ghidelli-Disse S., Rullas J., Angulo-Barturen I., et al. (2016). THPP target assignment reveals EchA6 as an essential fatty acid shuttle in mycobacteria. Nat. Microbiol. 1, 1–10. doi: 10.1038/nmicrobiol.2015.6
De Chiara C., Homšak M., Prosser G. A., Douglas H. L., Garza-Garcia A., Kelly G., et al. (2020). D-Cycloserine destruction by alanine racemase and the limit of irreversible inhibition. Nat. Chem. Biol. 16, 686–694. doi: 10.1038/s41589-020-0498-9
De Jager V. R., Vanker N., Van Der Merwe L., Van Brakel E., Muliaditan M., Diacon A. H. (2020). Optimizing β-Lactams against Tuberculosis. Am. J. Respiratory Crit. Care Med. 201, 1155–1157. doi: 10.1164/rccm.201911-2149LE
De Smet K. A., Kempsell K. E., Gallagher A., Duncan K., Young D. B. (1999). Alteration of a single amino acid residue reverses fosfomycin resistance of recombinant MurA from Mycobacterium tuberculosis. Microbiology 145, 3177–3184. doi: 10.1099/00221287-145-11-3177
De Wet T. J., Winkler K. R., Mhlanga M. M., Mizrahi V., Warner D. F. (2020). Arrayed CRISPRi and quantitative imaging describe the morphotypic landscape of essential mycobacterial genes. bioRxiv 000372. doi: 10.1101/2020.03.20.000372
Degiacomi G., Belardinelli J. M., Pasca M. R., Rossi E. D., Riccardi G., Chiarelli L. R. (2020). Promiscuous targets for antitubercular drug discovery: The paradigm of DprE1 and MmpL3. Appl. Sci. 10:623. doi: 10.3390/app10020623
Dessen A., Quemard A., Blanchard J. S., Jacobs W. R., Sacchettini J. C. (1995). Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 267, 1638–1641. doi: 10.1126/science.7886450
Dulberger C. L., Rubin E. J., Boutte C. C. (2019). The mycobacterial cell envelope—a moving target. Nat. Rev. Microbiol. 18 47–59. doi: 10.1038/s41579-019-0273-7
Dupont C., Chen Y., Xu Z., Roquet-Banères F., Blaise M., Witt A.-K., et al. (2019). A piperidinol-containing molecule is active against Mycobacterium tuberculosis by inhibiting the mycolic acid flippase activity of MmpL3. J. Biol. Chem. 294, 17512–17523. doi: 10.1074/jbc.RA119.010135
Faion L., Djaout K., Frita R., Pintiala C., Cantrelle F.-X., Moune M., et al. (2020). Discovery of the first Mycobacterium tuberculosis MabA (FabG1) inhibitors through a fragment-based screening. Eur. J. Med. Chem. 200, 112440. doi: 10.1016/j.ejmech.2020.112440
Flint L., Korkegian A., Parish T. (2020). InhA inhibitors have activity against non-replicating Mycobacterium tuberculosis. bioRxiv 2020, 257782. doi: 10.1101/2020.08.19.257782
Forsman L. D., Giske C., Bruchfeld J., Schön T., Juréen P., Ängeby K. (2015). Meropenem-clavulanate has high in vitro activity against multidrug-resistant Mycobacterium tuberculosis. Int. J. Mycobacteriol. 4, 80–81. doi: 10.1016/j.ijmyco.2014.10.018
Gautam A., Vyas R., Tewari R. (2011). Peptidoglycan biosynthesis machinery: a rich source of drug targets. Crit. Rev. Biotechnol. 31, 295–336. doi: 10.3109/07388551.2010.525498
Gold B., Smith R., Nguyen Q., Roberts J., Ling Y., Lopez Quezada L., et al. (2016). Novel cephalosporins selectively active on nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 59, 6027–6044. doi: 10.1021/acs.jmedchem.5b01833
Gonzalo X., Drobniewski F. (2013). Is there a place for β-lactams in the treatment of multidrug-resistant/extensively drug-resistant tuberculosis? Synergy between meropenem and amoxicillin/clavulanate. J. Antimicrob. Chemother. 68, 366–369. doi: 10.1093/jac/dks395
Grzegorzewicz A. E., Pham H., Gundi V. A., Scherman M. S., North E. J., Hess T., et al. (2012). Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8, 334–341. doi: 10.1038/nchembio.794
Grzegorzewicz A. E., Eynard N., QuéMard A., North E. J., Margolis A., Lindenberger J. J., et al. (2015). Covalent modification of the Mycobacterium tuberculosis FAS-II dehydratase by Isoxyl and Thiacetazone. ACS Infect. Dis. 1, 91–97. doi: 10.1021/id500032q
Grzelak E. M., Choules M. P., Gao W., Cai G., Wan B., Wang Y., et al. (2019). Strategies in anti-Mycobacterium tuberculosis drug discovery based on phenotypic screening. J. Antibiotics 72, 719–728. doi: 10.1038/s41429-019-0205-9
Harrison J., Lloyd G., Joe M., Lowary T. L., Reynolds E., Walters-Morgan H., et al. (2016). Lcp1 is a phosphotransferase responsible for ligating arabinogalactan to peptidoglycan in Mycobacterium tuberculosis. MBio 7 (4), e00972–00916. doi: 10.1128/mBio.00972-16
Hering J., Dunevall E., Ek M., Brändén G. (2018). Structural basis for selective inhibition of antibacterial target MraY, a membrane-bound enzyme involved in peptidoglycan synthesis. Drug Discov. Today 23, 1426–1435. doi: 10.1016/j.drudis.2018.05.020
Hett E. C., Rubin E. J. (2008). Bacterial growth and cell division: a mycobacterial perspective. Microbiol. Mol. Biol. Rev. 72, 126–156. doi: 10.1128/MMBR.00028-07
Holas O., Ondrejcek P., Dolezal M. (2015). Mycobacterium tuberculosis enoyl-acyl carrier protein reductase inhibitors as potential antituberculotics: development in the past decade. J. Enzyme Inhibition Med. Chem. 30, 629–648. doi: 10.3109/14756366.2014.959512
Hugonnet J.-E., Tremblay L. W., Boshoff H. I., Barry C. E., Blanchard J. S.. (2009). Meropenem-clavulanate is effective against extensively drug-resistant. Mycobacterium Tuberculosis 323, 1215–1218. doi: 10.1126/science.1167498
Huszár S., Singh V., Polčicová A., Baráth P., Barrio M. B., Lagrange S., et al. (2017). N-Acetylglucosamine-1-phosphate transferase, WecA, as a validated drug target in Mycobacterium tuberculosis. Antimicrobial Agents Chemother. 61 (11), e01310–01317. doi: 10.1128/AAC.01310-17
Jankute M., Cox J. A., Harrison J., Besra G. S. (2015). Assembly of the mycobacterial cell wall. Annual. Rev. Microbiol. 69, 405–423. doi: 10.1146/annurev-micro-091014-104121
Jeon A. B., Obregon-Henao A., Ackart D. F., Podell B. K., Belardinelli J. M., Jackson M., et al. (2017). 2-aminoimidazoles potentiate ß-lactam antimicrobial activity against Mycobacterium tuberculosis by reducing ß-lactamase secretion and increasing cell envelope permeability. PloS One 12, e0180925. doi: 10.1371/journal.pone.0180925
Jeon A. B., Ackart D. F., Li W., Jackson M., Melander R. J., Melander C., et al. (2019). 2-aminoimidazoles collapse mycobacterial proton motive force and block the electron transport chain. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-018-38064-7
Kaur P., Rausch M., Malakar B., Watson U., Damle N. P., Chawla Y., et al. (2019). LipidII interaction with specific residues of Mycobacterium tuberculosis PknB extracytoplasmic domain governs its optimal activation. Nat. Commun. 10, 1231. doi: 10.1038/s41467-019-09223-9
Kieser K. J., Rubin E. J. (2014). How sisters grow apart: mycobacterial growth and division. Nat. Rev. Microbiol. 12, 550–562. doi: 10.1038/nrmicro3299
Kim M. G., Strych U., Krause K., Benedik M., Kohn H. (2003). N (2)-substituted D, L-cycloserine derivatives. J. Antibiot. 56, 160–168. doi: 10.7164/antibiotics.56.160
Konyariková Z., Savková K., Kozmon S., Mikušová K. (2020). Biosynthesis of Galactan in Mycobacterium tuberculosis as a viable TB Drug Target? Antibiotics 9:20. doi: 10.3390/antibiotics9010020
Kouidmi I., Levesque R. C., Paradis-Bleau C. (2014). The biology of Mur ligases as an antibacterial target. Mol. Microbiol. 94, 242–253. doi: 10.1111/mmi.12758
Kremer L., Guérardel Y., Gurcha S. S., Locht C., Besra G. S. (2002). Temperature-induced changes in the cell-wall components of Mycobacterium thermoresistibile. Microbiology 148, 3145–3154. doi: 10.1099/00221287-148-10-3145
Krishnan N., Malaga W., Constant P., Caws M., Chau T. T. H., Salmons J., et al. (2011). Mycobacterium tuberculosis lineage influences innate immune response and virulence and is associated with distinct cell envelope lipid profiles. PloS One 6, e23870. doi: 10.1371/journal.pone.0023870
Kumar P., Arora K., Lloyd J. R., Lee I. Y., Nair V., Fischer E., et al. (2012). Meropenem inhibits D,D-carboxypeptidase activity in M ycobacterium tuberculosis. Mol. Microbiol. 86, 367–381. doi: 10.1111/j.1365-2958.2012.08199.x
Kumar P., Chauhan V., Silva J. R. A., Lameira J., D’andrea F. B., Li S.-G., et al. (2017). Mycobacterium abscessus L,D-transpeptidases are susceptible to inactivation by carbapenems and cephalosporins but not penicillins. Antimicrobial Agents Chemother. 61, e00866–00817. doi: 10.1128/AAC.00866-17
Laddomada F., Miyachiro M. M., Jessop M., Patin D., Job V., Mengin-Lecreulx D., et al. (2019). The MurG glycosyltransferase provides an oligomeric scaffold for the cytoplasmic steps of peptidoglycan biosynthesis in the human pathogen. Bordetella Pertussis 9, 1–17. doi: 10.1038/s41598-019-40966-z
Lehmann J., Cheng T. Y., Aggarwal A., Park A. S., Zeiler E., Raju R. M., et al. (2018). An antibacterial β-lactone kills Mycobacterium tuberculosis by disrupting mycolic acid biosynthesis. Angewandte Chem. Int. Ed. 57, 348–353. doi: 10.1002/anie.201709365
Levefaudes M., Patin D., De Sousa-D’auria C., Chami M., Blanot D., Hervé M., et al. (2015). Diaminopimelic acid amidation in corynebacteriales: new insights into the role of LtsA in peptidoglycan modification. J. Biol. Chem. 290, 13079–13094. doi: 10.1074/jbc.M115.642843
Li F. K., Rosell F. I., Gale R. T., Simorre J.-P., Brown E. D., Strynadka N. C. (2020). Crystallographic analysis of Staphylococcus aureus LcpA, the primary wall teichoic acid ligase. J. Biol. Chem. 295, 2629–2639. doi: 10.1074/jbc.RA119.011469
Lo M.-C., Men H., Branstrom A., Helm J., Yao N., Goldman R., et al. (2000). A new mechanism of action proposed for ramoplanin. J. Am. Chem. Soc. 122, 3540–3541. doi: 10.1021/ja000182x
Lopez Quezada L. A., Smith R., Lupoli T., Edoo Z., Li X., Gold B., et al. (2020). Activity-based protein profiling reveals that cephalosporins selectively active on nonreplicating Mycobacterium tuberculosis bind multiple protein families and spare peptidoglycan transpeptidases. Front. Microbiol. 11:1248. doi: 10.3389/fmicb.2020.01248
Maitra A., Munshi T., Healy J., Martin L. T., Vollmer W., Keep N. H., et al. (2019). Cell wall peptidoglycan in Mycobacterium tuberculosis: An Achilles’ heel for the TB-causing pathogen. FEMS Microbiol. Rev. 43, 548–575. doi: 10.1093/femsre/fuz016
Martínez-Hoyos M., Perez-Herran E., Gulten G., Encinas L., Álvarez-Gómez D., Alvarez E., et al. (2016). Antitubercular drugs for an old target: GSK693 as a promising InhA direct inhibitor. EBioMedicine 8, 291–301. doi: 10.1016/j.ebiom.2016.05.006
Mashalidis E. H., Lee S.-Y. (2020). Structures of bacterial MraY and human GPT provide insights into rational antibiotic design. J. Mol. Biol. 432, 4946–4963. doi: 10.1016/j.jmb.2020.03.017
Mathew B., Hobrath J. V., Ross L., Connelly M. C., Lofton H., Rajagopalan M., et al. (2016). Screening and development of new inhibitors of FtsZ from Mycobacterium tuberculosis. PloS One 11, e0164100. doi: 10.1371/journal.pone.0164100
Mavrici D., Marakalala M. J., Holton J. M., Prigozhin D. M., Gee C. L., Zhang Y. J., et al. (2014). Mycobacterium tuberculosis FtsX extracellular domain activates the peptidoglycan hydrolase, RipC. Proc. Natl. Acad. Sci. 111, 8037–8042. doi: 10.1073/pnas.1321812111
Mehaffy C., Islam M. N., Fitzgerald B., Belisle J., Prenni J., Dobos K. (2018). Biochemical characterization of isoniazid-resistant Mycobacterium tuberculosis: can the analysis of clonal strains reveal novel targetable pathways? Mol. Cell. Proteomics 17, 1685–1701. doi: 10.1074/mcp.RA118.000821
Mir M., Asong J., Li X., Cardot J., Boons G.-J., Husson R. N. (2011). The extracytoplasmic domain of the Mycobacterium tuberculosis Ser/Thr kinase PknB binds specific muropeptides and is required for PknB localization. PloS Pathog. 7, e1002182. doi: 10.1371/journal.ppat.1002182
Mishra S., Shukla P., Bhaskar A., Anand K., Baloni P., Jha R. K., et al. (2017). Efficacy of β-lactam/β-lactamase inhibitor combination is linked to WhiB4-mediated changes in redox physiology of Mycobacterium tuberculosis. Elife 6, e25624. doi: 10.7554/eLife.25624
Moopanar K., Mvubu N. (2020). Lineage-specific differences in lipid metabolism and its impact on clinical strains of Mycobacterium tuberculosis. Microbial Pathogen. 146, 104250. doi: 10.1016/j.micpath.2020.104250
Münch D., Roemer T., Lee S. H., Engeser M., Sahl H. G., Schneider T. (2012). Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus. PloS Pathog. 8, e1002509. doi: 10.1371/journal.ppat.1002509
Ostash B., Doud E., Fedorenko V. (2010). The molecular biology of moenomycins: towards novel antibiotics based on inhibition of bacterial peptidoglycan glycosyltransferases. Biol. Chem. 391, 499–504. doi: 10.1515/bc.2010.053
Patel H., Karpoormath R., Palkar M. (2020). Exploring MDR-TB inhibitory potential of 4-amino quinazolines as Mycobacterium tuberculosis N-acetylglucosamine-1-phosphate uridyltransferase (GlmU MTB) inhibitors. Chem. Biodivers. 17, e2000237. doi: 10.1002/cbdv.202000237
Pazos M., Peters K. (2019). Peptidoglycan. Bacterial Cell Walls Membranes 92, 127–168. doi: 10.1007/978-3-030-18768-2_5
Pidgeon S. E., Apostolos A. J., Nelson J. M., Shaku M., Rimal B., Islam M. N., et al. (2019). L,D-Transpeptidase Specific Probe Reveals Spatial Activity of Peptidoglycan Cross-Linking. ACS Chem. Biol. 14, 2185–2196. doi: 10.1021/acschembio.9b00427
Prosser G. A., De Carvalho L. P. S. (2013). Metabolomics reveal d-alanine: d-alanine ligase as the target of d-cycloserine. Mycobacterium Tuberculosis 4, 1233–1237. doi: 10.1021/ml400349n
Rego E. H., Audette R. E., Rubin E. J. (2017). Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157. doi: 10.1038/nature22361
Rock J. (2019). Tuberculosis drug discovery in the CRISPR era. PloS Pathog. 15, e1007975. doi: 10.1371/journal.ppat.1007975
Sassetti C. M., Boyd D. H., Rubin E.J.J.M.M. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Seepe P. M. (2011). Differential expression of genes in clinical strains of Mycobacterium tuberculosis in response to isoniazid. Doctoral dissertation (University of Stellenbosch). Available at: https://scholar.sun.ac.za/handle/10019.1/6774.
Senior S. J., Illarionov P. A., Gurcha S. S., Campbell I. B., Schaeffer M. L., Minnikin D. E., et al. (2003). Biphenyl-based analogues of thiolactomycin, active against Mycobacterium tuberculosis mtFabH fatty acid condensing enzyme. Bioorg. Med. Chem. Lett. 13, 3685–3688. doi: 10.1016/j.bmcl.2003.08.015
Singh A., Gupta R., Vishwakarma R., Narayanan P., Paramasivan C., Ramanathan V., et al. (2005). Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J. Bacteriol. 187, 4173–4186. doi: 10.1128/JB.187.12.4173-4186.2005
Singh V., Dhar N., Pató J., Kolly G. S., Korduláková J., Forbak M., et al. (2017). Identification of aminopyrimidine-sulfonamides as potent modulators of Wag31-mediated cell elongation in mycobacteria. Mol. Microbiol. 103, 13–25. doi: 10.1111/mmi.13535
Siricilla S., Mitachi K., Wan B., Franzblau S. G., Kurosu M. (2015). Discovery of a capuramycin analog that kills nonreplicating Mycobacterium tuberculosis and its synergistic effects with translocase I inhibitors. J. Antibiotics 68, 271–278. doi: 10.1038/ja.2014.133
Stanley S. A., Kawate T., Iwase N., Shimizu M., Clatworthy A. E., Kazyanskaya E., et al. (2013). Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. 110, 11565–11570. doi: 10.1073/pnas.1302114110
Story-Roller E., Lamichhane G. (2018). Have we realized the full potential of β-lactams for treating drug-resistant TB? IUBMB Life 70, 881–888. doi: 10.1002/iub.1875
Syson K., Stevenson C. E., Rashid A. M., Saalbach G., Tang M., Tuukkanen A., et al. (2014). Structural insight into how Streptomyces coelicolor maltosyl transferase GlgE binds α-maltose 1-phosphate and forms a maltosyl-enzyme intermediate. Biochemistry 53, 2494–2504. doi: 10.1021/bi500183c
Tanino T., Al-Dabbagh B., Mengin-Lecreulx D., Bouhss A., Oyama H., Ichikawa S., et al. (2011). Mechanistic analysis of muraymycin analogues: a guide to the design of MraY inhibitors. J. Med. Chem. 54, 8421–8439. doi: 10.1021/jm200906r
Thanna S., Sucheck S. J. (2016). Targeting the trehalose utilization pathways of Mycobacterium tuberculosis. MedChemComm 7, 69–85. doi: 10.1039/C5MD00376H
Tooke C. L., Hinchliffe P., Bragginton E. C., Colenso C. K., Hirvonen V. H., Takebayashi Y., et al. (2019). β-Lactamases and β-lactamase inhibitors in the 21st century. J. Mol. Biol. 431, 3472–3500. doi: 10.1016/j.jmb.2019.04.002
Turapov O., Forti F., Kadhim B., Ghisotti D., Sassine J., Straatman-Iwanowska A., et al. (2018). Two faces of CwlM, an essential PknB substrate, in Mycobacterium tuberculosis. Cell Rep. 25, 57–67. e55. doi: 10.1016/j.celrep.2018.09.004
Veeravarapu H., Malkhed V., Mustyala K. K., Vadija R., Malikanti R., Vuruputuri U., et al. (2020). Structure-based drug design, synthesis and screening of MmaA1 inhibitors as novel anti-TB agents. Mol. Diversity 24, 1–16. doi: 10.1007/s11030-020-10107-0
Vilchèze C. (2020). Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Appl. Sci. 10:2278. doi: 10.3390/app10072278
Viljoen A., Richard M., Nguyen P. C., Fourquet P., Camoin L., Paudal R. R., et al. (2018). Cyclipostins and cyclophostin analogs inhibit the antigen 85C from Mycobacterium tuberculosis both in vitro and in vivo. J. Biol. Chem. 293, 2755–2769. doi: 10.1074/jbc.RA117.000760
Warrier T., Tropis M., Werngren J., Diehl A., Gengenbacher M., Schlegel B., et al. (2012). Antigen 85C inhibition restricts Mycobacterium tuberculosis growth through disruption of cord factor biosynthesis. Antimicrobial Agents Chemother. 56, 1735–1743. doi: 10.1128/AAC.05742-11
WHO (2019a). Global tuberculosis report 2019. Available at: https://www.who.int/tb/global-report-2019 (Accessed 03 September 2020).
WHO (2019b). WHO consolidated guidelines on drug-resistant tuberculosis treatment. Available at: https://www.who.int/tb/publications/2019/consolidated-guidelines-drug-resistant-TB-treatment/en/ (Accessed 03 September 2020).
Wilson R., Kumar P., Parashar V., Vilchèze C., Veyron-Churlet R., Freundlich J. S., et al. (2013). Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 9, 499–506. doi: 10.1038/nchembio.1277
Xu Z., Meshcheryakov V. A., Poce G., Chng S.-S. (2017). MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. 114, 7993–7998. doi: 10.1073/pnas.1700062114
Yang X., Hu T., Yang X., Xu W., Yang H., Guddat L. W., et al. (2020). Structural basis for the inhibition of mycobacterial MmpL3 by NITD-349 and SPIRO. J. Mol. Biol. 432, 4426–34. doi: 10.1016/j.jmb.2020.05.019
Zhang R., Pan Y. T., He S., Lam M., Brayer G. D., Elbein A. D., et al. (2011). Mechanistic analysis of trehalose synthase from Mycobacterium smegmatis. J. Biol. Chem. 286, 35601–35609. doi: 10.1074/jbc.M111.280362
Zhang D., Wang Y., Lu J., Pang Y. (2016). In vitro activity of β-lactams in combination with β-lactamase inhibitors against multidrug-resistant Mycobacterium tuberculosis isolates. Antimicrobial Agents Chemother. 60, 393–399. doi: 10.1128/AAC.01035-15
Zhang W., Lun S., Wang S.-H., Jiang X.-W., Yang F., Tang J., et al. (2018). Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against Mycobacterium tuberculosis. J. Med. Chem. 61, 791–803. doi: 10.1021/acs.jmedchem.7b01319
Keywords: tuberculosis drugs, cell surface, peptidoglycan, arabinogalactan, mycolic acids
Citation: Shaku M, Ealand C and Kana BD (2020) Cell Surface Biosynthesis and Remodeling Pathways in Mycobacteria Reveal New Drug Targets. Front. Cell. Infect. Microbiol. 10:603382. doi: 10.3389/fcimb.2020.603382
Received: 06 September 2020; Accepted: 20 October 2020;
Published: 12 November 2020.
Edited by:
Vinayak Singh, University of Cape Town, South AfricaReviewed by:
Babak Javid, University of California, San Francisco, United StatesAmit Singh, Indian Institute of Science (IISc), India
Copyright © 2020 Shaku, Ealand and Kana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bavesh D. Kana, YmF2ZXNoLmthbmFAbmhscy5hYy56YQ==