 Wei Wang1
Wei Wang1 Yong Nie
Yong Nie Bo Huang
Bo Huang- 1Anhui Provincial Key Laboratory for Microbial Pest Control, Anhui Agricultural University, Hefei, China
- 2College of Life Sciences, Shandong Normal University, Jinan, China
Sarocladium terricola is a species of ascomycete fungus that has been recognized as a biocontrol agent for managing animal and plant pathogens, and exhibits significant potential as a feed additive. In this study, we utilized a combination of short-read Illumina sequencing and long-read PacBio sequencing to sequence, assemble, and analyze the genome of S. terricola. The resulting genome consisted of 11 scaffolds encompassing 30.27 Mb, with a GC content of 54.07%, and 10,326 predicted protein coding gene models. We utilized 268 single-copy ortholog genes to reconstruct the phylogenomic relationships among 26 ascomycetes, and found that S. terricola was closely related to two Acremonium species. We also determined that the ergosterol content of S. terricola was synthesized to nearly double levels when cultured in potato dextrose media compared to bean media (4509 mg/kg vs. 2382 mg/kg). Furthermore, transcriptome analyses of differentially expressed genes suggested that the ergosterol synthesis genes ERG3, ERG5, and ERG25 were significantly up-regulated in potato dextrose media. These results will help us to recognize metabolic pathway of ergosterol biosynthesis of S. terricloa comprehensivelly.
Introduction
The genus Sarocladium W. Gams & D. Hawksw. (Sarocladiaceae, Hypocreales, Sordariomycetes, Ascomycota) is a diverse group of fungi proposed by Gams and Hawksworth in 1975, which includes important animal and plant pathogenic species (Gams and Hawksworth, 1975; Giraldo et al., 2015). Sarocladium comprises 30 species (http://www.indexfungorum.org, accessed on 13 November 13, 2022) with S. oryzae (Sawada) W. Gams & D. Hawksw as its type species (Gams and Hawksworth, 1975). Notably, S. terricola (J.H. Mill., Giddens & A.A. Foster) A. Giraldo, Gené & Guarro, originally classified as Acremonium, was transferred based on molecular evidence and morphological features (Giraldo et al., 2015).
Sarocladium terricola culture (well-known as Acremonium terricola culture or ATC) is an important feed additive, possessing various biological properties such as antioxidant, immunomodulatory, and anti-inflammatory activities (Li et al., 2016; Li et al., 2018). Previous studies suggested that ATC plays anti-inflammatory and antioxidant roles in lipopolysaccharide-induced mastitis in rats (Li et al., 2020), and improved rumen fermentation, decreased somatic cell counts in milk, and enhanced milk yield in cows or sheep when used as an animal feed additive (Jiang et al., 2018; Li et al., 2018; Kong et al., 2022). S. terricola is known to synthesize various metabolites, including D-mannitol, galactomannan, ergosterol, cordycepin, and essential amino acids (Li et al., 2020). Among these, ergosterol, an essential component of fungal cell membranes that plays an important role in maintaining structure and function, is the main focus of this study (White et al., 1998; Abe et al., 2009; Guan et al., 2009). Ergosterol is a clinically available target for most antifungal agents, and it exhibits anti-inflammatory and antioxidant properties (Dopont et al., 2012; Dhingra and Cramer, 2017). Additionally, the content of ergosterol in S. terricola is higher than other metabolites, making it a potential main contributor to the anti-inflammatory activity of ATC.
As the cost of sequencing has decreased, the number of species with genome sequence assemblies has rapidly increased. For example, the NCBI database contains more than 2,770 genomes from Ascomycota (https://www.ncbi.nlm.nih.gov/genome/, accessed on November 13, 2022). Genome and transcriptome sequences have provided novel insights into fungal phylogenomics, biosynthesis of metabolites, mechanisms of symbiosis, and others (Zhao et al., 2010; Ren et al., 2021; Sun et al., 2022; Wu et al., 2022; Zhao et al., 2022b; Zhao et al., 2022a). Although genome and mitochondrial genome sequences in the genus Sarocladium have been published (Hittalmani et al., 2016; Yao et al., 2016; Tian et al., 2022), phylogenomic relationships, transcriptome sequences, and characterization of the ergosterol biosynthesis pathway have not been studied. Transcriptome analyses mainly study gene transcription and regulation, revealing the molecular mechanisms of specific biological processes, such as symbiotic interactions between fungi and plants (Ren et al., 2021).
In this study, we generated a high-quality genome sequence assembly of S. terricola and characterized the S. terricola ergosterol biosynthesis pathway using transcriptome analyses combined with high performance liquid chromatography (HPLC).
Materials and methods
Strains, media, and fermentation
In this study, Sarocladium terricola was isolated from soil in Anhui Province, China, and deposited in the Anhui Agricultural University with the accession number RCEF 6201. The morphological trait is mainly as followed: Mycelia hyaline, 1.5–1.9 µm wide; Phialides subulate, solitary, 12–24 µm long, 2–3 µm wide at the base; Conidia fusiform, hyaline, sharply, 7–9 × 3–4 µm; Chlamydospores not observed.
Strains were incubated with potato dextrose agar media (PDA: potato 200 g/L, glucose 20 g/L, agar 20 g/L, and 1 L distilled water) at 25°C for 1 week to produce spores and then collected using sterile water. The spore suspension was adjusted to a concentration of 1 × 106/mL, then 1 mL spore suspension was inoculated into a 500 mL flask containing either 200 mL potato dextrose media (potato 200 g/L, glucose 20 g/L, and 1 L distilled water) or bean media (bean 30 g/L, glucose 20 g/L, and 1 L distilled water). Finally, the two types of media including HCBe (cluturing in bean media) and HCPo (cluturing in potato dextrose media) with difference in nitrogen source were incubated at 25°C with 150 rpm shaking for three days, and ergosterol was then detected using high performance liquid chromatography (HPLC). Three biological replicates were performed for each type of media.
Ergosterol measurement
We referred and improved the previous study (Nout et al., 1987): after three days, fresh mycelium was collected using a circulating water multi-purpose vacuum pump and placed in an oven at 45°C and allowed to equilibrate. To extract ergosterols, 0.5 g mycelium was mixed with 50 mL methanol in a 100 mL flask, disrupted with ultrasonic waves at 60°C for 30 min, and then centrifuged at 4000 g for 5 min to obtain supernatant. As a standard, 0.01 g ergosterol was placed in a 100 mL volumetric flask with methanol solvent added until volume reached 100 mL.
The ergosterol profiles were assayed using HPLC with a C18 column (250 mm × 4.6 mm × 5 µm). The analytical conditions were as follows: solvent system, anhydrous methanol; temperature, 35°C; injection volume, 10 µL; flow rate, 1 mL/min; and UV wavelength, 268 nm. Three technical replicates were assayed. The amount of ergosterol in the sample was determined by mass fraction (ω, unit is expressed in mg/kg), and the formula used for calculation was as follows:
A: peak area of ergosterol in sample.
As: peak area of ergosterol in standard working solution.
: mass concentration of ergosterol in standard working solution (unit: μg/mL).
V: final constant volume of sample solution (unit: mL).
m: mass of the test portion (unit: g).
f: sample dilution ratio.
Genome sequencing and assembly, gene prediction, and functional annotation
S. terricola was incubated in potato dextrose agar media (PDA: potato 200 g/L, glucose 20 g/L, agar 20 g/L, and 1 L distilled water) and cultured for two weeks. Total cell DNA was extracted from mycelia using methods as described in a previous study (Zhao et al., 2022b) and detected using the DNA/Protein Analyzer and 1% agarose gel electrophoresis. Two approaches were used for sequencing: paired-end short reads (300 bp) using the Illumina NovaSeq 6000 platform and long reads (more than 20 kb) using the PacBio Sequel II platform. Low-quality reads were removed (end-polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing, and short reads less than 1000 bp for PacBio sequencing), leaving only high-quality reads. The high-quality reads were de novo assembled using SMRT (version 5.1.0) downloads from https://www.pacb.com/support/software-downloads with default parameters (Chin et al., 2013) and assessed using BUSCO (version 5.2.2) with Ascomycota gene set downloaded from https://busco-data.ezlab.org/v4/data/lineages/ascomycota_odb10.2020-09-10.tar.gz (Simão et al., 2015).
Gene prediction was performed using four tools, Augustus (version 3.2.1) (Stanke et al., 2008), Genemark-ES (version 4.21) (Ter-Hovhannisyan et al., 2008), Genewise (version 2.20) (Birney et al., 2004), and SNAP (version 2010-07-28) (Johnson et al., 2008), and EvidenceModeler (version 1.1.1) (Haas et al., 2008) was then used to make integrated gene models. A total of ten databases were used to functionally annotate genes, namely Gene Ontology (GO) (Ashburner et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2016), Cluster of Orthologous Groups of proteins (COG) (Galperin et al., 2015), Swiss-Prot (Consortium, 2015), nr (https://www.ncbi.nlm.nih.gov/protein/), Pathogen Host Interactions (PHI) (Torto-Alalibo et al., 2009), Fungal Cytochrome P450 (Fischer et al., 2007), Carbohydrate-Active Enzymes (CAZy) (Levasseur et al., 2013), Virulence Factor Database (VFDB) (Chen et al., 2016), and Type III secretion system Effector protein (T3SS) (Vargas et al., 2012) using Diamond with an e-value less than 1 × 10−5. In addition, antiSMASH (fungal version) (Medema et al., 2011), Extensive de novo TE Annotator (EDTA) pipeline (version 1.9.5) (Ou et al., 2019), RNAmmer (version 1.2) (Lagesen et al., 2007), and tRNAscan-SE (version 2.0.5) (Lowe and Eddy, 1997) were used to predict gene clusters of secondary metabolites, repetitive elements, rRNAs, and tRNAs, respectively.
Phylogenomic analysis
In this study, a total of 26 species of ascomycetes (Table S1), including 25 genomes downloaded from genomic databases on the NCBI website and the strain of S. terricola (RCEF 6201) sequenced in this study, were used for phylogenomic analysis. This was performed using single copy ortholog genes that were identified using OrthoFinder (version 2.5.4) (Emms and Kelly, 2019) and aligned using MAFFT (version 7) with default parameters (Katoh and Standley, 2013). A Maximum Likelihood (ML) tree was reconstructed with RaxML (version 8.2.12) (Stamatakis, 2014) using the best optimal model of PROTGAMMAILGF with 100 bootstrap replicates.
Transcriptome sequencing and assembly
Six samples (three biological replicates from both types of growth media) of S. terricola were collected, and total RNA was extracted and purified using methods as described in a previous study (Ren et al., 2021). The transcriptome libraries were constructed and sequenced using the Illumina NovaSeq 6000 platform. After quality control and cleaning of raw reads, the high-quality reads were aligned and assembled using HISAT2 (version 0.1.6-beta) (Kim et al., 2015) and Cufflinks (version 2.2.1) (Trapnell et al., 2012), respectively.
Differential gene expression and enrichment analyses
Bowtie2 (version 2.2.5) (Langmead and Salzberg, 2012) was used to count the proportion of high-quality reads of each sample gene compared to the genome of S. terricola, and RSEM (version 1.2.12) (Li and Dewey, 2011) was then used to convert to standardized gene expression levels, viz. reads per kilobase of exon model per million mapped reads (FPKM values). DESeq2 (Wang et al., 2009) was used to identify differentially expressed genes (DEGs), using the criteria of fold change (FC) ≥ 2 and false discovery rate (FDR)< 0.05. Functional enrichments of DEGs were performed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway databases.
Results
Ergosterol content in different media
The present study measured the ergosterol content in Sarocladium terricola grown in two different growth media: potato dextrose media and bean media. The study found that ergosterol synthesis was more efficient in potato dextrose media, which resulted in a significantly higher content of ergosterol compared to bean media. Specifically, the ergosterol content in potato dextrose media was 4509 mg/kg, while in bean media, it was only 2382 mg/kg. This difference in ergosterol content between the two growth media was statistically significant with a p-value less than 0.01, as shown in Figure 1.
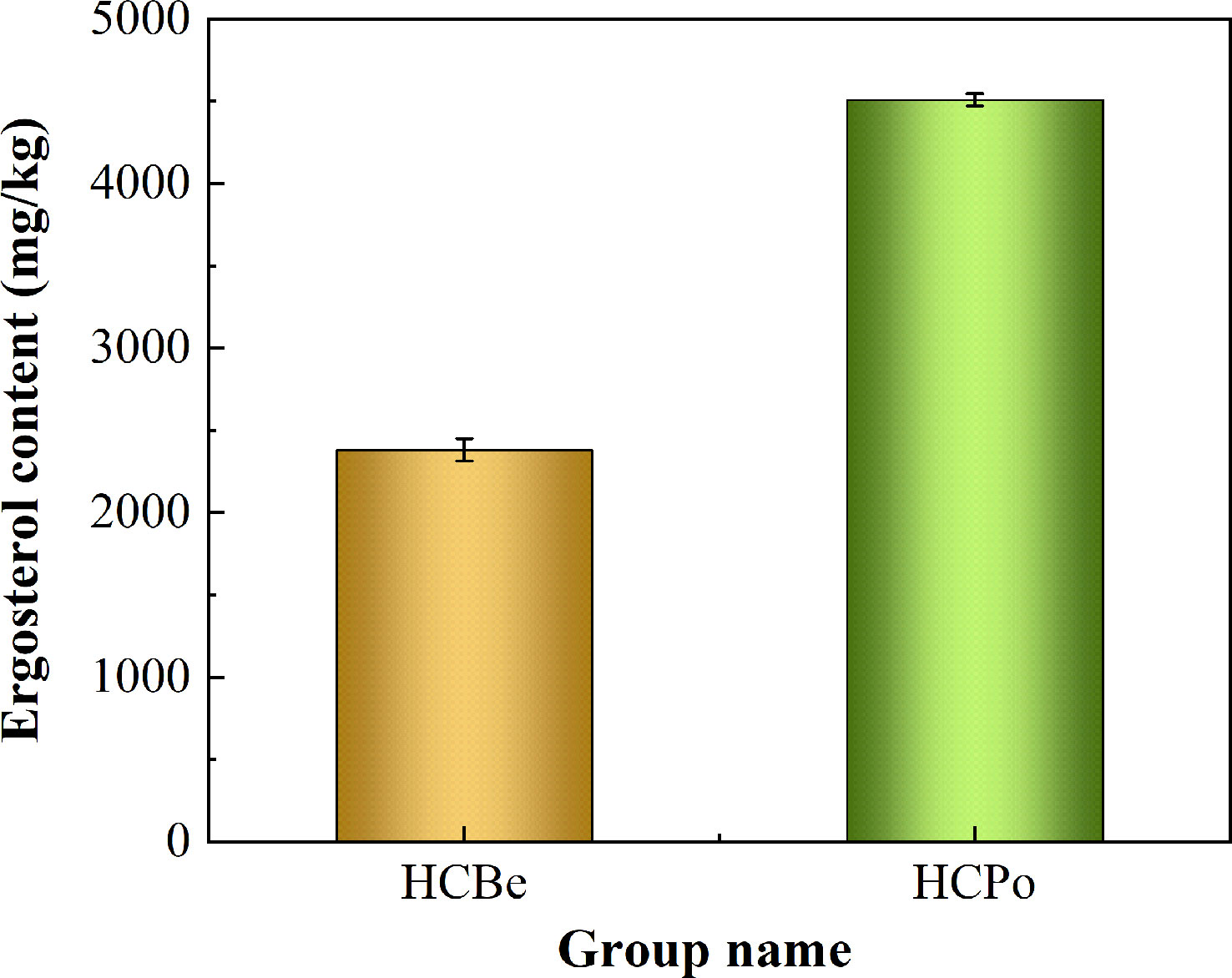
Figure 1 The ergosterol content in two types of media. Notes: HCBe, bean media; HCPo, potato dextrose media.
Genomic characteristics of the S. terricola assembly
The genome assembly of S. terricola spans 11 scaffolds and contains 30.27 Mb with a GC content of 54.07% (Table 1; Figure 2). We predicted a total of 10,326 protein-coding gene (PCG) models using four gene prediction tools. Among these gene models, 5,926, 4,220, 1,277, 3,044, 9,055, 1,425, 124, 321, 42, and 3,661 were functionally annotated using GO, KEGG, COG, Swiss-Prot, nr, PHI, P450, CAZy, VFDB, and T3SS databases, respectively. In addition, we predicted 41 gene clusters of secondary metabolites, including 9 NRPS-like, 11 NRPS, 12 T1PKS, 2 T2PKS, 5 terpene, and a single cluster of both beta-lactone and phosphonate. Repetitive elements accounted for 0.81% of the whole genome. Moreover, we detected 282 non-coding RNAs (ncRNAs) in this genome.
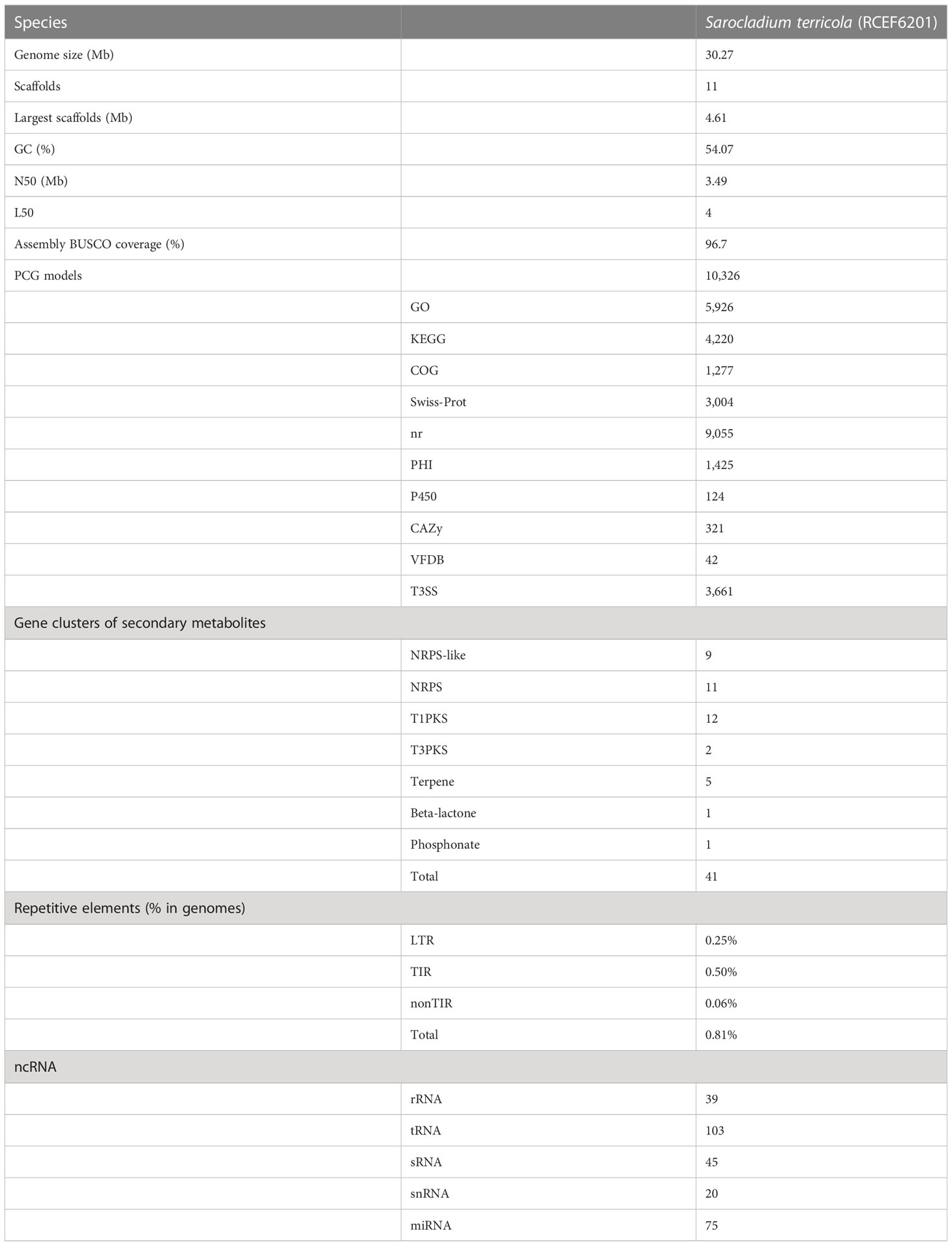
Table 1 Genomic features of Sarocladium terricola sequence assembly.
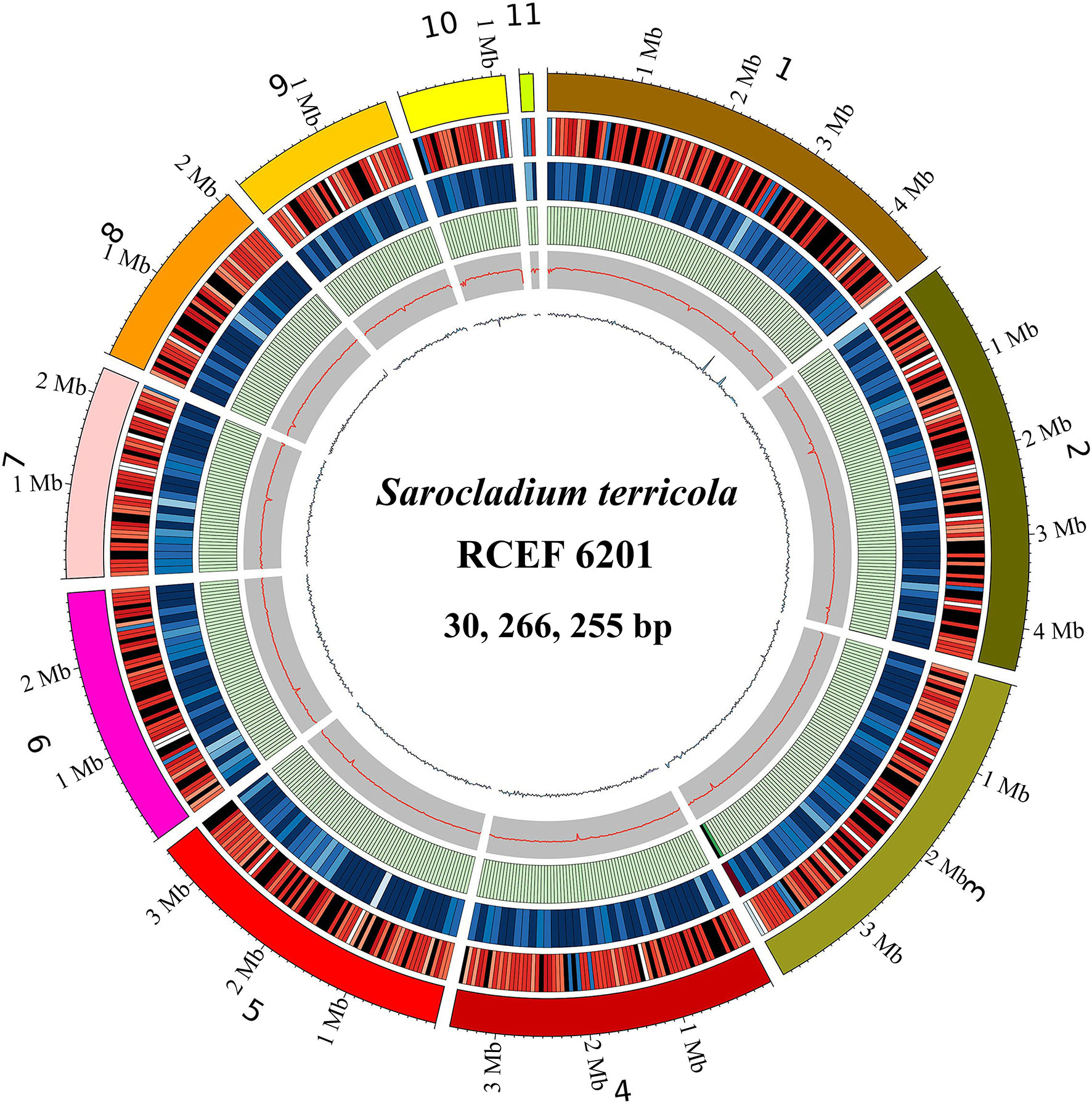
Figure 2 Genome features of Sarocladium terricola (RCEF6201). From outer to inner: 1: Genome scaffolds (sorted by length); 2: Gene density (in 50000bp nonoverlapping windows); 3: ncRNA density (in 100000bp nonoverlapping windows); 4: Repeats coverage (in 50000bp nonoverlapping windows); 5: GC content (in 20000bp nonoverlapping windows); 6: GC skew (in 20000bp nonoverlapping windows).
Phylogenomic analysis
A phylogenomic tree was reconstructed using single copy ortholog genes from 26 species, with Agaricus bisporus, Mortierella alpina, and Backusella circina serving as outgroups. A total of 268 single copy ortholog genes were identified through OrthoFinder (Emms and Kelly, 2019), and were used to create an alignment dataset with a length of 339,589 characters. In the Maximum Likelihood phylogenomic tree, S. terricola of the family Sarocladiaceae was found to be within a clade that included members of the family Bionectriaceae, such as Acremonium chrysogenum and A. citrinum (refer to Figure 3).
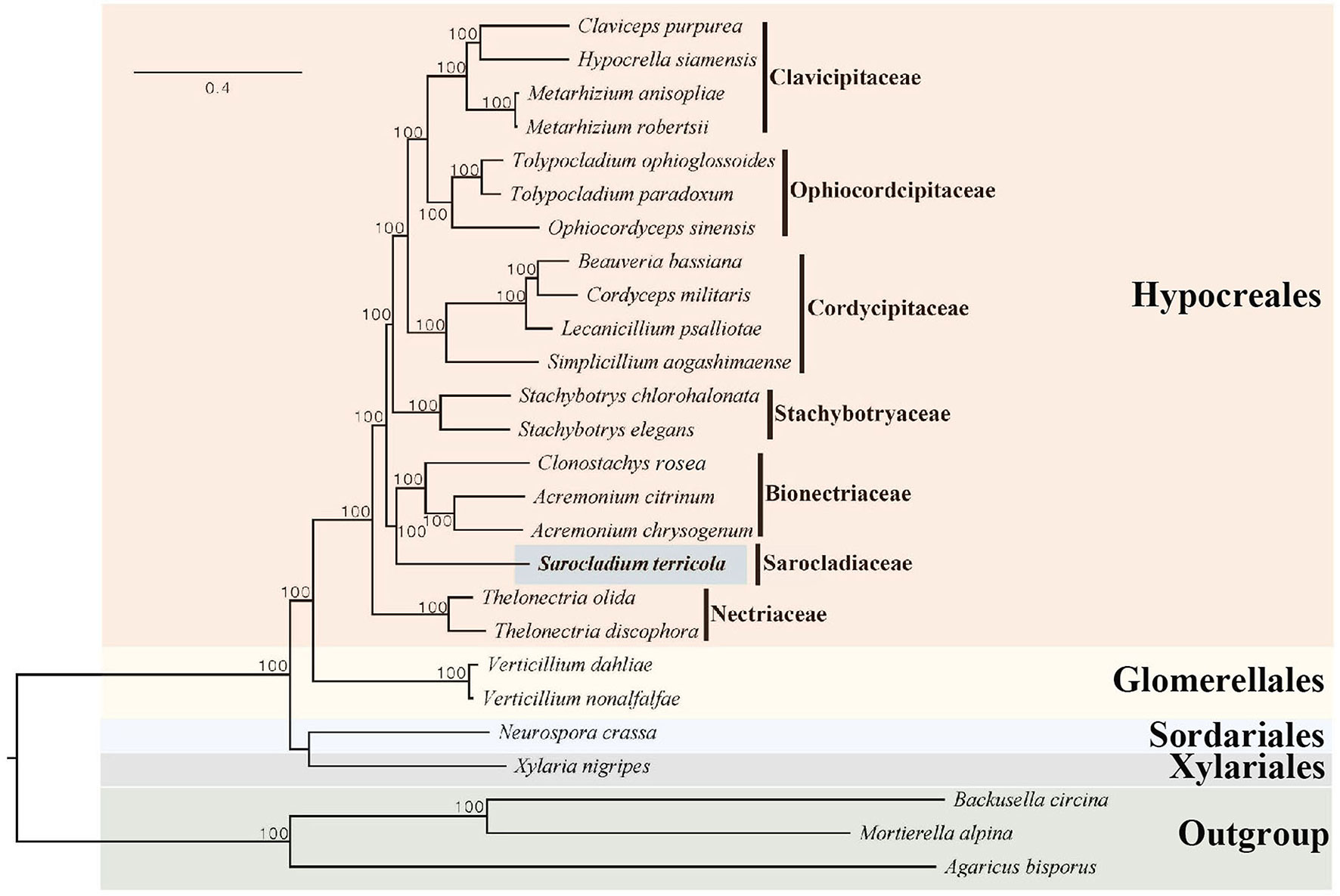
Figure 3 Maximum Likelihood phylogenomic tree based on the amino acids encoded by 268 single copy ortholog genes from 26 ascomycetes. The new genome sequence from the present study is highlighted in blue. Maximum Likelihood bootstrap values (≥ 95%) are indicated along branches. A scale bar in the upper left indicates substitutions per site.
Transcriptome sequence analyses and differentially expressed genes
To investigate the impact of ergosterol biosynthesis in different media, we performed transcriptome analyses of S. terricola. We sequenced six transcriptome libraries, three from both bean and potato dextrose media, resulting in a total of 37,043,298 to 42,033,580 clean reads per sample. A total of 5.56 Gb to 6.31 Gb of clean reads were generated, with more than 95.93% of clean reads meeting the Q30 quality threshold (Table S2). Furthermore, over 97.65% of the clean reads mapped to the genome of S. terricola. In each sample, a total of 9,176-9,738 genes (88.86-94.31% of the total number of genes) were found to be expressed.
The differential expression analysis indicated that the HCBe group had a slightly higher number of down-regulated genes compared to up-regulated genes (2221 vs. 1919) when compared to the HCPo group. Furthermore, 5880 genes did not exhibit any significant differences in their expression levels between the two groups, as depicted in Figure 4. To gain insights into the biological functions of the differentially expressed genes, we performed GO and KEGG pathway enrichment analyses, which are shown in Figures 5, S1. Nineteen GO categories were significantly enriched with over 100 DEGs, including cellular processes, metabolic processes, membrane, membrane parts, catalytic activity, and binding, as illustrated in Figures 5A, S1A. Additionally, the KEGG pathway enrichment analysis identified several major pathways associated with the set of DEGs, including global and overview maps, carbohydrate metabolism, signal transduction, cell growth and death, and amino acid metabolism, as shown in Figures 5B, S1B.
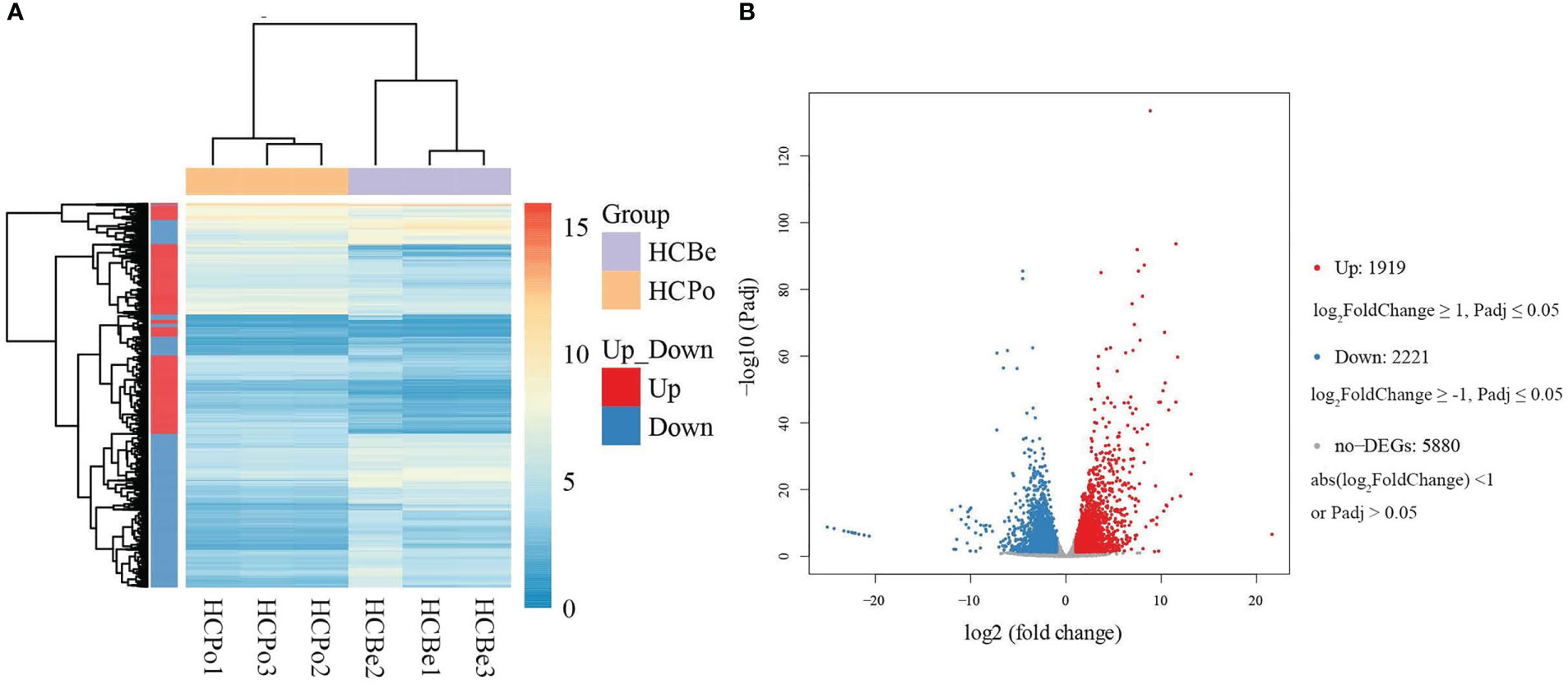
Figure 4 The differentially expressed genes of S. terricola from two types of media. (A) The heat map of expression levels of differentially expressed genes, (B) Volcano plot for HCBe vs. HCPo based on the DESeq2 method. “Up” and “Down” refer to levels in potato dextrose media as compared to bean media.
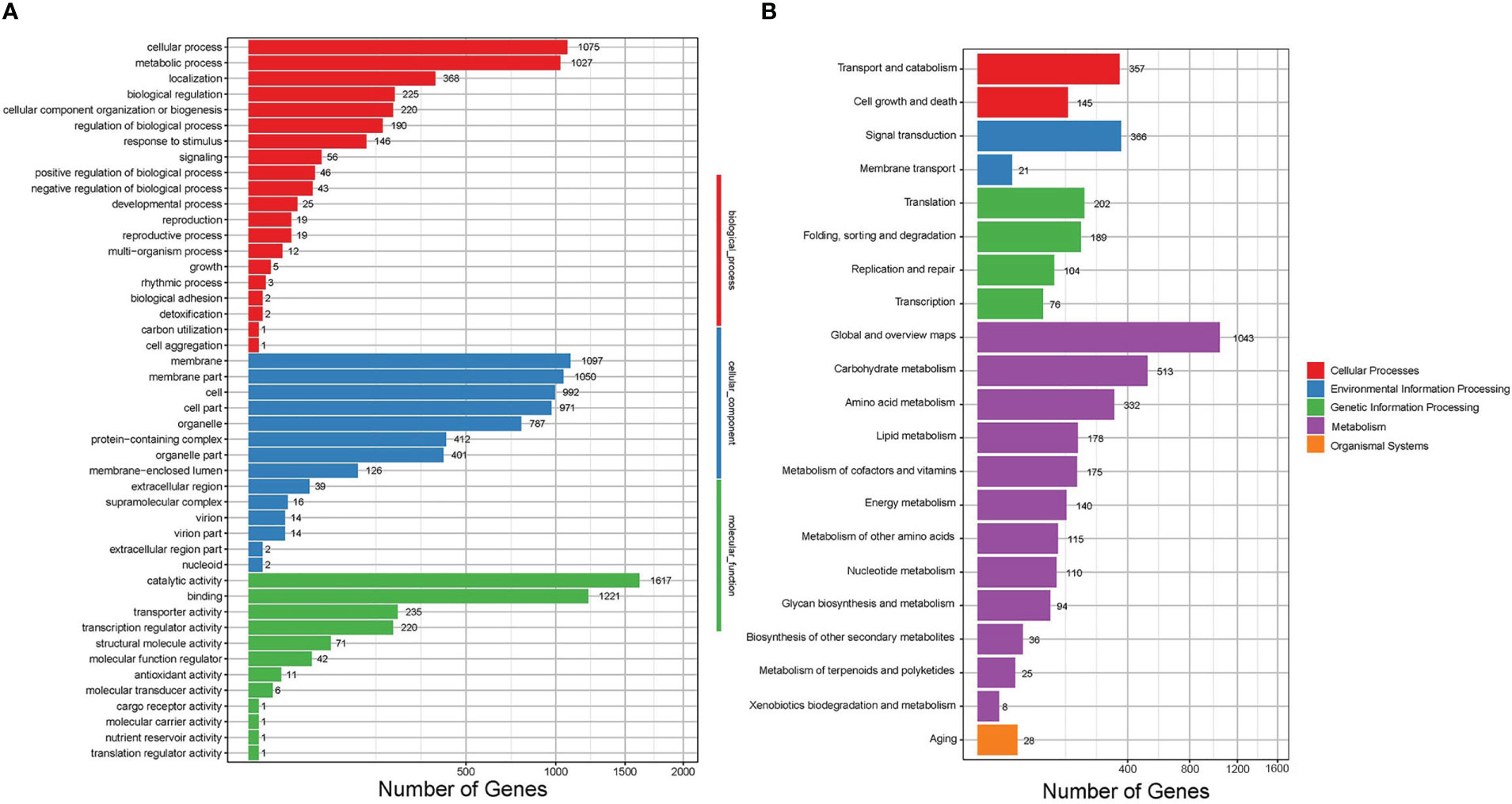
Figure 5 GO function and KEGG pathway enrichment analyses for DEGs of Sarocladium terricola cultured in two types of media. (A) GO analysis, (B) KEGG analysis. Differentially expressed genes were classified into biological process, cellular component, and molecular function categories in the GO analysis, and cellular processes, environmental information processing, genetic information processing, metabolism and organismal systems in the KEGG analysis.
Ergosterol biosynthesis in S. terricola
In this study, we annotated eleven genes (Figure 5) associated with ergosterol biosynthesis from the KEGG database, which were lanosterol synthase (ERG7; S2G02520), sterol 14alpha-demethylase (CYP51; S1G01142), delta7-sterol 5-desaturase (ERG24; S3G03213), methylsterol monooxygenase (ERG25; S5G06091), sterol-4alpha-carboxylate 3-dehydrogenase (ERG26; S2G02191), 3-keto steroid reductase (ERG27; S7G08361), sterol 24-C-methyltransferase (ERG6-1: S10G10199 and ERG6-2: S1G00302), Delta7-sterol 5-desaturase (ERG3; S7G08143), sterol 22-desaturase (ERG5; S5G06474), and delta24(24(1))-sterol reductase (ERG4; S1G00326). Out of these genes, five genes (S3G03213, S2G02191, S1G00302, S1G00326, and S7G08361) were significantly down-regulated in the HCPo group, whereas three genes (S5G06474, S5G06091, and S7G08143) were significantly up-regulated, as shown in Figure 6.
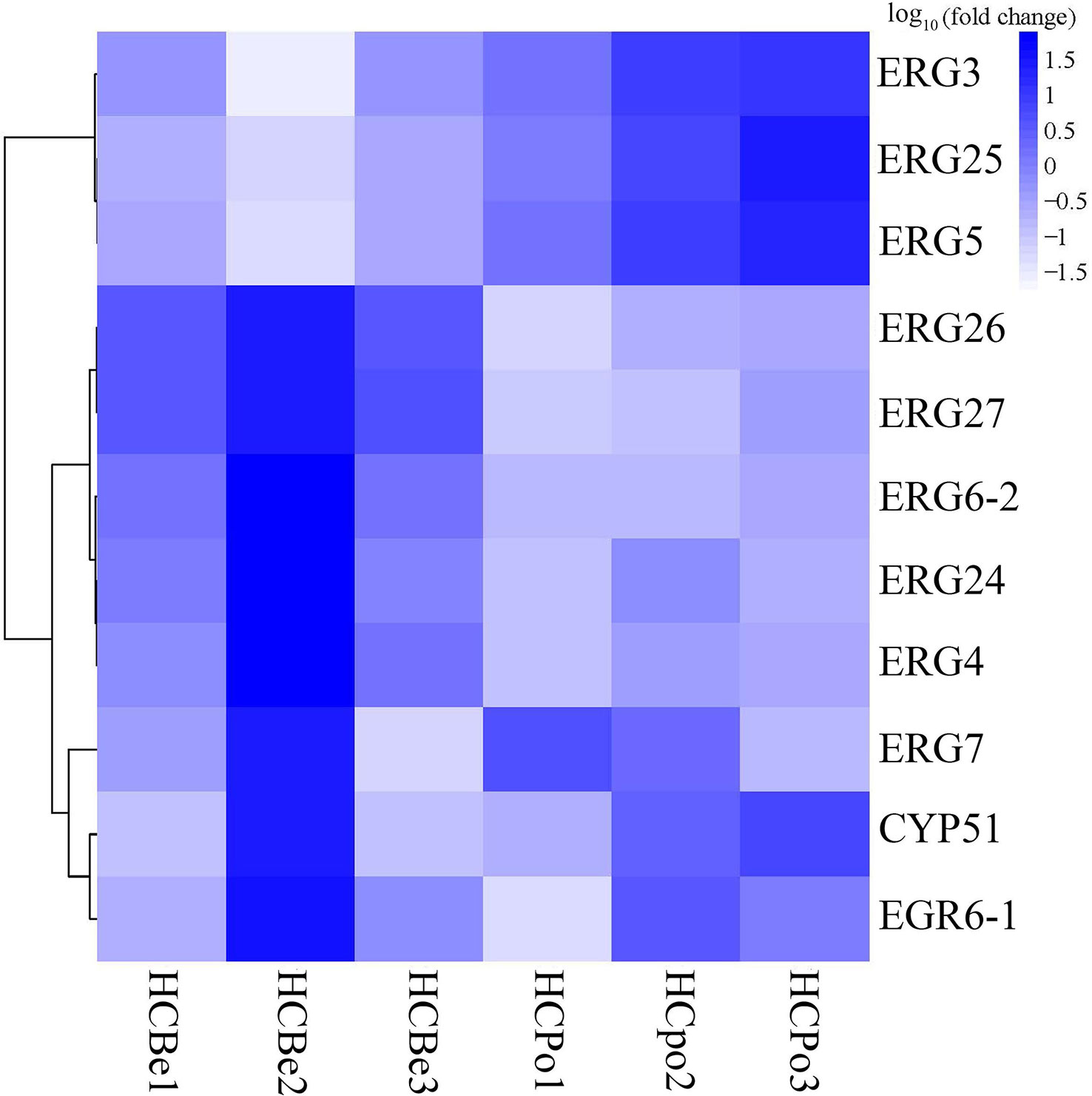
Figure 6 The heat map of expression levels of genes associated with ergosterol biosynthesis in Sarocladium terricola. ERG3 (S7G08143, delta7-sterol 5-desaturase), ERG25 (S5G06091, methylsterol monooxygenase), ERG5 (S5G06474, sterol 22-desaturase), ERG26 (S2G02191, sterol-4alpha-carboxylate 3-dehydrogenase), ERG27 (S7G08361, 3-keto steroid reductase), ERG6-2 (S1G00302, sterol 24-C-methyltransferase), ERG24 (S3G03213, delta14-sterol reductase), ERG4 (S1G00326, delta24 (24(1))-sterol reductase), ERG7 (S2G02520, lanosterol synthase), CYP51 (S1G01142, sterol 14alpha-demethylase), and ERG6-1 (S10G10199, delta24(24(1))-sterol reductase). Fold change refers to expression levels in potato dextrose media as compared to bean media.
Discussion
With the advent of genome and transcriptome sequencing, omics technology has been increasingly utilized to investigate various biological processes, discover novel natural products, and reconstruct phylogenomic relationships (Hittalmani et al., 2016; Ren et al., 2021; Tian et al., 2022; Wu et al., 2022; Zhao et al., 2022b; Zhao et al., 2022a). Although the Sarocladium genus encompasses 30 species (http://www.indexfungorum.org, accessed on 13 November 2022), genome-scale studies of Sarocladium remain scarce. The first genome sequence analysis of Sarocladium oryzae, a pathogen causing sheath rot disease in rice, identified genes associated with helvolic acid and cerulenin biosynthesis pathways and identified approximately 9.37% of S. oryzae genes as pathogenicity genes (Hittalmani et al., 2016). In the present study, we identified 1,425 genes from the S. terricola genome assembly in the Pathogen Host Interactions (PHI) database, accounting for 13.8% of predicted protein-coding gene models (Table 1). This finding implies that Sarocladium species harbor a vast array of pathogenic genes. Moreover, S. oryzae and S. terricola have been reported to contain numerous natural biosynthetic gene clusters (Hittalmani et al., 2016; Tian et al., 2022). For example, 33 secondary metabolite gene clusters were discovered in the S. terricola strain TR, including ten NRPS clusters, thirteen PKS clusters, one PKS-NRPS hybrid cluster, and five terpene clusters (Tian et al., 2022). This study identified slightly more gene clusters in the S. terricola genome assembly (Table 1).
Ergosterol is a crucial component of fungal cell membranes and has numerous biological and physiological functions, including delaying aging, preventing cancer, reducing inflammation and fever, and exhibiting anti-oxidative and antibacterial activities (Fernandes and Cabral, 2007; Abe et al., 2009; Guan et al., 2009; Caspeta et al., 2014; Dhingra and Cramer, 2017). Although fungi can accumulate ergosterol, its content is influenced by several factors, such as biomass, growth temperature, oxygen concentration, and the availability of carbon and nitrogen sources (Shang et al., 2006; He et al., 2007). In this study, significant differences in ergosterol content were observed when S. terricola was cultured in different media, including bean and potato dextrose media. To investigate the mechanism of ergosterol biosynthesis, we analyzed gene expression using transcriptome sequences from cultures grown in different media. Our analysis revealed that ERG3, ERG5, and ERG25 were up-regulated in potato dextrose media compared to bean media, while ERG4, ERG6, ERG24, ERG26, and ERG27 were down-regulated. In Saccharomyces cerevisiae, the enzymes essential for ergosterol biosynthesis include ERG9, ERG1, ERG7, ERG11, ERG24, ERG25, ERG26, ERG27, ERG6, ERG2, ERG3, ERG4, and ERG5 (Hu et al., 2018). The catalytic steps of ERG3, ERG5, and ERG25 require oxygen (Onyewu et al., 2003). Therefore, the up-regulation of ERG3, ERG5, and ERG25 in S. terricola grown in potato media may promote the synthesis of ergosterol. We also found no significant difference in the expression of three genes (S2G02520, S1G01142, and S10G10199). The three genes with significantly increased expression in potato dextrose media were ERG3, ERG25, and ERG5, which participate in the final steps of ergosterol synthesis and require the participation of oxygen. The down-regulated genes, ERG26, ERG27, and ERG6, have similar functions to ERG25, catalyzing the synthesis of fecosterol from 4,4-dimethylzymosterol. ERG4 and ERG5 participate in the synthesis of ergosta-5,7,24(28)-stienol, which is a precursor to ergosterol. Thus, the down-regulated genes, ERG26, ERG27, ERG6, and ERG4, have a limited effect in potato media.
To reconstruct the evolutionary history of S. terricola, we performed a maximum likelihood (ML) analysis using 268 single-copy orthologous genes from 26 genomes. The resulting ML tree revealed that S. terricola did not form a clade with Acremonium chrysogenum and A. citrinum, which supported the transfer of A. terricola to the genus Sarocladium (Giraldo et al., 2015).
Conclusion
In the present study, we sequenced, assembled, and annotated the genome of S. terricola. Our HPLC results indicated that the ergosterol levels of S. terricola were almost twice as high when cultured in potato dextrose media compared to bean media. Furthermore, our transcriptome analyses suggested that ERG3, ERG5, and ERG25 were significantly up-regulated in potato dextrose media, which may contribute to the increased ergosterol levels. Our phylogenomic analysis demonstrated that S. terricola is closely related to the family Bionectriaceae. The findings of this study hold great promise for the application of S. terricola as an animal feed additive. Additionally, we plan to conduct further studies to explore the relative genes involved in ergosterol biosynthesis, with the aim of further enhancing the ergosterol content of this strain in future work.
Data availability statement
The genomic and transcriptomic sequences have been deposited in GenBank with accession numbers: JAHKSO000000000 and PRJNA949268, respectively.
Author contributions
WW; conceptualization, writing—original draft preparation, and writing—review, YN; formal analysis, X-YL; investigation, BH; funding acquisition, projection administration, and writing—review. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China, Grant No. 32172473.
Acknowledgments
We thank Dr. Heng Zhao (Beijing Forestry University) for data analyses. We would like to thank Dr. Joseph Elliot at the University of Kansas for his assistance with English language and grammatical editing of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1181287/full#supplementary-material
Supplementary Figure 1 | GO function and KEEG pathway enrichment analyses in responding to the two type media of S. terricola. (A) differentially expressed genes number of the most enriched GO term, (B) differentially expressed genes number of the most enriched KEGG pathway.
References
Abe, F., Usui, K., Hiraki, T. (2009). Fluconazole modulates membrane rigidity, heterogeneity, and water penetration into the plasma membrane in saccharomyces cerevisiae. Biochemistry 48 (36), 8494–8504. doi: 10.1021/bi900578y
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25 (1), 25–29. doi: 10.1038/75556
Birney, E., Clamp, M., Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14 (5), 988–995. doi: 10.1101/gr.1865504
Caspeta, L., Chen, Y., Ghiaci, P., Feizi, A., Buskov, S., Hallström, B. M., et al. (2014). Altered sterol composition renders yeast thermotolerant. Science 346 (6205), 75–78. doi: 10.1126/science.1258137
Chen, L., Zheng, D., Liu, B., Yang, J., Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44 (D1), D694–D697. doi: 10.1093/nar/gkv1239
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10 (6), 563–569. doi: 10.1038/nmeth.2474
Consortium, U. (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43 (D1), D204–D212. doi: 10.1093/nar/gku989
Dhingra, S., Cramer, R. A. (2017). Regulation of sterol biosynthesis in the human fungal pathogen aspergillus fumigatus: opportunities for therapeutic development. Front. Microbiol. 8, 92. doi: 10.3389/fmicb.2017.00092
Dopont, S., Lemetais, G., Ferreira, T., Cayot, P., Gervais, P., Beney, L. (2012). Ergosterol biosynthesis: a fungal pathway for life on land? Evolution: Int. J. Organic Evol. 66 (9), 2961–2968. doi: 10.1111/j.1558-5646.2012.01667.x
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi: 10.1186/s13059-019-1832-y
Fernandes, P., Cabral, J. (2007). Phytosterols: applications and recovery methods. Bioresource Technol. 98 (12), 2335–2350. doi: 10.1016/j.biortech.2006.10.006
Fischer, M., Knoll, M., Sirim, D., Wagner, F., Funke, S., Pleiss, J. (2007). The cytochrome P450 engineering database: a navigation and prediction tool for the cytochrome P450 protein family. Bioinformatics 23 (15), 2015–2017. doi: 10.1093/bioinformatics/btm268
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43 (D1), D261–D269. doi: 10.1093/nar/gku1223
Gams, W., Hawksworth, D. L. (1975). The identity of acrocylindrium oryzae sawada and a similar fungus causing sheath-rot of rice. Kavaka 3, 57–61.
Giraldo, A., Gené, J., Sutton, D., Madrid, H., De Hoog, G., Cano, J., et al. (2015). Phylogeny of sarocladium (Hypocreales). Persoonia-Molecular Phylogeny Evol. Fungi 34 (1), 10–24. doi: 10.3767/003158515X685364
Guan, X. L., Souza, C. M., Pichler, H., Dewhurst, G., Schaad, O., Kajiwara, K., et al. (2009). Functional interactions between sphingolipids and sterols in biological membranes regulating cell physiology. Mol. Biol. Cell 20 (7), 2083–2095. doi: 10.1091/mbc.e08-11-1126
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9 (1), 1–22. doi: 10.1186/gb-2008-9-1-r7
He, X., Guo, X., Liu, N., Zhang, B. (2007). Ergosterol production from molasses by genetically modified saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 75 (1), 55–60. doi: 10.1007/s00253-006-0807-6
Hittalmani, S., Mahesh, H., Mahadevaiah, C., Prasannakumar, M. K. (2016). De novo genome assembly and annotation of rice sheath rot fungus sarocladium oryzae reveals genes involved in helvolic acid and cerulenin biosynthesis pathways. BMC Genomics 17 (1), 1–13. doi: 10.1186/s12864-016-2599-0
Hu, C., Zhou, M., Wang, W., Sun, X., Yarden, O., Li, S. (2018). Abnormal ergosterol biosynthesis activates transcriptional responses to antifungal azoles. Front. Microbiol. 9, 9. doi: 10.3389/fmicb.2018.00009
Jiang, X., Xia, L., Chen, L., Wu, D., Zhu, H. (2018). “In Effects of acremonium terricola culture on the fattening performance of hu sheep,” in The Proceedings of the Fifteenth Congress of China Sheep Industry Development Sponsored by the China Animal Husbandry Association in 2018, Henan, China, 10-11 October, 2018, Chinese Academy of Agricultural Sciences (CAAS), Scientech Documentation and …: 2018.
Johnson, A. D., Handsaker, R. E., Pulit, S. L., Nizzari, M. M., O'Donnell, C. J., De Bakker, P. I. (2008). SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24 (24), 2938–2939. doi: 10.1093/bioinformatics/btn564
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 (D1), D457–D462. doi: 10.1093/nar/gkv1070
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12 (4), 357–360. doi: 10.1038/nmeth.3317
Kong, F., Zhang, Y., Wang, S., Cao, Z., Liu, Y., Zhang, Z., et al. (2022). Acremonium terricola culture’s dose–response effects on lactational performance, antioxidant capacity, and ruminal characteristics in Holstein dairy cows. Antioxidants 11 (1), 175. doi: 10.3390/antiox11010175
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35 (9), 3100–3108. doi: 10.1093/nar/gkm160
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Levasseur, A., Drula, E., Lombard, V., Coutinho, P. M., Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6 (1), 1–14. doi: 10.1186/1754-6834-6-41
Li, B., Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinf. 12 (1), 323. doi: 10.1186/1471-2105-12-323
Li, Y., Jiang, X., Xu, H., Lv, J., Zhang, G., Dou, X., et al. (2020). Acremonium terricola culture plays anti-inflammatory and antioxidant roles by modulating MAPK signaling pathways in rats with lipopolysaccharide-induced mastitis. Food Nutr. Res. 64, 3649. doi: 10.29219/fnr.v64.3649
Li, Y., Sun, Y.-K., Li, X., Zhang, G.-N., Xin, H.-S., Xu, H.-J., et al. (2018). Effects of Acremonium terricola culture on performance, milk composition, rumen fermentation and immune functions in dairy cows. Anim. Feed Sci. Technol. 240, 40–51. doi: 10.1016/j.anifeedsci.2018.03.015
Li, Y., Wang, Y.-Z., Ding, X., Zhang, Y.-G., Xue, S.-C., Lin, C., et al. (2016). Effects of Acremonium terricola culture on growth performance, antioxidant status and immune functions in weaned calves. Livestock Sci. 193, 66–70. doi: 10.1016/j.livsci.2016.09.009
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25 (5), 955–964. doi: 10.1093/nar/25.5.955
Medema, M. H., Blin, K., Cimermancic, P., De Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39 (suppl_2), W339–W346. doi: 10.1093/nar/gkr466
Nout, M. J. R., Laarhoven, B. V., de Jongh, P., de Koster, P. G. (1987). Ergosterol content of Rhizopus oligosporus NRRL 5905 grown in liquid and solid substrates. Appl. Microbiol. Biotechnol. 2, 456–461. doi: 10.1007/BF00253532
Onyewu, C., Blankenship, J. R., Del Poeta, M., Heitman, J. (2003). Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against candida albicans, candida glabrata, and candida krusei. Antimicrobial Agents chemotherapy 47 (3), 956–964. doi: 10.1128/AAC.47.3.956-964.2003
Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J. R., Hellinga, A. J., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20 (1), 1–18. doi: 10.1186/s13059-019-1905-y
Ren, L.-Y., Zhao, H., Liu, X.-L., Zong, T.-K., Qiao, M., Liu, S.-Y., et al. (2021). Transcriptome reveals roles of lignin-modifying enzymes and abscisic acid in the symbiosis of mycena and gastrodia elata. Int. J. Mol. Sci. 22 (12), 6557. doi: 10.3390/ijms22126557
Shang, F., Wen, S., Wang, X., Tan, T. (2006). Effect of nitrogen limitation on the ergosterol production by fed-batch culture of saccharomyces cerevisiae. J. Biotechnol. 122 (3), 285–292. doi: 10.1016/j.jbiotec.2005.11.020
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Diekhans, M., Baertsch, R., Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve. novo Gene finding. Bioinf. 24 (5), 637–644. doi: 10.1093/bioinformatics/btn013
Sun, Y.-F., Lebreton, A., Xing, J.-H., Fang, Y.-X., Si, J., Morin, E., et al. (2022). Phylogenomics and comparative genomics highlight specific genetic features in ganoderma species. J. Fungi 8 (3), 311. doi: 10.3390/jof8030311
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18 (12), 1979–1990. doi: 10.1101/gr.081612.108
Tian, X., Sun, T., Lin, R., Liu, R., Yang, Y., Xie, B., et al. (2022). Genome sequence resource of sarocladium terricola TR, an endophytic fungus as a potential biocontrol agent against meloidogyne incognita. Mol. Plant-Microbe Interact. 35 (6), 505–508. doi: 10.1094/MPMI-11-21-0284-A
Torto-Alalibo, T., Collmer, C. W., Gwinn-Giglio, M. (2009). The plant-associated microbe gene ontology (PAMGO) consortium: community development of new gene ontology terms describing biological processes involved in microbe-host interactions. BMC Microbiol. 9 (1), 1–5. doi: 10.1186/1471-2180-9-S1-S1
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat. Protoc. 7 (3), 562–578. doi: 10.1038/nprot.2012.016
Vargas, W. A., Martín, J. M. S., Rech, G. E., Rivera, L. P., Benito, E. P., Díaz-Mínguez, J. M., et al. (2012). Plant defense mechanisms are activated during biotrophic and necrotrophic development of colletotricum graminicola in maize. Plant Physiol. 158 (3), 1342–1358. doi: 10.1104/pp.111.190397
Wang, L., Feng, Z., Wang, X., Wang, X., Zhang, X. (2009). DEGseq: an r package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26 (1), 136–138. doi: 10.1093/bioinformatics/btp612
White, T. C., Marr, K. A., Bowden, R. A. (1998). Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin. Microbiol. Rev. 11 (2), 382–402. doi: 10.1128/CMR.11.2.382
Wu, G., Miyauchi, S., Morin, E., Kuo, A., Drula, E., Varga, T., et al. (2022). Evolutionary innovations through gain and loss of genes in the ectomycorrhizal boletales. New Phytol. 233 (3), 1383–1400. doi: 10.1111/nph.17858
Yao, Y., Lin, R., Tian, X., Shen, B., Mao, Z., Xie, B. (2016). The complete mitochondrial genome of the nematophagous fungus acremonium implicatum. Mitochondrial DNA Part A 27 (5), 3246–3247. doi: 10.3109/19401736.2015.1007367
Zhao, H., Dai, Y.-C., Liu, X.-Y. (2022a). Outline and divergence time of subkingdom mucoromyceta: two new phyla, five new orders, six new families and seventy-three new species. bioRxiv.
Zhao, J., Lin, W., Ma, X., Lu, Q., Ma, X., Bian, G., et al. (2010). The protein kinase Hal5p is the high-copy suppressor of lithium-sensitive mutations of genes involved in the sporulation and meiosis as well as the ergosterol biosynthesis in saccharomyces cerevisiae. Genomics 95 (5), 290–298. doi: 10.1016/j.ygeno.2010.02.010
Keywords: Sarocladium terricola, Phylogenomic analyses, genome, metabolites, transcriptome
Citation: Wang W, Nie Y, Liu X-Y and Huang B (2023) The genome and transcriptome of Sarocladium terricola provide insight into ergosterol biosynthesis. Front. Cell. Infect. Microbiol. 13:1181287. doi: 10.3389/fcimb.2023.1181287
Received: 07 March 2023; Accepted: 28 March 2023;
Published: 14 April 2023.
Edited by:
Jia-Jia Chen, Jiangsu Vocational College of Agriculture and Forestry, ChinaReviewed by:
Yang Gao, Jiangxi Agricultural University, ChinaQiumei Zhou, First Affiliated Hospital of Anhui University of Traditional Chinese Medicine, China
Copyright © 2023 Wang, Nie, Liu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Huang, Ymh1YW5nQGFoYXUuZWR1LmNu