 Takako Araki1*
Takako Araki1* Ning-Ai Liu2
Ning-Ai Liu2- 1Division of Diabetes, Endocrinology and Metabolism, Department of Medicine, University of Minnesota, Minneapolis, MN, United States
- 2Department of Medicine, Pituitary Center, Cedars-Sinai Medical Center, Los Angeles, CA, United States
Cell cycle proteins are critical to pituitary development, but their contribution to lineage-specific tumorigenesis has not been well-elucidated. Emerging evidence from in vitro human tumor analysis and transgenic mouse models indicates that G1/S-related cell cycle proteins, particularly cyclin E, p27, Rb, and E2F1, drive molecular mechanisms that underlie corticotroph-specific differentiation and development of Cushing disease. The aim of this review is to summarize the literature and discuss the complex role of cell cycle regulation in Cushing disease, with a focus on identifying potential targets for therapeutic intervention in patients with these tumors.
Introduction
Pituitary corticotrophs play critical physiologic roles in hypothalamic–pituitary–adrenal axis functions, including the acute stress response, regulation of body metabolism and energy expenditures, and immune function (1). Patients with Cushing disease caused by a pituitary corticotroph adenoma can manifest obesity, diabetes, susceptibility to infections, psychosis, and coagulopathy, which contributes to increased mortality (2). Surgical tumor resection is the primary therapy for Cushing disease, but persistent/recurrent disease is seen in 12–40% of patients depending on surgical expertise, the definition of remission, and the duration of follow-up (3, 4). Pituitary-directed radiation and bilateral adrenalectomy to inhibit adrenal cortisol production are effective at inducing biochemical control but are infrequently used. Response to radiation therapy is slow and risk of hypopituitarism is high. Following adrenalectomy, lifelong replacement glucocorticoid and mineralocorticoid is needed, and the loss of negative feedback on pituitary adrenocorticotropic hormone (ACTH) can lead to tumor growth (5, 6).
Safe and effective medical management of Cushing disease has also proven challenging. The steroidogenesis inhibitors ketoconazole and metyrapone normalize cortisol in about 50% of cases, but with a risk of inducing adrenal insufficiency or hepatotoxicity (7, 8). The investigational steroidogenesis inhibitor osilodrostat showed improved responses, with up to 80% achieving biochemical remission, but still carries the risk of adrenal insufficiency and hepatotoxicity, and the loss of feedback led to a 4-fold increase in ACTH (9). The glucocorticoid receptor blocker mifepristone, approved for treatment of diabetes due to Cushing's syndrome, improves the systemic effects of excess cortisol on glycemia and weight, but is associated with risk of adrenal insufficiency, hypokalemia, and endometrial thickening, and may induce tumor growth (10). Importantly, none of these agents directly target the tumor.
Currently, two tumor-targeting agents are used in Cushing disease (11). The dopamine agonist cabergoline targets D2 receptors in the tumor and biochemical control is seen in 30–40% of patients after treatment with relatively high doses of 2–3 mg/week for at least 2 years (12, 13). However, cabergoline is not approved for use in this disease. The somatostatin receptor ligand pasireotide, which targets somatostatin receptors, is currently the only tumor-targeting agent approved for use in patients with Cushing disease. In the phase 3 trial, 26% of patients achieved urinary free cortisol normalization after the 6 months of treatment. However, 73% showed hyperglycemic-related events (14).
An alternative approach to medical therapy for Cushing disease is to more specifically target the corticotroph lineage (15). Corticotroph differentiation occurs following expression of the corticotroph-specific transcription factor Tpit (16); these cells do not express differentiation transcription factors, such as Prop1 and Pit1, that are required for somatotroph, lactotroph, and thyrotroph lineage development (17, 18). Some cell cycle proteins involved in pituitary development are also involved in tumorigenesis (19), specifically serine-threonine cyclin-dependent kinases (CDKs), cyclins, CDK inhibitors, retinoblastoma (Rb) and its complex with transcription factor (Rb/E2F), and pituitary tumor transforming gene (PTTG) (20). However, the contribution of cell cycle proteins to development of lineage-specific tumors has not been well-elucidated.
In this review, we discuss molecular mechanisms underlying the corticotroph lineage-specific cell cycle proteins cyclin E, the CDK inhibitor p27, Rb, and E2F1 in Cushing disease tumorigenesis and consider how cell cycle regulation affects pituitary biology. Then, we present updated data from studies evaluating corticotroph lineage targeting therapy for Cushing disease.
Overview of Cell Cycle Proteins and Pituitary Tumorigenesis
Cell division is divided into four phases: S phase (synthesis of DNA), M phase (mitosis), and G1 and G2 (gap) phases. G1 phase occurs before S phase, and G2 precedes M phase. In mammalian cells, this process is driven by CDKs that regulate progression through the phases of the cell cycle (21). Cyclin D (D1, D2, and D3) activates CDK4 and CDK6 and facilitates progression during G1. CDK2/cyclin E (E1 and E2) complexes become active at the end of G1 and participate in the transition from G1 to S phase. At the end of S phase and during G2, cyclin E is substituted by cyclin A (A1, A2) to activate CDK2 and CDK1. Finally, CDK1/cyclin B (mostly B1 and B2) complex is involved in progression through G2 and entry into M phase.
Cell cycle progression is also under the control of negative regulators. Specifically the CDK inhibitors INK4 and Cip/Kip families (22). The INK4 family, including p16, p15, p18, and p19, targets the CDK4/6/cyclin D complexes, while the Cip/Kip family, which consists of p21, p27, and p57, targets the CDK2/cyclin E complex (23).
Finally, the tumor suppressor protein Rb negatively regulates entry into the cell cycle and G1/S progression (24–26) by binding to the transcription factor family E2F to target cell cycle-specific genes for repression, while PTTG, part of the securin family, is associated with cell cycle proteins in G1/S phase and chromosomal instability (27–29).
The Rb heterozygous knockout mouse was the first model linking cell cycle proteins with pituitary tumorigenesis, with nearly 100% developing pituitary tumors by age 12 months (30–32). Several CDK inhibitor knockout models, including p18 and p27, also exhibited pituitary tumors (23, 33), while models of combined CDK inhibitor knockout shortened latency of tumor formation or increased size of pituitary tumors, including knockout of p21/Rb (34), p27/Rb (35), p16/p18 (36), p27/cyclin E (37), p27/p18 (33), p21/p18 (38), and CDK4/p27 (39). Germline, but rarely somatic, mutations (40–42) as well as underexpression or DNA methylation of CDK inhibitors (43) have also been reported in human pituitary tumors.
Cell Cycle Proteins and Corticotroph Lineage
Corticotrophs represent 10–15% of pituitary anterior lobe cells. Corticotroph is the earliest pituitary lineage to initiate cell development and reaches terminal differentiation by expressing lineage-specific transcription factors such as Tpit (16, 44). Cell cycle proteins cyclin E, p27, Rb, and E2F1 exhibit different levels of lineage-specific expression patterns in embryonic cells, adult normal pituitary, and corticotroph adenoma (Table 1).

Table 1. Expression of cell cycle proteins in corticotrophs.
Cyclin E and Corticotroph Lineage Specificity
Cyclin E is upregulated in late G1 and is maintained into S phase, forming a complex with and activating CDK2 at the restriction point shortly prior to entry into S phase. Unique transient expression patterns of cyclin E have been reported in early stages of pituitary cell development. Pituitary progenitor cells in cell cycle progression express cyclin A, D1, D2, and D3, but cyclin E and p57 are only expressed once progenitor cells exit from the cell cycle (23, 46). By the time Tpit is expressed, cyclin E can no longer be detected in corticotroph progenitors (23).
Cyclin E expression is significantly increased in corticotroph adenomas compared to normal pituitary as well as to somatotroph, lactotroph, and non-functional adenomas (45). In a zebrafish model with PTTG-driven corticotroph adenomas, we found cyclin E was significantly upregulated, while cyclin D, p27, and Rb was unchanged (47). By contrast, others have shown that cyclin D is upregulated in non-functioning adenomas and in aggressive non-functioning pituitary adenomas (45, 60, 61).
Transgenic mice overexpressing cyclin E in cells expressing the adrenocorticotrophin (ACTH) precursor proopiomelanocortin (POMC) show abnormal reentry into the cell cycle as well as centrosome instability (37, 62). Molecular analysis suggests that cyclin E levels inversely correlate with expression of the tumor suppressor Brahma-related gene 1 (Brg1), which exerts negative feedback on the glucocorticoid receptor through the rPomc promoter (37, 62). Brg1 forms a complex with histone deacetylase 1, the glucocorticoid receptor, and orphan nuclear receptor growth factor 1B; loss of Brg1 disrupts this complex, inducing loss of glucocorticoid negative feedback, as is evident in the clinical phenotype of Cushing disease (62). Indeed, disordered Brg1 and/or cyclin E expression was found in about half of corticotroph tumors derived from 25 patients with Cushing disease, indicating that loss of Brg1 tumor suppression combined with cyclin E upregulation may contribute to corticotroph tumorigenesis (37). Transgenic mice overexpressing cyclin E exhibit pituitary hyperplasia but no pituitary tumors (37), and cyclin E overexpression in a p27 knockout model known to induce pituitary tumor show increased tumor size, further confirming the contribution of cyclin E to corticotroph tumor development and proliferation (37).
Nevertheless, as noted above, about half of 25 Cushing tumors showed an inverse correlation between Brg1 and cyclin E, but the other half did not fit the pattern (37). Also, study of 48 human prolactinoma specimens showed increased expression of both cyclin D1 and cyclin E on immunostaining, and that co-expression correlated with invasiveness (63). These data suggest additional mechanisms of cyclin E regulation in corticotroph lineage specific tumorigenesis.
p27 and Corticotroph Lineage Specificity
p27, a member of the Cip/Kip family of CDK inhibitors, targets G1/S progression. p27 knockout mice develop enlarging pituitaries by as early as 10 weeks, and tumors in the intermediate lobe show positive POMC expression by 12 months (48–50). Combined Rb/p27 knockout results in even shorter latency of pituitary tumors (35), suggesting two separate pathways for p27 and Rb in G1/S phase deregulation. On immunohistochemistry, p27 labeling is suppressed in corticotroph adenomas and carcinomas (51), and recurrent human pituitary adenomas show lower p27 protein levels than do non-recurrent adenomas (52, 64), supporting the contribution of p27 to corticotroph tumorigenesis. In human specimens, p27 germline mutations are reported as MEN type 4, which manifests as neuroendocrine tumors as well as pituitary tumors, including, but not limited to, Cushing disease (65–67). p27 somatic mutations in human specimens are even rarer (40, 41, 68) and p27 mRNA levels are not altered in corticotroph tumors. Rather, post-translational modifications such as proteolysis or ubiquitination may be involved in downregulating p27 protein levels (69, 70).
Rb and Potential Corticotroph Lineage Specificity
Rb regulation of cell cycle progression was initially studied in Rb heterozygous knockout mice as homozygous knockout is lethal (30–32). In humans, individuals who inherit one defective copy of Rb have an ~90% risk of developing retinoblastoma at an early age (71). Mice heterozygous for Rb do not develop retinoblastoma, but instead exhibit nearly 100% penetrance of pituitary tumors by 12 months (30–32). Pituitary tumors have been reported in the intermediate lobe in Rb heterozygous mice (54) and in POMC-specific conditional Rb heterozygous mice in which the reporter gene was restricted to the intermediate and anterior lobes (57). Tumors in the former knockout model stained positive for α-melanocyte–stimulating hormone, and neither reported on the presence of tumors in the anterior lobe.
In humans, pituitary neoplasms have not been reported in those with familial retinoblastoma (72). However, loss of an Rb allele has been reported in case reports and a few small series of corticotroph adenomas. No allelic loss of Rb was seen in a study of 12 human pituitary tumors, including one corticotroph microadenoma and one macroadenoma (53), but among 13 highly invasive human pituitary adenomas or metastatic carcinomas tumors, all showed loss of Rb, suggesting a preference for loss of Rb in more aggressive corticotroph tumors (55). Indeed, in a patient with adjacent pituitary benign adenoma and carcinoma, Rb expression was significantly reduced in the corticotroph carcinoma but not in the adenoma (56). Given the potential association between aggressive corticotroph tumors and Rb loss, as well as the tendency for Rb to complex with the corticotroph-specific E2F1 (59), it is tempting to speculate that Rb-related dysregulation of the cell cycle might have corticotroph preferential pattern, but further investigation is still needed.
E2F1 and Corticotroph Lineage Specificity
The E2F family of cell cycle proteins, numbered E2F1 through E2F8, includes those that interact with Rb and act on G1 to S phase progression (73). E2F family proteins are expressed ubiquitously, particularly in association with cancer cell cycles and tumorigenesis. Free E2F1 unbound from Rb, the active form of E2F1, binds to target gene promoters and may target cell cycle regulators such as cyclin E and cyclin D (74).
We studied E2F1 and POMC gene regulation in ectopic ACTH-secreting tumors (58). In addition to its cell cycle effects, E2F1 also directly binds to the POMC promoter (58). Co-transfection of E2F1 and its heterodimerization partner DP1 enhanced POMC promoter activities as well as POMC mRNA levels, while knocking down E2F1 by siRNA-suppressed POMC. E2F1 direct binding and dissociation from POMC promoter region is controlled by site-specific phosphorylation/de-phosphorylation of E2F1 serine 337 (58).
Of note, we found that E2F1 expression is highly specific to corticotrophs in human pituitary tissue (59). By co-staining with human pituitary hormones, we found that E2F1 co-localizes with POMC in normal human corticotrophs, but not with prolactin or growth hormone in lactotrophs or somatotrophs, respectively (59). E2F1 corticotroph specificity also seems to be subclass specific: E2F1 but not E2F3 enhanced POMC promoter activities by deletion mutant hPOMC luciferase assays using ectopic ACTH-secreting tumor cells derived from human small cell lung cancer DMS79 cells (58). Others similarly found that overexpression of E2F3 is not sufficient to produce pituitary tumors, even though it leads to pituitary hyperplasia (75).
Whether E2F1 is involved in pituitary development or tumorigenesis is unknown. Double Rb and E2F1 gene knockout mice show fewer pituitary tumors than do Rb knockout mice with intact E2F1 (76), suggesting a role of E2F1 in pituitary tumorigenesis, but Rb heterozygous mice with double E2F4 knockout also show increased tumor incidence (77).
E2F1 as Part of Downstream Signaling of EGFR
Epidermal growth factor receptor (EGFR) is expressed to varying degrees in human pituitary tissue, including corticotroph adenomas (78), and EGFR regulates POMC transcription and ACTH production (79). However, mechanisms for corticotroph-specific tumor induction by EGFR or for EGFR upregulation of ACTH/POMC expression have not been clearly elucidated.
rPomc promoter-driven EGFR transgenic mice show pituitary corticotroph tumors in the anterior and intermediate lobes and demonstrate phenotypes similar to those in human Cushing disease, including obesity, glucose intolerance, and adrenal hyperplasia (59). E2F1 and phosphorylated serine 337-E2F1 were both upregulated in these tumors, but were attenuated with EGFR inhibition. Although EGFR is expressed in other lineages of aggressive pituitary tumors (80), our findings suggest that EGFR signaling induces E2F1-mediated POMC transcription and corticotroph adenoma pathogenesis. This pathway may therefore be a candidate for corticotroph-specific targeted therapy in patients with Cushing disease.
Recently, gain-of-function somatic mutations in USP8 were reported in 30–50% of Cushing tumors (81, 82). USP8 is a deubiquitinase that protects EGFR from lysosomal degradation. These mutations lead to a higher rate of USP8-induced EGFR deubiquitination, increasing EGFR pathway stimulation, and ultimately increasing ACTH secretion (81, 82). However, the mechanisms and influence of USP8 mutations on Cushing tumors are not fully understood. EGFR expression is unchanged comparing mutated vs. non-mutated tumors (83), and phenotypic features of aggressive tumors, particularly larger tumor size, are not consistently seen (81, 83, 84). Further study of these mechanisms and the clinical implications of USP8-mutated Cushing tumors is ongoing.
G1/S Phase Is the Key in Corticotroph Tumors
The cyclin E promoter has binding sites for E2F1, which, in turn, upregulates cyclin E mRNA (74, 85). E2F1 also binds Rb, while p27 targets the CDK2/cyclin E complex. This suggests cyclin E, p27, E2F1, and Rb could interact to regulate cell cycle progression through G1 (74).
Dysregulation of G1/S transition is often seen in human cancers. Our data suggest that lineage-specific amplification of cyclin E/E2F1 signals contribute to uncontrolled POMC transcription and autonomous ACTH production in corticotroph tumors (86), and cyclin E, p27, Rb, and E2F1 have also been shown to affect G1/S transition in corticotrophs (23, 58, 59, 62). Targeting G1/S could therefore be a reasonable therapeutic approach in patients with corticotroph tumors (Figure 1).
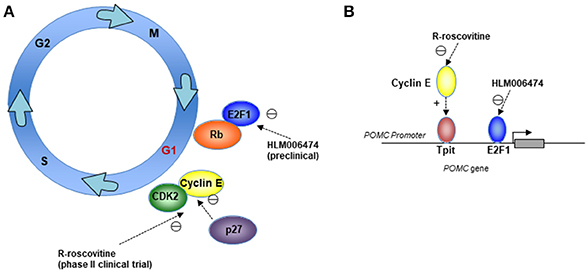
Figure 1. Mechanisms for therapeutically targeting the cell cycle G1/S phase in corticotroph adenomas. Cartoons depicting (A) Cell-cycle targets for the CDK2/cyclin E inhibitor R-roscovitine, which is being investigated in a phase II clinical trial, and the E2F inhibitor HLM006474, which is being investigated in preclinical models; and (B) Suppression of the POMC promoter through cyclin E and E2F1 by R-roscovitine and HLM006474.
Therapeutic Targeting of Cell Cycle Proteins in Corticotroph Tumors
Several small molecule CDK inhibitors are being evaluated in different pituitary tumor types (87, 88). To date, such agents have proven effective in preclinical studies (39, 47, 86). The first study treated CDK4/p27 double knockout mice harboring anterior pituitary tumors with flavopiridol, an inhibitor of CDK1, CDK2, CDK4, CDK6, and CDK7 that causes cell-cycle arrest at G1 and G2, and showed shrinkage of pituitary tumor size as well as prolonged survival (39). Using the small molecule E2F inhibitor HLM006474, originally developed as a therapy for melanoma (89, 90), we showed dose-dependent suppression of POMC mRNA expression in primary cultures of surgically resected Cushing tumor tissue, but no suppression of growth hormone used as a control (59). Despite its lack of specificity for E2F1, the corticotroph-specific effects suggest this agent may be useful in POMC-producing tumors.
Using a germline transgenic zebrafish model overexpressing PTTG in pituitary corticotrophs to recapitulate human Cushing disease, we tested several small molecule CDK inhibitors on corticotroph adenomas in vivo, including flavopiridol, R-roscovitine (seliciclib; primarily an inhibitor of CDK1 and CDK2 in late G1 but not an inhibitor of CDK4 and CDK6), olomoucine (CDK1, CDK2), PD-0332991 (CDK4/6 in early G1), and CAY10572 (CDK7 in S phase). Addition of PD-0332991 or CAY10572 to the culture medium of double transgenic embryos generated by breeding the POMC:PTTG model with transgenic zebrafish expressing green fluorescent protein (eGFP) resulted in no significant change in pituitary expression of POMC-eGFP compared with controls, while a modest reduction of ~20% was observed in the olomoucine-treated group (47). By contrast, R-roscovitine-treated embryos exhibited ~40% reduction in pituitary POMC-eGFP expression compared with controls (P < 0.02) (47).
R-roscovitine is a second-generation CDK inhibitor that interrupts ATP binding of CDK. It has a relatively broad range of activity, but primarily targets the CDK2/cyclin E complex (91). R-roscovitine has been studied in patients with nasopharyngeal cancer, non-small lung cancer, and B-cell malignancies, but few clinical trial data have been reported (92). Side effects include mild to moderate fatigue, nausea/vomiting, constipation, fever, cough, and elevated liver enzymes, which typically resolved after drug discontinuation (93).
In AtT20 cells, a murine corticotroph adenoma cell line commonly used as a model for Cushing disease, as well as in PTTG zebrafish models, R-roscovitine treatment significantly suppressed POMC expression both in vitro and in vivo (47). Plasma ACTH, corticosterone levels, and tumor size were significantly reduced in AtT20 cells xenografted in mice with R-roscovitine treatment (47), and POMC mRNA and ACTH levels were dose dependently suppressed in primary cultures derived from human corticotroph adenomas (86). In addition to its cell cycle effects, R-roscovitine also has direct inhibitory effects on the POMC promoter (58, 86). Deletion mutant and point mutant rPomc luciferase assays showed that R-roscovitine suppressed the rPomc promoter by targeting the Tpit binding region (TCACACC) and suppressed protein expression of cyclin E and E2F1 in a dose-dependent manner (86), suggesting that suppression of Tpit expression is mediated by cyclin E/E2F1 reciprocal regulation. Importantly, viable tumor cell numbers were largely unchanged despite decreased ACTH concentration in the culture medium of primary cultures (86). In ectopic ACTH-secreting tumor xenografted mice, R-roscovitine similarly suppressed POMC/ACTH secretion, but it did not alter tumor proliferation in DMS79 cells (58, 94). The inhibitory effect of this agent in human corticotroph tumors preferentially targets ACTH expression rather than tumor cell growth suggests other corticotroph mechanisms independent of cell cycle regulation may be present. A phase II study of R-roscovitine (seliciclib) is currently underway for the treatment of patients with de novo, recurrent, or persistent Cushing disease (ClinicalTrials.gov NCT02160730).
Conclusions
Corticotrophs are sensitive to changes in cell cycle regulation, and evidence suggests involvement of corticotroph lineage-specific cell cycle regulators such as cyclin E, p27, Rb, and E2F1 in tumorigenesis. Currently, only R-roscovitine, a cyclin E/E2F1 inhibitor, is in clinical development, but it is likely that other agents targeting these factors will prove attractive as novel medical therapy options for patients with Cushing disease.
Author Contributions
TA and N-AL conceived and designed the project. TA collected and analyzed the evidence and prepared the draft manuscript. TA and N-AL contributed to manuscript revisions and approved the submitted version.
Funding
This work was supported by NIH grants R21DK103198 (N-AL) and T32DK007770 (TA). The funding sources had no role in study design, data analysis, or decision to publish.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Shira Berman for technical advice and assistance.
References
1. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. (2009) 5:374–81. doi: 10.1038/nrendo.2009.106
2. Raff H, Carroll T. Cushing's syndrome: from physiological principles to diagnosis and clinical care. J Physiol. (2015) 593:493–506. doi: 10.1113/jphysiol.2014.282871
3. Petersenn S, Beckers A, Ferone D, van der Lely A, Bollerslev J, Boscaro M, et al. Therapy of endocrine disease: outcomes in patients with Cushing's disease undergoing transsphenoidal surgery: systematic review assessing criteria used to define remission and recurrence. Eur J Endocrinol. (2015) 172:R227–39. doi: 10.1530/EJE-14-0883
4. Fleseriu M, Hamrahian AH, Hoffman AR, Kelly DF, Katznelson L, Neuroendocrine A, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Disease state clinical review: diagnosis of recurrence in cushing disease. Endocr Pract. (2016) 22:1436–48. doi: 10.4158/EP161512.DSCR
5. Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. (2008) 93:2454–62. doi: 10.1210/jc.2007-2734
6. Nieman LK, Biller BMK, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing's syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. (2015) 100:2807–31. doi: 10.1210/jc.2015-1818
7. Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, et al. Ketoconazole in Cushing's disease: is it worth a try? J Clin Endocrinol Metab. (2014) 99:1623–30. doi: 10.1210/jc.2013-3628
8. Daniel E, Aylwin S, Mustafa O, Ball S, Munir A, Boelaert K, et al. Effectiveness of metyrapone in treating Cushing's syndrome: a retrospective multicenter study in 195 patients. J Clin Endocrinol Metab. (2015) 100:4146–54. doi: 10.1210/jc.2015-2616
9. Fleseriu M, Pivonello R, Young J, Hamrahian AH, Molitch ME, Shimizu C, et al. Osilodrostat, a potent oral 11 beta-hydroxylase inhibitor: 22-week, prospective, phase II study in Cushing's disease. Pituitary (2016) 19:138–48. doi: 10.1007/s11102-015-0692-z
10. Fleseriu M, Molitch ME, Gross C, Schteingart DE, Vaughan TB,III, and Biller BM. A new therapeutic approach in the medical treatment of Cushing's syndrome: glucocorticoid receptor blockade with mifepristone. Endocr Pract. (2013) 19:313–26. doi: 10.4158/ep12149.ra
11. Langlois F, Chu J, Fleseriu M. Pituitary-directed therapies for Cushing's disease. Front Endocrinol. (2018) 9:164. doi: 10.3389/fendo.2018.00164
12. Pivonello R, De Martino MC, Cappabianca P, De Leo M, Faggiano A, Lombardi G, et al. The medical treatment of Cushing's disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab. (2009) 94:223–30. doi: 10.1210/jc.2008-1533
13. Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing's disease. Eur J Endocrinol. (2010) 163:709–16. doi: 10.1530/eje-10-0382
14. Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. A 12-month phase 3 study of pasireotide in Cushing's disease. N Engl J Med. (2012) 366:914–24. doi: 10.1056/NEJMoa1105743
15. Fukuoka H. New potential targets for treatment of Cushing's disease: epithelial growth factor receptor and cyclin-dependent kinases. Pituitary (2015) 18:274–8. doi: 10.1007/s11102-015-0637-6
16. Lamolet B, Pulichino A-M, Lamonerie T, Gauthier Y, Brue T, Enjalbert A, et al. A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell (2001) 104:849–59. doi: 10.1016/S0092-8674(01)00282-3
17. Camper SA, Saunders TL, Katz RW, Reeves RH. The Pit-1 transcription factor gene is a candidate for the murine Snell dwarf mutation. Genomics (1990) 8:586–90.
18. Li S, Crenshaw EB,III, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature (1990) 347:528–33. doi: 10.1038/347528a0
19. Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. (2003) 112:1603–18. doi: 10.1172/jci20401
20. Quereda V, Malumbres M. Cell cycle control of pituitary development and disease. J Mol Endocrinol. (2009) 42:75–86. doi: 10.1677/jme-08-0146
21. Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. (2005) 30:630–41. doi: 10.1016/j.tibs.2005.09.005
22. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer (2009) 9:153. doi: 10.1038/nrc2602
23. Bilodeau S, Roussel-Gervais A, Drouin J. Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol Cell Biol. (2009) 29:1895–908. doi: 10.1128/mcb.01885-08
24. DeCaprio JA, Ludlow JW, Lynch D, Furukawa Y, Griffin J, Piwnica-Worms H, et al. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell (1989) 58:1085–95.
25. Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. (2000) 14:3051–64.
26. Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. (2000) 14:3037–3050.
27. Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol Endocrinol. (1997) 11:433–41. doi: 10.1210/mend.11.4.9911
28. Romero F, Multon M-C, Ramos-Morales F, Domínguez Á, Bernal JA, Pintor-Toro JA, et al. Human securin, hPTTG, is associated with Ku heterodimer, the regulatory subunit of the DNA-dependent protein kinase. Nucleic Acids Res. (2001) 29:1300–7.
29. Tong Y, Tan Y, Zhou C, Melmed S. Pituitary tumor transforming gene interacts with Sp1 to modulate G1/S cell phase transition. Oncogene (2007) 26:5596–605. doi: 10.1038/sj.onc.1210339
30. Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, et al. Requirement for a functional Rb-1 gene in murine development. Nature (1992) 359:328–30. doi: 10.1038/359328a0
31. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature (1992) 359:295–300. doi: 10.1038/359295a0
32. Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature (1992) 359:288–94. doi: 10.1038/359288a0
33. Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. (1998) 12:2899–911.
34. Brugarolas J, Bronson RT, Jacks T. p21 Is a critical CDK2 regulator essential for proliferation control in Rb-deficient cells. J Cell Biol. (1998) 141:503–14.
35. Park MS, Rosai J, Nguyen HT, Capodieci P, Cordon-Cardo C, Koff A. p27 and Rb are on overlapping pathways suppressing tumorigenesis in mice. Proc Natl Acad Sci USA. (1999) 96:6382–7. doi: 10.1073/pnas.96.11.6382
36. Ramsey MR, Krishnamurthy J, Pei XH, Torrice C, Lin W, Carrasco DR, et al. Expression of p16Ink4a compensates for p18Ink4c loss in cyclin-dependent kinase 4/6-dependent tumors and tissues. Cancer Res. (2007) 67:4732–41. doi: 10.1158/0008-5472.can-06-3437
37. Roussel-Gervais A, Bilodeau S, Vallette S, Berthelet F, Lacroix A, Figarella-Branger D, et al. Cooperation between cyclin E and p27(Kip1) in pituitary tumorigenesis. Mol Endocrinol. (2010) 24:1835–45. doi: 10.1210/me.2010-0091
38. Franklin DS, Godfrey VL, O'Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. (2000) 20:6147–58.
39. Sotillo R, Renner O, Dubus P, Ruiz-Cabello J, Martin-Caballero J, Barbacid M, et al. Cooperation between Cdk4 and p27kip1 in tumor development: a preclinical model to evaluate cell cycle inhibitors with therapeutic activity. Cancer Res. (2005) 65:3846–52. doi: 10.1158/0008-5472.can-04-4195
40. Takeuchi S, Koeffler HP, Hinton DR, Miyoshi I, Melmed S, Shimon I. Mutation and expression analysis of the cyclin-dependent kinase inhibitor gene p27/Kip1 in pituitary tumors. J Endocrinol. (1998) 157:337–41.
41. Komatsubara K, Tahara S, Umeoka K, Sanno N, Teramoto A, Osamura RY. Immunohistochemical analysis of p27 (Kip1) in human pituitary glands and in various types of pituitary adenomas. Endocr Pathol. (2001) 12:181–8. doi: 10.1385/ep:12:2:181
42. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. (2009) 94:1826–34. doi: 10.1210/jc.2008-2083
43. Ogino A, Yoshino A, Katayama Y, Watanabe T, Ota T, Komine C, et al. The p15(INK4b)/p16(INK4a)/RB1 pathway is frequently deregulated in human pituitary adenomas. J Neuropathol Exp Neurol. (2005) 64:398–403. doi: 10.1093/jnen/64.5.398
44. Japon MA, Rubinstein M, Low MJ. In situ hybridization analysis of anterior pituitary hormone gene expression during fetal mouse development. J Histochem Cytochem. (1994) 42:1117–25. doi: 10.1177/42.8.8027530
45. Jordan S, Lidhar K, Korbonits M, Lowe DG, Grossman AB. Cyclin D and cyclin E expression in normal and adenomatous pituitary. Eur J Endocrinol. (2000) 143:R1–6.
46. Davis SW, Mortensen AH, Camper SA. Birthdating studies reshape models for pituitary gland cell specification. Dev Biol. (2011) 352:215–27. doi: 10.1016/j.ydbio.2011.01.010
47. Liu NA, Jiang H, Ben-Shlomo A, Wawrowsky K, Fan XM, Lin S, et al. Targeting zebrafish and murine pituitary corticotroph tumors with a cyclin-dependent kinase (CDK) inhibitor. Proc Natl Acad Sci USA. (2011) 108:8414–9. doi: 10.1073/pnas.1018091108
48. Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell (1996) 85:733–44.
49. Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell (1996) 85:721–32. doi: 10.1016/S0092-8674(00)81238-6
50. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell (1996) 85:707–20.
51. Nakazumi H, Sasano H, Iino K, Ohashi Y, Orikasa S. Expression of cell cycle inhibitor p27 and Ki-67 in human adrenocortical neoplasms. Mod Pathol. (1998) 11:1165–70.
52. Lidhar K, Korbonits M, Jordan S, Khalimova Z, Kaltsas G, Lu X, et al. Low expression of the cell cycle inhibitor p27Kip1 in normal corticotroph cells, corticotroph tumors, and malignant pituitary tumors. J Clin Endocrinol Metab. (1999) 84:3823–30. doi: 10.1210/jcem.84.10.6066
53. Cryns VL, Alexander JM, Klibanski A, Arnold A. The retinoblastoma gene in human pituitary tumors. J Clin Endocrinol Metab. (1993) 77:644–6. doi: 10.1210/jcem.77.3.7690360
54. Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene (1994) 9:1021–7.
55. Pei L, Melmed S, Scheithauer B, Kovacs K, Benedict WF, Prager D. Frequent loss of heterozygosity at the retinoblastoma susceptibility gene (RB) locus in aggressive pituitary tumors: evidence for a chromosome 13 tumor suppressor gene other than RB. Cancer Res. (1995) 55:1613–6.
56. Hinton DR, Hahn JA, Weiss MH, Couldwell WT. Loss of RB expression in an ACTH-secreting pituitary carcinoma. Cancer Lett. (1998) 126:209–14. doi: 10.1016/S0304-3835(98)00013-5
57. Vooijs M, van der Valk M, te Riele H, Berns A. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene (1998) 17:1–12. doi: 10.1038/sj.onc.1202169
58. Araki T, Liu NA, Tone Y, Cuevas-Ramos D, Heltsley R, Tone M, et al. E2F1-mediated human POMC expression in ectopic Cushing's syndrome. Endocr Relat Cancer (2016) 23:857–70. doi: 10.1530/erc-16-0206
59. Araki T, Liu X, Kameda H, Tone Y, Fukuoka H, Tone M, et al. EGFR Induces E2F1-mediated corticotroph tumorigenesis. J Endocr Soc. (2017) 1:127–43. doi: 10.1210/js.2016-1053
60. Saeger W, Schreiber S, Lüdecke DK. Cyclins D1 and D3 and topoisomerase IIα in inactive pituitary adenomas. Endocr Pathol. (2001) 12:39–47. doi: 10.1385/EP:12:1:39
61. Simpson DJ, Frost SJ, Bicknell JE, Broome JC, McNicol AM, Clayton RN, et al. Aberrant expression of G 1/S regulators is a frequent event in sporadic pituitary adenomas. Carcinogenesis (2001) 22:1149–54. doi: 10.1093/carcin/22.8.1149
62. Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, et al. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. (2006) 20:2871–86. doi: 10.1101/gad.1444606
63. Liu C, Xie W, Wu D, Li Z, Li C, Zhang Y. Expression of cell-cycle regulators is associated with invasive behavior and poor prognosis in prolactinomas. Int J Clin Exp Pathol. (2016) 9:3245–55.
64. Bamberger CM, Fehn M, Bamberger AM, Ludecke DK, Beil FU, Saeger W, et al. Reduced expression levels of the cell-cycle inhibitor p27Kip1 in human pituitary adenomas. Eur J Endocrinol. (1999) 140:250–5.
65. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. (2006) 103:15558–63. doi: 10.1073/pnas.0603877103
66. Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. (2007) 92:3321–5. doi: 10.1210/jc.2006-2843
67. Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. (2013) 9:e1003350. doi: 10.1371/journal.pgen.1003350
68. Tanaka C, Yoshimoto K, Yang P, Kimura T, Yamada S, Moritani M, et al. Infrequent mutations of p27Kip1 gene and trisomy 12 in a subset of human pituitary adenomas. J Clin Endocrinol Metab. (1997) 82:3141–7. doi: 10.1210/jcem.82.9.4202
69. Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science (1995) 269:682–5.
70. Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol. (2003) 13:41–7. doi: 10.1016/S1044-579X(02)00098-6
71. Matsunaga E. On estimating penetrance of the retinoblastoma gene. Hum Genet. (1980) 56:127–8. doi: 10.1007/BF00281583
72. Draper GJ, Sanders BM, Kingston JE. Second primary neoplasms in patients with retinoblastoma. Br J Cancer (1986) 53:661–71.
73. Helin K, Lees JA, Vidal M, Dyson N, Harlow E, Fattaey A. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell (1992) 70:337–50.
74. Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci USA. (1995) 92:12146–50.
75. Lazzerini Denchi E, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol. (2005) 25:2660–72. doi: 10.1128/mcb.25.7.2660-2672.2005
76. Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E, Jacks T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/–)mice. Nat Genet. (1998) 18:360–4. doi: 10.1038/ng0498-360
77. Lee EY, Cam H, Ziebold U, Rayman JB, Lees JA, Dynlacht BD. E2F4 loss suppresses tumorigenesis in Rb mutant mice. Cancer Cell (2002) 2:463–472.
78. Theodoropoulou M, Arzberger T, Gruebler Y, Jaffrain-Rea ML, Schlegel J, Schaaf L, et al. Expression of epidermal growth factor receptor in neoplastic pituitary cells: evidence for a role in corticotropinoma cells. J Endocrinol. (2004) 183:385–94. doi: 10.1677/joe.1.05616
79. Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, et al., EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest. (2011) 121:4712–21. doi: 10.1172/jci60417
80. Liu X, Kano M, Araki T, Cooper O, Fukuoka H, Tone Y, et al. ErbB receptor-driven prolactinomas respond to targeted lapatinib treatment in female transgenic mice. Endocrinology (2015) 156:71–9. doi: 10.1210/en.2014-1627
81. Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, et al. Recurrent gain-of-function USP8 mutations in Cushing's disease. Cell Res. (2015) 25:306–17. doi: 10.1038/cr.2015.20
82. Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet. (2015) 47:31–8. doi: 10.1038/ng.3166
83. Hayashi K, Inoshita N, Kawaguchi K, Ibrahim Ardisasmita A, Suzuki H, Fukuhara N, et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing's disease. Eur J Endocrinol. (2016) 174:213–26. doi: 10.1530/eje-15-0689
84. Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernandez-Ramirez LC, Settas N, et al. Somatic USP8 gene mutations are a common cause of pediatric Cushing disease. J Clin Endocrinol Metab. (2017) 102:2836–43. doi: 10.1210/jc.2017-00161
85. Herrera RE, Sah VP, Williams BO, Makela TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. (1996) 16:2402–7.
86. Liu NA, Araki T, Cuevas-Ramos D, Hong J, Ben-Shlomo A, Tone Y, et al. Cyclin E-mediated human proopiomelanocortin regulation as a therapeutic target for Cushing disease. J Clin Endocrinol Metab. (2015) 100:2557–64. doi: 10.1210/jc.2015-1606
87. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. (2015) 14:130–46. doi: 10.1038/nrd4504
88. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. (2016) 13:417. doi: 10.1038/nrclinonc.2016.26
89. Ma Y, Kurtyka CA, Boyapalle S, Sung SS, Lawrence H, Guida W, et al. A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. (2008) 68:6292–9. doi: 10.1158/0008-5472.can-08-0121
90. Kurtyka CA, Chen L, Cress WD. E2F inhibition synergizes with paclitaxel in lung cancer cell lines. PLoS ONE (2014) 9:e96357. doi: 10.1371/journal.pone.0096357
91. Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. (1997) 243:527–36.
92. Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I, et al. Roscovitine in cancer and other diseases. Ann Transl Med. (2015) 3:135. doi: 10.3978/j.issn.2305-5839.2015.03.61
93. Yeo W, Goh B, Le Tourneau C, Green S, Chiao J, Siu L. A phase II randomized study of oral seliciclib in patients with previously treated nasopharyngeal carcinoma. ASCO Meeting Abstracts (2009) 27:6026.
Keywords: pituitary tumor, Cushing disease, cell cycle, cyclin E, E2F1, POMC
Citation: Araki T and Liu N-A (2018) Cell Cycle Regulators and Lineage-Specific Therapeutic Targets for Cushing Disease. Front. Endocrinol. 9:444. doi: 10.3389/fendo.2018.00444
Received: 22 March 2018; Accepted: 18 July 2018;
Published: 10 August 2018.
Edited by:
Maria Fleseriu, Oregon Health & Science University, United StatesReviewed by:
Leila Warszawski, Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione, BrazilRoberto Salvatori, Johns Hopkins University, United States
Copyright © 2018 Araki and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takako Araki, dGFyYWtpQHVtbi5lZHU=