 Liam D. Hurley
Liam D. Hurley Hugo Lee
Hugo Lee Gina Wade
Gina Wade Judith Simcox
Judith Simcox Feyza Engin
Feyza Engin- 1Department of Biomolecular Chemistry, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 2Department of Biochemistry, College of Agricultural and Life Sciences, University of Wisconsin-Madison, Madison, WI, United States
- 3Division of Endocrinology, Diabetes & Metabolism, Department of Medicine, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 4Department of Cell and Regenerative Biology, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
Chronic elevation of sphingolipids contributes to β-cell failure. ORMDL3 has been identified as a key regulator of sphingolipid homeostasis, however, its function in pancreatic β-cell pathophysiology remains unclear. Here, we generated a mouse model lacking Ormdl3 within pancreatic β-cells (Ormdl3β-/-). We show that loss of β-cell Ormdl3 does not alter glucose tolerance, insulin sensitivity, insulin secretion, islet morphology, or cellular ceramide levels on standard chow diet. When challenged with a high fat diet, while Ormdl3β-/- mice did not exhibit any alteration in metabolic parameters or islet architecture, lipidomics analysis revealed significantly higher levels of very long chain ceramides in their islets. Taken together, our results reveal that loss of Ormdl3 alone is not sufficient to impinge upon β-cell function or whole-body glucose and insulin homeostasis, however, β-cell-specific loss of Ormdl3 does significantly alter levels of specific sphingolipid species in islets upon high fat feeding.
1 Introduction
In obesity, chronically elevated levels of circulating free fatty acids contribute to the de novo production of cellular lipids, including sphingolipids. Left unchecked, elevated sphingolipid production can lead to the accumulation of sphingolipid species such as ceramide within the cell (1, 2). In pancreatic β-cells, obesity-directed sphingolipid accumulation contributes to β-cell dysfunction through induction of apoptotic, inflammatory, and cellular stress (1, 3, 4).
Orosomucoid-like proteins (ORMDLs) are an ER-resident protein family that inhibits serine palmitoyltransferase (SPT), the rate limiting enzyme in de novo sphingolipid biosynthesis (5–7). ORMDL3 is one of three isoforms of this protein family, and genome-wide association studies (GWAS) have identified Ormdl3 as an obesity-related gene whose expression negatively correlates with body mass index (8). While this suggests that ORMDL3 plays a role in obesity, the role of ORMDL3 in β-cell physiology and pathology remains unknown.
In the current study, we designed a β-cell-specific Ormdl3 knockout mouse and investigated the consequences of its loss of function under non-stressed (chow or low fat diet) and high fat diet (HFD) challenged conditions. Our results showed that Ormdl3 is dispensable for β-cell function regardless of diet. Indeed, β-cell loss of Ormdl3 did not alter fasting blood glucose, body weight, islet morphology, glucose tolerance, insulin sensitivity, or insulin secretion. Lipidomics of isolated islets identified significantly elevated levels of very long chain ceramides in HFD challenged knockout animals. Taken together, our results suggest that Ormdl3 is not required for β-cell function and survival under physiological or surplus nutrition conditions.
2 Results
2.1 Chow-fed Ormdl3β-/- mice do not exhibit altered β-cell function or glucose homeostasis
To examine the function of ORMDL3 in β-cells, we generated β-cell specific Ormdl3 knockout mice (KO; Ormdl3β-/-) by mating mice harboring floxed (exons 2-4) Ormdl3 (Ormdlfl/fl) with mice expressing Cre recombinase under the control of the Ins1 promotor (Ins1tm1.1 Cre) (9). To determine the efficiency of deletion, we performed qPCR analysis in pancreatic islets isolated from 7-week-old Ormdl3β-/- mice and found approximately 80% reduction in Ormdl3 mRNA levels (Figure 1A). We also investigated the expression of Ormdl1 and Ormdl2 in these islets and found no significant differences in gene expression level relative to wild type (Wt) mice, suggesting that the deletion of Ormdl3 in β-cells did not trigger compensation by either Ormd1 or Ormdl2 at the mRNA level (Figure 1A).
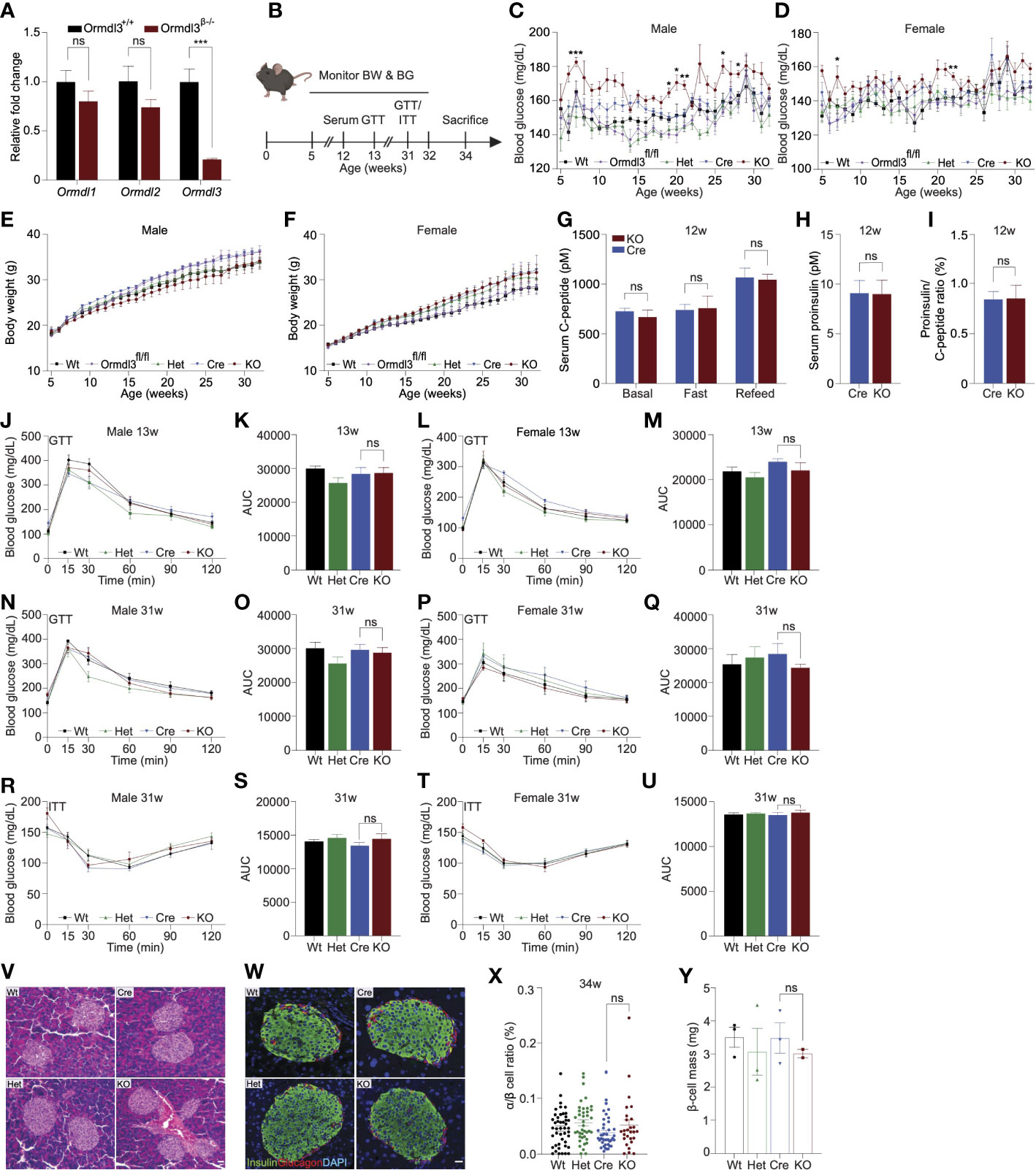
Figure 1 Chow-fed Ormdl3β-/- mice do not exhibit altered β-cell function or glucose homeostasis. (A) Fold change gene expression of Ormdl isoforms in isolated islets of 7-week-old wild-type (Wt) and knockout (KO; Ormdl3β-/-) mice (n=3/group; Student’s t-test). (B) Schematic representation of mouse monitoring. (C) Weekly blood glucose measurements in Wt, Ormdl3fl/fl, Cre, Heterozygous (Het; Ormdlβ+/-), and KO male (n=5-8/group) and (D) female (n=7-8/group) mice (Two-way ANOVA with Tukey’s post-hoc test; stars indicate significant differences between Cre and KO groups). (E) Weekly body weight measurements in Wt, Ormdl3fl/fl, Cre, Het, and KO male (n=5-8/group) and (F) female (n=7-8/group) mice (Two-way ANOVA with Tukey’s post-hoc test). (G-I) ELISAs on serum samples from 12-week-old Cre and KO mice (Student’s t-test). (G) ELISA for C-peptide before/after a 6 hour fast and after 1 hour refeeding (n=5-6/group). (H) ELISA for proinsulin after 1 hour refeeding (n=5-6/group). (I) Ratio of proinsulin/C-peptide after 1 hour refeeding (n=5-6/group). (J-Q) Glucose tolerance and area under the curve (AUC) calculations for male and female Wt, Het, CO, and KO mice. (J) Glucose tolerance and (K) total area under the curve (AUC) of 13-week-old male mice (n=5-6/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (L) Glucose tolerance and (M) total AUC of 13-week-old female mice (n=5-6/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (N) Glucose tolerance and (O) total AUC of 31-week-old male mice (n=5-6/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (P) Glucose tolerance and (Q) total AUC of 31-week-old female mice (n=4/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (R-U) Insulin tolerance and AUC calculations for 31-week-old male and female Wt, Het, Cre, and KO mice (R) Insulin tolerance and (S) total AUC of male mice (n=5-6/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (T) Insulin tolerance and (U) total AUC of female (n=4/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (V) Representative images of hematoxylin and eosin staining of 34-week-old male Wt, Cre, Het, and KO mice. (W) Co-staining for insulin and glucagon in pancreatic sections from 34-week-old male Wt, Cre, Het, and KO mice. (X) Ratio of α:β cells in co-stained insulin and glucagon sections (n=3/group; One-way ANOVA with Tukey’s post-hoc test). (Y) β-cell mass in insulin-stained sections (n=2-3/group; One-way ANOVA with Tukey’s post-hoc test). Data are presented as means ± SEM. One- and two-way ANOVA with Tukey’s post-hoc test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001) and Student’s t-test (***p<.001) are applied where indicated. BW, body weight; BG, blood glucose; GTT, glucose tolerance test; ITT, insulin tolerance test; Wt, wild type; Ormdl3fl/fl, Ormdl3 floxed mouse not expressing cre recombinase; Het, Ormdl3β+/- heterozygous knockout; Cre, mouse expressing Ins1tm1.1(cre)Thor/J with wild type Ormdl3 alleles; KO, Ormdl3β-/- homozygous knockout; AUC, area under the curve; DAPI, 4′,6-diamidino-2-phenylindole; w, weeks; ns, not significant; ANOVA, analysis of variance. Scale bars are 20 µm.
Next, we monitored chow-fed male and female Wt, Ormdl3fl/fl, and Cre mice (as control conditions) as well as Ormdlβ+/- (β-cell Ormdl3 heterozygous knockout; Het), and KO mice starting from 5 weeks of age for 27 weeks (Figure 1B). During this 27-week period, we measured fasting blood glucose and body weights weekly. We observed a trend towards elevation of blood glucose levels only in male KO mice when compared to Cre control mice (Figures 1C, D) while body weight remained comparable between knockout and Cre control mice (Figures 1E, F).
To determine if Ormdl3 ablation would result in impairment of β-cell insulin secretion, we performed fast-refeed experiments in male mice at 12 weeks of age and analyzed serum C-peptide and proinsulin levels (Figure 1G). Knockout mice did not exhibit altered levels of C-peptide in the ad libitum basal condition, after six hours of fasting, or during the refeed condition. Proinsulin levels were also unchanged during the refeed condition (Figure 1H). The ratio of proinsulin to C-peptide during the refeed condition (Figure 1I) was unaffected. Taken together, these data suggest that loss of Ormdl3 in β-cells does not significantly alter insulin secretion.
Next, we examined if the loss of β-cell Ormdl3 would result in aberrant glucose homeostasis. We performed glucose and insulin tolerance tests in both male and female mice. Our results showed that there were no significant alterations in glucose tolerance in male and female mice at 13 (Figures 1J-M) or 31 weeks of age (Figures 1N-Q). We also measured insulin tolerance in 31-week-old male (Figures 1R, S) and female mice (Figures 1T, U) and found no change in insulin sensitivity between KO mice and controls (Wt, Het, and Cre). These results suggest that Ormdl3 deletion in β-cells does not impair whole-body insulin sensitivity or glucose metabolism.
To determine the impact of loss of Ormdl3 on islet morphology and composition, we stained pancreatic sections from male 34-week-old KO, het, and control (Wt and Cre) mice either with hematoxylin and eosin (H&E) or co-stained sections with insulin and glucagon. Our analysis did not reveal any changes in gross morphology with H&E staining (Figure 1V), alterations in islet architecture, or α:β cell ratio in Ormdl3-deficient mice (Figures 1W, X). Lastly, we examined β-cell mass in insulin-stained sections and did not find a significant change in KO mice as compared to control (Figure 1Y). Collectively, our data suggest that under chow-fed conditions, Ormdl3 deletion is dispensable for β-cell function and glucose homeostasis.
2.2 High fat diet feeding in Ormdl3β-/- mice does not impact β-cell function or glucose homeostasis
Our initial results revealed that deletion of Ormdl3 in the β-cells of mice fed chow diet did not contribute to β-cell dysfunction or impair glucose homeostasis. The ORMDL proteins are known to inhibit serine palmitoyltransferase, the enzyme responsible for the condensation of palmitoyl-CoA and serine into 3-ketosphinganine (10). Since this condensation reaction requires palmitic acid-derived palmitoyl-CoA, we reasoned that low levels of palmitic acid present in chow diet may not lead to sufficient accumulation of sphingolipids underlying β-cell dysfunction. Therefore, we decided to nutritionally challenge these mice with HFD, a model of diet induced obesity. To this end, we placed male mice on HFD or control low fat diet (LFD) starting at 6 weeks of age for 24 weeks (Figure 2A) and measured fasting blood glucose levels and body weights weekly. We did not observe any significant changes in fasting blood glucose levels in the LFD-fed Wt, Het, Cre, or KO animals (Figure 2B). When challenged with a HFD, there were also no changes observed in fasting blood glucose levels (Figure 2C). Additionally, besides the expected weight gain resulting from high fat diet, there was no significant difference in body weights (Figure 2D) or food consumption (Figures 2E, F) between Wt, Het, Cre, or KO mice on either diet.
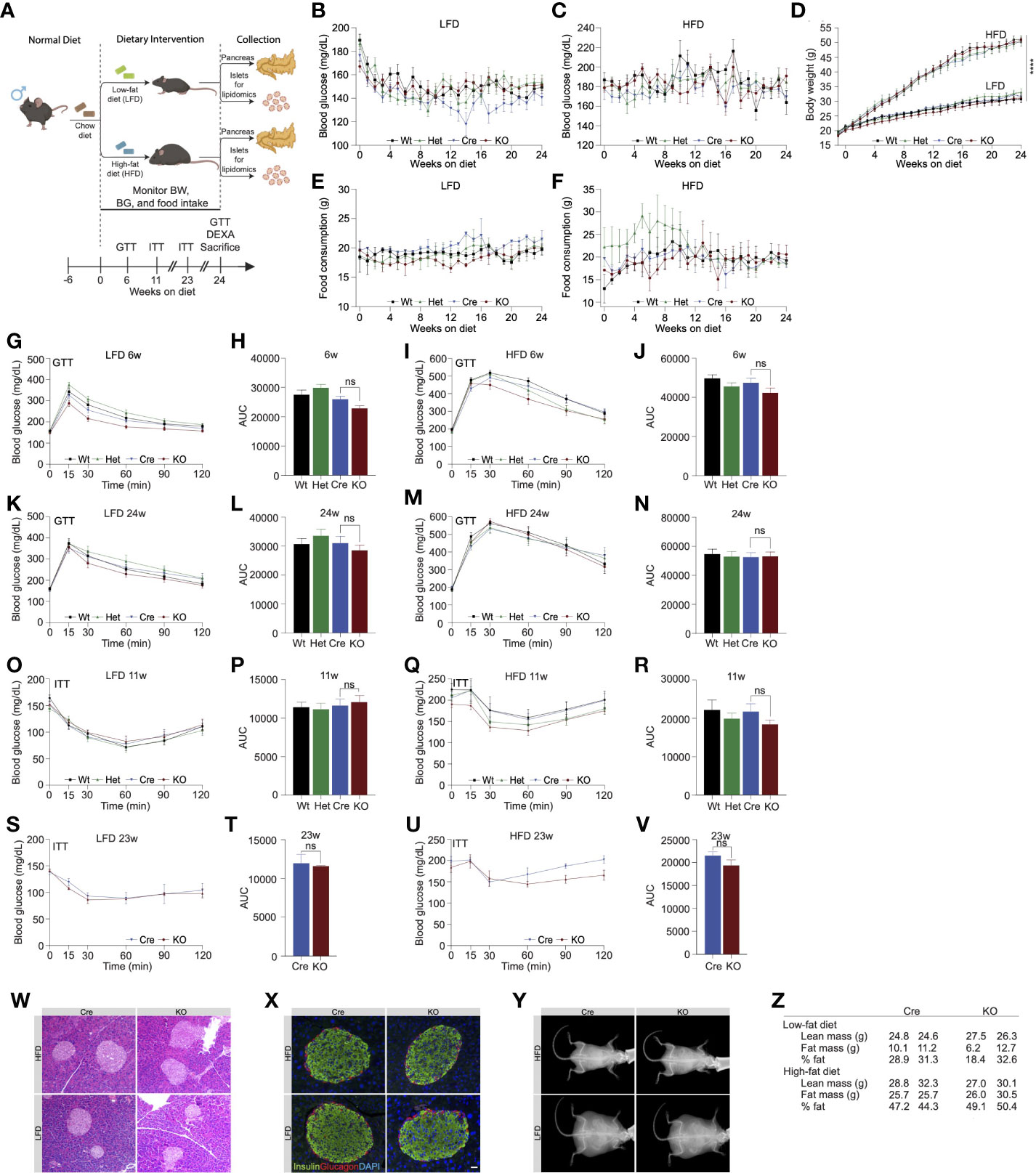
Figure 2 High fat diet feeding in Ormdl3β-/- mice does not impact β-cell function or glucose homeostasis. (A) Schematic representation of mouse monitoring and study design. (B) Weekly fasting blood glucose assessment of Wt, Het, Cre, and KO mice under LFD and (C) HFD conditions (n=9/group; two-way ANOVA with Tukey’s post-hoc test). (D) Weekly body weight measurements in LFD and HFD-fed Wt, Het, Cre, and KO mice (n=9/group; Two-way ANOVA with Tukey’s post-hoc test). (E) Weekly food consumption in Wt, Het, Cre, and KO LFD and (F) HFD-fed mice (grams of food eaten/mouse/week; Two-way ANOVA with Tukey’s post-hoc test). (G-N) Glucose tolerance and area under the curve (AUC) calculations for LFD and HFD-fed male Wt, Het, Cre, and KO mice. (G) Glucose tolerance and (H) total AUC of 6-weeks-on-diet LFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (I) Glucose tolerance and (J) total AUC of 6-weeks-on-diet HFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (K) Glucose tolerance and (L) total AUC of 24-weeks-on-diet LFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (M) Glucose tolerance and (N) total AUC of 24-weeks-on-diet HFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (O-V) Insulin tolerance and AUC calculations for LFD and HFD-fed male Wt, Het, Cre, and KO mice. (O) Insulin tolerance and (P) total AUC of 11-weeks-on-diet LFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (Q) Insulin tolerance and (R) total AUC of 11-weeks-on-diet HFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (S) Insulin tolerance and (T) total AUC of 23-weeks-on-diet LFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (U) Insulin tolerance and (V) total AUC of 23-weeks-on-diet HFD-fed mice (n=9/group; Two- and one-way ANOVA with Tukey’s post-hoc test, respectively). (W) Representative images of H&E staining of 24-weeks-on-diet male LFD and HFD-fed Cre and KO mice. (X) Representative images from co-staining for insulin and glucagon in pancreatic sections from 24-weeks-on-diet LFD and HFD-fed male Cre and KO mice. (Y) Representative images of DEXA assessment in 24-weeks-on-diet LFD and HFD-fed male Cre and KO mice. (Z) Table of DEXA assessment of lean mass and fat mass (n=2/group). Data are presented as means ± SEM. One- and two-way ANOVA with Tukey’s post-hoc test (****p<0.0001) are applied where indicated. BW, body weight; BG, blood glucose; GTT, glucose tolerance test; ITT, insulin tolerance test; DEXA, dual-energy x-ray absorptiometry; Wt, wild type; Het, Ormdl3β+/- heterozygous knockout; Cre, mouse expressing Ins1tm1.1(cre)Thor/J with wild type Ormdl3 alleles; KO, Ormdl3β-/- homozygous knockout; LFD, low fat diet; HFD, high fat diet; AUC, area under the curve; DAPI, 4′,6-diamidino-2-phenylindole; w, weeks; ns, not significant; ANOVA, analysis of variance. Scale bars are 20 µm.
We next examined glucose and insulin tolerance throughout the course of the study. Our results indicated that KO mice did not display any significant difference in glucose tolerance compared to Cre mice following 6 (Figures 2G-J) or 24 weeks of LFD or HFD feeding (Figures 2K-N). In addition, we did not observe any significant effects of Ormdl3 deletion on insulin tolerance following 11 (Figures 2O-R) and 23 weeks of LFD or HFD feeding (Figures 2S-V).
To investigate islet morphology, we performed H&E staining of pancreatic sections on LFD and HFD-fed control (Cre) and knockout mice that had been on diet for 24 weeks (Figure 2W). Further, insulin and glucagon immunofluorescence co-staining showed no significant alterations in islet morphology or composition between HFD- or LFD-fed Cre and KO mice (Figure 2X).
Next, we asked whether loss of Ormdl3 would result in changes to lean and fat mass using Dual-energy X-ray absorptiometry (DEXA) assessment of Cre and KO mice. Following 24 weeks of LFD or HFD feeding, there were no significant changes in lean or fat mass in mice on either diet (Figures 2Y, Z). Taken together, our results suggest that Ormdl3 deletion does not impinge upon β-cell function and is dispensable for glucose and insulin homeostasis even under obesogenic conditions.
2.3 Lipidomics analysis reveals significant upregulation of very long chain ceramide species in islets of high fat diet-fed Ormdl3β-/- mice
Given the inhibitory role of ORMDL proteins in SPT-mediated sphingolipid catabolism (Figure 3A), we hypothesized that the levels of sphingolipid species downstream of SPT could be altered. To test this, we performed targeted lipidomic analysis (Figure 3A; stars indicate species measured) in isolated islets of chow-fed, LFD-fed, and HFD-fed Cre and Ormdl3β-/- mice. We first examined fold change between 10-week-old chow-fed Ormdl3β-/- and Cre mice, but we did not observe any significantly upregulated or downregulated ceramides species between groups (Figure 3B). We next analyzed the levels of ceramides from LFD-fed or HFD-challenged Ormdl3β-/- mice after 24 weeks of feeding. Initial fold change comparison between LFD-fed Ormdl3β-/- and Cre mice revealed upregulation and downregulation of d18:1/24:1 and d18:2/24:0 ceramides, respectively (Figure 3C). However, fold change comparison between HFD counterparts revealed striking upregulation of islet long chain ceramide species d18:1/16:0 as well as very long chain ceramide species d18:1/24:1, d18:1/24:0, d18:1/22:0, d18:0/24:0, m18:1/22:0, and d18:1/26:1 as compared to Cre control mice (Figure 3D). There were no significant differences between total very long chain ceramide species (C22-26) in chow-fed (Figure 3E) and LFD-fed (Figure 3F) KO mice as compared to control. Interestingly, levels of total very long chain ceramides species were increased in HFD-fed KO mice compared to Cre mice (Figure 3G). Furthermore, analysis of individual sphingolipid species confirms these changes are indeed observed between HFD-fed Ormdl3β-/- mice as compared to Cre control mice (Figures 3H-N). Taken together, our results suggest that while loss of β-cell Ormdl3 alone does not impinge upon systemic metabolism or β-cell function in nutritionally unstressed conditions, deletion of Ormdl3 results in a significant increase in very long chain ceramide species in obesity.
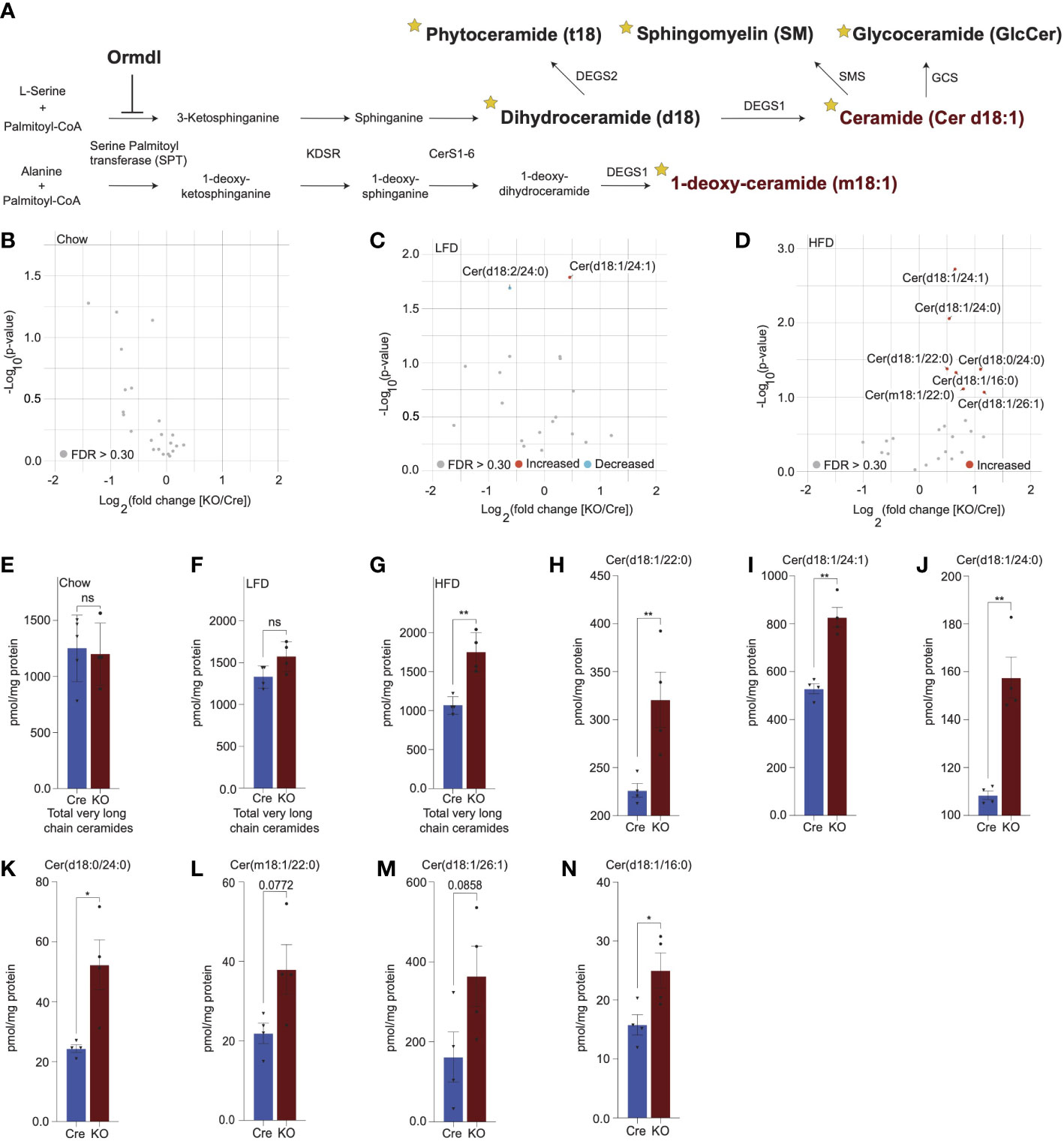
Figure 3 Lipidomics analysis reveals significant upregulation of very long chain ceramide species in islets of high fat diet-fed Ormdl3β-/- mice. (A) Schematic illustration of de novo sphingolipid biosynthetic pathway (stars denote lipid classes measured). (B) Volcano plot of fold change in lipid species between 10-week-old chow-fed Cre and KO mice. (C) Volcano plot of fold change in lipid species between LFD-fed Cre and KO mice after 24-weeks-on-diet (30 weeks of age). (D) Volcano plot of fold change in lipid species between HFD-fed Cre and KO mice after 24-weeks-on-diet (30 weeks of age). (E-G) pmol total very long chain ceramide species (C22-26) normalized to total protein content (mg) in (Welch’s t-test) (E) 10-week-old chow-fed Cre (n=5) and KO (n=4) mice. (F) LFD-fed Cre (n=4) and KO (n=4) after 24-weeks-on-diet (30 weeks of age). (G) HFD-fed Cre (n=4) and KO (n=4) mice after 24-weeks-on-diet (30 weeks of age). (H-N) pmol ceramide species for HFD-fed mice normalized to total protein content (mg) for (Welch’s t-test) (H) Cer(d18:1/22:0), (I) Cer(d18:1/24:1), (J) Cer(d18:1/24:0), (K) Cer(d18:0/24:0), (L) Cer(m18:1/22:0), (M) Cer(d18:1/26:1), and (N) Cer(d18:1/16:0). Data are presented as means ± SEM. Unpaired t-test with Welch correction (*p<0.05, **p<0.01) is applied where indicated. KDSR, 3-keto-dihydrosphingosine reductase; CerS1-6, Ceramide synthase 1-6; DEGS1, Δ4-dihydroceramide desaturase 1; DEGS2, Δ4-dihydroceramide desaturase 2; GCS, glycoceramide synthase; SMS, sphingomyelin synthase. FDR, false discovery rate; FDR q >.3, grey dots; FDR q <.3, red dots represent upregulated species and blue dots represent downregulated species; Cre, mouse expressing Ins1tm1.1(cre)Thor/J with wild type Ormdl3 alleles; KO, Ormdl3β-/- homozygous knockout; ns, not significant.
3 Discussion
ORMDL3 has been implicated in a variety of disorders including asthma, inflammatory bowel disease, and obesity (8, 11–13). Recently, it was reported that a whole-body knockout of Ormdl3 when challenged with cold exposure or HFD exhibit impaired regulation of brown adipose tissue thermogenesis, white adipose tissue browning, and insulin resistance (14). Yet the contribution of β-cells to this phenotype remained unknown. We previously showed that islets from overweight/obese human donors displayed significant downregulation of ORMDL3 expression compared with islets from lean donors (15). In contrast, Ormdl3 was substantially upregulated in the islets of leptin-deficient obese (ob/ob) mice compared with lean mice (15). Ormdl3 knockdown in a murine β-cell line induced expression of pro-apoptotic markers suggesting a role for Ormdl3 in β-cell apoptosis (15). In this study, we found that genetic ablation of Ormdl3 did not affect glucose metabolism, insulin sensitivity, insulin secretion, or islet architecture. However, despite seemingly unaltered β-cell health and function, targeted lipidomic determination of ceramide levels revealed increases in very long chain ceramides (C22-C26) and long chain C16 ceramide when Ormdl3β-/- mice were fed HFD. While our results suggest that β-cell expression of Ormdl3 is dispensable for normal islet function and glycemic control, they also suggest that ORMDL3 regulates sphingolipid levels in the β-cell.
Ormdl3 ablation has generated conflicting reports on SPT regulation and sphingolipid metabolism (16, 17). For instance, sphingolipid levels were found to be unchanged in transgenic Ormdl3 overexpression and whole-body Ormdl3 KO mice, and this observation was recapitulated in HEK cells (17). However, many studies report some effect of Ormdl3 overexpression or deletion on systemic sphingolipid levels with minor changes to rodent health measures (14, 18, 19). For example, while absence of Ormdl3 has been associated with increased levels of sphingolipids within the brain, Ormdl3 knockout mice appeared metabolically healthy and similar to control mice (19). Our results are in line with these reports demonstrating increased ceramide levels following Ormdl3 ablation.
Our findings revealed that β-cell loss of Ormdl3 during obesity leads to increased generation of islet very long chain ceramide species, hinting at a potential regulatory axis in which ORMDL3 contributes to control of this sphingolipid class. In mammals, CERS2 catalyzes the N-acylation of the sphingoid base with very long chain fatty acyl-CoAs during de novo sphingolipid production to produce very long chain ceramides species (C22-26) (20, 21). Additionally, reports suggest that the ratio of very long chain:long chain ceramides is important for proper cellular function (20, 22). For instance, in BALB/c primary mouse hepatocytes overexpressing Cers2, the ratio of very long chain:long chain ceramides was increased, but despite the increase in overall ceramide abundance, insulin signal transduction was improved while markers of ER stress and gluconeogenesis were reduced (22). In addition, a recent report proposes an obesity-independent CERS2-dependent lipid signature of imbalanced very long chain:long chain ceramides as contributing to β-cell failure through impaired proinsulin processing (23). Our findings suggest that very long chain ceramide species may be neither beneficial nor deleterious within the β-cell. However, future work including feeding with a ketogenic diet containing a higher fat content could be performed to determine the optimal abundance and ratio of very long acyl chain-containing lipids for normal β-cell health. Additionally, while the 60% HFD model used in this study simulates rapid weight gain, we cannot rule out that the impact of β-cell Ormdl3-deficiency may be more pronounced after a longer dietary intervention period, in aged mice, or upon feeding with other diets that can impair lipid metabolism such as high fat/sucrose/fructose western diet.
Taken together, our results suggest that β-cell ORMDL3 regulates sphingolipid production but does not alter metabolic health. While we did not observe a distinct role for ORMDL3 in β-cell health and glucose homeostasis, our results and others suggest that ORMDL3 contributes to the regulation of very long chain sphingolipid generation (14).
4 Materials and methods
4.1 Animals
The animal care and experimental procedures were carried out in accordance with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol (#M005064-R01-A02 by F.E. for mice) was approved by the University of Wisconsin-Madison Institutional Animal Care and Use Committee. Mice were bred and maintained under specific pathogen-free conditions at University of Wisconsin-Madison under approved protocols and were housed at 20–24°C on a 12 h light/12 h dark cycle. Animals were observed daily for health status, any mice that met IACUC criteria for euthanasia were immediately euthanized.
4.2 Generation of β-cell specific Ormdl3 knockout mice
β-cell specific Ormdl3 knockout mice (Ormdl3β-/-) were generated by Cyagen Biosciences on C57BL/6N background. Briefly, a targeting vector containing mouse Ormdl3 was generated, containing LoxP sites surrounding exons 2-4 of the Ormdl3 gene and a Neo selection cassette flanked by Frt motifs. This construct was then electroporated into embryonic stem cells (ESC) of C57BL/6N background, and the resulting cells were then screened for homologous recombination. The ESCs were then validated, and neo-excision was achieved in vitro by electroporation with an Flp-O expression plasmid. The resulting neo-excised ESC clones were then screened for successful deletion by PCR and injected into blastocysts isolated from pregnant B6 albino B6(Cg)-Tyrc-2J/J females. This resulted in generation of Ormdlfl/fl mice on C57BL/6N background. We then bred these mice with commercially available mice expressing a Cre construct expressed under the control of the insulin promotor (B6(Cg)-Ins1tm1.1(cre)Thor/J) to delete Ormdl3 specifically from β-cells.
4.3 Diets, feeding regimen, and weekly measurements
Chow, low fat diet, and high fat diet (Envigo 2919, Research Diets D12450J, and Research diets D12492, respectively) fed animals had ad libitum access to food and water unless otherwise specified. Mice that were fed a 60% high fat diet (Research Diets D12492) and 10% low fat diet (Research Diets D12492) began feeding on diet at 6 weeks of age and extending for a period of 24 weeks. Experiments on chow fed mice were performed on male and female mice between 5 and 34 weeks of age. Experiments on low-fat diet and high-fat diet fed mice were performed on male mice between 5 and 30 weeks of age. Weekly assessment of blood glucose and body weights was done after 6 hours of fasting. Blood glucose was analyzed by glucometer (Contour Next EZ 9628) after tail snip.
4.4 Histology
Pancreata from mice were fixed with 10% zinc formalin overnight and paraffin embedded. 5-μm sections of the pancreata were generated, and staining was performed after blocking with 5% normal goat serum with anti-Insulin (Linco) and anti-Glucagon (Cell Signaling) antibodies using established protocols. Antigen retrieval was prepared by using citrate buffer pH 6.0. After staining, slides were mounted with antifade mounting medium containing 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Immunofluorescent images of pancreatic sections were obtained using a Nikon Storm/Tirf/Epifluorescence. Images used for β-cell mass calculations were obtained with an EVOS FL Auto imaging system. The images of hematoxylin and eosin (H&E)-stained pancreatic sections were obtained using an AmScope light microscope. For analysis of β-cell mass and α:β cell ratio, the images were analyzed by using the Nikon Elements Advanced Research software program.
4.5 Islet isolation
Islets were isolated using the standard collagenase/protease digestion method as previously described (15, 24). Briefly, the pancreatic duct was cannulated and distended with 4°C collagenase/protease solution using Collagenase P (Sigma-Aldrich, USA) in 1x Hank’s balanced salt solution and 0.02% bovine serum albumin. The protease reaction was stopped using RPMI 1640 with 10% fetal bovine serum. Islets were separated from the exocrine tissue using Histopaque-1077 (Sigma-Aldrich, USA). Hand-picked islets were spun briefly at 1000 rpm for 1 minute before snap freezing in liquid nitrogen and storage at -80°C.
4.6 C-peptide and proinsulin ELISA
For measurement of serum C-peptide and proinsulin, blood was collected from mice via tail snip. Whole blood was then spun at 9000 rpm for 7 minutes and serum was collected and stored at -80°C. Frozen serum was thawed and measured by ELISA for proinsulin (Mercodia, 10-1232-01) and C-peptide (Alpco 80-CPTMS-E01). Samples were analyzed in duplicate.
4.7 Glucose and insulin tolerance tests
Glucose tolerance tests were performed on wild-type, heterozygous, knockout, and Cre control mice on chow, low fat, and high fat diets after a 6-hour morning fast. Blood glucose levels were measured at 0, 15, 30, 60, 90, and 120 minutes after an intraperitoneal administration of glucose at dose of 2g/kg body weight or insulin at dose of .75 U/kg body weight. Blood glucose measurements were measured using a glucometer (Contour Next EZ 9628). Blood glucose readings above the limit of detection were input as 600 mg/dL.
4.8 RT-qPCR
Total RNA was extracted from Ormdl3β+/+ and Ormdl3β-/- mouse islets using TRIzol reagent (Invitrogen) according to manufacturer’s instructions. cDNAs were synthesized from extracted RNA by using Superscript III First Strand RT-PCR kit (Invitrogen). Real-time quantitative PCR amplifications were performed on CFX96 Touch Real-time PCR detection system (Bio-Rad). β-Actin was used as internal control for the quantity of the cDNAs in real time PCR assays.
4.9 DEXA measurement
DEXA assessment of lean and fat mass was performed in 24-week-old low fat and high fat diet fed male mice. Mice were anesthetized with isoflurane before determination of body composition using a Faxitron Ultrafoucus in DXA mode. All measurements of body composition were performed in the University of Wisconsin – Madison Small Animal Imaging Facility Core.
4.10 Lipid extraction
70-90 isolated pancreatic islets were homogenized in 215 μL MeOH with internal standard using a Qiagen TissueLyzer II (9244420) for 4x40s cycles using chilled (4°C) blocks. 750 μL MTBE was added followed by 250 μL water. Samples were mixed by inversion and phase separation was carried out by centrifugation at 4°C for 10 min at 16,000 g. The top organic phase was transferred to a new 1.5 mL tube and extracts were dried in a SpeedVac. Samples were resuspended in 50:50 ACN/MeOH. A processed blank was prepared in the same way without internal standards added. Samples were stored at -20°C for 1 week before analysis.
4.11 LC-MS parameters
LC-MS analysis was performed on an Agilent 1290 Infinity II UHPLC system coupled to an Agilent 6495C triple quadrupole MS. Lipid extracts were separated on an Acquity BEH C18 column (Waters 186009453; 1.7 μm 2.1 × 100 mm) with a VanGuard BEH C18 precolumn (Waters 18003975) maintained at 60°C. Samples were held at 4°C in a multisampler prior to analysis. Sphingolipids were detected with multiple reaction monitoring (MRM) in positive ion mode. The gas temperature was 210°C with flow of 11 l/min and the sheath gas temperature was 400°C with a flow of 12 l/min. The nebulizer pressure was 30 psi, the capillary voltage was set at 4000 V, and the nozzle voltage at 500 V. High pressure RF was 190 and low-pressure RF was 120. Sample injection volume was 10 μL and the injection order was randomized. The chromatography gradient consisted of mobile phase A containing 60:40 ACN/H2O in 10 mM ammonium formate and 0.1% formic acid and mobile phase B containing 90:9:1 IPA/ACN/H2O in 10 mM ammonium formate and 0.1% formic acid at a flow rate of 0.500 mL/min. The gradient began with 30% B, increasing to 60% over 1.8 min, then increasing to 80% until 7 min, and 99% until 7.14 min held until 10 minutes.
Collision energies, retention times, and scanning windows were optimized based on standards and pooled plasma lipid extracts. Sphingolipid class MRM transitions from are found in Supplementary Table 1. Retention times for sphingolipids without standards were adjusted using a standard of similar acyl chain length and full scan analysis with a matching chromatography gradient.
4.12 Data processing
Quantification was performed in the Agilent MassHunter Workstation. Volcano plots and bar graphs were made using the ggpubr package in R version 4.1.2. Data were normalized to protein content as measured by BCA assay (Thermo Scientific 23225) on the pellet in the aqueous phase following lipid extraction. Data is considered “semi-quantitative” because standards were not available for all compounds detected.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by The University of Wisconsin-Madison School of Medicine and Public Health Institutional Animal Care and Use Committee.
Author contributions
LH and HL designed and performed the in vitro and in vivo experiments, analyzed the data, and prepared the figures. LH wrote the manuscript. GW performed lipidomics analysis and prepared the figures. JS interpreted lipidomics data. FE conceived, supervised and supported the project, designed experiments, interpreted results and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
HL was supported by the NIH National Research Service Award T32 GM007215 and a University of Wisconsin Stem Cell and Regenerative Medicine Center Graduate Fellowship. JS is supported by grants from the Juvenile Diabetes Research Foundation (JDRF201309442), the Glenn Foundation for Medical Research (GFMR) and American Federation for Aging Research (AFAR) (22068), the National Institutes of Health (R01DK133479), and the University of Wisconsin-Madison College of Agricultural and Life Science’s Hatch Grant (WIS04000). FE is supported by grants from the National Institutes of Health (DK130919 and DK128136), the Juvenile Diabetes Research Foundation (3-SRA-2023-1315-S-B), Greater Milwaukee Foundation, and startup funds from the University of Wisconsin-Madison.
Acknowledgments
We thank Dr. William Holland for providing the protocol and guidance on islet lipidomic studies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1170461/full#supplementary-material
References
1. Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocrine Rev (2008) 29(4):381–402. doi: 10.1210/er.2007-0025
2. Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res (2006) 45(1):42–72. doi: 10.1016/j.plipres.2005.11.002
3. Ye R, Onodera T, Scherer PE. Lipotoxicity and Beta; cell maintenance in obesity and type 2 diabetes. J Endocrine Soc (2019) 3(3):617–31. doi: 10.1210/js.2018-00372
4. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res (2016) 57(12):2099–114. doi: 10.1194/jlr.R066514
5. Davis D, Kannan M, Wattenberg B. Orm/Ormdl proteins: gate guardians and master regulators. Adv Biol Regul (2018) 70:3–18. doi: 10.1016/j.jbior.2018.08.002
6. Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, et al. Orm family proteins mediate sphingolipid homeostasis. Nature (2010) 463(7284):1048–53. doi: 10.1038/nature08787
7. Siow DL, Wattenberg BW. Mammalian ormdl proteins mediate the feedback response in ceramide biosynthesis. J Biol Chem (2012) 287(48):40198–204. doi: 10.1074/jbc.C112.404012
8. Pan DZ, Garske KM, Alvarez M, Bhagat YV, Boocock J, Nikkola E, et al. Integration of human adipocyte chromosomal interactions with adipose gene expression prioritizes obesity-related genes from gwas. Nat Commun (2018) 9(1):1512. doi: 10.1038/s41467-018-03554-9
9. Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkila E, Ferrer J. Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia (2015) 58(3):558–65. doi: 10.1007/s00125-014-3468-5
10. Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta (2003) 1632(1-3):16–30. doi: 10.1016/s1388-1981(03)00059-3
11. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A Large-scale, consortium-based genomewide association study of asthma. N Engl J Med (2010) 363(13):1211–21. doi: 10.1056/NEJMoa0906312
12. Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating Ormdl3 expression contribute to the risk of childhood asthma. Nature (2007) 448(7152):470–3. doi: 10.1038/nature06014
13. McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet (2010) 42(4):332–7. doi: 10.1038/ng.549
14. Song Y, Zan W, Qin L, Han S, Ye L, Wang M, et al. Ablation of Ormdl3 impairs adipose tissue thermogenesis and insulin sensitivity by increasing ceramide generation. Mol Metab (2022) 56:101423. doi: 10.1016/j.molmet.2021.101423
15. Lee H, Fenske RJ, Akcan T, Domask E, Davis DB, Kimple ME, et al. Differential expression of ormdl genes in the islets of mice and humans with obesity. iScience (2020) 23(7):101324. doi: 10.1016/j.isci.2020.101324
16. Li S, Xie T, Liu P, Wang L, Gong X. Structural insights into the assembly and substrate selectivity of human spt-Ormdl3 complex. Nat Struct Mol Biol (2021) 28(3):249–57. doi: 10.1038/s41594-020-00553-7
17. Zhakupova A, Debeuf N, Krols M, Toussaint W, Vanhoutte L, Alecu I, et al. Ormdl3 expression levels have no influence on the activity of serine palmitoyltransferase. FASEB J (2016) 30(12):4289–300. doi: 10.1096/fj.201600639R
18. Debeuf N, Zhakupova A, Steiner R, Van Gassen S, Deswarte K, Fayazpour F, et al. The Ormdl3 asthma susceptibility gene regulates systemic ceramide levels without altering key asthma features in mice. J Allergy Clin Immunol (2019) 144(6):1648–59 e9. doi: 10.1016/j.jaci.2019.06.041
19. Clarke BA, Majumder S, Zhu H, Lee YT, Kono M, Li C, et al. The ormdl genes regulate the sphingolipid synthesis pathway to ensure proper myelination and neurologic function in mice. Elife (2019) 8:e51067. doi: 10.7554/eLife.51067
20. Raichur S. Ceramide synthases are attractive drug targets for treating metabolic diseases. Front Endocrinol (Lausanne) (2020) 11:483. doi: 10.3389/fendo.2020.00483
21. Garic D, De Sanctis JB, Shah J, Dumut DC, Radzioch D. Biochemistry of very-Long-Chain and long-chain ceramides in cystic fibrosis and other diseases: the importance of side chain. Prog Lipid Res (2019) 74:130–44. doi: 10.1016/j.plipres.2019.03.001
22. Montgomery MK, Brown SH, Lim XY, Fiveash CE, Osborne B, Bentley NL, et al. Regulation of glucose homeostasis and insulin action by ceramide acyl-chain length: a beneficial role for very long-chain sphingolipid species. Biochim Biophys Acta (2016) 1861(11):1828–39. doi: 10.1016/j.bbalip.2016.08.016
23. Griess K, Rieck M, Muller N, Karsai G, Hartwig S, Pelligra A, et al. Sphingolipid subtypes differentially control proinsulin processing and systemic glucose homeostasis. Nat Cell Biol (2023) 25(1):20–9. doi: 10.1038/s41556-022-01027-2
Keywords: islet, knockout mouse, lipid, lipidomics, high fat diet, Ormdl3, beta cell
Citation: Hurley LD, Lee H, Wade G, Simcox J and Engin F (2023) Ormdl3 regulation of specific ceramides is dispensable for mouse β-cell function and glucose homeostasis under obesogenic conditions. Front. Endocrinol. 14:1170461. doi: 10.3389/fendo.2023.1170461
Received: 20 February 2023; Accepted: 31 March 2023;
Published: 14 April 2023.
Edited by:
Clemens Fürnsinn, Medical University of Vienna, AustriaReviewed by:
David Hill, Lawson Health Research Institute, CanadaEwa Gurgul-Convey, Hannover Medical School, Germany
Copyright © 2023 Hurley, Lee, Wade, Simcox and Engin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feyza Engin, ZmVuZ2luQHdpc2MuZWR1
†These authors have contributed equally to this work