 Annayya R. Aroor1,2
Annayya R. Aroor1,2 Vincent G. DeMarco1,2,3
Vincent G. DeMarco1,2,3 Guanghong Jia1,2
Guanghong Jia1,2 Zhe Sun4
Zhe Sun4 Ravi Nistala5
Ravi Nistala5 Gerald A. Meininger3,4
Gerald A. Meininger3,4 James R. Sowers1,2,3,4*
James R. Sowers1,2,3,4*- 1Department of Internal Medicine, Division of Endocrinology, Diabetes and Metabolism, University of Missouri Columbia School of Medicine, Columbia, MO, USA
- 2Harry S. Truman Memorial Veterans Hospital, Columbia, MO, USA
- 3Department of Medical Pharmacology and Physiology, University of Missouri Columbia School of Medicine, Columbia, MO, USA
- 4Dalton Cardiovascular Research Center, Columbia, MO, USA
- 5Department of Internal Medicine, Division of Nephrology, University of Missouri Columbia School of Medicine, Columbia, MO, USA
Epidemiological studies support the notion that arterial stiffness is an independent predictor of adverse cardiovascular events contributing significantly to systolic hypertension, impaired ventricular-arterial coupling and diastolic dysfunction, impairment in myocardial oxygen supply and demand, and progression of kidney disease. Although arterial stiffness is associated with aging, it is accelerated in the presence of obesity and diabetes. The prevalence of arterial stiffness parallels the increase of obesity that is occurring in epidemic proportions and is partly driven by a sedentary life style and consumption of a high fructose, high salt, and high fat western diet. Although the underlying mechanisms and mediators of arterial stiffness are not well understood, accumulating evidence supports the role of insulin resistance and endothelial dysfunction. The local tissue renin-angiotensin-aldosterone system (RAAS) in the vascular tissue and immune cells and perivascular adipose tissue is recognized as an important element involved in endothelial dysfunction which contributes significantly to arterial stiffness. Activation of vascular RAAS is seen in humans and animal models of obesity and diabetes, and associated with enhanced oxidative stress and inflammation in the vascular tissue. The cross talk between angiotensin and aldosterone underscores the importance of mineralocorticoid receptors in modulation of insulin resistance, decreased bioavailability of nitric oxide, endothelial dysfunction, and arterial stiffness. In addition, both innate and adaptive immunity are involved in this local tissue activation of RAAS. In this review we will attempt to present a unifying mechanism of how environmental and immunological factors are involved in this local tissue RAAS activation, and the role of this process in the development of endothelial dysfunction and arterial stiffness and targeting tissue RAAS activation.
Introduction
Arterial stiffness is now considered an independent risk factor for the progression of cardiovascular and chronic kidney disease (CKD) (1). Arterial stiffness increases with aging and is associated with isolated systolic hypertension which occurs in most elderly persons (2). However, the process is accelerated in the presence of obesity and diabetes and occurs at earlier ages (1, 3). Given the association between arterial stiffness and obesity, it is likely that the prevalence of arterial stiffness has been increasing proportionately to the obesity epidemic, which is driven by consumption of a high fat, high fructose, and high salt western diet and further aggravated by a sedentary life style in adults and children in the Unites States and around the globe (4–7). This underscores the importance of arterial stiffness not only as a biomarker for the evaluation of progression of cardiovascular disease (CVD) and kidney disease, but also an important therapeutic target for improved cardiovascular and renal outcomes in obesity and diabetes.
Arterial Stiffness as a Risk Factor for Cardiovascular and Kidney Disease
Arterial stiffness is associated with obesity, insulin resistance, and activation of the renin-angiotensin-aldosterone system (RAAS) in individuals with the cardiorenal syndrome (CRS) and even in obese children (1, 2, 5, 8). Increased arterial stiffness is also seen in normotensive subjects predisposed to develop hypertension and in pre-hypertensive subjects (9, 10). In the Atherosclerosis Risk in Communities analysis, incident hypertension was more robustly predicted when subjects were in the highest quartile of arterial stiffness. For each standard deviation decrease in elasticity, there was a 15% increase in developing hypertension (11). Arterial stiffness increases with age, metabolic abnormalities, and increased sodium intake, all of which are associated with CVD, including heart failure (12, 13). Furthermore, arterial stiffness itself is associated with left ventricular diastolic dysfunction (14). Increased arterial stiffness is a marker of vasculopathy in CKD patients, suggesting significant cardiovascular damage (15). Arterial stiffness increases with worsening renal function (16). A significant link between aortic pulse wave velocity (PWV) and vascular calcification burden has also been described in CKD patients (17).
Measurement of Arterial Stiffness: In vivo, Ex vivo, and In vitro
The evaluation of arterial stiffness in vivo in the clinical setting is accomplished by measurement of arterial compliance and distensibility by ultrasound, determination of PWV by measuring the velocity of the pressure wave traveling between two arterial segments, and augmentation index by measuring the augmentation pressure divided by blood pressure (1, 18). PWV closely relates to arterial wall stiffness whereas augmentation index is related to arterial wall stiffness, as well as wave reflection that is dependent on peripheral resistance and affected by heart rate variation (1, 18). The measurement of tissue and cell stiffness ex vivo and in vitro is greatly enhanced by use of atomic force microscopy (AFM) which can be performed on vascular tissues, endothelial cells, and vascular smooth muscle cells (VSMC) and complimented by confocal imaging (2, 3, 19, 20). Actin can be fluorescently labeled with Alexa 568-phalloidin and cell images, topography, and stiffness recorded with an integrated AFM-confocal microscope system. Furthermore, studies employing AFM probes that have been bio-conjugated with extracellular matrix (ECM) proteins can be used to assess the role of β1-integrin binding and cell adhesion to the ECM. These studies provided a novel concept that both β1-integrin and α-smooth muscle actin play significant role in increased stiffness of VSMCs (2, 3, 20).
Endothelial Dysfunction, Arterial Stiffness, and Insulin Resistance
Endothelial Dysfunction and Arterial Stiffness
Arterial intima consists of an endothelial cell layer and underlying layer of smooth muscle cells. It is separated from media by internal elastic lamina. In larger conduit vessels, the medial layer consists of concentric layers of elastic lamina interspersed with collagen and smooth muscle cells (18, 21). The adventitial layer is rich in fibroblasts, macrophages, lymphocytes, adipocytes, dendritic cells, and collagen (22). Arterial stiffness is regulated by a variety of factors including those from endothelial cells, VSMC alterations, cytokines, and inflammatory signals from the adventitia, and characteristic alterations in the ECM. The role of the ECM in modulation of vascular stiffness is well-recognized, and the high elastin to collagen ratio contributes to the elasticity of healthy large arteries (22). With advancing age, there is progressive thickening of arterial walls – predominantly in the intimal layer – with marked increases in the intimal to medial thickness ratio (23). There is also increased fragmentation and depletion of arterial elastin coupled with greater medial deposition of matrix metalloproteins and collagen (18, 21). Collectively, this leads to thicker and stiffer arteries, and is more predominant in the central elastic arteries compared to the peripheral, more muscular arteries. However, the relationships between stiffness in central arteries and more muscular arteries have not been clearly elucidated. The pre-diabetic state is associated with increased arterial stiffness but stiffness was unrelated to vessel wall thickness suggesting mechanisms distinct from ECM remodeling contributing to arterial stiffness (24). In this regard, accumulating evidence suggests a role for the vascular endothelium and provides new insights into the regulation of arterial stiffness (25–27). Endothelial cells regulate several arterial properties including arterial vascular tone and permeability, angiogenesis, and the vascular inflammatory response (25–28). Recently, increased intrinsic stiffness of VSMC has also been implicated in aging (2, 3, 20) and spontaneously hypertensive rats (2, 3, 20, 29). Modulation of transglutaminase 2 (TGM2) by endothelial nitric oxide (NO) (30), identification of vascular smooth muscle cytoskeletal proteins as substrates of TGM2 (31) and inhibition of smooth muscle metalloproteinase expression by NO (32) suggest the role of endothelial and smooth muscle cross talk in modulating arterial stiffness.
Insulin and RAAS Signaling and Imbalance of Metabolic and Growth Signaling in the Development of Endothelial Dysfunction and Arterial Stiffness
The effects of insulin in the vasculature involve metabolic signaling through the insulin receptor substrate-1 (IRS-1)/phosphatidylinositol 3-kinase (PI3 kinase) AKT/endothelial nitric oxide synthase (eNOS) pathway, as well as growth factor signaling through the ERK1/2/endothelin-1 (ET-1) pathway (28, 33–36). Regulation of endothelial function by insulin metabolic signaling is critical for normal endothelial function and vascular stiffness (1, 8, 33, 34). This insulin metabolic signaling is inhibited by both angiotensin II (Ang II) and aldosterone in vascular endothelial cells and VSMCs (Figure 1). The local vascular effect of insulin beyond systemic effects regulates endothelial activation of eNOS and other signaling pathways (28, 33, 35). In vascular endothelial cells, insulin stimulates production of the vasodilator NO via activation of IRS-1/PI3K signaling (Figure 1) (34, 35). In contrast, growth signaling pathway leads to activation of ERK1/2 and production of the vasoconstrictor ET-1. ET-1 as well as Ang II and aldosterone cause vascular stiffness (1, 8, 28, 36) and increased serum levels of ET-1 are seen in conditions associated with arterial stiffness (36). Activation of the RAAS also leads to impaired IRS-1/PI3K signaling and blunts downstream antioxidant, anti-inflammatory effects of insulin metabolic signaling (22, 34). This, in turn, further impairs insulin-induced vasodilation, capillary recruitment, and augments increases in arterial stiffness (33, 34, 37).
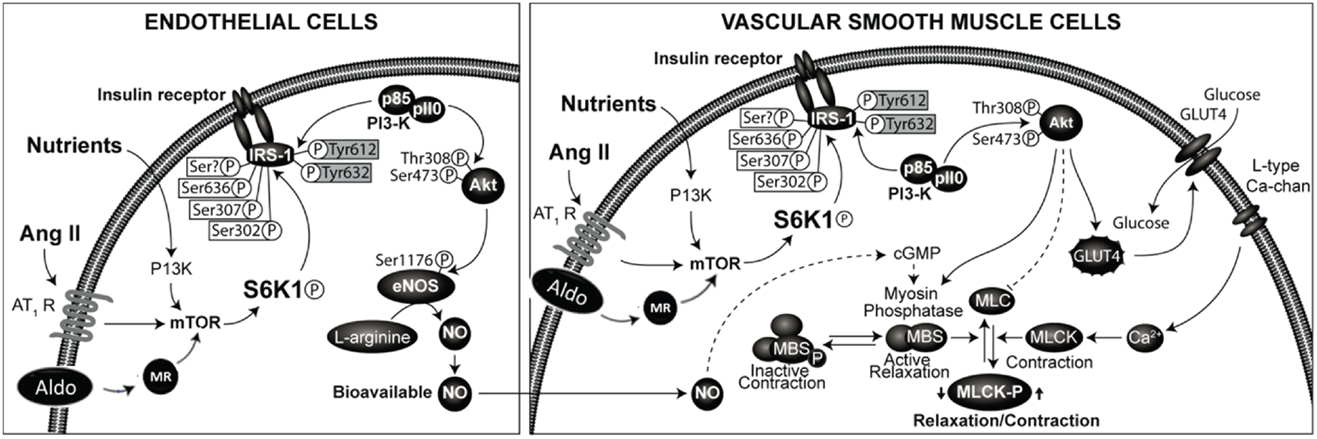
Figure 1. Renin-angiotensin-aldosterone system and regulation of insulin signaling phosphorylation of docking protein insulin receptor substrate -1 (IRS-1) is the major converging point in insulin signaling. The phosphorylation of serine residues of IRS-1 by mammalian target of ribosomal p70 S6 kinase (S6K1) acts as a convergence point for the regulation of IRS-1 phosphorylation by nutrients, hormones, and cytokines. Activation of RAAS in endothelial cells and vascular smooth muscle cells leads to inhibition of insulin signaling though phosphorylation of serine residues of IRS-1. This results in impaired signaling though attenuation of phosphatidylinsositol-3 kinase (PI3- kinase)/protein kinase B (Akt) signaling pathway linked to metabolic insulin signaling. This leads to reduced production of nitric oxide and endothelial dysfunction and altered vascular smooth muscle function.
Insulin Resistance, Endothelial Dysfunction, and Arterial Stiffness as an Early Event in Progression of CVD and CKD
Endothelial dysfunction is strongly associated with insulin resistance, arterial stiffness, and progression to CVD and CKD (24, 25, 33). Arterial stiffness may also be seen in the absence of insulin resistance in conditions such as hyperglycemia of diabetes mellitus and accumulation of advanced glycation end products (AGE) (37, 38). Individuals with obesity are likely to have an increase in aortic stiffness, independent of blood pressure level. Obesity and arterial stiffness are also independent factors for diastolic dysfunction (38, 39). The occurrence of arterial stiffness and diastolic dysfunction in the absence of elevated blood pressure suggest that arterial stiffness is an early event in the progression to CVD and CKD. In this regard, arterial stiffness is also associated with insulin resistance and an activated RAAS in obesity (38, 39), and insulin resistance alone in the absence of hypertension and coronary heart disease is also associated with diastolic dysfunction (obesity cardiomyopathy) (34). Insulin resistance precedes the development of vascular, cardiac, and renal complications associated with obesity (35). Reduction of aortic dilation to insulin, but not acetylcholine, prior to the onset of hypertension in the spontaneously hypertensive rats (40) and in aged rats (41) provides evidence that insulin resistance is an early event in the development of hypertension.
Role of Tissue RAAS in Vascular Cells
Beyond Classical and Circulating RAAS
Inappropriate activation of RAAS is being increasingly recognized as a major factor in determining endothelial dysfunction, arterial stiffness, and progression to CVD and CKD (37, 38, 42–44). The RAAS is considered as an endocrine system with kidney-derived renin regulating the production of Ang II. In the blood, renin acts on liver-derived angiotensinogen to form Angiotensin I (a decapeptide). Angiotensin I is converted to biologically active Ang II (octapeptide) by the action of endothelial (mainly pulmonary endothelium) derived angiotensin converting enzyme (ACE) (45–49). Ang II acts on adrenals to stimulate the production of aldosterone and on cardiovascular and other tissues to regulate cardiovascular remodeling and blood pressure, in part by inhibiting insulin metabolic signaling in cardiovascular tissues (33, 34, 45) (Figure 1).
In addition to the conventional circulating RAAS, the presence of RAAS components have been detected in tissues such as heart, kidney, vasculature, adipose tissue immune cells, and brain (44–49). Recent studies have shown that VSMCs synthesize angiotensin II intracellularly. Intracellular Ang II regulates the expression of angiotensinogen and renin, generating a feedback loop. The first reaction of intracellular Ang II synthesis is catalyzed by renin or cathepsin D, depending on the cell type, and chymase, not ACE, catalyzes the second step (46, 47). The increased production of Ang II in vascular tissue in conditions of high glucose suggests this component may be of significance in diabetes (46, 47). In additional to the classical Ang II system, the role of non-classical angiotensin peptides generated by tissue ACE2 comprising Ang-(1–9) and Ang-(1–7) which generally antagonize the actions of Ang II are increasingly recognized for their bioactivity (46–49). Ang-(1–7) is also converted to Ang-(1–5) by ACE. Ang III, Ang IV, Ang-(3–7) are other peptides formed from Ang II (46–49). The role for these peptides in vascular tissue is not well understood.
Although the precise role of aldosterone-induced vascular insulin resistance has not been fully elucidated, improved endothelial function in various disease models following treatment with mineralocorticoid receptor (MR) antagonists has been reported (34, 50–53). Blockade of MR by spironolactone decreases local inflammation and vascular stiffness in rodent models of hypertension and insulin resistance (50, 52–54). The contribution of MR signaling to insulin resistance is also supported by insulin resistance in patients with primary hyperaldosteronism (55) and correlation of plasma aldosterone levels with BMI and insulin resistance in normotensive subjects (56).
Cellular and Molecular Mechanisms of Vascular RAAS-Induced Insulin Resistance, Endothelial Dysfunction, and Arterial Stiffness
Molecular mechanisms underlying RAAS-mediated endothelial dysfunction and arterial stiffness in aging, obesity, CRS, and diabetes is not well understood. The role of increased serine phosphorylation of IRS-1 in Ang II and aldosterone-mediated impaired insulin signaling has been demonstrated (33, 34, 57) but the role of mammalian target of rapamycin (mTOR)/S6 kinase (S6K) mediated IRS-1 serine phosphorylation in endothelial cells are not well characterized. We have recently examined the signaling pathways mediating insulin resistance by enhanced activation of tissue RAAS in cardiovascular tissue (57). The serine phosphorylation of IRS-1 was increased and insulin-stimulated phosphorylation of eNOS was decreased by Ang II treatment. Moreover, rapamycin, an inhibitor of (mTOR) activation attenuated Ang II-stimulated phosphorylation of p70S6K and IRS-1 and blocked the ability of Ang II to impair insulin-stimulated phosphorylation of eNOS and NO-dependent arteriole vasodilation. These results suggest the role for activation of mTOR/p70S6K by Ang II in vascular endothelium in mediating impairment of insulin-stimulated vasodilation through phosphorylation of IRS-1 (57). However, MR-dependent effects on endothelial insulin signaling have not been examined.
The role of cross talk between Ang II and aldosterone signaling is increasingly recognized in the development of insulin resistance, endothelial dysfunction, and arterial stiffness (35, 50, 58–60) (Figure 1) and MR blockade attenuates Ang II-induced vascular damage (35, 50, 58, 59). Aldosterone activates NADPH oxidase, thereby promoting oxidative stress and decreased NO bioavailability (34, 50, 61). This is further supported by decreased reactive oxygen species production and agonist-mediated vasoconstriction by specific deletion of VSMC MR in aged mice (59). Aldosterone-induced MR activation increases expression of the intracellular cell adhesion molecule 1 (ICAM-1) (34). Moreover, aldosterone was shown to increase epithelial Na+ channel expression on the endothelial cell surface that correlated with increased cortical stiffness of the cytoskeleton in endothelial cells (62). Of potential importance is that the increase in endothelial cell stiffness was associated with a reduced release of NO (62), which in turn could impact stiffness of VSMC. These observations suggest that inhibition of MR might be a beneficial therapeutic approach for preventing vascular stiffening.
Up Regulation of Local Intracrine RAAS in Obesity, CRS, and Diabetes: Role of Maladaptive Immune and Inflammatory Response
Although the significance of local RAAS may not be fully understood, the increased expression of RAAS components in vascular tissues in animal models of obesity (63, 64), and direct modulation of vascular RAAS in the vasculature in vivo and in vitro by insulin (33, 63), uric acid (65), and estrogens (66), favors the role of vascular RAAS modulating endothelial dysfunction and arterial stiffness. Importantly, these factors also cause dysregulation of immune function and a pro-inflammatory response in the vasculature that contribute to endothelial dysfunction and arterial stiffness associated with the consumption of western diet or increased cardiovascular risk in women in the setting of obesity and diabetes.
Maladaptive Immunity and Low Grade Systemic Inflammatory Response
Accumulating evidence suggests the association of inappropriate activation of RAAS and maladaptive immune and inflammatory responses in modulating endothelial dysfunction and vascular stiffness in obesity and diabetes (67–71). Increased levels of cytokines in the plasma due mainly to visceral adipocyte dysfunction, may contribute significantly to the activation of RAAS in the vascular tissue (38, 68, 69). Moreover, oxidative stress has been shown to cause increased expression of the angiotensin II type-1 (AT1) receptor (68, 69, 71). Decreased levels of interleukin (IL)-10 and impaired function of T-regulatory cells, result in activation of endothelial NADPH oxidase (68, 69, 71). Therefore, an inappropriate activation of RAAS causes cytokine imbalance in plasma and inappropriate activation of RAAS in vascular tissues by cytokines results in a feed forward loop of persistent activation of vascular RAAS in obesity and diabetes (68, 69, 72).
Perivascular Adipocyte Dysfunction
The role of perivascular adipose tissue contributing to inflammation, insulin resistance, endothelial dysfunction, and vascular stiffness is increasingly recognized (69, 72–74). In lean mice, perivascular fat exerts protective vasoregulatory effects, but this protective effect is lost in obese mice (74). Endothelial dysfunction in obesity is associated with a significant infiltration of macrophages and T cells in perivascular adipose tissue (72–74). Moreover, perivascular adipose tissue is also a source of Ang II and increased production of Ang II by perivascular fat may also account for impairment of vascular function (75).
High Fructose Diet, Uric Acid, and Vascular RAAS
Elevated serum uric acid level is a frequent finding in persons with obesity, hypertension, cardiovascular, and kidney disease. Increased consumption of a fructose-rich western diet also results in elevations in uric acid (6, 7). Elevated serum levels of uric acid appear to contribute to maladaptive immune and inflammatory responses (65, 69, 76), activation of angiotensin system in the vascular cells (65), impaired NO production/endothelial dysfunction (77), and increased vascular stiffness (78, 79).
Sex Differences: Abrogation of Cardiovascular Protective Effects of Estradiol in Obesity and Diabetes in Premenopausal Women
Females of reproductive age have fewer cardiovascular events however this protection is lost after menopause, suggesting cardio-protective effects of estradiol. The cardio-protective effect of estradiol is also lost in the setting of obesity and diabetes in premenopausal women (69, 80–83). In this regard, arterial stiffness is substantially higher in women than in age-matched men, and is associated with cardiac diastolic dysfunction (82). In a community-based cohort study, increased arterial stiffness was associated with reduced left ventricular diastolic function in both men and women. However, the greater arterial stiffness observed in women was associated with higher incidence of diastolic dysfunction (83–85). Estrogen modulates both Ang II signaling and immune and inflammatory responses. Estradiol normally suppresses actions of Ang II by inhibiting the expression of AT1 (86, 87). However, under the conditions of inhibition of NO synthase and high salt, estradiol increases the expression of AT1 receptor (66, 87). Moreover, GPR-30 which also mediates estradiol effects, increases the expression of ACE2 and decreases the expression of AT1 receptor (88, 89). Estrogen receptor alpha and GPR-30 have been shown to exert an anti-inflammatory effect via modulation of T-cell immune response (90, 91). In addition, estrogen receptor alpha-mediated signaling in macrophages contributes to enhanced insulin sensitivity (92). These findings suggest that a crosstalk between estrogen and Ang II signaling may be one of the factors contributing to sex differences in altered immune and inflammatory responses, endothelial dysfunction, and arterial stiffness, in obesity and diabetes. Furthermore, a recent study demonstrating arterial stiffness in obese pre-menopausal women underscores the role of obesity in abrogating cardiovascular protection in those women (93).
Conclusion
Arterial stiffness is an independent factor promoting the progression of CVD and renal disease in obesity and diabetes. Inappropriate activation of vascular RAAS in humans and animal models of obesity and diabetes is associated with endothelial dysfunction and arterial stiffness. However, accumulating evidence suggests the role of local tissue RAAS in the vascular tissue, immune cells, and perivascular adipose tissue in endothelial dysfunction contributes significantly to arterial stiffness. The cross talk between angiotensin and aldosterone underscores the importance of the MR in modulation of oxidative stress, insulin resistance, decreased bioavailability of NO, endothelial dysfunction, and arterial stiffness. In addition, both innate and adaptive immunity are involved in local tissue activation of RAAS and in turn are modulated by environmental factors such as high fat/sucrose western diet. Moreover, arterial stiffness is reported in pre-menopausal obese women and estrogen mediated cardiovascular protection is lost in obese or diabetic pre-menopausal women. Taken together, targeting endothelial function and arterial stiffness by modulating tissue RAAS system appears to be an attractive therapeutic strategy to reduce the CVD and CKD complications associated with obesity and diabetes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported in part by NIH grant awards (NIH P01HL095486) to Gerald A. Meininger (HL73101-01A1 and R01 HL107910-01) to James R. Sowers and Veterans Affairs Merit System 0018 to James R. Sowers. The authors wish to thank Brenda Hunter for her editorial assistance.
References
1. Cavalcante JL, Lima JA, Redheuil A, Al-Mallah MH. Aortic stiffness: current understanding and future directions. J Am Coll Cardiol (2011) 57:1511–22. doi:10.1016/j.jacc.2010.12.017
2. Qiu H, Zhu Y, Vatner DE, Gansner M, Peppas AP, Depre C, et al. Vascular smooth muscle stiffness as a mechanism for increased aortic stiffness with aging. Circ Res (2010) 107:615–9. doi:10.1161/CIRCRESAHA.110.221846
3. Zhu Y, Qiu H, Trzeciakowski JP, Sun Z, Li A, Hong Z, et al. Temporal analysis of vascular smooth muscle elasticity and adhesion reveals time-dependent oscillations that differ with aging. Aging Cell (2012) 11:1–10. doi:10.1111/j.1474-9726.2012.00840.x
4. Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA (2010) 303:235–41. doi:10.1001/jama.2009.2014
5. Sowers JR. Diabetes mellitus and vascular disease. Hypertension (2013) 61:943–7. doi:10.1161/HYPERTENSIONAHA.111.00612
6. Khitan Z, Kim DH. Fructose: a key factor in the development of metabolic syndrome and hypertension. J Nutr Metab (2013) 2013:682673. doi:10.1155/2013/682673
7. Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang DH, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, cardiovascular disease. Am J Clin Nutr (2007) 86:899–906.
8. Fernhall B, Agiovlasitis S. Arterial function in youth: window into cardiovascular risk. J Appl Physiol (2008) 105:325–33. doi:10.1152/japplphysiol.00001.2008
9. Graettinger WF. Proceedings of the American Society of hypertension. Presented at the Annual Scientific Meeting of the American Society of Hypertension. New York: (1987).
10. Femia R, Kozakova M, Nannipieri M, Gonzales-Villalpando C, Stern MP, Haffner SM, et al. Carotid intima-media thickness in confirmed prehypertensive subjects: predictors and progression. Arterioscler Thromb Vasc Biol (2007) 27:2244–2249. doi:10.1161/ATVBAHA.107.149641
11. Liao D, Arnett DK, Tyroler HA, Riley WA, Chambless LE, Szklo M, et al. Arterial stiffness and the development of hypertension. The study. Hypertension (1999) 34:201–6. doi:10.1161/01.HYP.34.2.201
12. Marti CN, Gheorghiade M, Kalogeropoulos AP, Pedersen LR, Simonsen L, Rasmusen H, et al. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol (2012) 60:1455–69. doi:10.1016/j.jacc.2011.11.082
13. Mottram PM, Haluska BA, Leano R, Carlier S, Case C, Marwick TH. Relation of arterial stiffness to diastolic dysfunction in hypertensive heart disease. Heart (2005) 91:1551–6. doi:10.1136/hrt.2004.046805
14. Hu Y, Li L, Shen L, Gao H. The relationship between arterial wall stiffness and left ventricular dysfunction. Neth Heart J (2013) 21:222–7. doi:10.1007/s12471-012-0353-z
15. Wang MC, Tsai WC, Chen JY, Huang JJ. Stepwise increase in arterial stiffness corresponding with the stages of chronic kidney disease. Am J Kidney Dis (2005) 45:494–501. doi:10.1053/j.ajkd.2004.11.011
16. Ford ML, Tomlinson LA, Chapman TPE, Rajkumar C, Holt SG. Aortic stiffness is independently associated with rate of renal function decline in chronic kidney disease stages 3 and 4. Hypertension (2010) 55:1110–5. doi:10.1161/HYPERTENSIONAHA.109.143024
17. Nitta K, Akiba T, Uchida K, Otsubo S, Otsubo Y, Takei T, et al. Left ventricular hypertrophy is associated with arterial stiffness and vascular calcification in hemodialysis patients. Hypertens Res (2004) 27:47–52. doi:10.1291/hypres.27.47
18. Shirwany NA, Zou MH. Arterial stiffness: a brief review. Acta Pharmacol Sin (2010) 31:1267–76. doi:10.1038/aps.2010.123
19. Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep (2012) 29:1259–71. doi:10.1016/j.celrep.2012.09.018
20. Hong Z, Sun Z, Li Z, Mesquitta WT, Trzeciakowski JP, Meininger GA. Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc Res (2012) 96:73–80. doi:10.1093/cvr/cvs239
21. Luft FC. Molecular mechanisms of arterial stiffness: new insights. J Am Soc Hypertens (2012) 6:436–8. doi:10.1016/j.jash.2012.10.004
22. Stenmark KR, Yeager MF, El Kasmi KC, Nozik-Grayck E, Gerrasimovskaya EV, Li M, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol (2013) 75:23–47. doi:10.1146/annurev-physiol-030212-183802
23. Steppan J, Barodka V, Berkowitz DE, Nyhan D. Vascular stiffness and increased pulse pressure in the aging cardiovascular system. Cardiol Res Pract (2011) 2011:263585. doi:10.4061/2011/263585
24. Sliem H, Nasr G. Aortic stiffness in prediabetic adults: relationship to insulin resistance. J Clin Med Res (2010) 20:62–7. doi:10.4021/jocmr2010.03.269w
25. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol (2005) 25:932–43. doi:10.1161/01.ATV.0000160548.78317.29
26. Ghiadoni L, Taddei S, Virdis A. Hypertension and endothelial dysfunction: therapeutic approach. Curr Vasc Pharmacol (2012) 10:42–60. doi:10.2174/157016112798829823
27. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J (2010) 4:302–12. doi:10.2174/1874192401004010302
28. Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord (2013) 14:5–12. doi:10.1007/s11154-012-9229-1
29. Sehgel NL, Zhu Y, Sun Z, Trzeciakowski JP, Hong Z, Hunter WC, et al. Increased vascular smooth muscle cell stiffness; a novel mechanism for aortic stiffness in hypertension. Am J Physiol Heart Circ Physiol (2013). doi:10.1152/ajpheart.00232.2013
30. Jandu SK, Webb AK, Pak A, Sevinc B, Nyhan D, Belkin AM, et al. Nitric oxide regulates tissue transglutaminase localization and function in the vasculature. Amino Acids (2013) 44:261–9. doi:10.1007/s00726-011-1090-0
31. van den Akker J, VanBavel E, van Geel R, Matlung HL, Guvenc Tuna B, Janssen GM, et al. The redox state of transglutaminase 2 controls arterial remodeling. PLoS ONE (2011) 6:e23067. doi:10.1371/journal.pone.0023067
32. Upchurch GR Jr, Ford JW, Weiss SJ, Knipp BS, Peterson DA, Thompson RW, et al. Nitric oxide inhibition increases matrix metalloproteinase-9 expression by rat aortic smooth muscle cells in vitro. J Vasc Surg (2001) 34(76–83):2001. doi:10.1067/mva.2001.115598
33. Brillante DG, O’Sullivan AJ, Howes LG. Arterial stiffness in insulin resistance: the role of nitric oxide and angiotensin II receptors. Vasc Health Risk Manag (2009) 5:73–8.
34. Aroor AR, Mandavia CH, Sowers JR. Insulin resistance and heart failure: molecular mechanisms. Heart Fail Clin (2012) 8:609–17. doi:10.1016/j.hfc.2012.06.005
35. Bender SB, McGraw AP, Jaffe IZ, Sowers JR. Mineralocorticoid receptor-mediate vascular insulin resistance: an early contributor to diabetes-related vascular disease? Diabetes (2013) 62:313–9. doi:10.2337/db12-0905
36. Nar G, Soylu K, Akcay M, Gulel O, Yuksel S, Meric M, et al. Evaluation of the relationship between arterial blood pressure, aortic stiffness and serum endothelin-1 levels in patients with essential hypertension. Clin Exp Hypertens (2013). doi:10.3109/10641963.2013.776565
37. Yki-Järvinen H, Westerbacka J. Insulin resistance, arterial stiffness and wave reflection. Adv Cardiol (2007) 44:252–60. doi:10.1159/000096746
38. Targosz-Korecka M, Brzezinka GD, Malek KE, Stepien E, Szymonski M. Stiffness memory of EA.hy926 endothelial cells in response to chronic hyperglycemia. Cardiovasc Diabetol (2013) 12:96. doi:10.1186/1475-2840-12-96
39. Seifalian AM, Filippatos TD, Joshi J, Mikhailidis DP. Obesity and arterial compliance alterations. Curr Vasc Pharmacol (2010) 8:155–68. doi:10.2174/157016110790886956
40. Li R, Zhang H, Wang W, Wang X, Huang Y, Huang C, et al. Vascular insulin resistance in prehypertensive rats: role of PI3-kinase/Akt/eNOS signaling. Eur J Pharmacol (2010) 628:140–7. doi:10.1016/j.ejphar.2009.11.038
41. Schulman IH, Zhou MS, Jaimes EA, Raij L. Dissociation between metabolic and vascular insulin resistance in aging. Am J Physiol Heart Circ Physiol (2007) 29:H853–9. doi:10.1152/ajpheart.00138.2007
42. Milan A, Tosello F, Fabbri A, Vairo A, Leone D, Chiarlo M, et al. Arterial stiffness: from physiology to clinical implications. High Blood Press Cardiovasc Prev (2011) 1:1–12. doi:10.2165/11588020-000000000-00000
43. Mahmud A, Feely J. Arterial stiffness and the renin-angiotensin-aldosterone system. J Renin Angiotensin Aldosterone Syst (2004) 5:102–8. doi:10.3317/jraas.2004.025
44. Underwood PC, Adler GK. The renin angiotensin aldosterone system and insulin resistance in humans. Curr Hypertens Rep (2013) 15:59–70. doi:10.1007/s11906-012-0323-2
45. Hayden MR, Sowers KM, Pulakat L, Joginpally T, Krueger B, Whaley-Connell A, et al. Possible mechanisms of local tissue renin-angiotensin system activation in the cardiorenal metabolic syndrome and type 2 diabetes mellitus. Cardiorenal Med (2011) 1:193–210. doi:10.1159/000329926
46. Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin-angiotensin system. Clin Sci (Lond) (2012) 123:273–84. doi:10.1042/CS20120089
47. Bader M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu Rev Pharmacol Toxicol (2010) 50:439–65. doi:10.1146/annurev.pharmtox.010909.105610
48. Sevá Pessôa B, van der Lubbe N, Verdonk K, Roks AJ, Hoorn EJ, Danser AH. Key developments in renin-angiotensin-aldosterone system inhibition. Nat Rev Nephrol (2013) 9:26–36. doi:10.1038/nrneph.2012.249
49. Nguyen Dinh Cat A, Touyz RM. A new look at the renin-angiotensin system-focusing on the vascular system. Peptides (2011) 32:2141–50. doi:10.1016/j.peptides.2011.09.010
50. Whaley-Connell A, Johnson MS, Sowers JR. Aldosterone: role in the cardiometabolic syndrome and resistant hypertension. Prog Cardiovasc Dis (2010) 52:401–9. doi:10.1016/j.pcad.2009.12.004
51. Resch M, Schmid P, Amann K, Fredersdorf S, Weil J, Schach C, et al. Eplerenone prevents salt-induced vascular stiffness in Zucker diabetic fatty rats: a preliminary report. Cardiovasc Diabetol (2011) 10:94. doi:10.1186/1475-2840-10-94
52. Kithas PA, Supiano MA. Spironolactone and hydrochlorothiazide decrease vascular stiffness and blood pressure in geriatric hypertension. J Am Geriatr Soc (2010) 58:1327–32. doi:10.1111/j.1532-5415.2010.02905.x
53. Baldo MP, Forechi L, Morra EA, Zaniqueli D, Machado RC, Lunz W, et al. Long-term use of low-dose spironolactone in spontaneously hypertensive rats: effects on left ventricular hypertrophy and stiffness. Pharmacol Rep (2011) 63:975–82.
54. Pojoga LH, Baudrand R, Adler GK. Mineralocorticoid receptor throughout the vessel: a key to vascular dysfunction in obesity. Eur Heart J (2013). doi:10.1093/eurheartj/eht158
55. Garg R, Adler GK. Role of mineralocorticoid receptor in insulin resistance. Curr Opin Endocrinol Diabetes Obes (2012) 19:168–75. doi:10.1097/MED.0b013e3283533955
56. Garg R, Hurwitz S, Williams GH, Hopkins PN, Adler GK. Aldosterone production and insulin resistance in healthy adults. J Clin Endocrinol Metab (2010) 95:1986–90. doi:10.1210/jc.2009-2521
57. Kim JA, Jang HJ, Martinez-Lemus LA, Sowers JR. Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab (2012) 302:E201–8. doi:10.1152/ajpendo.00497.2011
58. McGraw AP, McCurley A, Preston IR, Jaffe IZ. Mineralocorticoid receptors in vascular disease: connecting molecular pathways to clinical implications. Curr Atheroscler Rep (2013) 15:340. doi:10.1007/s11883-013-0340-x
59. McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med (2012) 18:1429–33. doi:10.1038/nm.2891
60. Rautureau Y, Paradis P, Schiffrin EL. Cross-talk between aldosterone and angiotensin signaling in vascular smooth muscle cells. Steroids (2011) 76:834–9. doi:10.1016/j.steroids.2011.02.015
61. Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, et al. Mineralocorticoid receptors modulate vascular endothelial function in human obesity. Clin Sci (Lond) (2013) 125:513–20. doi:10.1042/CS20130200
62. Drüppel V, Kusche-Vihrog K, Grossmann C, Gekle M, Kasprzak B, Brand E, et al. Long-term application of the aldosterone antagonist spironolactone prevents stiff endothelial cell syndrome. FASEB J (2013) 27:3652–9. doi:10.1096/fj.13-228312
63. Nyby MD, Abedi K, Smutko V, Eslami P, Tuck ML. Vascular Angiotensin type 1 receptor expression is associated with vascular dysfunction, oxidative stress and inflammation in fructose-fed rats. Hypertens Res (2007) 30:451–7. doi:10.1291/hypres.30.451
64. Shinozaki K, Ayajiki K, Nishio Y, Sugaya T, Kashiwagi A, Okamura T. Evidence for a causal role of the renin-angiotensin system in vascular dysfunction associated with insulin resistance. Hypertension (2004) 43:255–62. doi:10.1161/01.HYP.0000111136.86976.26
65. Filiopoulos V, Hadjiyannakos D, Vlassopoulos D. New insights into uric acid effects on the progression and prognosis of chronic kidney disease. Ren Fail (2012) 34:510–20. doi:10.3109/0886022X.2011.653753
66. Ricchiuti V, Lian CG, Oestreicher EM, Tran L, Stone JR, Yao T, et al. Estradiol increases angiotensin II type 1 receptor in hearts of ovariectomized rats. J Endocrinol (2009) 200:75–84. doi:10.1677/JOE-08-0199
67. Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens (2006) 15:152–8.
68. Aroor A, McKarns S, Nistala R, Demarco V, Gardner M, Garcia-Touza M, et al. DPP-4 inhibitors as therapeutic modulators of immune cell function and associated cardiovascular and renal insulin resistance in obesity and diabetes. Cardiorenal Med (2013) 3:48–56. doi:10.1159/000348756
69. Aroor AR, McKarns S, Demarco VG, Jia G, Sowers JR. Maladaptive immune and inflammatory pathways lead to cardiovascular insulin resistance. Metabolism (2013) 62(11):1543–52. doi:10.1016/j.metabol.2013.07.001
70. van Bussel BC, Schouten F, Henry RM, Schalkwijk CG, de Boer MR, Ferreira I, et al. Endothelial dysfunction and low-grade inflammation are associated with greater arterial stiffness over a 6-year period. Hypertension (2011) 58:588–95. doi:10.1161/HYPERTENSIONAHA.111.174557
71. Wassmann S, Nickenig G. Pathophysiological regulation of the AT1-receptor and implications for vascular disease. J Hypertens Suppl (2006) 24:S15–21. doi:10.1097/01.hjh.0000220402.53869.72
72. Campia U, Tesauro M, Cardillo C. Human obesity and endothelium-dependent responsiveness. Br J Pharmacol (2012) 165:561–73. doi:10.1111/j.1476-5381.2011.01661.x
73. Bailey-Downs LC, Tucsek Z, Toth P, Sosnowska D, Gautam T, Sonntag WE, et al. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: a paracrine mechanism contributing to vascular redox dysregulation and inflammation. J Gerontol A Biol Sci Med Sci (2013) 68:780–92. doi:10.1093/gerona/gls238
74. Eringa EC, Bakker W, van Hinsbergh VW. Paracrine regulation of vascular tone, inflammation and insulin sensitivity by perivascular adipose tissue. Vascul Pharmacol (2012) 56:204–9. doi:10.1016/j.vph.2012.02.003
75. Szasz T, Bomfim GF, Webb RC. The influence of perivascular adipose tissue on vascular homeostasis. Vasc Health Risk Manag (2013) 9:105–16. doi:10.2147/VHRM.S33760
76. Puddu P, Puddu GM, Cravero E, Vizioli L, Muscari A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: molecular mechanisms and clinical implications. J Cardiol (2012) 59:235–42. doi:10.1016/j.jjcc.2012.01.013
77. Park JH, Jin YM, Hwang S, Cho DH, Kang DH, Jo I. Uric acid attenuates nitric oxide production by decreasing the interaction between endothelial nitric oxide synthase and calmodulin in human umbilical vein endothelial cells: a mechanism for uric acid-induced cardiovascular disease development. Nitric Oxide (2013) 32:36–42. doi:10.1016/j.niox.2013.04.003
78. Ishizaka N, Ishizaka Y, Toda E, Hashimoto H, Nagai R, Yamakado M. Higher serum uric acid is associated with increased arterial stiffness in Japanese individuals. Atherosclerosis (2007) 192:131–7. doi:10.1016/j.atherosclerosis.2006.04.016
79. Tsioufis C, Kyvelou S, Dimitriadis K, Syrseloudis D, Sideris S, Skiadas I, et al. The diverse associations of uric acid with low-grade inflammation, adiponectin and arterial stiffness in never-treated hypertensives. J Hum Hypertens (2011) 25:554–9. doi:10.1038/jhh.2010.98
80. Manrique C, Demarco VG, Aroor AR, Mugerfeld I, Garro M, Habibi J, et al. Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a western diet. Endocrinology (2013) 154:3632–42. doi:10.1210/en.2013-1256
81. Manrique C, Lastra G, Habibi J, Mugerfeld I, Garro M, Sowers JR. Loss of estrogen receptor α signaling leads to insulin resistance and obesity in young and adult female mice. Cardiorenal Med (2012) 2:200–10. doi:10.1159/000339563
82. Berry KL, Cameron JD, Dart AM, Dewar EM, Gatzka CD, Jennings GL, et al. Large-artery stiffness contributes to the greater prevalence of systolic hypertension in elderly women. J Am Geriatr Soc (2004) 52:368–73. doi:10.1111/j.1532-5415.2004.52107.x
83. Russo C, Jin Z, Palmieri V, Homma S, Rundek T, Elkind MS, et al. Arterial stiffness and wave reflection: sex differences and relationship with left ventricular diastolic function. Hypertension (2012) 60:362–8. doi:10.1161/HYPERTENSIONAHA.112.191148
84. Duprez D, Jacobs DR Jr. Arterial stiffness and left ventricular diastolic function: does sex matter? Hypertension (2012) 60:283–4. doi:10.1161/HYPERTENSIONAHA.112.197616
85. Coutinho T, Borlaug BA, Pellikka PA, Turner ST, Kullo IJ. Sex differences in arterial stiffness and ventricular-arterial interactions. J Am Coll Cardiol (2013) 61:96–103. doi:10.1016/j.jacc.2012.08.997
86. Yung LM, Wong WT, Tian XY, Leung FP, Yung LH, Chen ZY, et al. Inhibition of renin-angiotensin system reverses endothelial dysfunction and oxidative stress in estrogen deficient rats. PLoS ONE (2011) 6:e17437. doi:10.1371/journal.pone.0017437
87. Maric-Bilkan C, Manigrasso MB. Sex differences in hypertension: contribution of the renin-angiotensin system. Gend Med (2012) 9:287–91. doi:10.1016/j.genm.2012.06.005
88. Lindsey SH, Chappell MC. Evidence that the G protein-coupled membrane receptor GPR30 contributes to the cardiovascular actions of estrogen. Gend Med (2011) 8:343–54. doi:10.1016/j.genm.2011.10.004
89. Lindsey SH, Yamaleyeva LM, Brosnihan KB, Gallagher PE, Chappell MC. Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2.Lewis rat. Hypertension (2011) 58:665–71. doi:10.1161/HYPERTENSIONAHA.111.175174
90. Foryst-Ludwig A, Kintscher U. Metabolic impact of estrogen signalling through ERalpha and ERbeta. J Steroid Biochem Mol Biol (2010) 122:74–81. doi:10.1016/j.jsbmb.2010.06.012
91. Meyer MR, Clegg DJ, Prossnitz ER, Barton M. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol (Oxf) (2011) 203:259–69. doi:10.1111/j.1748-1716.2010.02237.x
92. Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, et al. Myeloid-specific estrogen receptor alpha deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Natl Acad Sci U S A (2011) 108:16457–62. doi:10.1073/pnas.1104533108
Keywords: renin-angiotensin-aldosterone system, arterial stiffness, insulin resistance, endothelial dysfunction, obesity, diabetes
Citation: Aroor AR, DeMarco VG, Jia G, Sun Z, Nistala R, Meininger GA and Sowers JR (2013) The role of tissue renin-angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front. Endocrinol. 4:161. doi: 10.3389/fendo.2013.00161
Received: 30 August 2013; Paper pending published: 25 September 2013;
Accepted: 11 October 2013; Published online: 29 October 2013.
Edited by:
Walmor De Mello, University of Puerto Rico, USAReviewed by:
Rudy M. Ortiz, University of California Merced, USAHonoo Satake, Suntory Institute for Bioorganic Research, Japan
Copyright: © 2013 Aroor, DeMarco, Jia, Sun, Nistala, Meininger and Sowers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James R. Sowers, University of Missouri Columbia School of Medicine, D109 Diabetes Center HSC, One Hospital Drive, Columbia, MO 65212, USA e-mail:c293ZXJzakBoZWFsdGgubWlzc291cmkuZWR1