 Hongyang Wang1,2†
Hongyang Wang1,2† Dongfang Liu
Dongfang Liu Lian Duan
Lian Duan- 1Department of Endocrinology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, China
- 2The Infirmary, Chongqing Mechanical Senior Technician School (Chongqing Mechanical Technician College), Chongqing, China
- 3Department of Hematology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China
- 4Department of Endocrinology, The Third Affiliated Hospital of Chongqing Medical University (Jie er Hospital), Chongqing, China
Background and Objectives: It is currently controversial whether subclinical hyperthyroidism is associated with PRKAR1A gene variants. We describe a man with subclinical hyperthyroidism and a PRKAR1A gene variant who was diagnosed with Carney complex (CNC), and we performed a systematic review of published studies to assess the association between PRKAR1A gene variants and the risk of subclinical hyperthyroidism.
Design and Methods: The PubMed, EMBASE, OVID, Science Direct, and gray literature electronic databases were searched for articles published from January 2002 to May 2021 using predefined keywords and inclusion and exclusion criteria. Data on thyroid function from selected studies were extracted and analyzed.
Results: We identified a CNC patient with a subclinical hyperthyroidism phenotype combined with multiple components and genetic sequenced data. In a subsequent systematic review, twenty selected studies (14 case studies and 6 series studies) enrolling 23 individuals were included in the final analysis. The patient’s thyroid function data were qualitative in 11 cases and quantitative in 12 cases. The prevalence of subclinical hyperthyroidism in the CNC patients with a PRKAR1A gene variant, including our patient, was markedly higher than that in the normal population (12.5% vs. 2%)
Conclusions: The findings of this systematic review provide helpful evidence that PRKAR1A gene variants and subclinical hyperthyroidism are related and suggest that subclinical hyperthyroidism may be a neglected phenotype of PRKAR1A gene variants and a novel component of CNC patients.
Systematic Review Registration: https://www.crd.york.ac.uk/PROSPERO, identifier CRD42021197655.
Introduction
Subclinical hyperthyroidism, with normal thyroxine and/or triiodothyronine levels and suppressed thyroid-stimulating hormone levels, includes progression to overt hyperthyroidism, cardiovascular conditions, bone loss, fractures, and dementia (1). Carney complex (CNC) is a rare multiple endocrine and nonendocrine neoplasia syndrome, described for the first time in 1985 by J Aidan Carney as “the complex of myxomas, spotty pigmentation and endocrine overactivity” (2). According to the CNC diagnostic criteria in 2001, the syndrome can affect the thyroid gland, manifesting as thyroid carcinoma or multiple hypoechoic nodules on thyroid ultrasonography in young patients (3), but the thyroid functions of these patients are rarely reported and controversial. Stratakis CA et al. reported that all patients had normal results of physical and biochemical examinations of the thyroid gland (total and free thyroxine, triiodothyronine, and thyrotropin levels) (4); however, some researchers observed thyrotoxicosis as a clinical finding of the syndrome (5). Seventy percent of CNC cases are caused by a PRKAR1A variant (3, 4).
The effect of PRKAR1A variants on thyroid function remains unknown. An animal study found that thyroid-specific ablation of mouse PRKAR1A caused hyperthyroidism and follicular carcinoma (6). We also reported a laboratory result with subclinical hyperthyroidism as the first diagnosis in a CNC patient whose variant was confirmed by PRKAR1A gene sequencing. These results led us to speculate that PRKAR1A gene variants may be related to thyroid function. To date, however, thyroid function has not been implicated in CNC patients with PRKAR1A variants.
CNC is a rare disease. To expand the number of patients, we performed a systematic review with strict inclusion and exclusion criteria to obtain original data on thyroid function in CNC patients with a PRKAR1A gene variant and compared the prevalence of subclinical hyperthyroidism between CNC patients with a PRKAR1A gene variant and the normal population. We searched the PubMed, EMBASE, OVID, Science Direct, and gray literature databases and systematically reviewed the thyroid function of CNC patients affected by a PRKAR1A gene variant.
Methods
Study participant and registration
The patient has signed informed consent forms and consented to the publication of this case report. This systematic review was registered with PROSPERO (CRD42021197655). Details of the protocol for this systematic review can be accessed at www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=197655. It was reported based on the Joanna Briggs Institute’s approach, for a systematic review of etiology (7).
Search strategy
Electronic databases, including the PubMed, EMBASE, OVID, Science Direct, and gray literature databases, were searched for articles published from January 2002 to May 2021. The included research was strictly human research. There were no limits regarding the language of publication. The article type was limited to case and series studies. The search strategies used are shown in the Supplementary Material.
Inclusion and exclusion criteria
Inclusion criteria: CNC patients (as diagnosed using the diagnostic criteria) (3) with thyroid function data and a PRKAR1A gene variant.
Exclusion criteria: CNC patients with a history of thyroid diseases (such as Graves’ disease, Hashimoto’s thyroiditis, subacute thyroiditis and so on) and pituitary or thyroid surgery.
Data extraction
The first selection was performed by filtering duplicates with EndNote ×9 and manual filtering. Eligible studies were selected according to a multistep approach (title reading, abstract reading and full-text assessment) by two researchers working independently. Disagreements between the two researchers were resolved by a third researcher. The extracted data included age, sex, thyrotropin (TSH), free thyroxine (FT4), adrenocorticotropic hormone (ACTH), PRKAR1A gene variant, cortisol, and other relevant components of CNC.
Risk of bias assessment
To control the risk of bias, gray literature databases were used. Because the original data of thyroid function were extracted, the bias control tool was not involved.
Strategy for data synthesis
A meta-analysis was not performed because there were no three or more thyroid function data of CNC patients with PRKAR1A variant in enrolled reports that had been extracted.
Research outcome
The main outcome was the prevalence of subclinical hyperthyroidism in CNC patients with thyroid function data and a PRKAR1A gene variant.
Case presentation
A 36-year-old Chinese male was admitted to our endocrine ward in January 2018 with a 2-month history of repeated fatigue, decreased TSH (0.01 mIU/L), and normal free triiodothyronine (FT3), FT4, and thyroid color Doppler ultrasound. He was treated with methimazole 5 mg QD. He had a history of hypertension for 2 years and was being treated with metoprolol succinate, valsartan, and amlodipine tablets. Cardiac ultrasound suggested thickening of the ventricular septum. He had moderate sleep apnea syndrome. He was married and had one child. Many people in his family had pigmentation.
His heart rate was 119 times/min, his blood pressure was 122/95 mmHg, his height was 167 cm, his weight was 70.3 kg, and his body mass index was 25.2 kg/m2. A physical examination at the time of admission revealed bruising at the elbow skin, purple streaks on the lower abdomen (Figure 1A), spotty pigmentation of the lip mucosa and multiple skin areas (Figures 1B, C), skin myxoma of the posterior neck (Figure 1D), and no thyroid enlargement.
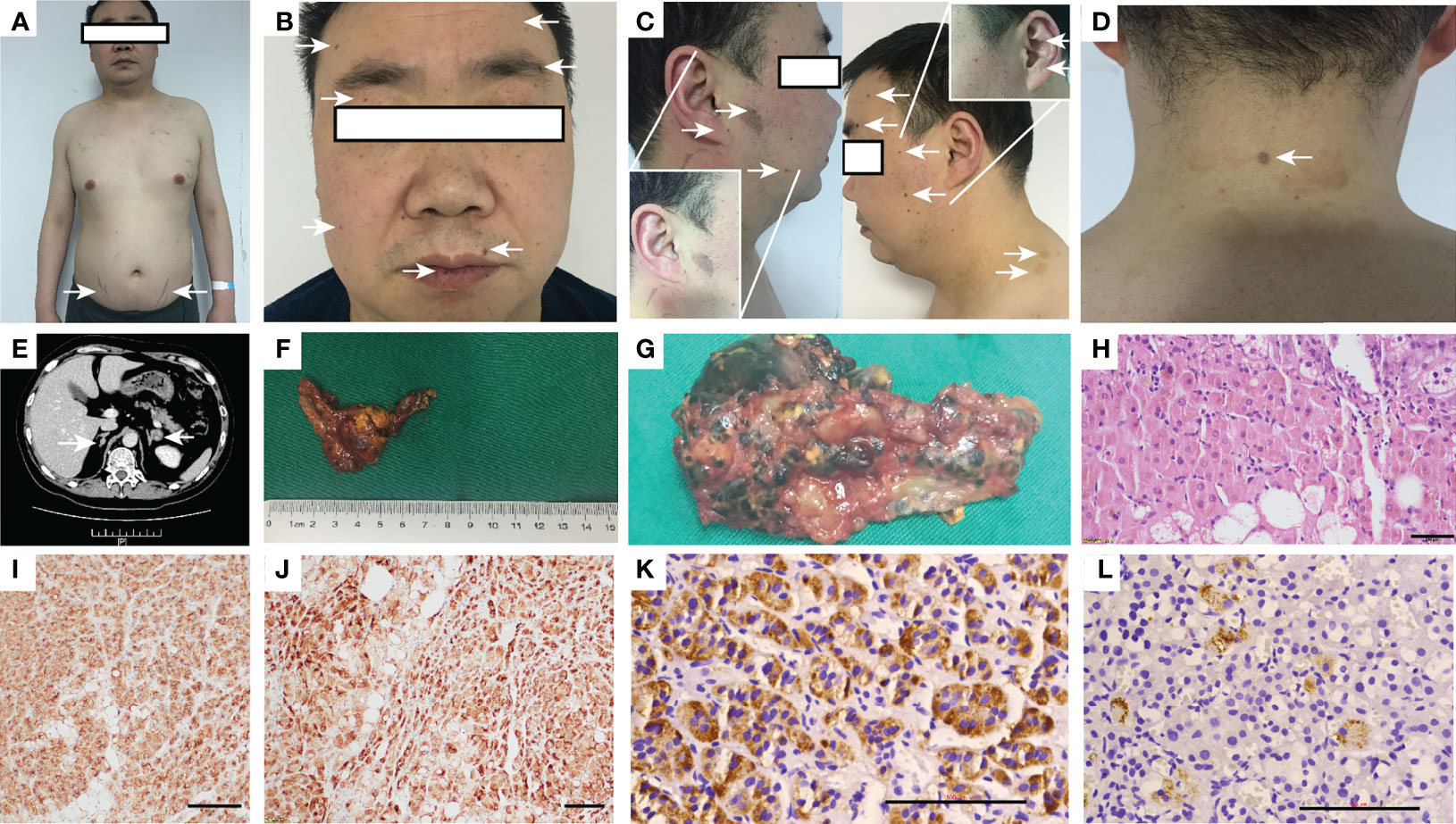
Figure 1 Appearance of the patient, adrenal CT, postoperative gross, HE and histochemical staining. (A) Appearance and lower abdomen purple streaks; (B, C) Lip mucosa and multiple skin spot-like pigmentation; (D) Posterior neck skin myxoma; (E) Adrenal CT: bilateral adrenal nodules, the left side diameter is 1.9 cm, the enhanced CT value is approximately 40 HU; (F, G) Postoperative gross after left adrenalectomy: The diseased tissue is grayish-yellow–gray–brown, 4.5•2.5•2 cm, multinodular, some cells have pigmentation, and the surrounding cortex is squeezed and thinned; (H) After hematoxylin-eosin staining, brown are seen particles •400; (I–L) Immunohistochemistry of SYN (•200), Inhibin-α (•200), CYP11B1and CYP11B2 (•400). CT, Computed tomography; HE, hematoxylin-eosin.
His white blood cell count was 10.15 × 10^9/L, his potassium was 3.09 mmol/l, his oral glucose tolerance test was 6.42 mmol/l at 0 h and 9.3 mmol/l at 2 h, and his growth hormone was 0.067 ng/ml. The patient’s TSH was repeatedly lower than the lower limit of the normal reference value when he was treated at other hospitals in the previous 2 months and before he was admitted to our hospital, and the fluctuation was between 0.01 and 0.02 IU/ml (Table 1). Preoperative assessments of the adrenal gland and thyroid function are shown in Table 1.
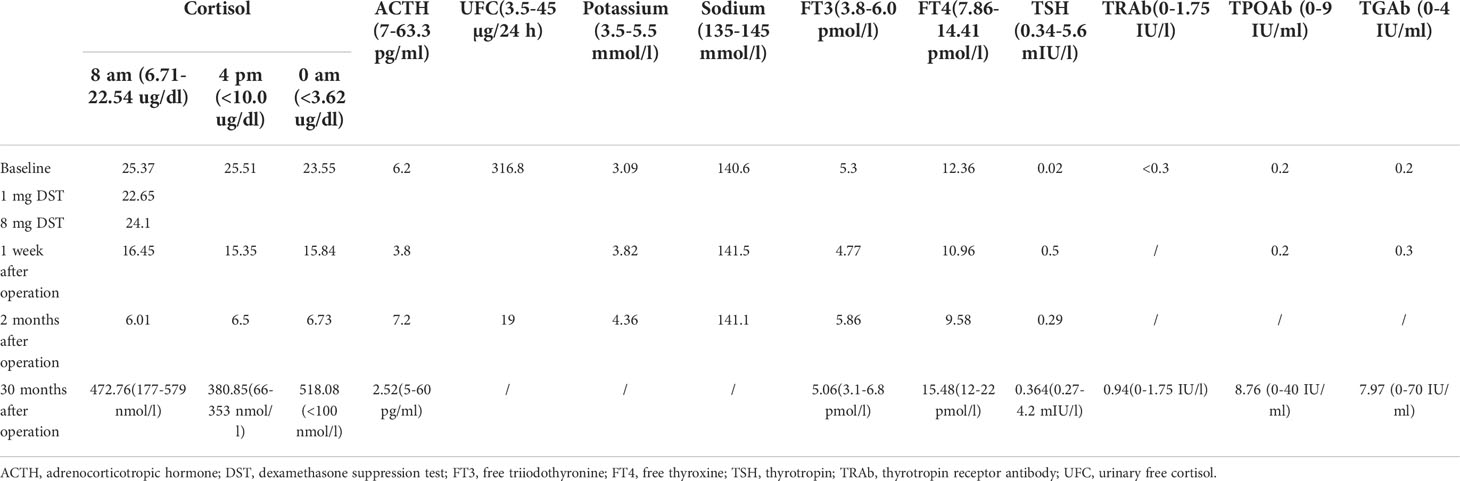
Table 1 Preoperative assessment and postoperative follow-up of the patient.
Because cortisol is not suppressed by low-dose dexamethasone and low levels of ACTH, the patient was diagnosed with ACTH-independent Cushing syndrome. Computed tomography (CT) of the adrenal glands showed nodules on the left lateral adrenal branch and nodular thickening of the medial adrenal branch on the right. Enhanced scanning showed uniform enhancement (Figure 1E). The thyroid gland had a full shape, a smooth capsule, and a homogeneous glandular echo; color Doppler flow imaging (CDFI); the blood flow signal within the thyroid gland did not show an abnormal increase or decrease, and there was no significant difference in bilateral blood flow signals. No echogenic lesions were found in the glands. A thyroid scan was not performed because no thyroid nodules were found on thyroid ultrasound.
Pituitary magnetic resonance imaging, cardiac, breast and testicular ultrasound and bone density assessments showed no abnormalities.
The pathological diagnosis was primary pigmented nodular adrenocortical disease (PPNAD) after left adrenalectomy (Figures 1F–L), postoperative gross, hematoxylin-eosin (HE), and histochemical staining). After obtaining the patient’s informed consent, direct DNA sequencing of PRKAR1A was performed. Sequence analysis revealed a reported heterozygous point variant at codon 439 of exon 4 (c.439 A>G) of the PRKAR1A gene (Figure 2). A diagnosis of CNC was finally made. The patient refused right adrenalectomy at the follow-up 2 months after surgery and evaluation of family members due to privacy concerns. The postoperative follow-up of the patient is shown in Table 1.
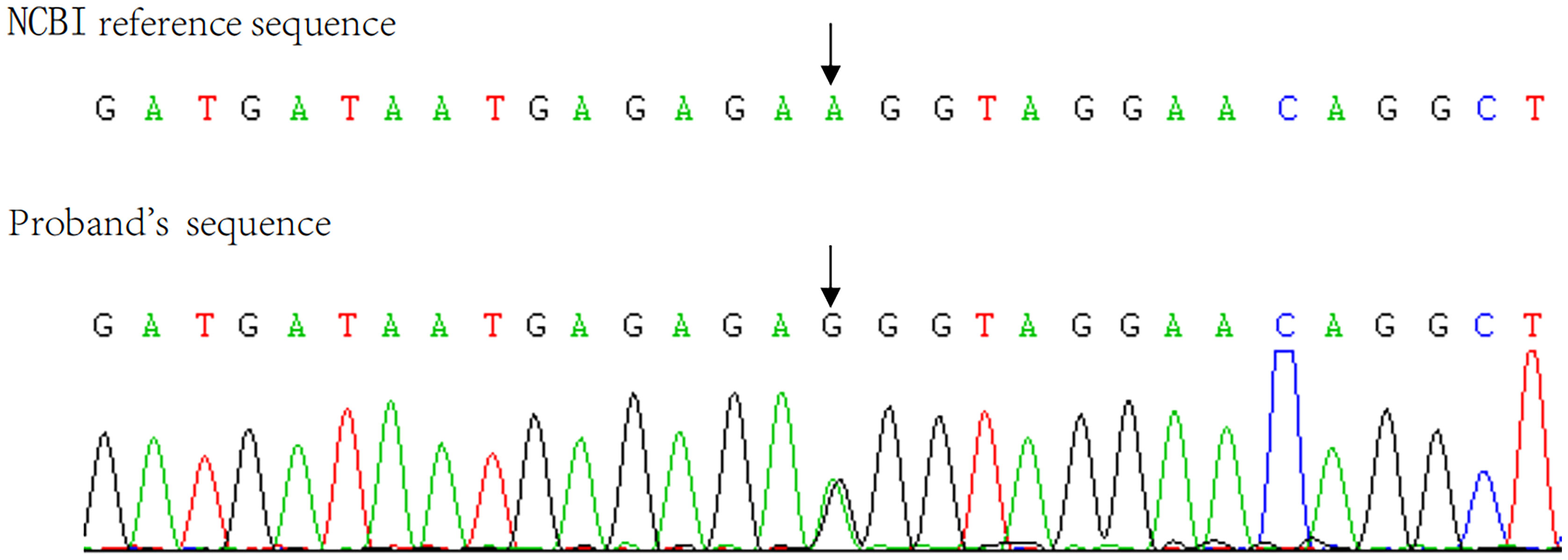
Figure 2 Sanger sequencing showed the patient’s C.439 A>G mutation in exon 4 of the PRKAR1A gene.
Results of the systematic review
Study identification
We identified 195 potentially eligible studies on patients with a PRKAR1A gene variant diagnosed with CNC. Twenty studies including patients with thyroid function test results were deemed eligible. Figure 3 describes the study selection process.
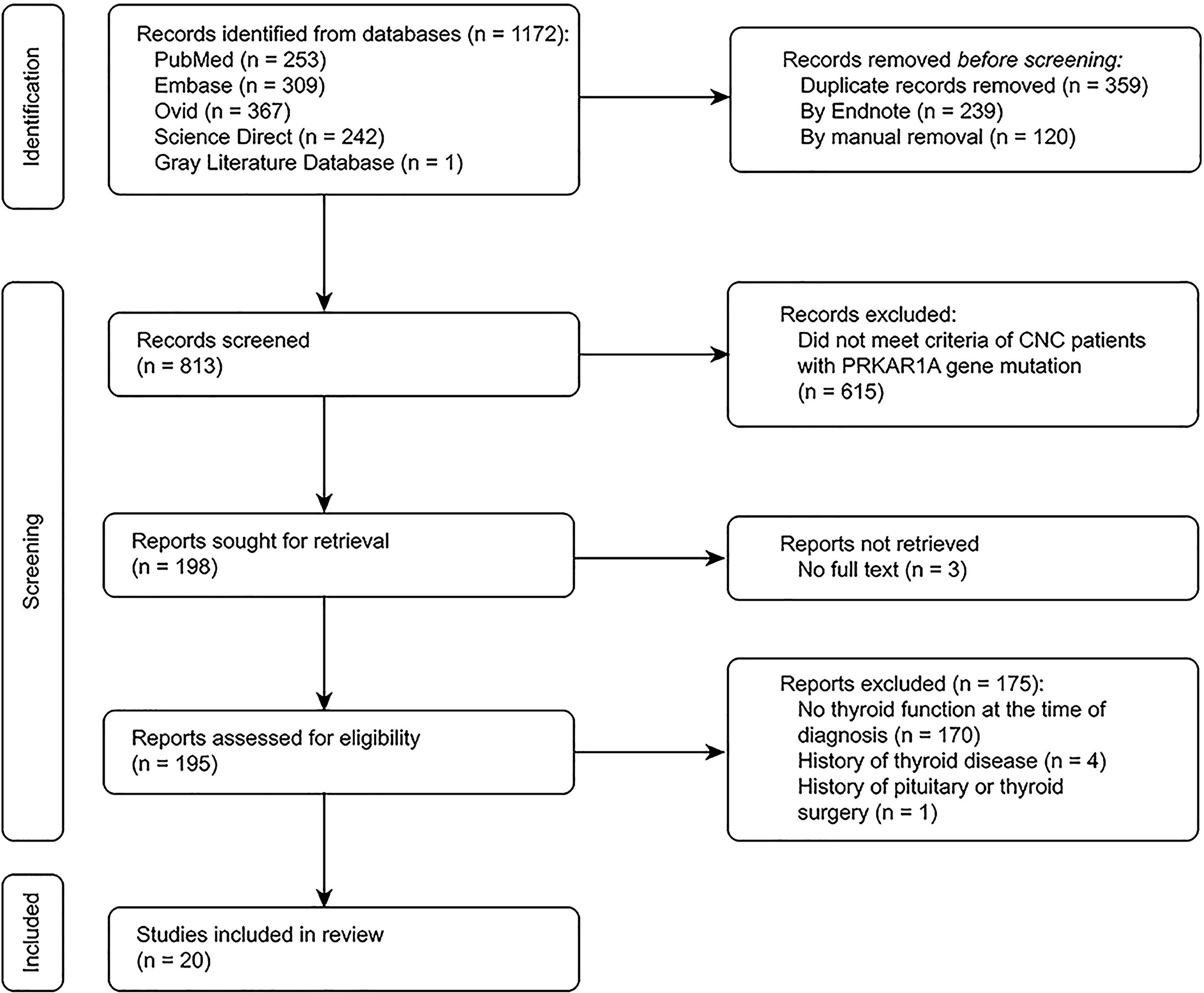
Figure 3 Flow chart for the systematic review.
Study characteristics
A total of 20 selected studies (14 case studies and 6 series studies) enrolling 23 individuals were included for final analysis (Table 2). The age of the patients ranged from infant age to 55 years. The ratio of male to female patients was 13:10. The rate of the merging PPNAD phenotype was 47.83% (11/23). At least 20 different PRKAR1A variants were described in the systematic review, covering six coding exons (exons 2-7), at least two intronic sequences (introns 2 and 7) and the initiator sequence (8–27).
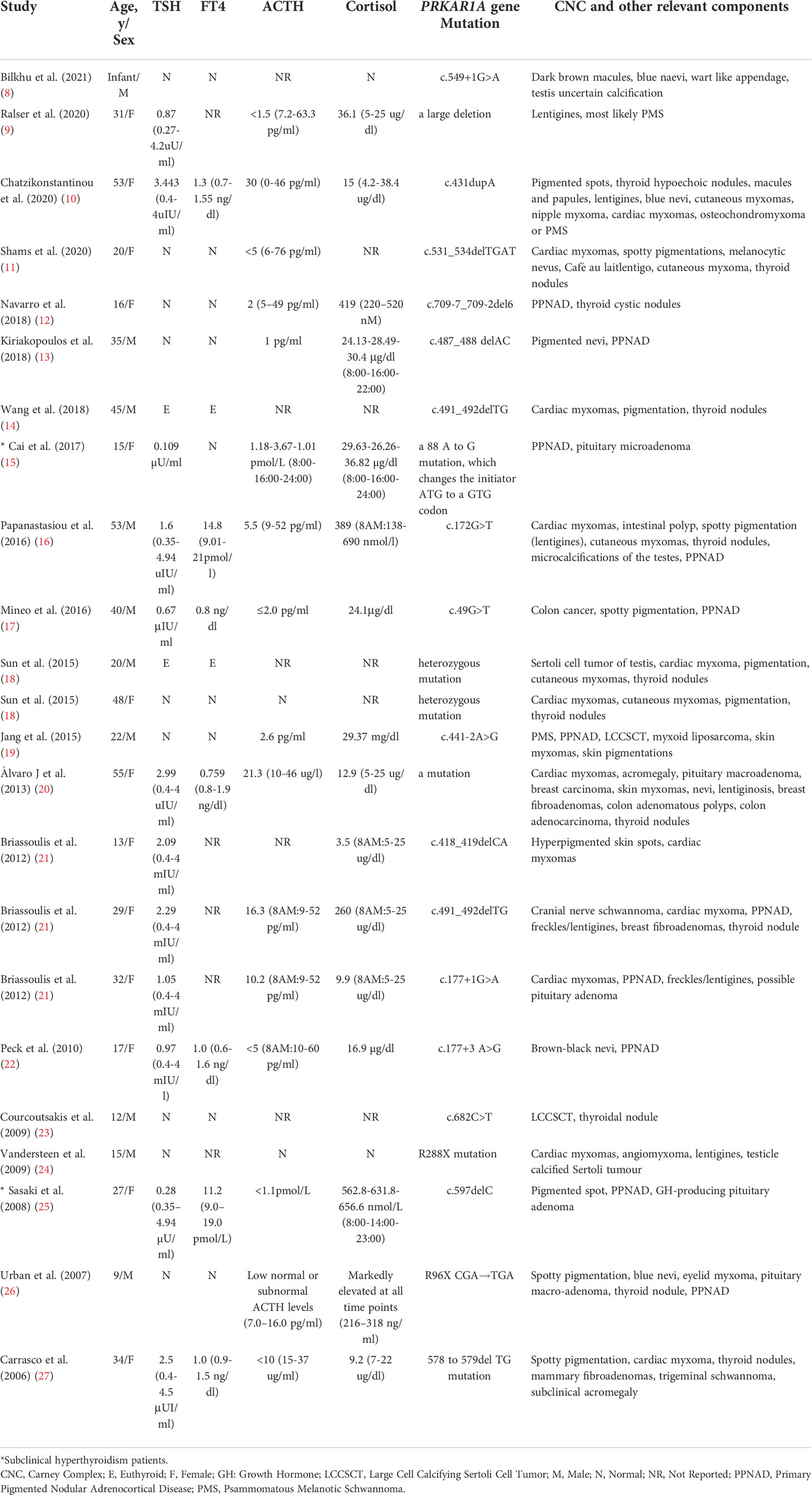
Table 2 Characteristics of the included studies.
Thyroid function
The patient’s thyroid function data were qualitative in 11 cases and quantitative in 12 cases. Among the patients, two (highlighted with * in Table 2) had subclinical hyperthyroidism (15, 25), and 21 had normal thyroid function (Table 2). With the addition of our patient, the prevalence of subclinical hyperthyroidism was 12.5% (3/24) in the CNC patients with thyroid function results and a confirmed PRKAR1A gene variant.
Discussion
We reviewed the thyroid hormone levels of CNC patients with thyroid function affected by PRKAR1A gene variants. The prevalence of subclinical hyperthyroidism in this populations is much higher than that in the general population, which is approximately 1% to 2% (28). This finding suggests that PRKAR1A gene variants may be related to subclinical hyperthyroidism, and this phenotype may be a common phenotype that is overlooked in CNC patients.
CNC is an autosomal dominant inherited multiple tumor syndrome with symptoms and signs including spotty skin pigmentation, heart and skin myxoma, endocrine overactivity, psammomatous melanotic schwannoma (PMS), etc. Spotty skin pigmentation is the most common clinical manifestation, appearing in approximately 77% of cases, and thyroid nodules or cancer occurs in 5% of cases (3). However, the thyroid function of these patients is rarely reported and remains controversial. We believe that PRKAR1A variant may be associated with abnormal thyroid function. First, subclinical hyperthyroidism was the first symptom in our patient associated with a PRKAR1A gene variant. Second, research has shown that mice with an ablated PRKAR1A gene and lower TSH levels have not only thyroid follicular carcinoma but also hyperthyroidism (6). Third, when J Aidan Carney summarized the spectrum of thyroid gland pathology in CNC patients, he also thought that thyrotoxicosis should be added to the clinical findings associated with the syndrome (5). However, he did not explore whether subclinical hyperthyroidism is related to PRKAR1A gene variants. These findings inspired us to explore the relationship between subclinical hyperthyroidism and the PRKAR1A gene. Because CNC patients are rare, a systematic review with strict inclusion and exclusion criteria was performed to gather all published studies worldwide to analyze the thyroid function of CNC patients affected by PRKAR1A gene variants. Considering that the confirmed diagnostic criteria for this syndrome were published in 2001, we excluded studies before 2001. To reduce bias and confounding factors, we excluded patients who had been diagnosed with thyroid disease and had affected thyroid function due to pituitary and thyroid surgery, as well as those with other CNC gene variants, such as PRKACA encoding PKA catalytic subunit α, PRKACB encoding PKA catalytic subunit β, PDE11A encoding phosphodiesterase expressed in the adrenal cortex and PDE8B encoding another phosphodiesterase (2), which may affect thyroid function.
Our systematic review is helpful for understanding the rare causes of hyperthyroidism. The main causes of hyperthyroidism are Graves’ disease, subacute thyroiditis, Hashimoto’s thyroiditis, excessive iodine intake, TSH tumor, thyroid hormone resistance, painless thyroiditis, thyroid follicular cancer metastasis invasion, etc. (29). The causes of subclinical hyperthyroidism are the same as the causes of overt hyperthyroidism (1). Even though we know that patients with CNC caused by PRKAR1A gene variants can have involvement of the thyroid gland, the rate of thyroid function testing in this population is low because it is not widely known that PRKAR1A gene variants may affect thyroid function. We describe a case of a Chinese man with a PRKAR1A gene variant and normal thyrotropin receptor antibody (TRAb), whose diagnosis of CNC was delayed, with a final diagnosis 2 months after the initial misdiagnosis and treatment for Graves’ disease. Because the patient had no typical symptoms of thyrotoxicosis and because of the lack of understanding that PRKAR1A gene variants may lead to subclinical hyperthyroidism, decreased TSH is easily considered by clinicians as method-specific interference and overlooked. This makes the diagnosis and treatment complicated, leading to the delayed diagnosis of CNC. The prevalence of subclinical hyperthyroidism in the specific population pooled in this systematic review was much higher than that in the general population, suggesting that PRKAR1A gene variants may be the cause of subclinical hyperthyroidism. Three patients in our systematic review all manifested decreased TSH but no thyroid nodules. Similar to Stergiopoulos et al., the biochemical indicators GH and IGF-1 were elevated, but no adenomas were found on imaging, which may be because hyperplasia most likely precedes the formation of GH-producing adenomas in CNC patients (30). However, we found that 11 patients in our systematic review with thyroid nodules did not manifest decreased TSH. The change in growth hormone axis status is not completely similar to subclinical hyperthyroidism in CNC patients, suggesting that their pathogenic mechanism needs further study.
The PKA holoenzyme is a heterotetramer composed of two regulatory subunits, each of which is bound to one catalytic subunit (31). Indeed, despite the existence of four regulatory subunits of PKA (PRKAR1A, PRKAR1B, PRKAR2A, and PRKAR2B), both cloning and gene knockout studies have demonstrated that PRKAR1A is critical for maintaining the PKA response to cAMP by regulating free catalytic subunits, especially in adrenocortical cells (32). More than 130 different variants of the PRKAR1A gene have been described to date in over 400 families of different ethnic origins with CNC (2). The CNC gene located at 17q22-24 was identified in 2000 as the tumor suppressor gene PRKAR1A encoding the regulatory subunit type 1α of protein kinase A (33, 34). When gene variants result in abnormal protein synthesis of regulatory subunit type 1α, they can lead to altered activity of protein kinase A, with consequently increased cell proliferation and tumorigenesis (35). The variant site in our case was in exon 4, and the variant sites in the other two subclinical hyperthyroidism patients were located in the promoter and exon 6. The variant sites and other components of CNC vary, suggesting that there may be no hot spot variant. How PRKAR1A gene variants cause subclinical hyperthyroidism remains unclear. Elevated levels of TSH stimulating PKA activity via the activation of adenylyl cyclase and the production of cAMP are associated with the development of thyroid cancer in humans (36). However, a mouse model of elevated TSH signaling in a genetic wild-type background did not develop thyroid cancer (37). Pringle, D. R. et al. observed that thyroid-specific ablation of PRKAR1A, exhibiting low levels of TSH, leads to hyperthyroidism and thyroid cancer and speculated that TSH may be suppressed by activating alternative pathways along with activated PKA that provide negative feedback on cell growth. This hypothesis may explain why mice with PKA activation develop FTC, while tumors driven by elevated TSH do not develop cancers (6). Whether the mechanism of PRKAR1A variant-induced low TSH levels in CNC is the same as the PRKAR1A tumorigenic mechanism needs further investigation.
Subclinical hyperthyroidism may be influenced by not only excessive glucocorticoid suppression but also PRKAR1A gene variants. It is well known that glucocorticoids at high levels, such as those in Cushing’s syndrome, induce the suppression of TSH secretion (38). Bilateral adrenalectomy used to be considered the treatment of choice for patients with overt Cushing’s syndrome (39). The suppression of TSH is dissolved along with decreases in the high cortisol level after adrenalectomy. Although there is no mention of PRKAR1A gene variants, ACTH-independent Cushing’s syndrome results in postoperative adrenal insufficiency with inappropriate secretion of TSH (high TSH) after complete adrenalectomy (40). Similar to this study (15), our patient after unilateral adrenalectomy showed decreases in high cortisol levels and increased ACTH, but TSH of our patient was still below the lower limit of normal reference values, suggesting that preoperative subclinical hyperthyroidism may be mainly caused by PRKAR1A gene variants, not by excessive glucocorticoid suppression. Unfortunately, no study has observed changes in TSH after bilateral adrenalectomy in subclinical hyperthyroidism CNC patients caused by PRKAR1A gene variants. This warrants confirmation that the suppression of TSH is mainly caused by variants of the PRKAR1A gene in a follow-up prospective observation.
Hyperthyroidism is a common endocrine dysfunction. The prevalence of hyperthyroidism is 0.8% in Europe and 1.3% in the USA general population (41). This study found that in CNC patients, PRKAR1A gene variants led to a significantly higher prevalence of subclinical hyperthyroidism than that in the general population. Even so, subclinical hyperthyroidism in this specific group may still be underestimated. 1. We found that many patients who may suffer from hyperthyroidism were excluded for different reasons, such as a history of pituitary or thyroid surgery and thyroid diseases. For example, Hernandez-Ramirez LC et al. (42) reported that a patient was diagnosed with central hypothyroidism and corticotropinoma before the subclinical hyperthyroidism diagnosis, and this patient was excluded due to a history of pituitary surgery. In another study (5), the diagnosis was thyrotoxicosis; however, Graves’ disease is mentioned in the following table, which makes us suspect that thyrotoxicosis was caused by Graves’ disease. Due to the lack of specific key thyroid function data, such as TSH and TRAb, to identify the cause of thyrotoxicosis, we excluded this patient after discussion. Piper, S. N. et al. (43) believed that transient hyperthyroidism may have occurred in a patient with CNC. We excluded this patient because the study was conducted before 2001. The exclusion of patients who may have had subclinical hyperthyroidism led to fewer thyroid function data available for the final analysis. 2. Autopsy research reports show that most CNC patients have pathological changes in the adrenal glands and other organs, indicating that many patients with abnormal findings that can only be diagnosed by endocrinological tests have been overlooked (25). Other low-incidence phenotypes of CNC may be temporarily asymptomatic (subclinical hyperthyroidism often has no typical hypermetabolic symptoms of overt hyperthyroidism) orinaccurate assessment of endocrine parameters (25), and many components can be tested only by endocrinology (such as thyroid function abnormalities that could be diagnosed through testing for thyroid function), causing these patients to be ignored. 3. Due to incomplete penetrance at the time of diagnosis and continued follow-up, subclinical hyperthyroidism may gradually manifest. For example, J Aidan Carney et al. (5) believed that thyroid disorder was a late-developing feature of CNC, and that abnormal thyroid function may appear after CNC diagnosis. Similar to this study (44), the accurate prevalence of subclinical hyperthyroidism in CNC patients with PRKAR1A in the real world should be investigated in a multicenter prospective long-term follow-up study.
Our study has some limitations that lead to a deviation from the real-world population prevalence of subclinical hyperthyroidism. 1. Due to the retrospective nature of the study, insufficient thyroid function data reduced the robustness of the results. 2. Even if thyroid function is checked, there are still qualitative data described by words such as “normal” and “euthyroid”. We did not contact the researchers to ask for patients’ concrete TSH and FT4 levels, so they could not be included in the meta-analysis, which weakened the performance of the results. 3. Studies for which the full text was not available were not included in the analysis, and there may be a bias that affects the performance of the results. Further prospective studies with a large number of patients are needed to confirm our findings.
In conclusion, the results of our systematic review showed that the prevalence of subclinical hyperthyroidism in CNC patients with PRKAR1A gene variants is higher than that in the normal population. To the best of our knowledge, this is the first discovery that PRKAR1A gene variants may be related to subclinical hyperthyroidism, and our results also suggest that subclinical hyperthyroidism may be a new component of CNC patients with a PRKAR1A gene variant; however, confirmatory prospective studies are needed.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
LD conceptualized the research question and study design. HW informed the search strategy. MM informed the analytic plan and drafted and edited the manuscript. LD and DL revised the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by grants from Chongqing medical scientific research project (Joint project of Chongqing Health Commission and Science and Technology Bureau) (2022GDRC016), Key Laboratory Incubation Project of the Third Affiliated Hospital of Chongqing Medical University (KY19025), and High-level Medical Reserved Personnel Training Project of Chongqing (CQSZQNYXGDRC201829).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.951133/full#supplementary-material
References
1. Biondi B, Cooper DS. Subclinical hyperthyroidism. N Engl J Med (2018) 378(25):2411–9. doi: 10.1056/NEJMcp1709318
2. Bouys L, Bertherat J. MANAGEMENT OF ENDOCRINE DISEASE: Carney complex: Clinical and genetic update 20 years after the identification of the CNC1 (PRKAR1A) gene. Eur J Endocrinol (2021) 184(3):R99–R109. doi: 10.1530/EJE-20-1120
3. Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab (2001) 86(9):4041–6. doi: 10.1210/jcem.86.9.7903
4. Stratakis CA, Courcoutsakis NA, Abati A, Filie A, Doppman JL, Carney JA, et al. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J Clin Endocrinol Metab (1997) 82(7):2037–43. doi: 10.1210/jcem.82.7.4079
5. Carney JA, Lyssikatos C, Seethala RR, Lakatos P, Perez-Atayde A, Lahner H, et al. The spectrum of thyroid gland pathology in Carney complex: The importance of follicular carcinoma. Am J Surg Pathol (2018) 42(5):587–94. doi: 10.1097/PAS.0000000000000975
6. Pringle DR, Yin Z, Lee AA, Manchanda PK, Yu L, Parlow AF, et al. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr Relat Cancer (2012) 19(3):435–46. doi: 10.1530/ERC-11-0306
7. Moola S, Munn Z, Sears K, Sfetcu R, Currie M, Lisy K, et al. Conducting systematic reviews of association (etiology): The Joanna Briggs institute’s approach. Int J Evid Based Healthc (2015) 13(3):163–9. doi: 10.1097/XEB.0000000000000064
8. Bilkhu AS, Sunderesan R. Newborn infant with congenital lentigines as a manifestation of Carney complex. BMJ Case Rep (2021) 14(1):e239259. doi: 10.1136/bcr-2020-239259
9. Ralser DJ, Strizek B, Kupczyk P, Stoffel-Wagner B, Altengarten J, Müller A, et al. Obstetric and neonatal outcome of pregnancy in Carney complex: A case report. Front Endocrinol (Lausanne) (2020) 11:296. doi: 10.3389/fendo.2020.00296
10. Chatzikonstantinou S, Kazis D, Giannakopoulou P, Poulios P, Pikou O, Geroukis T, et al. Carney Complex syndrome manifesting as cardioembolic stroke: a case report and review of the literature. Int J Neurosci (2022) 132(7):649–55. doi: 10.1080/00207454.2020.1834393
11. Shams S, Kyavar M, Sadeghipour P, Khesali H, Mozaffari K, Mahdieh N, et al. Carney Complex syndrome. Cardiovasc Pathol (2020) 49:107231. doi: 10.1016/j.carpath.2020.107231
12. Navarro Moreno C, Delestienne A, Marbaix E, Aydin S, Hörtnagel K, Lechner S, et al. Familial forms of cushing syndrome in primary pigmented nodular adrenocortical disease presenting with short stature and insidious symptoms: A clinical series. Horm Res Paediatr (2018) 89(6):423–33. doi: 10.1159/000488761
13. Kiriakopoulos A, Linos D. Carney Syndrome presented as a pathological spine fracture in a 35-Year-Old Male. Am J Case Rep (2018) 19:1366–9. doi: 10.12659/AJCR.911962
14. Wang L, Wang Q, Zhou Y, Xue Q, Sun X, Wang Z, et al. Recurrent left atrial myxoma in Carney complex: A case report of a familial pedigree. Med (Baltimore) (2018) 97(12):e0247. doi: 10.1097/MD.0000000000010247
15. Cai XL, Wu J, Luo YY, Chen L, Han XY, Ji LN, et al. A novel mutation of PRKAR1A caused carney complex in a Chinese patient. Chin (Engl) (2017) 130(24):3009–10. doi: 10.4103/0366-6999.220309
16. Papanastasiou L, Fountoulakis S, Voulgaris N, Kounadi T, Choreftaki T, Kostopoulou A, et al. Identification of a novel mutation of the PRKAR1A gene in a patient with carney complex with significant osteoporosis and recurrent fractures. Hormones (Athens) (2016) 15(1):129–35. doi: 10.14310/horm.2002.1627
17. Mineo R, Tamba S, Yamada Y, Okita T, Kawachi Y, Mori R, et al. A novel mutation in the type iα regulatory subunit of protein kinase a (PRKAR1A) in a cushing’s syndrome patient with primary pigmented nodular adrenocortical disease. Intern Med (2016) 55(17):2433–8. doi: 10.2169/internalmedicine.55.6605
18. Sun Y, Chen X, Sun J, Wen X, Liu X, Zhang Y, et al. A novel inherited mutation in PRKAR1A abrogates PreRNA splicing in a Carney complex family. Can J Cardiol (2015) 31(11):1393–401. doi: 10.1016/j.cjca.2015.05.018
19. Jang YS, Moon SD, Kim JH, Lee IS, Lee JM, Kim HS. A novel PRKAR1A mutation resulting in a splicing variant in a case of Carney complex. Korean J Intern Med (2015) 30(5):730–4. doi: 10.3904/kjim.2015.30.5.730
20. Álvaro JR, Martínez de Esteban JP, Pineda Arribas JJ, Ollero García-Agulló MD, Munárriz Alcuaz P. Acromegaly in a patient with carney’s complex. Endocrinol Nutr (2013) 60(5):277–8. doi: 10.1016/j.endonu.2012.06.003
21. Briassoulis G, Kuburovic V, Xekouki P, Patronas N, Keil MF, Lyssikatos C, et al. Recurrent left atrial myxomas in Carney complex: A genetic cause of multiple strokes that can be prevented. J Stroke Cerebrovasc Dis (2012) 21(8):914.e1–8. doi: 10.1016/j.jstrokecerebrovasdis.2012.01.006
22. Peck MC, Visser BC, Norton JA, Pasche L, Katznelson L. A novel PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease and the Carney complex. Endocr Pract (2010) 16(2):198–204. doi: 10.4158/EP09245.OR
23. Courcoutsakis N, Patronas N, Filie AC, Carney JA, Moraitis A, Stratakis CA. Ectopic thymus presenting as a thyroid nodule in a patient with the Carney complex. Thyroid (2009) 19(3):293–6. doi: 10.1089/thy.2008.0404
24. Vandersteen A, Turnbull J, Jan W, Simpson J, Lucas S, Anderson D, et al. Cutaneous signs are important in the diagnosis of the rare neoplasia syndrome Carney complex. Eur J Pediatr (2009) 168(11):1401–4. doi: 10.1007/s00431-009-0935-y
25. Sasaki A, Horikawa Y, Suwa T, Enya M, Kawachi S-i, Takeda J. Case report of familial Carney complex due to novel frameshift mutation c.597del c (p.Phe200LeufsX6) in PRKAR1A. Mol Genet Metab (2008) 95(3):182–7. doi: 10.1016/j.ymgme.2008.07.009
26. Urban C, Weinhäusel A, Fritsch P, Sovinz P, Weinhandl G, Lackner H, et al. Primary pigmented nodular adrenocortical disease (PPNAD) and pituitary adenoma in a boy with sporadic carney complex due to a novel, de novo paternal PRKAR1A mutation (R96X). J Pediatr Endocrinol Metab (2007) 20(2):247–52. doi: 10.1515/jpem.2007.20.2.247
27. Carrasco CA, Rojas-Salazar D, Chiorino R, Venega JC, Wohllk N. Melanotic nonpsammomatous trigeminal schwannoma as the first manifestation of Carney complex: Case report. Neurosurgery (2006) 59(6):E1334–5. doi: 10.1227/01.NEU.0000245608.07570.D2
28. Donangelo I, Suh SY. Subclinical hyperthyroidism: When to consider treatment. Am Fam Physician. (2017) 95(11):710–6.
29. Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, et al. American Thyroid association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid (2016) 26(10):1343–421. doi: 10.1089/thy.2016.0229
30. Stergiopoulos SG, Abu-Asab MS, Tsokos M, Stratakis CA. Pituitary pathology in Carney complex patients. Pituitary (2004) 7(2):73–82. doi: 10.1007/s11102-005-5348-y
31. Bossis I, Stratakis CA. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology (2004) 145(12):5452–8. doi: 10.1210/en.2004-0900
32. Amieux PS, McKnight GS. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann N Y Acad Sci (2002) 968:75–95. doi: 10.1111/j.1749-6632.2002.tb04328.x
33. Casey M, Vaughan CJ, He J, Hatcher CJ, Winter JM, Weremowicz S, et al. Mutations in the protein kinase a R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J Clin Invest (2000) 106(5):R31–8. doi: 10.1172/JCI10841
34. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase a type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet (2000) 26(1):89–92. doi: 10.1038/79238
35. Wilkes D, McDermott DA, Basson CT. Clinical phenotypes and molecular genetic mechanisms of Carney complex. Lancet Oncol (2005) 6(7):501–8. doi: 10.1016/S1470-2045(05)70244-8
36. Pitsava G, Stratakis CA, Faucz FR. PRKAR1A and thyroid tumors. Cancers (Basel) (2021) 13(15):3834. doi: 10.3390/cancers13153834
37. Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res (2007) 67(17):8002–6. doi: 10.1158/0008-5472.CAN-07-2471
38. Bartalena L, Martino E, Petrini L, Velluzzi F, Loviselli A, Grasso L, et al. The nocturnal serum thyrotropin surge is abolished in patients with adrenocorticotropin (ACTH)-dependent or ACTH-independent cushing’s syndrome. J Clin Endocrinol Metab (1991) 72(6):1195–9. doi: 10.1210/jcem-72-6-1195
39. Chevalier B, Vantyghem MC, Espiard S. Bilateral adrenal hyperplasia: Pathogenesis and treatment. Biomedicines (2021) 9(10):1397. doi: 10.3390/biomedicines9101397
40. Tamada D, Onodera T, Kitamura T, Yamamoto Y, Hayashi Y, Murata Y, et al. Hyperthyroidism due to thyroid-stimulating hormone secretion after surgery for cushing’s syndrome: A novel cause of the syndrome of inappropriate secretion of thyroid-stimulating hormone. J Clin Endocrinol Metab (2013) 98(7):2656–62. doi: 10.1210/jc.2013-2135
41. De Leo S, Lee SY, Braverman LE. Hyperthyroidism. Lancet (2016) 388:906–18. doi: 10.1016/S0140-6736(16)00278-6
42. Hernandez-Ramirez LC, Tatsi C, Lodish MB, Faucz FR, Pankratz N, Chittiboina P, et al. Corticotropinoma as a component of Carney complex. J Endocr Soc (2017) 1(7):918–25. doi: 10.1210/js.2017-00231
43. Piper SN, Maleck WH, Triem JG, Isgro F, Kaufmann V, Saggau W. Myxoma complex associated with transient hyperthyroidism: are diseases of the thyroid part of the complex? Thorac Cardiovasc Surg (1997) 45(5):245–7. doi: 10.1055/s-2007-1013736
Keywords: hyperthyroidism, PRKAR1A gene, Carney complex, case report, systematic review
Citation: Wang H, Mao M, Liu D and Duan L (2022) Association between subclinical hyperthyroidism and a PRKAR1A gene variant in Carney complex patients: A case report and systematic review. Front. Endocrinol. 13:951133. doi: 10.3389/fendo.2022.951133
Received: 23 May 2022; Accepted: 07 September 2022;
Published: 23 September 2022.
Edited by:
Terry Francis Davies, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Paraskevi Xekouki, University of Crete, GreeceGeorgia Pitsava, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NIH), United States
Copyright © 2022 Wang, Mao, Liu and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lian Duan, amFzb25kdWFuQGhvc3BpdGFsLmNxbXUuZWR1LmNu
†These authors have contributed equally to this work