 Lele Pan1,2,3,4,5†Lijian Wei1,2,3,4,5,6†Shihua Luo1,2,3,4,5Baoyan Ren7,8Miao Li9
Lele Pan1,2,3,4,5†Lijian Wei1,2,3,4,5,6†Shihua Luo1,2,3,4,5Baoyan Ren7,8Miao Li9 Lina Liang1,2,3,4,5*Xuebin Li3,4,5,8*
Lina Liang1,2,3,4,5*Xuebin Li3,4,5,8* Guijiang Wei1,2,3,4,5,8*
Guijiang Wei1,2,3,4,5,8*- 1Center for Medical Laboratory Science, Affiliated Hospital of Youjiang Medical University for Nationalities, Baise, Guangxi, China
- 2Key Laboratory of Research on Clinical Molecular Diagnosis for High Incidence Diseases in Western Guangxi of Guangxi Higher Education Institutions, Baise, Guangxi, China
- 3Baise Key Laboratory for Precise Genetic Testing of Long-dwelling Nationalities, Baise, Guangxi, China
- 4Engineering Research Center of Guangxi Higher Education Institutions for Precise Genetic Testing of Long-dwelling Nationalities, Baise, Guangxi, China
- 5Guangxi Engineering Research Center for Precise Genetic Testing of Long-dwelling Nationalities, Baise, Guangxi, China
- 6Department of Clinical Laboratory, Baise People’s Hospital, Affiliated Southwest Hospital of Youjiang Medical University for Nationalities, Baise, Guangxi, China
- 7Yaneng BlOscience (Shenzhen) Corporation, Shenzhen, Guangdong, China
- 8Modern Industrial College of Biomedicine and Great Health, Youjiang Medical University for Nationalities, Baise, Guangxi, China
- 9Clinical Genome Center, Guangxi KingMed Diagnostics, Nanning, Guangxi, China
Background: Klebsiella pneumoniae (KP) is a significant pathogenic bacterium responsible for severe infections in hospitals. However, existing traditional detection techniques, such as culture and PCR, are relatively inefficient. Therefore, this study aims to establish a rapid and convenient method for detecting KP.
Methods: This study developed a single-tube detection method combining recombinant polymerase amplification (RPA) and light-controlled CRISPR/Cas12a. RPA primers were designed and screened for the rcsA gene of KP to effectively amplify the target. A light-controlled CRISPR/Cas12a system was created using crRNA modified with a photocleavable group (NPOM). The two systems were integrated into a single tube. Following RPA amplification, UV light-controlled release of crRNA inhibition activates CRISPR-mediated target recognition and Cas12a trans-cleavage, detecting fluorescent signals (FD) in conjunction with UV analysis.
Results: The light-controlled RPA-CRISPR/Cas12a detection platform developed in this study uses a 15 μL reaction system. By optimizing key parameters such as RPA amplification time (20 min), primer concentration (400 nM), UV light activation time (30 s), and crRNA/Cas12a concentration (300 nM), the platform achieves optimal detection efficiency. The platform has a fluorescence detection limit of 4.072×102 copies/reaction and can specifically identify KP in seven common clinical strains. Clinical sample validation demonstrated that the method yields results fully consistent with PCR detection (30/30 agreement rate of 100%), showcasing excellent detection performance and clinical application potential.
Conclusion: We have successfully developed a light-controlled RPA-CRISPR/Cas12a detection system capable of rapidly and highly sensitively detecting KP. This system demonstrates significant advantages in terms of detection speed (completed in as little as 50 minutes), sensitivity (as low as 4.072×102 copies/reaction), and ease of use, providing an efficient and reliable solution for clinical pathogen detection.
1 Introduction
KP, a Gram-negative bacterium that belongs to the Enterobacteriaceae family ubiquitously found on mucosal surfaces of animals and in the environment (e.g., water, soil, etc.) (Adeolu et al., 2016; Paczosa and Mecsas, 2016). In humans, the bacterium mainly colonizes the gastrointestinal tract, and a few can be detected in the nasopharynx. As a commensal and opportunistic pathogen, KP is an important causative agent of community-acquired infections and can also trigger severe hospital-acquired infections. In immunocompromised patients, the bacterium can lead to a variety of serious diseases such as urinary tract infections, stroke-associated pneumonia, bacteremia, liver abscesses, and sepsis (Chen et al., 2013; Yao et al., 2015). Clinical surveillance data in China showed that KP accounted for 11.9% of pathogens from intensive care unit-acquired pneumonia and ventilator-associated pneumonia (Zhang et al., 2014). In addition, a multicenter study covering 25 hospitals in 14 provinces in China showed that KP was detected in up to 73.9% of 664 clinical samples of carbapenem-resistant Enterobacteriaceae (Zhang et al., 2018). This organism is also one of the main causative pathogens of neonatal sepsis, ranking among the top three in most case statistics (Okomo et al., 2019). Its high morbidity and lethality impose a heavy burden on our public health. Of concern is the spread of carbapenem-resistant KP in recent years, which has exacerbated the difficulty of clinical treatment (Zhang et al., 2015). Therefore, the timely identification of KP is particularly crucial.
The currently available detection approaches for KP exhibit several inherent constraints. Traditional bacterial culture methods, although reliable as the gold standard, demand no less than 24 hours of incubation time (Khan et al., 2014). Matrix-assisted laser desorption ionization time-of-flight mass spectrometry provides superior identification accuracy but still relies on the incubation step (Huang et al., 2022). Among the molecular detection techniques, polymerase chain reaction (PCR) reduces detection time, but reliance on expensive instruments and specialized personnel limits its application at the grassroots level (Dai et al., 2019).
In recent years, isothermal nucleic acid amplification technologies (for example, loop-mediated isothermal amplification (LAMP), recombinase polymerase amplification (RPA), and rolling circle amplification (RCA), etc.) have radically eliminated the reliance on complex thermal cycling equipment by utilizing a specific enzyme/primer system to complete the amplification at constant temperatures, dramatically simplifying the operation process and significantly reducing the reliance on specialized instruments and environments, which greatly expands the applicability and accessibility of the nucleic acids for rapid detection (Dong et al., 2015; Si et al., 2021). Compared to LAMP and RCA technologies, RPA offers advantages of simpler primer design and higher amplification efficiency. Specifically, LAMP requires multiple primer pairs and has a longer reaction time; RCA relies on circular templates and can only perform linear amplification, resulting in lower product yields (Srivastava and Prasad, 2023).
CRISPR/Cas system molecular diagnostic technology has become an emerging detection tool due to its high sensitivity, specificity, and convenience. The CRISPR/Cas system can specifically recognize the target nucleic acid sequences by guide RNAs (crRNAs) and activate collateral cleavage activity of Cas proteins, including Cas12 and Cas13. Collateral cleavage activity non-specifically degrades fluorescent reporter molecules and generates detection signals (Gootenberg et al., 2017; Chen et al., 2018). The RPA-CRISPR/Cas technology integrates the rapidity of isothermal amplification with the high specificity of CRISPR/Cas. Based on this advantage, researchers have developed various RPA-CRISPR/Cas diagnostic platforms, such as DNA Nucleic Acid Endonuclease Targeted CRISPR Reporter System (DETECTR), High Sensitivity Enzymatic Unlocking Reporter System (SHERLOCK), and High Efficiency and Low Consumption Multi-Purpose System (HOLMES), which are technologies with single-base resolution and high sensitivity at the molar level (Li et al., 2018b; Kellner et al., 2019; Lin et al., 2023; Lan et al., 2024; Sen et al., 2024; Xue et al., 2025). Therefore, “one-pot” assays that combine the two steps of traditional RPA-CRISPR/Cas technology into a single reaction tube are particularly important (Harrington et al., 2018; Li et al., 2018a).
Li et al. leveraged plant suction dynamics to isolate and link the RPA-CRISPR/Cas reaction mixture in the channel, enabling a one-pot assay; however, the technique is laborious and contamination-prone (Chen et al., 2025). Liu and Hu et al. proposed an improved approach by loading amplification reagents into the bottom of reaction tubes and fixing Cas12a/crRNA complexes on the tube caps to form a compartmentalized system, effectively avoiding premixing (Hu J.J, et al., 2023). Nevertheless, sensitivity still has room for improvement. Subsequently, Zhang, Tan, and Wang teams introduced high-viscosity additives (e.g., glycerol, paraffin, sucrose) to achieve spatial separation, enhancing simplicity and efficiency (Tan Q. et al., 2024; Wang et al., 2024; Zhang et al., 2024). However, the reproducibility of this method may be compromised by the disruptive effects of high-viscosity additives on the solution phase. Building on significant advances in “one-pot” nucleic acid detection technology, the Hu and Chen team. Employed light-cleavable linkers to block crRNA, achieving spatiotemporal separation in a single-tube closed system (Chen et al., 2022; Hu et al., 2022). However, the additional hybridization step limited the development of lyophilized reagents. To address this, Hu et al. Further optimized the approach by incorporating the photocleavable base NPOM-dt into crRNA, eliminating the pre-hybridization step and providing a feasible solution for future lyophilized reagent development (Hu et al., 2023). To date, the detection of KP using a light-controlled RPA-CRISPR/Cas12a using NPOM-dt has not yet been reported.
In this study, we plan to design RPA primers based on the conserved gene rcsA of KP and develop a single-tube rapid detection system based on light-controlled RPA-CRISPR/Cas12a using NPOM-dt. The method adopts a tube-closed reaction without the addition of extra reagents, and triggers the CRISPR/Cas12a system by ultraviolet light irradiation after RPA amplification, achieving simple, rapid, and highly specific detection of KP.
2 Materials and methods
2.1 Materials
The standard strains used in this study are as follows: KP (ATCC 700603), Escherichia coli (ATCC 25922), Pseudomonas aeruginosa (ATCC 27853), Streptococcus pneumoniae (ATCC 49619), Enterococcus faecalis (ATCC 29212), and Staphylococcus aureus (ATCC 29213). The above strains were provided by the Laboratory Strain Bank. Enterococcus faecalis (GDMCC 1.388) was procured from Guangdong Microbiology, and clinical strains of Haemophilus parainfluenzae were isolated and confirmed by the Microbiology Unit of the Department of Laboratory Medicine. Genomic DNA extraction for all strains was performed in a molecular biology laboratory. Details of strain sources are shown in Supplementary Table S1. The standard RPA kit (TwistDx Company Limited, UK) was used for nucleic acid amplification. Cas12a (Cpf1) protein (EnGen® Lba) and the corresponding NEBuffer r2.1 were procured from New England Biolabs (China branch). PCR reactions were performed using 2X Taq MasterMix (with dye) from Cwbio (Jiangsu, China). The TIANamp Bacterial DNA Kit (Tiangen, China) was employed for bacterial genomic DNA extraction. Supplementary Table S2 summarizes the equipment specifications for fluorescence recording, gel documentation, and UV-based detection. Most oligonucleotides in this study, including PCR primers, cRNA, RPA primers, and ssDNA (single-stranded DNA), were purchased and synthesized from Nanning GenSys Biotechnology Co. Caged crRNA was purchased from Bolles Biosciences (Guangzhou, China). Ultraviolet lamp (365 nm) was purchased from Zhonglian UV Optics Factory (Shenzhen, China).
2.2 Genomic DNA extraction
For long-term preservation, the reference strains were stored at -80°C in 20% glycerol. Before the experiment, the reference strain was streaked onto blood agar plates and incubated at 37 °C overnight (approximately 16–24 h). A single colony was placed in a 1.5 mL EP tube, and genomic DNA was extracted using the TIANamp Bacterial DNA Extraction Kit. Clinical strains were preserved in skimmed milk medium at -80 °C, and DNA extraction was also done using the TIANamp kit. The concentration of all extracted DNA samples was determined spectrophotometrically, and the OD260/280 ratio of eligible samples was between 1.8 and 2.0. Bacterial genomic DNA that was not used immediately was stored at -80 °C for backup, whereas DNA templates before the experiment were temporarily stored at -20 °C for use.
2.3 Primer, crRNA, and caged crRNA design
Based on the rcsA gene sequence of the KP strain obtained from the NCBI database (GenBank: AY059955.1), three pairs of RPA primers (rcsA Pair1, rcsA Pair3, and rcsA Pair4) using the NCBI online “Primer-BLAST” tool. The primer pairs designed by previous researchers (Tan et al., 2024a) were selected as rcsA Pair2 for screening. The primer design parameters were set as follows: length 28–35 bp, GC content 30-70%, amplicon length 150–300 bp, Tm value 50-100%, and maximum single-nucleotide repeat length not more than 5. After screening the primers and verifying specificity via 2% agarose gel electrophoresis, we determined the optimal RPA primer combinations. Subsequently, three sets of crRNAs were designed by identifying PAM (protospacer adjacent motif, TTTN) sites in the amplification region of the selected primers using CRISPR online tools (http://www.rgenome.net/cas-designer), and the most effective crRNAs were screened by Cas12a-mediated fluorescence signal detection. Finally, the photocleavable group 6-nitropiperonyloxymethyl-caged thymidine (NPOM-dt) was introduced into the spacer region of the selected optimal crRNA sequence, resulting in the construction of an activity-modulable caged crRNA (Hu et al., 2023). As a photocleavable group, NPOM-dt modification temporarily inactivates caged crRNA, inhibiting Cas12a activity. Upon UV irradiation of caged crRNA, Cas12a cleavage function will be activated. The details of primers, crRNA, ssDNA, and caged crRNA used in this work are provided in Supplementary Table S3.
2.4 RPA primer optimization and specificity validation
RPA reactions were performed in strict accordance with the Twist AmpTM Basic Kit instructions, and contained 2.4 μL (10 μM) of forward and reverse primers, 29.5 μL of buffer, 4 μL of DNA template, 2.5 μL of magnesium ions (incorporated at the end to avoid non-specific amplification), and lyophilized enzyme powders (polymerase, single-stranded binding protein, and recombinase, etc.), and the volume was made up using enzyme-free water. To avoid loss of enzyme activity, lyophilized enzyme powders should be centrifuged briefly before use, and repeated freezing and thawing should be avoided. The reaction tubes were mixed, centrifuged, and incubated in a PCR machine at 37 °C for 20 min. Amplification products were then resolved on a 2% agarose gel (130 V, 45 min) for analysis. Primer specificity verification was tested by using the same reaction system and only replacing different DNA templates.
2.5 Establishment of a light-controlled one-pot RPA-CRISPR/Cas12a method
Firstly, to screen the optimal crRNA, a Cas12a-mediated fluorescence detection assay was performed. The reaction system (20 μL) incorporated 2 μL of 10×NEBuffer, 500 nM FAM-BHQ1-tagged reporter molecule (5’-FAM-TTATT-BHQ1-3’), 50 nM Cas12a protein, 50 nM crRNA, and enzyme-free water, and 2 μL of RPA amplification product was added. 2 μL of RPA amplification product was followed by monitoring the fluorescence signal for 30 min at 37 °C (one acquisition per minute), and the data were analyzed by GraphPad Prism 9 to determine the optimal crRNA. Three NPOM-dt modifications were subsequently embedded in its spacer region to generate caged crRNA. Based on this, we designed the light-controlled single-tube RPA-CRISPR/Cas12a system to detect KP. The reaction system consisted of a 15 μL mixture (containing 10 μL of RPA solution: 5.9 μL of 2× buffer, 0.48 μL of forward/reverse primer (10 μM), 0.5 μL of magnesium acetate, and enzyme-free water to make up to 10 μL) and 5 μL of light-controlled CRISPR/Cas12a components (1 μL of 10× buffer, 1 μL of Cas12a protein (4 μM), 1 μL of caged crRNA (4 μM), 1 μL FAM-BHQ1 probe (10 μM) and enzyme-free water made up to 5 μL) were composed. After mixing and centrifugation, the reaction tubes were incubated at 37 °C for 20 min, and the crRNA was activated by ultraviolet light irradiation for 30 s. The fluorescence assay was then continued at 37 °C for 30 min (recorded every minute). The feasibility of the method was verified by deletion experiments (ultraviolet light irradiation, template, Cas12a, caged crRNA, or probe).
2.6 Optimization of the light-controlled one-pot RPA-CRISPR/Cas12a system
In order to achieve optimal amplification efficiency of the method, systematic optimization of reaction parameters was essential. First, the amplification conditions were systematically evaluated, including testing different amplification times (10 min, 15 min, 20 min, 30 min, 40 min) and primer concentration gradients (160 nm, 240 nm, 320 nm, 400 nm, 480 nm, 560 nm) to determine the optimal amplification efficiency. Meanwhile, the ultraviolet light irradiation time (10s, 15s, 20s, 30s, 60s) was finely regulated to ensure adequate activation of caged crRNA. In addition, the concentrations of Cas12a (100–500 nM) and caged crRNA (100–400 nM) were optimized under different concentration conditions to maximize the target cleavage efficiency and minimize non-specific cleavage by maintaining the initial reaction volume constant. All optimization experiments were performed with fluorescence intensity as the key assessment metric, a blank reaction was set up as a negative control, and real-time fluorescence monitoring was performed by a qPCR instrument, and each parameter combination was repeated three times to ensure data reliability.
2.7 Sensitivity and specificity evaluation of the light-controlled one-pot RPA-CRISPR/Cas12a method
The sensitivity and specificity of the light-controlled “one-pot” RPA-CRISPR/Cas12a system were evaluated. A 10-fold gradient dilution (4.072×106 copies/reaction to 4.072×102 copies/reaction) of KP genomic DNA was used as a template for sensitivity testing using ddH2O as diluent. Subsequently, system specificity was examined using standardized genomic DNA of KP and seven other common clinical pathogens, including Haemophilus parainfluenzae, Pseudomonas aeruginosa, Streptococcus pneumoniae, Enterococcus faecalis, Enterococcus aureus, and Escherichia coli. All experiments were repeated three times.
2.8 Clinical evaluation of the light-controlled one-pot RPA-CRISPR/Cas12a method
To evaluate diagnostic performance, 30 KP and 20 non-KP samples were obtained from clinical laboratories. After obtaining genomic DNA using a commercial DNA extraction kit, the light-controlled single-tube RPA-CRISPR/Cas12a system was simultaneously validated against conventional PCR techniques, with statistical analysis performed using GraphPad Prism software(Version 10.4, La Jolla, California, USA). The total volume of the PCR reaction system was 25 μL, which consisted of 12.5 μL of 2× Es Taq MasterMix (Dye), 0.4 μM upstream and downstream primers, and 2 μL of DNA template. The PCR amplification program was set up strictly according to the CWBIO 2×Es Tap MasterMix (Dye) reagent instructions. The amplification products of the two methods were finally analyzed by 2% agarose gel electrophoresis. All experiments were repeated three times.
3 Result
3.1 The workflow for the detection of KP by light-controlled one-pot RPA-CRISPR/Cas12a method
As illustrated in Figure 1, the workflow for the detection of KP by the light-controlled “one-pot” RPA-CRISPR/Cas12a method. First, a 20-minute isothermal amplification of RPA was performed without ultraviolet light irradiation, which was performed by a synergistic combination of recombinase, single-stranded binding proteins, and specific primers. After the target fragment was fully amplified, it was precisely irradiated by 360 nm ultraviolet light on the tube wall for 30 seconds to promote the efficient dissociation of the NPOM-dt protective group on the crRNA and realize the functional activation of crRNA. The activated crRNA-Cas12a complex first recognizes the PAM sequence of the target DNA and then binds specifically by complementary base pairing, triggering the double cleavage activity of Cas12a, which can both precisely cleave the DNA strand of interest and non-specifically cleave the ssDNA fluorescent reporter molecule in the system. The detection results are directly interpreted by real-time fluorescence signals, and the design realizes the spatiotemporal isolation of amplification and detection, providing a non-contact, one-step solution for the point-of-care detection of nucleic acids.
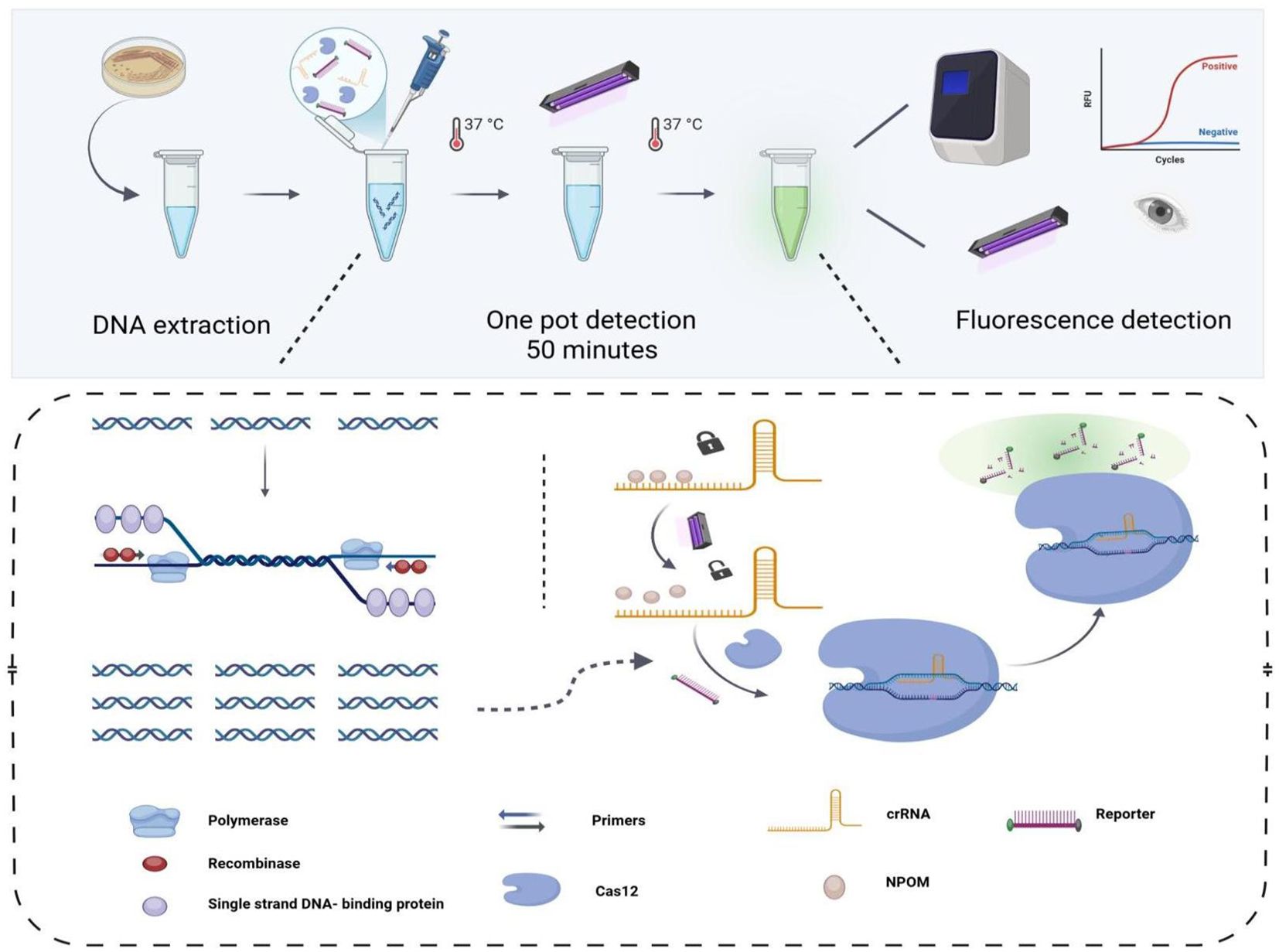
Figure 1. The workflow of the one-tube light-controlled RPA-CRISPR/Cas12a method. The template is introduced into a reaction tube containing both the RPA amplification system and the light-controlled CRISPR/Cas12a detection system. Subsequently, the CRISPR/Cas12a detection system is activated by ultraviolet light irradiation to perform specific and nonspecific cleavage of the template and the reporter DNA, respectively. Ultimately, the detection outcomes are interpretable using either a fluorescence detector or an ultraviolet (UV) detection system.
3.2 RPA primer screening and specificity validation
The rcsA gene is a specific gene of KP (Tan et al., 2024a). Based on the rcsA gene sequence, three pairs of RPA primers were designed in our research (Supplementary Table S3). Amplification reactions and electrophoresis analysis verified that each primer was effective in identifying the target sequence (Figure 2A). From the imaging results, it can be observed that rcsA Pair3 has the highest amplification product yield, and the specificity verification is satisfactory. The cross-reactivity experiment confirmed (Figure 2B) that the primer pair was only amplified to the target template, so the Pair3 amplification sequence was identified as critical for designing crRNA and downstream experiments.
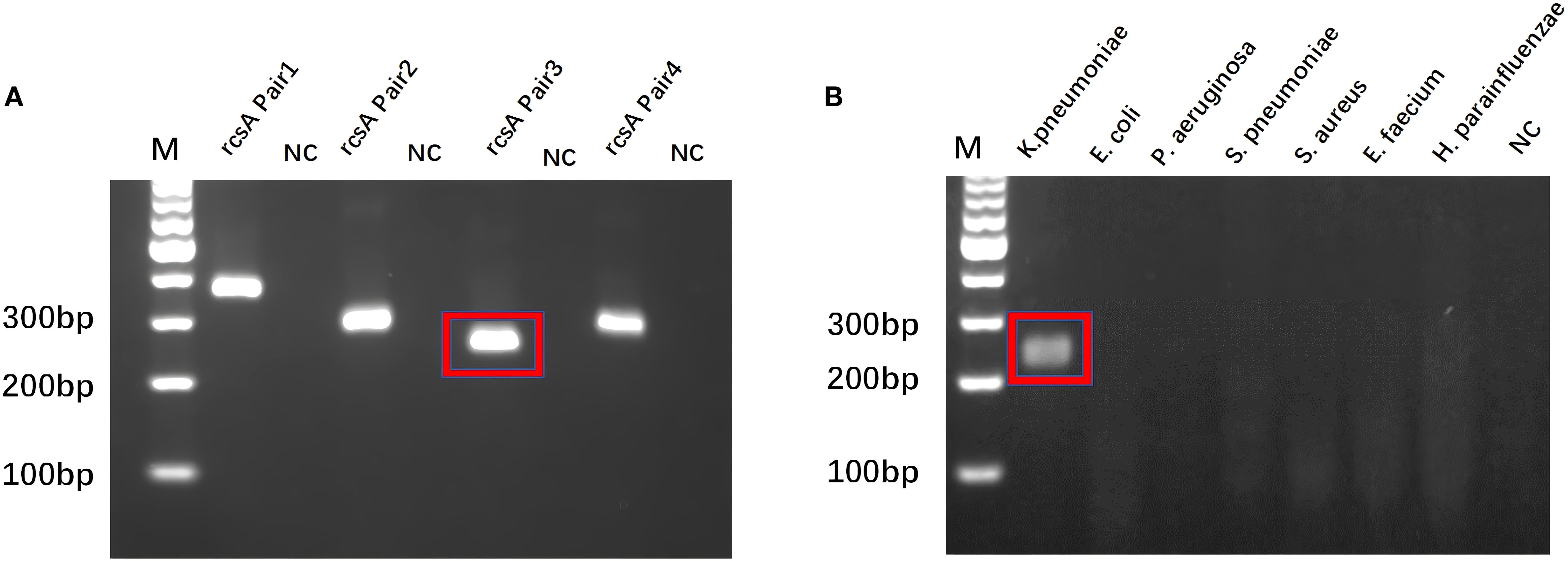
Figure 2. RPA primer screening and specificity validation. (A) Best RPA Primer Screening: rcsA Pair 3 is the best RPA primer. (B) Specificity verification of rcsA Pair 3. M, Marker; NC, Negative Control.
3.3 Establishment and performance evaluation of light control one-pot RPA-CRISPR/Cas12a method
Three crRNAs were designed based on the amplified fragments, and each crRNA successfully activated the specific recognition of Cas12a protein, but only rcsA-crRNA1 (Figures 3A, B) showed the highest non-specific cleavage efficiency at 30 min for designing caged crRNAs for subsequent experiments. We systematically excluded reaction components to assess the robustness of the light-regulated “one-pot” RPA-CRISPR/Cas12a assay. As shown in Figure 3C, the observed fluorescence signal is negligible in the absence of Cas12a, probes, target DNA, illumination, or caged crRNA, including the traditional “one-pot” method. However, only when all reaction components are present simultaneously and irradiated with ultraviolet light, the fluorescence signal intensity significantly increases, and specific and non-specific cleavage (trans-cleavage) functions are activated (Figures 3D, E).
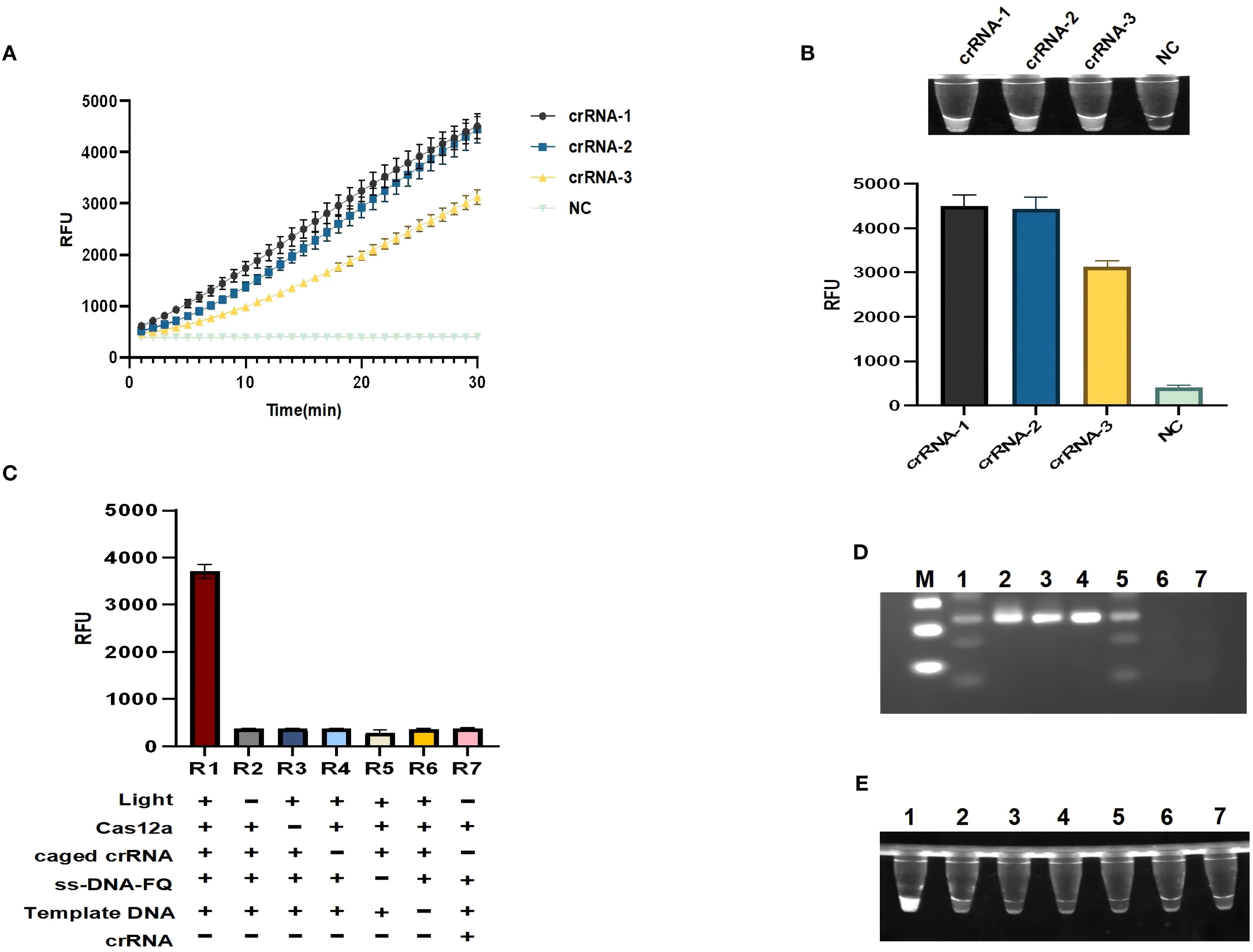
Figure 3. Establishment of a light-controlled one-pot RPA-CRISPR/Cas12a method. (A, B) Fluorescence curves and endpoint signals of three crRNAs designed for the rcsA amplicon. N = 3, error bars represent standard deviation. (C) Component omission experiments to validate the feasibility of the light-controlled one-pot RPA-CRISPR/Cas12a method.(+) represents the added reaction component, (-) represents the not added reaction component.R1:Light (+), Cas12a(+), caged crRNA(+), ss-DNA-FQ(+), Template DNA(+), crRNA (-); R2:Light (-), Cas12a(+), caged crRNA(+), ss-DNA-FQ(+), Template DNA(+), crRNA (-); R3:Light (-), Cas12a (-), caged crRNA(+), ss-DNA-FQ(+), Template DNA(+), crRNA (-); R4:Light (-), Cas12a(+), caged crRNA(-), ss-DNA-FQ(+), Template DNA(+), crRNA (-); R5:Light (-), Cas12a(+), caged crRNA(+), ss-DNA-FQ(-), Template DNA(+), crRNA (-); R6:Light (-), Cas12a(+), caged crRNA(+), ss-DNA-FQ(+), Template DNA(-), crRNA (-); R7:Light (-), Cas12a(+), caged crRNA (-), ss-DNA-FQ(+), Template DNA(-), crRNA (+). (D) Gel electrophoresis analysis under the same corresponding conditions as (C). (E) Ultraviolet imaging showing the nonspecific cleavage of the light-controlled one-pot RPA-CRISPR/Cas12a method. M, marker; NC, negative control.
3.4 Optimization of the light-controlled one-pot RPA-CRISPR/Cas12a method
To improve the sensitivity of the diagnostic method, the conditions were optimized. Firstly, the RPA amplification system was optimized. Optimization focused on testing different amplification times (10, 15, 20, 30 and 40 min) and primer concentrations (160, 240, 320, 400, 480 and 560 nM). When the primer concentration is 400 nM and amplified for 20 min, the amplification efficiency is the best, and the fluorescence efficiency is the highest (Figures 4A, C). The light-controlled Cas12a detection system was then optimized, and the fluorescence values showed that the fluorescence efficiency was best at 300 nM for the caged crRNA and 300 nM for the Cas protein (Figures 4D, E). In order to fully activate the caged crRNA, we optimized the light time. At 30 s of illumination, the fluorescence values were highest (Figure 4B).
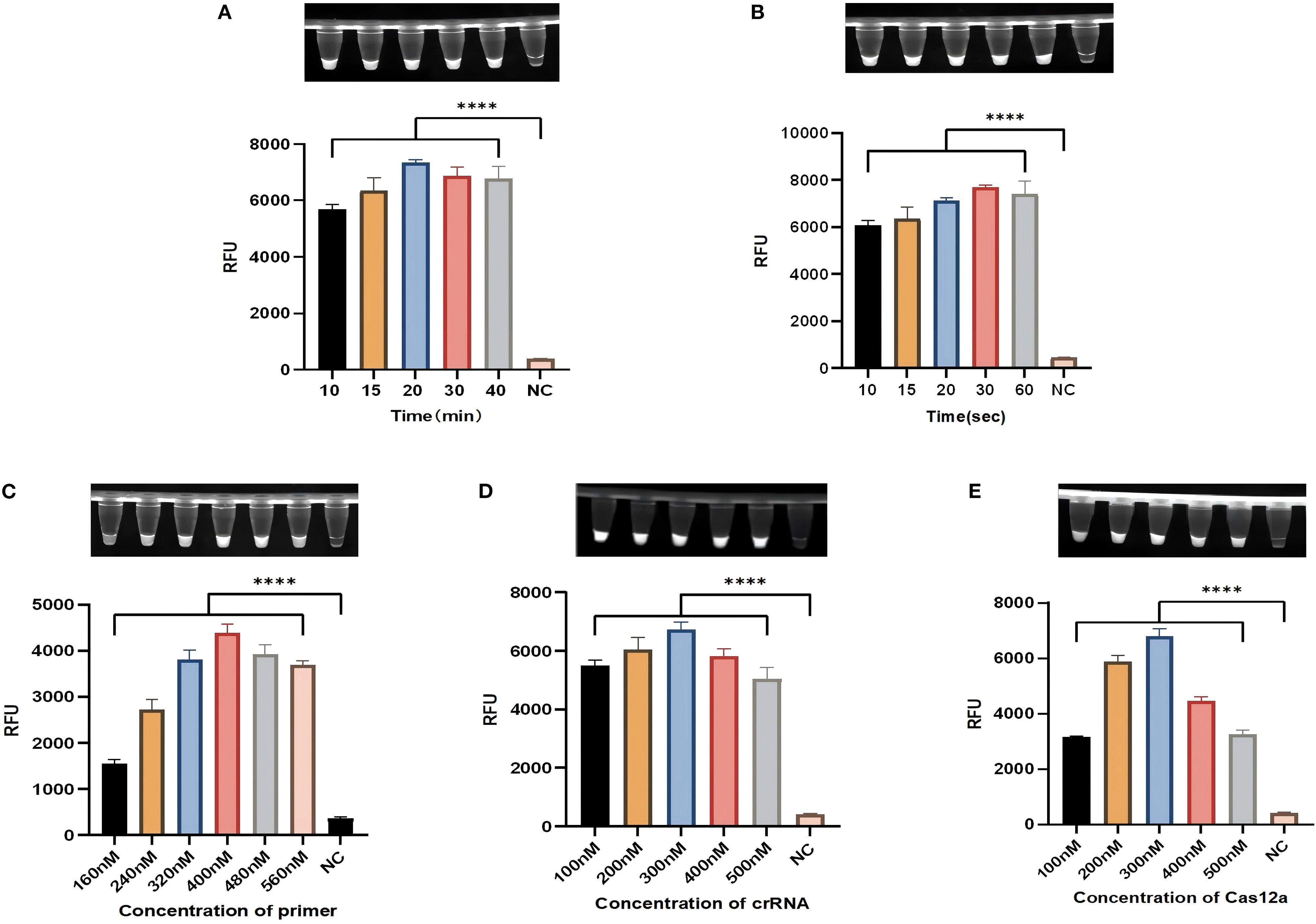
Figure 4. Optimization of the light-controlled one-pot RPA-CRISPR/Cas12a method. (A, C) The amplification time and primer concentration in the light-controlled one-pot RPA-CRISPR/Cas12a system. (B) The irradiation time in the light-controlled one-pot RPA-CRISPR/Cas12a system. (D, E) The caged crRNA concentration and Cas protein concentration in the light-controlled one-pot RPA-CRISPR/Cas12a system. N = 3, error bars represent the standard deviation. NC, negative control. Two-tailed Student’s t-test, ****p < 0.0001.
3.5 Sensitivity and specificity of the light-controlled one-pot RPA-CRISPR/Cas12a method
The sensitivity of the light-controlled one-tube RPA-CRISPR/Cas12a system was evaluated by serial dilutions of KP genomic DNA from 4.072×106 copies/reaction to 4.072×102 copies/reaction. The above research results indicate that the single-tube RPA-CRISPR/Cas12a system under light control may have higher sensitivity in detecting KP (Figure 5A). To determine the specificity of the light-controlled one-tube RPA-CRISPR/Cas12a system, we tested for common clinical pathogens, including KP, Haemophilus parainfluenzae, Pseudomonas aeruginosa, Streptococcus pneumoniae, Enterococcus faecium, Enterococcus faecalis, Staphylococcus aureus, and Escherichia coli. The results showed that KP exhibited a distinct fluorescent signal, while other strains showed almost no background fluorescence, confirming the specificity of the system (Figure 5B).
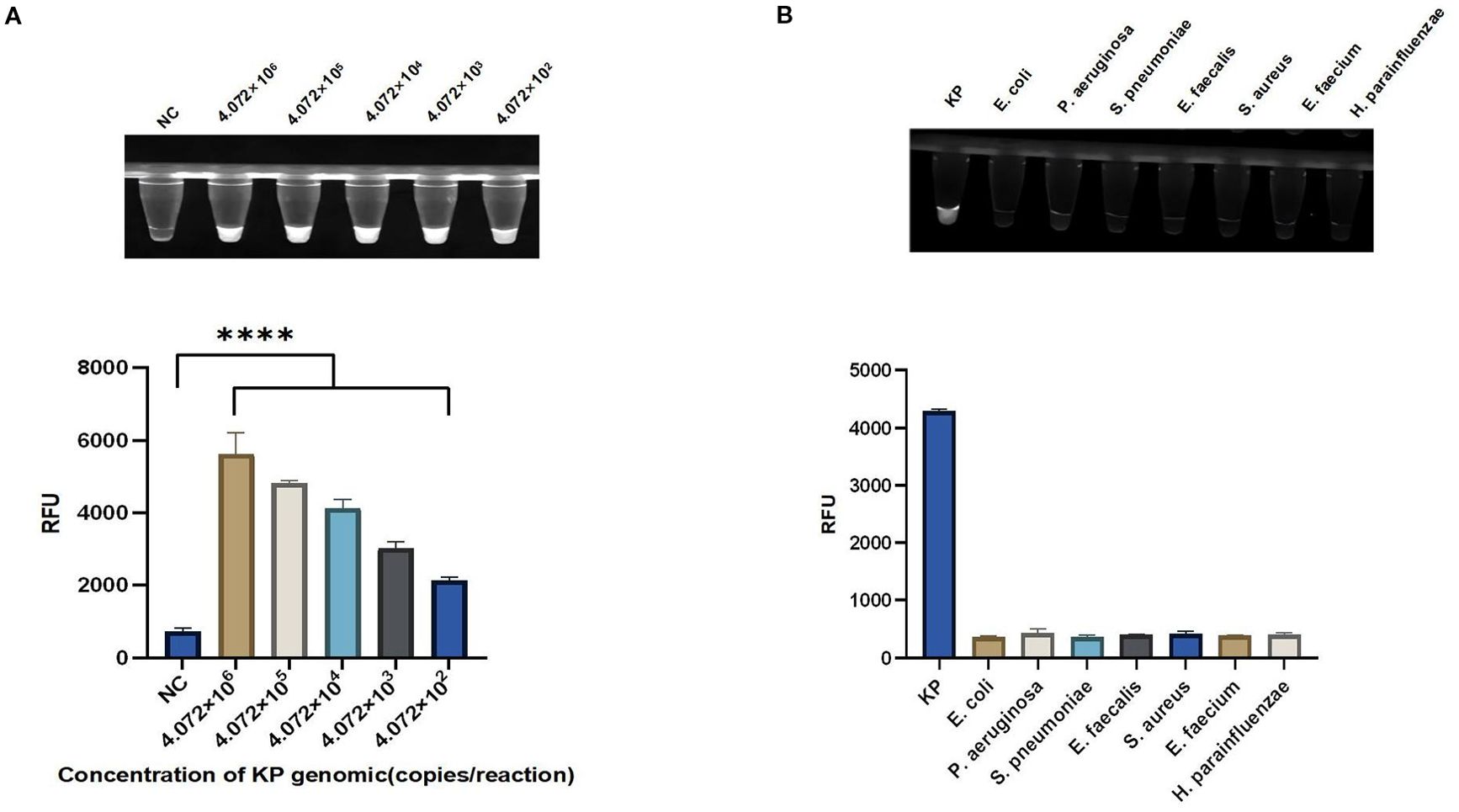
Figure 5. Sensitivity and specificity of the light-controlled one-pot RPA-CRISPR/Cas12a method. (A) Serial dilutions of the KP genomic DNA were used to validate the sensitivity of the light-controlled one-pot RPA-CRISPR/Cas12a system. (B) The specificity of the light-controlled one-pot RPA-CRISPR/Cas12a system was validated using seven common clinical pathogens. N = 3, error bars represent the standard deviation. NC, negative control. Two-tailed Student’s t-test, ****p < 0.0001.
3.6 The detection of clinical strains by light-controlled one-pot RPA-CRISPR/Cas12a method
In this study, 30 clinical strains of KP and 20 negative control strains were used as samples to compare and analyze the detection performance of the light-controlled RPA-CRISPR/Cas12a integrated detection system and the traditional PCR method, and the sensitivity and specificity of the technology in clinical application were systematically evaluated. The results of PCR analysis showed that among the 50 clinically isolated strains, 30 strains successfully amplified the rcsA gene fragment, and were identified and confirmed to be KP (positive detection rate of 60%). The remaining 20 strains were judged to be control colonies because the rcsA gene was not detected (Figure 6A). The light-controlled one-tube RPA-CRISPR/Cas12a detection system was further verified, and the ultraviolet signal analysis showed the detection results were in complete agreement with the PCR method (100% coincidence rate) (Figures 6B, C). The receiver operating characteristic (ROC) curve shows that at a fluorescence threshold of 2710, the area under the ROC curve (AUC) is 1.000, with sensitivity and specificity of 100% (95% confidence interval: 88.65–100%) and 100% (95% confidence interval: 83.89–100%), respectively (Supplementary Figure S1).
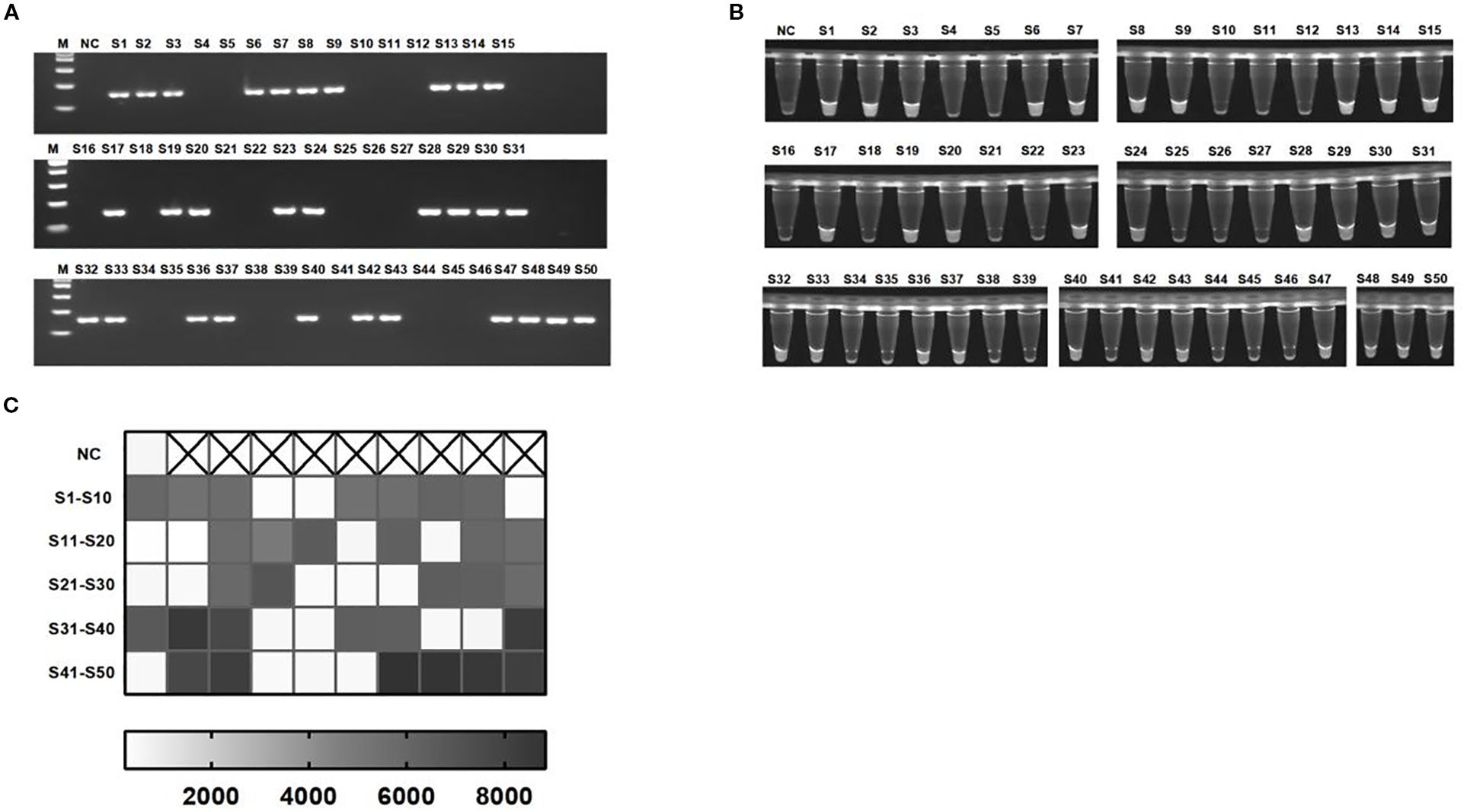
Figure 6. Detection of KP clinical strains. (A) Representative 2% agarose gel electrophoresis confirming PCR-based identification of clinical isolates. (B, C) Visualization signals and endpoint fluorescence value heatmap of 50 clinical strains detected using the light-controlled one-pot RPA-CRISPR/Cas12a method. NC, negative control. S1-S50, all strains are derived from clinical sources.
4 Discussion
KP is the main causative agent of pneumonia, urinary tract infections, and bacteremia, and is an important opportunistic healthcare-associated pathogen. In 2017, the World Health Organization recognized the prevalence of its multidrug-resistant strains as a serious public health threat (Stojowska-Swędrzyńska et al., 2021). This strain is not only an important vector for the global spread of antibiotic resistance, but also a major risk factor for infection due to colonization of the gastrointestinal tract, as well as a key hub for the spread of drug resistance (Lindstedt et al., 2022). Due to the high multidrug resistance of KP strains, which complicates infection treatment (Kim et al., 2022), there is an urgent need for rapid and accurate diagnostic methods to enable early prevention. This would effectively curb the spread of drug-resistant bacteria and improve clinical outcomes.
Although the traditional bacterial culture method is regarded as the gold standard for the diagnosis of KP, its diagnosis cycle is long, usually 48 to 72 hours, which is extremely detrimental to the timely treatment and disease control of patients. Although the detection time of the PCR method is shorter than that of traditional bacterial culture methods, it has higher requirements for instruments and equipment and needs to be performed by professionally trained operators, which limits its wide application to a certain extent. RPA is an isothermal nucleic acid amplification technology that has attracted much attention in recent years. This technology is characterized by easy operation, fast amplification speed (usually completed in 20–30 min), and mild reaction temperature (37-42 °C) (Tan et al., 2022; Liao et al., 2024). As a result, rapid amplification of the gene of interest can be achieved. The CRISPR/Cas12a system has the advantages of high specificity and high sensitivity. Therefore, the integration of RPA and CRISPR systems can achieve rapid and efficient detection of target genes (Aman et al., 2020, 2021; Zhang et al., 2023; Ji et al., 2025). This study successfully constructed a “one-pot” RPA detection platform based on photo-controlled CRISPR/Cas12a. By introducing NPOM modified crRNA, rapid detection of KP was achieved (detection time of 50 minutes, sensitivity of 4.072 × 102 copies/reaction). As a photocleavable group, NPOM-dt modification temporarily inactivates crRNA (caged crRNA), inhibiting Cas12a activity. Upon UV irradiation of caged crRNA, NPOM-dt dissociates, restoring crRNA activity to guide Cas12a in recognizing target DNA and triggering its cleavage function. The entire detection process enables a “one-pot” operation without requiring additional reagents. This feature is particularly advantageous for developing freeze-dried reagents, significantly enhancing the portability of detection kits.
Compared to the traditional two-step RPA-CRISPR/Cas12a detection system, this method employs a single-tube reaction format, significantly reducing the risk of aerosol contamination associated with the transfer of amplified products (Tan et al., 2024b; Wu et al., 2025). Experimental results demonstrate that the light-controlled “one-pot” assay exhibits comparable performance to the standard two-step method, achieving identical sensitivity (10–6 ng/μL) and specificity (100%) (Zhou et al., 2024; Wei et al., 2025). These findings indicate that the performance of the developed detection system is unaffected by single-tube operation. In the traditional one-step method, the CRISPR system activates and cleaves the template early in the RPA amplification, which affects the accumulation of target amplification products, thus reducing the sensitivity of the detection system (Gao et al., 2024; Zhang et al., 2024). This study successfully avoided premature activation of the CRISPR system by employing a light-controlled temporal regulation mechanism, thereby achieving high efficiency and reliability in single-tube reactions.
Compared to recent studies, this detection system demonstrates significant advantages in operational convenience, cost-effectiveness, and detection efficiency. The method developed by Fu et al. enables detection within 31 minutes (with a detection limit as low as 101 CFU/μL) (Fu et al., 2025). However, it requires screening multiple suboptimal PAM crRNAs, increasing detection costs. In contrast, our system requires only a 15 μL reaction volume (reducing standard RPA volume by four-fifths) and utilizes standard UV lamp equipment costing just $15, making the overall cost more competitive. Recently, Wu et al. reported that they have achieved rapid detection of KP (40 minutes, detection limit 100 copies/μL) through a extraction-free, tube-cap, “one-pot” RPA-CRISPR/Cas12a method (Wu et al., 2025). The simplicity and rapidity of the assay are augmented by a straightforward sample processing without extraction. In contrast, this study does not require separate regulation of RPA and CRISPR steps, but DNA extraction increases the time cost and operational complexity. Furthermore, Wang et al.’s PCR-CRISPR/Cas12a detection method takes approximately 2 hours (Wang et al., 2023); Zhang et al.’s LAMP-CRISPR/Cas12b system (Zhang et al., 2025) requires constant 58 °C incubation and multiple primer sets, further increasing instrumentation requirements and reagent costs.
During experimental optimization, we successfully enhanced detection performance through systematic adjustments to RPA primer concentration, illumination duration, Cas12a protein concentration, and caged crRNA concentration. The experiment of missing reaction components has demonstrated the feasibility of a one-pot RPA CRISPR/Cas12a detection system controlled by light, which only triggers the reaction under illumination. Following the method described by Hu et al. (Hu M. et al., 2023), we employed a 35 W, 365 nm UV lamp for 30 seconds of irradiation to ensure complete shedding of NPOM-dt. Given the brief exposure duration and high DNA concentration post-isothermal amplification, any potential DNA damage from UV exposure is negligible within the detection system.
Nevertheless, this study has the following limitations and directions for future improvement: Firstly, clinical applicability of UV irradiation: Although a commercial LED UV lamp with a fixed wavelength (365 nm) was used, activation still relies on manual timing control. Future development will focus on integrating a portable UV-regulated module into the fluorescent detection device to enhance operational standardization and clinical applicability. Secondly, sample size limitation: Although results from 50 clinical samples showed complete concordance with PCR, the small sample size still indicated that this study was a proof-of-concept and needs to be validated in a larger sample size before being put into clinical application in the future. Thirdly, DNA extraction dependency: Sample pretreatment is required, limiting point-of-care testing applications. Although DNA extraction-free methods exist (Wu et al., 2025), they involve complex procedures and higher costs. Future development will focus on developing a simple, low-cost, light-controlled, extraction-free, single-pot detection system to advance point-of-care testing applications.
In conclusion, our study proposes a one-tube detection system for the detection of KP using light-controlled CRISPR/CAS12a combined with RPA, which has significant advantages in preventing aerosol contamination and speed, sensitivity, and portability. This platform facilitates the rapid detection of the rcsA gene in KP. Its applicability in the early and rapid diagnosis of KP infection is expected to curb its potential for widespread spread, particularly in hospital settings. In addition, the system provides a cost-effective screening method tailored to resource-constrained areas.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
LP: Conceptualization, Validation, Writing – original draft. LW: Conceptualization, Writing – original draft, Visualization. SL: Data curation, Writing – review & editing. BR: Data curation, Writing – review & editing. ML: Data curation, Writing – review & editing. LL: Conceptualization, Writing – review & editing. XL: Supervision, Writing – review & editing. GW: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was funded by the Natural Science Foundation of China (82260418), Natural Science Foundation of Guangxi (2024JJH140215, 2024JJH140265), First Batch of High-level Talent Scientific Research Projects of the Affiliated Hospital of Youjiang Medical University for Nationalities (R202011701), Baise Scientific Research and Technology Development Project (20232031, 20241534, 20250344), and Research Project of Guangxi Zhuang Autonomous Region Disease Prevention and Control Bureau (GXJKKJ24C010).
Acknowledgments
We sincerely appreciate the technical support and facilities provided by the Center for Medical Laboratory Science, Affiliated Hospital of Youjiang Medical University for Nationalities.
Conflict of interest
Author BR was employed by the company Yaneng BlOscience Shenzhen Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1669860/full#supplementary-material
References
Adeolu, M., Alnajar, S., Naushad, S., and S Gupta, R. (2016). Genome-based phylogeny and taxonomy of the 'Enterobacteriales': proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 66, 5575–5599. doi: 10.1099/ijsem.0.001485
Aman, R., Mahas, A., Marsic, T., Hassan, N., and Mahfouz, M. M. (2020). Efficient, rapid, and sensitive detection of plant RNA viruses with one-pot RT-RPA-CRISPR/Cas12a assay. Front. Microbiol. 11, e610872. doi: 10.3389/fmicb.2020.610872
Aman, R., Marsic, T., Sivakrishna Rao, G., Mahas, A., Ali, Z., Alsanea, M., et al. (2021). iSCAN-V2: A one-pot RT-RPA-CRISPR/Cas12b assay for point-of-care SARS-CoV-2 detection. Front. Bioeng Biotechnol. 9, e800104. doi: 10.3389/fbioe.2021.800104
Chen, L.-F., Chang, C.-Y., Hsu, L.-C., Tsai, P.-H., Chang, S.-J., Chang, S.-C., et al. (2013). Bacterial pneumonia following acute ischemic stroke. J. Chin. Med. Assoc. 76, 78–82. doi: 10.1016/j.jcma.2012.10.005
Chen, L., Hu, M., and Zhou, X. (2025). Trends in developing one-pot CRISPR diagnostics strategies. Trends Biotechnol. 43, 98–110. doi: 10.1016/j.tibtech.2024.07.007
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 360, 436–439. doi: 10.1126/science.aar6245
Chen, Y., Xu, X., Wang, J., Zhang, Y., Zeng, W., Liu, Y., et al. (2022). Photoactivatable CRISPR/Cas12a strategy for one-pot DETECTR molecular diagnosis. Anal. Chem. 94, 9724–9731. doi: 10.1021/acs.analchem.2c01193
Dai, T., Yang, X., Hu, T., Jiao, B., Xu, Y., Zheng, X., et al. (2019). Comparative evaluation of a novel recombinase polymerase amplification-lateral flow dipstick (RPA-LFD) assay, LAMP, Conventional PCR, and leaf-disc baiting methods for detection of Phytophthora sojae. Front. Microbiol. 10, e1884. doi: 10.3389/fmicb.2019.01884
Dong, D., Liu, W., Li, H., Wang, Y., Li, X., Zou, D., et al. (2015). Survey and rapid detection of Klebsiella pneumoniae in clinical samples targeting the rcsA gene in Beijing, China. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00519
Fu, J., Mo, R., Li, Z., Xu, S., Cheng, X., Lu, B., et al. (2025). An extraction-free one-pot assay for rapid detection of Klebsiella pneumoniae by combining RPA and CRISPR/Cas12a. Biosens Bioelectron. 267, e116740. doi: 10.1016/j.bios.2024.116740
Gao, Y., Ang, Y. S., and Yung, L. Y. L. (2024). One-pot detection of proteins using a two-way extension-based assay with Cas12a. ACS Sens. 9, 3928–3937. doi: 10.1021/acssensors.4c00370
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 356, 438–442. doi: 10.1126/science.aam9321
Harrington, L. B., Burstein, D., Chen, J. S., Paez-Espino, D., Ma, E., Witte, I. P., et al. (2018). Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 362, 839–842. doi: 10.1126/science.aav4294
Hu, J. J., Liu, D., Cai, M. Z., Zhou, Y., Yin, W. X., and Luo, C. X. (2023). One-pot assay for rapid detection of Benzimidazole resistance in Venturia carpophila by combining RPA and CRISPR/Cas12a. J. Agric. Food Chem. 71, 1381–1390. doi: 10.1021/acs.jafc.2c06549
Hu, M., Liu, R., Qiu, Z., Cao, F., Tian, T., Lu, Y., et al. (2023). Light-start CRISPR-Cas12a reaction with caged crRNA enables rapid and sensitive nucleic acid detection. Angew Chem. Int. Ed Engl. 62, e202300663. doi: 10.1002/anie.202300663
Hu, M., Qiu, Z., Bi, Z., Tian, T., Jiang, Y., and Zhou, X. (2022). Photocontrolled crRNA activation enables robust CRISPR-Cas12a diagnostics. Proc. Natl. Acad. Sci. U. S. A. 119, e2202034119. doi: 10.1073/pnas.2202034119
Huang, Y., Li, J., Wang, Q., Tang, K., and Li, C. (2022). Rapid detection of KPC-producing Klebsiella pneumoniae in China based on MALDI-TOF MS. J. Microbiol. Methods 192, e106385. doi: 10.1016/j.mimet.2021.106385
Ji, T., Fang, X., Gao, Y., Yu, K., and Gao, X. (2025). Research progress on the application of RPA-CRISPR/Cas12a in the rapid visual detection of pathogenic microorganisms. Front. Cell. Infect. Microbiol. 15, e1640938. doi: 10.3389/fcimb.2025.1640938
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O., and Zhang, F. (2019). SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012. doi: 10.1038/s41596-019-0210-2
Khan, F. Y., Abukhattab, M., AbuKamar, M., and Anand, D. (2014). Adult Klebsiella pneumoniae meningitis in Qatar: clinical pattern of ten cases. Asian Pac. J. Trop. Biomed. 4, 669–672. doi: 10.12980/apjtb.4.201414b100
Kim, H., Jang, J. H., Jung, I. Y., and Cho, J. H. (2022). A novel peptide as a specific and selective probe for Klebsiella pneumoniae detection. Biosensors (Basel). 12, e153. doi: 10.3390/bios12030153
Lan, H., Shu, W., Jiang, D., Yu, L., and Xu, G. (2024). Cas-based bacterial detection: recent advances and perspectives. Analyst. 149, 1398–1415. doi: 10.1039/d3an02120c
Li, S.-Y., Cheng, Q. X., Liu, J. K., Nie, X. Q., Zhao, G. P., and Wang, J. (2018a). CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 28, 491–493. doi: 10.1038/s41422-018-0022-x
Li, S. Y., Cheng, Q. X., Wang, J. M., Li, X. Y., Zhang, Z. L., Gao, S., et al. (2018b). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4, e20. doi: 10.1038/s41421-018-0028-z
Liao, C., Pan, L., Tan, M., Zhou, Z., Long, S., Yi, X., et al. (2024). A dual RPA-LFD assay for the simultaneous detection of Salmonella typhimurium and Salmonella enteritidis. Front. Bioeng Biotechnol. 12, e1379939. doi: 10.3389/fbioe.2024.1379939
Lin, K., Guo, J., Guo, X., Li, Q., Li, X., Sun, Z., et al. (2023). Fast and visual detection of nucleic acids using a one-step RPA-CRISPR detection (ORCD) system unrestricted by the PAM. Anal. Chim. Acta 1248, e340938. doi: 10.1016/j.aca.2023.340938
Lindstedt, K., Buczek, D., Pedersen, T., Hjerde, E., Raffelsberger, N., Suzuki, Y., et al. (2022). Detection of Klebsiella pneumoniae human gut carriage: a comparison of culture, qPCR, and whole metagenomic sequencing methods. Gut Microbes 14, e2118500. doi: 10.1080/19490976.2022.2118500
Okomo, U., Akpalu, E. N. K., Le Doare, K., Roca, A., Cousens, S., Jarde, A., et al. (2019). Aetiology of invasive bacterial infection and antimicrobial resistance in neonates in sub-Saharan Africa: a systematic review and meta-analysis in line with the STROBE-NI reporting guidelines. Lancet Infect. Dis. 19, 1219–1234. doi: 10.1016/s1473-3099(19)30414-1
Paczosa, M. K. and Mecsas, J. (2016). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80, 629–661. doi: 10.1128/mmbr.00078-15
Sen, A., Masetty, M., Weerakoon, S., Morris, C., Yadav, J. S., Apewokin, S., et al. (2024). Paper-based loop-mediated isothermal amplification and CRISPR integrated platform for on-site nucleic acid testing of pathogens. Biosens. Bioelectron. 257, e116292. doi: 10.1016/j.bios.2024.116292
Si, Y., Zhang, T., Chen, N., Cheng, Y., Wang, L., Yuan, J., et al. (2021). A LAMP-based system for rapid detection of eight common pathogens causing lower respiratory tract infections. J. Microbiol. Methods 190, e106339. doi: 10.1016/j.mimet.2021.106339
Srivastava, P. and Prasad, D. (2023). Isothermal nucleic acid amplification and its uses in modern diagnostic technologies. 3 Biotech. 13, 200. doi: 10.1007/s13205-023-03628-6
Stojowska-Swędrzyńska, K., Łupkowska, A., Kuczyńska-Wiśnik, D., and Laskowska, E. (2021). Antibiotic heteroresistance in Klebsiella pneumoniae. Int. J. Mol. Sci. 23, e449. doi: 10.3390/ijms23010449
Tan, M., Liang, L., Liao, C., Zhou, Z., Long, S., Yi, X., et al. (2024a). A rapid and ultra-sensitive dual readout platform for Klebsiella pneumoniae detection based on RPA-CRISPR/Cas12a. Front. Cell. Infect. Microbiol. 14, e1362513. doi: 10.3389/fcimb.2024.1362513
Tan, M., Liao, C., Liang, L., Yi, X., Zhou, Z., and Wei, G. (2022). Recent advances in recombinase polymerase amplification: principle, advantages, disadvantages and applications. Front. Cell. Infect. Microbiol. 12, e1019071. doi: 10.3389/fcimb.2022.1019071
Tan, Q., Shi, Y., Duan, C., Li, Q., Gong, T., Li, S., et al. (2024). Simple, sensitive, and visual detection of 12 respiratory pathogens with one-pot-RPA-CRISPR/Cas12a assay. J. Med. Virol. 96, e29624. doi: 10.1002/jmv.29624
Tan, M., Yi, X., Liao, C., Zhou, Z., Ren, B., Liang, L., et al. (2024b). Establishment of a platform based on dual RPA combined with CRISPR/Cas12a for the detection of Klebsiella pneumoniae and its KPC resistance gene. Front. Bioeng Biotechnol. 12, e1447963. doi: 10.3389/fbioe.2024.1447963
Wang, Y., Tang, Y., Chen, Y., Yu, G., Zhang, X., Yang, L., et al. (2024). Ultrasensitive one-pot detection of monkeypox virus with RPA and CRISPR in a sucrose-aided multiphase aqueous system. Microbiol. Spectrum. 12, e0226723. doi: 10.1128/spectrum.02267-23
Wang, S., Wang, S., Tang, Y., Peng, G., Hao, T., Wu, X., et al. (2023). Detection of Klebsiella pneumonia DNA and ESBL positive strains by PCR-based CRISPR-LbCas12a system. Front. Microbiol. 14, e1128261. doi: 10.3389/fmicb.2023.1128261
Wei, L., Luo, S., Zhou, W., Ren, B., Li, M., Liang, L., et al. (2025). Rapid detection of Pseudomonas aeruginosa by glycerol one-pot RAA/CRISPR-Cas12a method. Front. Chem. 13, e1654270. doi: 10.3389/fchem.2025.1654270
Wu, Z., Xu, Y., Zhou, W., Shi, L., Shi, W., Pu, L., et al. (2025). Rapid detection of Klebsiella pneumoniae based on one-tube RPA-CRISPR/Cas12a system. Clin. Chim. Acta 573, e120281. doi: 10.1016/j.cca.2025.120281
Xue, P., Peng, Y., Wang, R., Wu, Q., Chen, Q., Yan, C., et al. (2025). Advances, challenges, and opportunities for food safety analysis in the isothermal nucleic acid amplification/CRISPR-Cas12a era. Crit. Rev. Food Sci. Nutr. 65, 2473–2488. doi: 10.1080/10408398.2024.2343413
Yao, B., Xiao, X., Wang, F., Zhou, L., Zhang, X., and Zhang, J. (2015). Clinical and molecular characteristics of multi-clone carbapenem-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in a tertiary hospital in Beijing, China. Int. J. Infect. Dis. 37, 107–112. doi: 10.1016/j.ijid.2015.06.023
Zhang, Y., Liu, T., Zhang, P., Ni, B., Wang, X., Bai, L., et al. (2025). A rapid and accurate method for Helicobacter pylori detection via integrating LAMP assay with CRISPR/Cas12b detection by one-step in one-pot. Front. Cell. Infect. Microbiol. 15, e1611134. doi: 10.3389/fcimb.2025.1611134
Zhang, J., Qin, L., Chang, Y., He, Y., Zhao, W., Zhao, Y., et al. (2024). One-pot assay for rapid detection of Stenotrophomonas maltophilia by RPA-CRISPR/Cas12a. ACS Synth. Biol. 13, 3400–3412. doi: 10.1021/acssynbio.4c00481
Zhang, K., Sun, Z., Shi, K., Yang, D., Bian, Z., Li, Y., et al. (2023). RPA-CRISPR/Cas12a-based detection of Haemophilus parasuis. Anim. (Basel). 13, 3317. doi: 10.3390/ani13213317
Zhang, Y., Wang, Q., Yin, Y., Chen, H., Jin, L., Gu, B., et al. (2018). Epidemiology of Carbapenem-resistant Enterobacteriaceae infections: report from the China CRE network. Antimicrob. Agents Chemother. 62, e01882–e01817. doi: 10.1128/aac.01882-17
Zhang, Y., Yao, Z., Zhan, S., Yang, Z., Wei, D., Zhang, J., et al. (2014). Disease burden of intensive care unit-acquired pneumonia in China: a systematic review and meta-analysis. Int. J. Infect. Dis. 29, 84–90. doi: 10.1016/j.ijid.2014.05.030
Zhang, Y., Zeng, J., Liu, W., Zhao, F., Hu, Z., Zhao, C., et al. (2015). Emergence of a hypervirulent carbapenem-resistant Klebsiella pneumoniae isolate from clinical infections in China. J. Infect. 71, 553–560. doi: 10.1016/j.jinf.2015.07.010
Keywords: Klebsiella pneumoniae, recombinase polymerase amplification, one-pot, CRISPR/Cas12a, light-controlled
Citation: Pan L, Wei L, Luo S, Ren B, Li M, Liang L, Li X and Wei G (2025) Klebsiella pneumoniae detection by a light-controlled one-pot RPA-CRISPR/Cas12a method. Front. Cell. Infect. Microbiol. 15:1669860. doi: 10.3389/fcimb.2025.1669860
Received: 05 August 2025; Accepted: 15 September 2025;
Published: 01 October 2025.
Edited by:
Rodolfo García-Contreras, National Autonomous University of Mexico, MexicoReviewed by:
Concha Ortiz Cartagena, Institute of Biomedical Research of A Coruña (INIBIC), SpainReza Mohammadhassan, University of Otago, New Zealand
Copyright © 2025 Pan, Wei, Luo, Ren, Li, Liang, Li and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lina Liang, bGlhbmdsaW5hMjAyMTAxMDFAMTYzLmNvbQ==; Xuebin Li, MDAwMjVAeW11bi5lZHUuY24=; Guijiang Wei, d2VpZ3VpamlhbmcyMDIxQDE2My5jb20=
†These authors have contributed equally to this work