 Xabier Erdocia
Xabier Erdocia Raquel Prado
Raquel Prado Jalel Labidi
Jalel Labidi- Chemical and Environmental Engineering Department, University of the Basque Country, San Seabastian, Spain
Organosolv lignin, obtained from olive tree pruning under optimized conditions, was subjected to a hydrothermal depolymerization process catalyzed by sodium hydroxide. The depolymerization of lignin was carried out at 300°C using different reaction times (20, 40, 60, 70, 80, 90, and 100 min) in order to study the influence of this parameter on lignin depolymerization. The resulting products (oil and residual lignin) were measured and analyzed by different techniques (GC/MS, high-performance size-exclusion chromatography, and pyrolysis–GC/MS) in order to determine their nature and composition. Coke was also formed, at a lower quantity, uncompetitive repolymerization reactions during the lignin hydrothermal treatment. The maximum oil yield and concentration of monomeric phenolic compounds was obtained after 80 min of reaction time. The highest reaction time studied (100 min) had the worst results with the lowest oil yield and highest coke production.
Introduction
Lignocellulosic biomass is said to be one of the most promising renewable raw materials since it can be transformed into a wide variety of products and by-products such as energy, materials, and chemicals. Lignocellulosic biomass is mainly composed of cellulose, hemicelluloses, and lignin. Among the main constituents of lignocellulosic biomass, lignin is one of the most interesting components since its aromatic nature and the broad variety of functional groups present in its chemical structure make it a unique and promising source of renewable products and commodity chemicals.
Lignin is primarily a structural material that adds strength and rigidity to cell walls and constitutes between 25 and 35% of the organic matter of woody plants (Kleinert and Barth, 2008). Lignin is an amorphous polyphenolic polymer composed principally of guaiacylpropane (G), syringylpropane (S), and p-hydroxyphenylpropane (H) units that form a randomized structure in a three-dimensional network inside the cell wall (Faix, 1991). Carbon–carbon and carbon–oxygen bonds between monomers are found in lignin. However, β-O-4 aryl ether bonds are the most frequent dimeric linkages (49–65%) probably due to the low steric hindrance at the β-carbon (Amen-Chen et al., 2001). The compositional ratios of these three units (H, G, S) can vary significantly with the feedstock. Guaiacylpropane is the predominant lignin monomer found in coniferous wood (softwoods) with minor amounts of H units. In hardwoods, G and S units are present in different ratios, but S units are the most typical (Gosselink et al., 2010). All three units (H, G, S) are usually present in grass or herbaceous lignins (Buranov and Mazza, 2008).
In addition to being a structural material, a further role of lignin in the plant is to provide protection against microbial attacks and external agents. Lignin is interconnected by polysaccharides and helps to bind the cellulose/hemicelluloses matrix. Lignin’s amorphous structure provides flexibility to the mixture and confers impermeability.
Biomass pretreatment is an essential key for lignin valorization. There are several different treatments in which lignin is produced as a product or by-product. Lignin is considered as a by-product in the pulp and paper industries (i.e., kraft or lignosulfonate) and is usually burned to fulfill the energy needs of the process. However, lignin will also be produced in large amounts in the new biorefinery schemes (i.e., organosolv, steam explosion). In the case of organosolv treatments, different organic solvents, such as ethanol, can be employed. In addition, the existing organosolv processes use different conditions and degradation techniques – including various pressures, temperatures, solvents, and pH ranges – that uniquely modify the chemical structure and linkages of the lignin (Zakzeski et al., 2010).
Depolymerizing lignin to low-molecular-weight (LMW) aromatic and phenolic compounds likely offers the greatest opportunity to truly expand the spectrum of lignin applications (Zhang et al., 2011). Several studies have been performed in order to convert lignin to value-added products. In these studies, organosolv lignins have been found to be an appropriate raw material for producing LMW compounds (González Alriols et al., 2009). Among many thermochemical methods, base-catalyzed depolymerization (BCD) has been widely studied and has been shown to produce phenolic monomeric compounds (Thring, 1994; Miller et al., 1999, 2002; Nenkova et al., 2008; Yuan et al., 2010; Roberts et al., 2011). Thring (1994) studied the alkaline degradation of lignin by means of the combined effect of reaction time and temperature concluding that the identifiable monomeric products decreased with increasing severity of treatment, indicative of the increasing importance of pyrolytic and recondensation reactions occurring in the lignin structure under these conditions. In this work, the predominant identifiable liquid phenolic products were guaiacol and syringol at low severities and catechol and its derivatives at high severities.
Alkaline hydrolysis of Alcell® lignin has also been studied by other authors (Miller et al., 1999, 2002). The reaction was enhanced by strong bases, and combinations of bases provided either positive synergistic effects, such as with NaOH and Ca(OH)2, or negative synergistic effects, such as with LiOH or CsOH with Ca(OH)2 (Miller et al., 1999). Model-compound studies indicated that the principle route for lignin depolymerization was through solvolysis of the ether linkages (Zakzeski et al., 2010). Several years later, BCD of lignin was carried out using water as the solvent (Miller et al., 2002). In this study, the authors observed that the base concentration was one of the most important factors governing lignin depolymerization. They found that a molar excess of a strong base (NaOH) gave better results but also that a small amount of a strong base together with a larger amount of a less-expensive base (CaO) produced good results.
In another study, it was shown that the optimal conditions required for lignin’s transition were 180°C, 5% NaOH, a hydromodule of 1:10 and treatment duration of 6 h (Nenkova et al., 2008). Yuan et al. (2010) studied alkaline lignin degradation in hot-compressed water and ethanol. Alkaline lignin was successfully degraded into oligomers with NaOH as a catalyst in the presence of phenol as the capping agent. However, complete suppression of char formation could not be achieved.
In the most recent studies, Roberts et al. (2011) concluded that the formation of monomers is directly proportional to the concentration of sodium hydroxide in the aqueous medium. In addition, a mechanism for the NaOH-catalyzed breakdown of the ether bonds of lignin was proposed. The preferential formation of derivatives of syringol can be explained by the stabilizing effect of the methoxy groups on the transition states of the carbenium ions. They also concluded that the production of monomers is limited by the oligomerization and polymerization reactions of the products formed.
The aim of this work was to study the influence of reaction time on the lignin depolymerization process. For this purpose, organosolv lignin samples were subjected to high temperatures and pressures with sodium hydroxide as a catalyst in an aqueous medium for different reaction times. The resulting products (oil and residual lignin) were measured and analyzed by different techniques [gas chromatography/mass spectrometry (GC/MS), high-performance size-exclusion chromatography (HPSEC), and pyrolysis (Py)–GC/MS] to determine the changes that occurred in both their nature and their quantity. Coke that was formed during a competitive repolymerization reaction was also measured.
Materials and Methods
Lignin Production
Olive tree pruning (Olea eurpaea, variety Arróniz) was used as the raw material. Lignin was extracted from the raw material by an organosolv treatment consisting of ethanol–water (70% w/w) at 200°C for 90 min in a pressure reactor using previously optimized conditions (Toledano et al., 2011). The liquid fraction in which lignin was dissolved was separated from the solid fraction by filtration. Dissolved lignin was isolated by precipitation by adding two acidified water volumes (pH around 2). The suspension was centrifuged at 4500 rpm for 20 min to recover the lignin. After that, lignin was dried at 50°C.
Base-Catalyzed Depolymerization
The reactions were conducted in a stirred batch reactor (5500 Parr reactor) with a 4848 Reactor controller. The volume of the reactor vessel was 100 mL. The reaction conditions were 300°C reaching pressures of about 9 MPa. The lignin:solvent (water) ratio was 1:20 (w/w). The catalyst (sodium hydroxide) concentration was set at 4% (w/w) (Toledano et al., 2014). The reaction conditions were chosen considering the reactor limitations, our previous experience, and the conditions reported in the literature (Thring, 1994; Roberts et al., 2011; Toledano et al., 2012). Three experiments were carried out, each using the reaction times 20, 40, 60, 70, 80, 90, and 100 min.
Product Separation Process
The liquid solution recovered after the reaction time in the batch microreactor was treated in order to separate the products. Firstly, HCl at 37% was added until pH 1 was reached. In this way, residual lignin and coke precipitated and were separated from the liquid by filtration using MN 640 w filters and washed with acidified water (water at pH 1 with HCl as the acidic agent) to remove residual liquid.
This liquid fraction was subjected to a liquid–liquid extraction process with ethyl acetate. Anhydrous sodium sulfate was added to the obtained organic phase in order to remove the traces of water and then it was filtrated to remove the added sodium sulfate. Then, this organic phase was vacuum evaporated at 0.02 MPa in order to obtain oil with the depolymerized products.
The solid phase was washed with tetrahydrofuran (THF) and was stirred for 3 h in a beaker. The THF solution was filtrated, and the undissolved solid (coke) was oven-dried at 50°C. The THF solution was vacuum evaporated to recover the unconverted lignin (Toledano et al., 2014).
Analysis of the Depolymerization Products
Oil was characterized in order to establish the nature of the monomeric phenolic compound. The oil was dissolved in HPLC-grade ethyl acetate in a metric flask. The solution was injected into a GC (7890A)–MS (5975C inert MSD with Triple-Axis Detector) Agilent equipped with a capillary column HP-5MS [(5%-phenyl)-methylpolysiloxane, 60 m × 0.25 mm]. The temperature program started at 50°C and then the temperature was raised to 120°C at 10°C/min, held for 5 min, raised to 280°C at 10°C/min, held for 8 min, raised to 300°C at 10°C/min, and held for 2 min. Helium was used as the carrier gas. Calibration was done using pure compounds obtained from Sigma-Aldrich: phenol, o-cresol, m-cresol, p-cresol, guaiacol, catechol, 3-methylcatechol, 4-methylcatechol, 4-ethylcatechol, 3-methoxycatechol, syringol, 4-hydroxybenzaldehyde, acetovanillone, veratrol, 4-hydroxybenzoic acid, 4-hydroxy-3-methoxyphenylacetone, vanillin, vanillic acid, syringaldehyde, 3,5-dimethoxy-4-hydroxyacetophenone, syringic acid, and ferulic acid (Erdocia et al., 2012).
Residual lignin was subjected to HPSEC to evaluate lignin molecular weight (MW) and molecular weight distribution (MWD) using a JASCO instrument equipped with an interface (LC-NetII/ADC) and a refractive index detector (RI-2031Plus). Two PolarGel-M columns (300 mm × 7.5 mm) and PolarGel-M guard (50 mm × 7.5 mm) were employed. Dimethylformamide solution containing 0.1% lithium bromide was used as the solvent. The flow rate was 0.7 mL/min, and the analyses were carried out at 40°C. Calibration was made using polystyrene standards (Sigma-Aldrich) ranging from 266 to 70,000 g/mol (Erdocia et al., 2012).
In order to elucidate the changes produced in the structure of residual lignin with respect to the raw lignin, Py–GC/MS analysis was performed. The pyrolysis was carried out using a CDS analytical Pyroprobe 5150. The pyrolysis temperature was set at 400°C for 15 s with a heating rate of 2°C/ms. Then, the products were analyzed by the GC–MS instrument described above. The oven program started at 50°C and was held for 2 min at this temperature, after which the temperature was raised to 120°C at 10°C/min and held for 5 min, raised to 280°C at 10°C/min, held for 8 min, and finally raised to 300°C at 10°C/min and held for 10 min.
Results and Discussion
Olive Tree Pruning Lignin Characterization
Organosolv olive tree pruning lignin presented the following composition: acid-insoluble lignin 71.90 ± 0.79%, acid-soluble lignin 1.63 ± 0.08%, total sugars 2.94 ± 0.14% (glucose 1.75 ± 0.12%, xylose 1.10 ± 0.03%, and arabinose 0.09 ± 0.01%), and ash content 0.39 ± 0.01%.
Yield of Depolymerized Products
Three main products were obtained after lignin depolymerization at any reaction time: oil, residual lignin, and coke. The obtained data were statistically analyzed and according to Table 1, all product yield values (oil, residual lignin, and coke) were significant for time factor (p < 0.05).
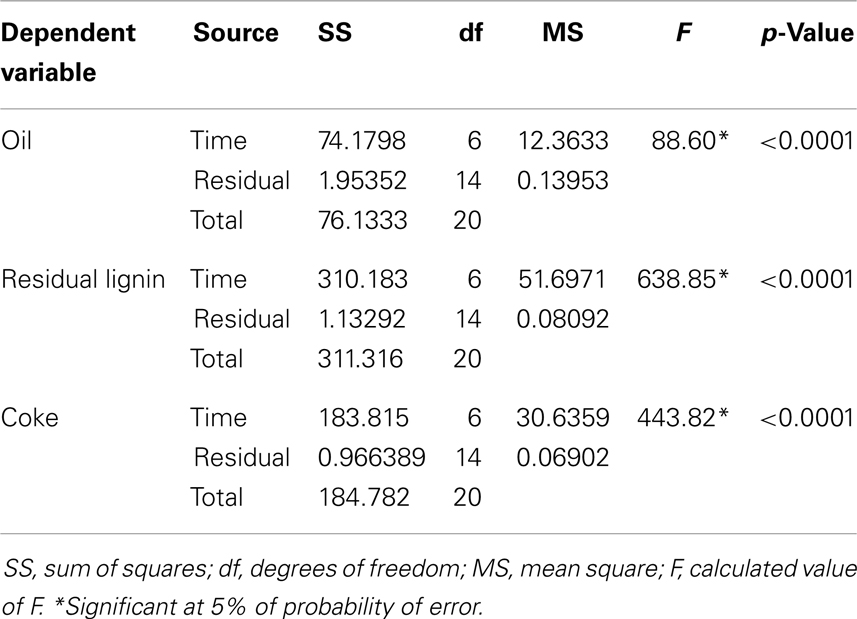
Table 1. Summary of the one-way ANOVA analysis for the oil, residual lignin, and coke yields with reaction time as factor.
In Figure 1 are presented the yields of the products obtained after base catalyzed depolymerization of the lignin at different reaction times. Regarding the oil yield, 80 min gave the best result for oil production (22.28 ± 0.37%) while the maximum time studied (100 min) presented the lowest yield of the desired product (16.57 ± 0.40%). These results are in agreement with other studies that showed that increasing the reaction severity decreases the concentration of monomeric compounds (oil production) as the importance of pyrolytic and recondensation reactions are enhanced (Miller et al., 1999). As can be observed, oil yield increased with the reaction time until reaching a maximum at 80 min and then decreased dramatically because of the recondensation reactions occurring at longer reaction times.
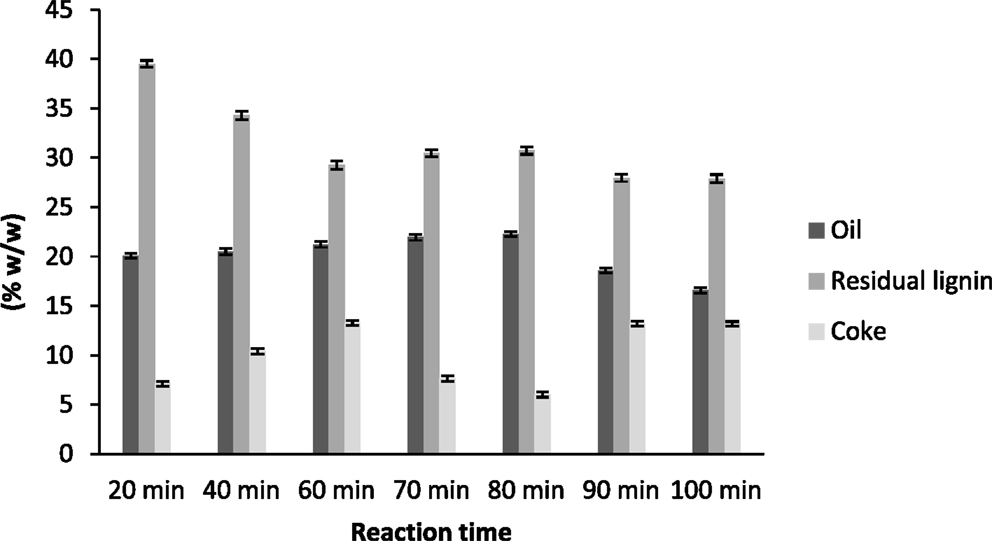
Figure 1. Yields of the products obtained after base-catalyzed depolymerization of lignin at different reaction times.
The residual lignin yield showed an opposite trend. At 20 min of reaction time, residual lignin was the main product (40% of the original amount of lignin). As the reaction time increased, this residual lignin decreased to a value of 30% of the initial lignin concentration, which remained almost constant after 60 min of reaction time. The increase of reaction time did not affect the residual lignin yield, which means that at high severities (more than 60 min of reaction time) the reactions taking place only affected oil production (hydrolysis and demethoxylation reactions) or coke production (pyrolytic and recondensation reactions).
Regarding coke production, it had a different behavior from the oil or residual lignin. Its production increased with reaction time but then decreased at 70 and 80 min (7.65 ± 0.30 and 6.04 ± 0.25%, respectively) and for the last two reaction times it increased again and produced the highest values. This behavior has already been reported by other authors who claimed that char formation from lignin in aqueous media was promoted by long reaction times (Yokoyama et al., 1998).
The yields of depolymerized products showed that, at 70 and 80 min reaction times, the main reactions were hydrolysis and demethoxylation reactions, and so oil yield increased and coke production decreased. However, at longer reaction times, depolymerization reactions were not as significant as pyrolytic or recondensation reactions, and so coke production was maximal.
Oil Characterization
The characterization of the oil obtained in different depolymerization reactions by GC–MS showed differences in the concentration of the obtained phenolic compounds but not in their nature. In all cases, the same compounds were produced but in different quantities as can be observed in Table 2. Among the different linkages present in the lignin structure, β-O-4 is said to be the first to be cleaved during hydrothermal degradation of lignin (Li et al., 2007). Roberts et al. (2011) stated that the cleavage of the β-O-4 ether bond takes place heterolytically via the formation of a sodium phenolate derivative and a carbenium-ion-like transition state, which is instantly neutralized by a hydroxide ion. The sodium cations catalyze the reaction by forming cation adducts with lignin and, thus, polarizing the ether bond. These findings suggest that the polarization of the base is a key step and will govern the kinetics and the mechanism followed in the depolymerization reaction. This is the main reason that makes the nature of phenolic compounds obtained in all experiments to be the same as the employed catalyst was NaOH in all cases.
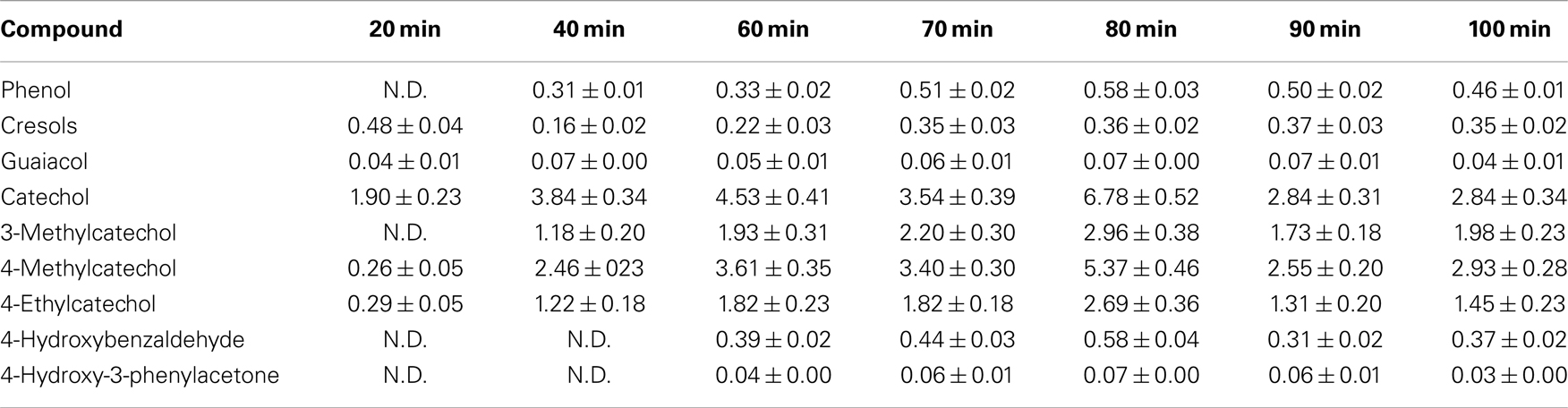
Table 2. Phenolic monomers concentration in the oil analyzed by GC–MS (% w/w).
The main products present in the oil were catechol and its derivatives: 3-methylcatechol, 4-methylcatechol, and 4-ethylcatechol. Concentrations of these products increased with reaction time (severity) until 80 min. High severities promoted the production of phenol, cresols, and catechol (Wahyudiono et al., 2007) but at very high severities, the concentration of monomeric compounds decreased (Thring, 1994). This was also observed here in the experiments with highest severities (90 and 100 min), the concentration of phenolic monomeric compounds dropped compared to shorter reaction times.
Residual Lignin
The changes produced in the residual lignins were analyzed by HPSEC with the raw lignin as a reference. As can be observed in Figure 2, all the residual lignins had two main peaks: one related to a fraction with higher MW than raw lignin and the other one related to a lower MW fraction. The first peak of the experiments appeared earlier than the raw lignin main peak, meaning higher MW fractions. The reason for this behavior was the polymerization reactions that have been reported by other authors (Yuan et al., 2010) occurring between unstable lignin fragments and the original lignin producing unconverted lignin with higher MW than the initial lignin. Repolymerization phenomena affected all samples regardless of the applied severity parameter. Alkaline condensations typically involve the activation of the unsubstituted aromatic C-5 position. Formaldehyde, formed as a by-product of the cleavage of the Cγ, may participate in the reaction (Valenzuela, 2011). However, residual lignin from experiments at 90 and 100 min presented the highest MW repolymerized fractions, which indicate that higher reaction times enhanced repolymerization reactions (Yuan et al., 2010).
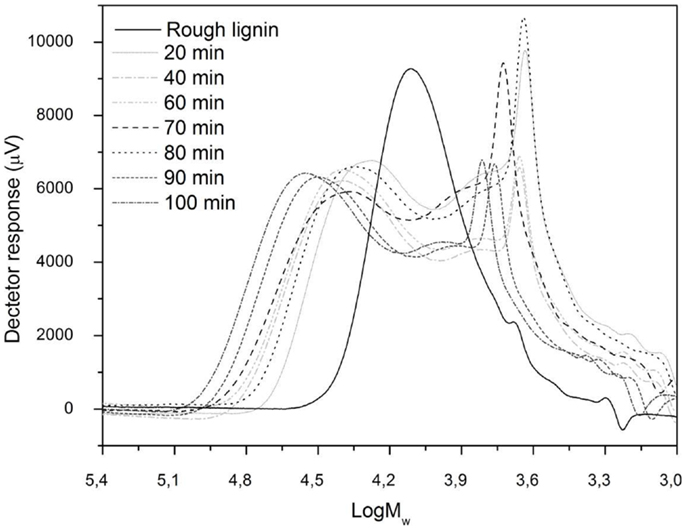
Figure 2. Molecular weight distribution of raw lignin and residual lignins samples.
Peaks associated with lower MW lignin fractions than the raw lignin could also be observed in all reaction mixtures containing residual lignin. These peaks show that depolymerization reactions occurred during the hydrothermal treatment of the lignin. In this case, after 80 min reaction time, residual lignin was the main fraction of the lowest MW peak, which means that, at this reaction time, depolymerization of lignin was enhanced. This is in agreement with the results discussed above in which the 80 min reaction time gave the highest yield of depolymerized products.
Residual lignin was also subjected to Py–GC/MS in order to analyze the changes in its nature and structure. As can be seen in Figure 3, raw lignin and all residual lignins were subjected to this method. Chromatograms of all residual lignins presented the same pattern, which indicates that the structure of all residual lignins was the same. This behavior also indicates that the mechanism that formed this residual lignin was identical in all the experiments regardless of the time employed for the reaction. Furthermore, this fact confirmed that the base concentration and its nature are the main factors governing depolymerization of lignin (Miller et al., 2002; Toledano et al., 2012).
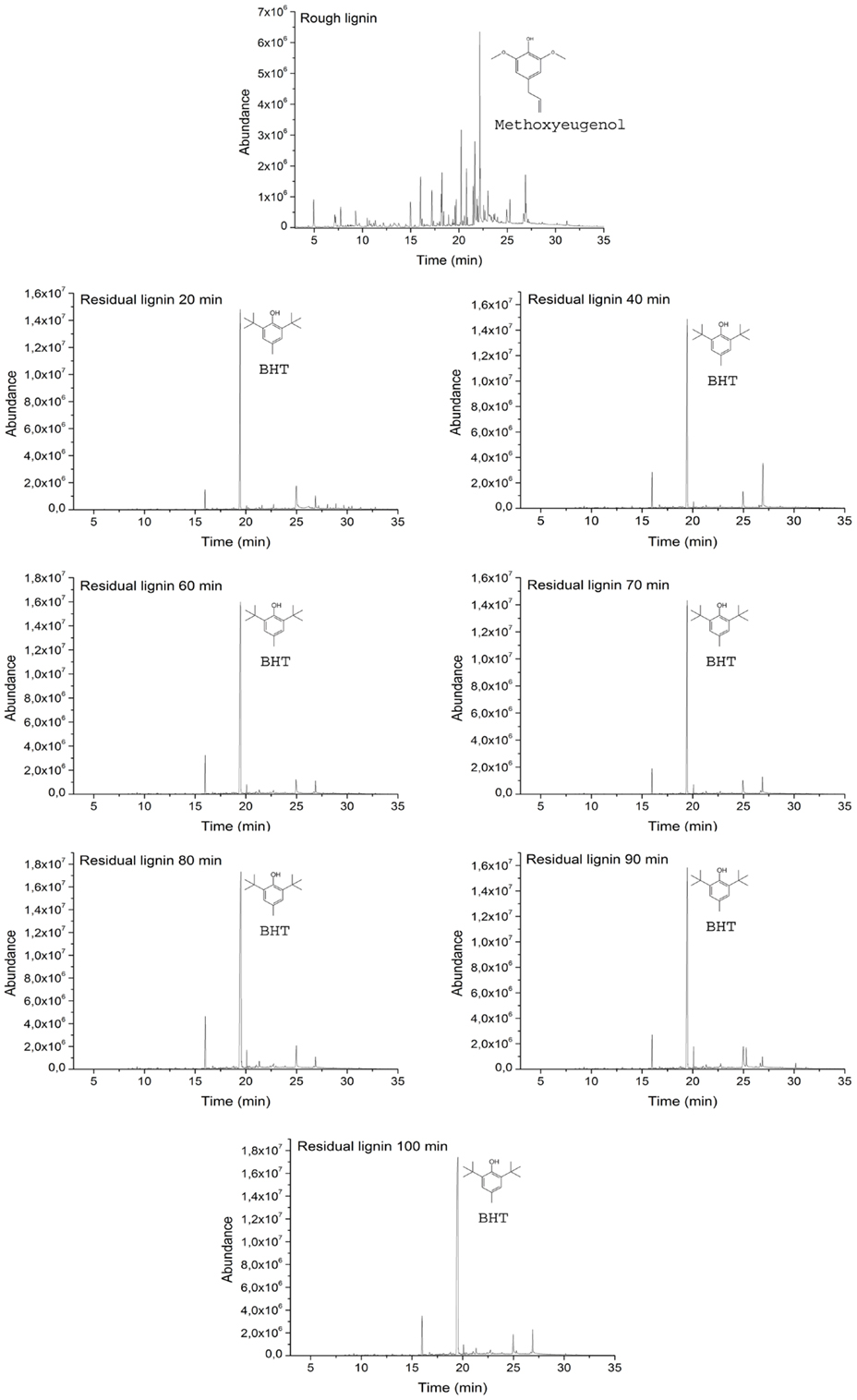
Figure 3. Chromatograms of raw and residual lignins pyrolysis.
The differences between the raw lignin and the residual lignins confirmed that residual lignins were not unconverted lignins they were new lignins created by repolymerization reactions that occurred between unstable fragments and depolymerization reactions of raw lignin. Figure 3 shows that the main peak in all residual lignin samples at 19.45 min was related to butylated hydroxytoluene while, in the raw lignin, this peak did not appear. Butylated hydroxytoluene occupied more than 65% of the area of the chromatograms of residual lignin. Other main compounds from residual lignin pyrolysis, but in lower quantity, were 2-tert-butyl-p-cresol and n-hexadecanoic acid.
The chromatogram of raw lignin was more heterogeneous than the chromatograms of residual lignins. Several compounds were obtained after the pyrolysis of the lignin sample and all of them were different from those obtained from the residual lignins. The main compound appeared at 22.18 min and was related to methoxyeugenol, which represented only 16.7% of the total area of the chromatogram. Other compounds obtained in a significant quantity were vanillin, eugenol, acetophenone, 4-hydroxy-3,5-dimethoxy-benzaldehyde, and octadecanoic acid.
Conclusion
The influence of reaction time on the lignin depolymerization process was studied in this work. The best results in terms of phenolic monomeric compounds production were found for 80 min of reaction time. At this time, coke production was also minimized, and the residual lignin MW was lowest. It was also concluded that, at highest reaction times, coke production and the MW of residual lignin increased because of pyrolytic and recondensation reactions. Moreover, the production of phenolic monomeric compounds dropped dramatically at the longest reaction times. It can also be concluded that the reaction time did not affect the reaction mechanism as all the obtained products were of the same nature and the structure of residual lignin was the same in all cases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors would like to thank the Department of Education, Universities and Investigation, and Department of Agriculture, Fishing and Food of the Basque Government (scholarship of young researchers training and project IT672-13) for financially supporting this work.
References
Amen-Chen, C., Pakdel, H., and Roy, C. (2001). Production of monomeric phenols by thermochemical conversion of biomass: a review. Bioresour. Technol. 79, 277–299. doi:10.1016/S0960-8524(00)00180-2
Buranov, A. U., and Mazza, G. (2008). Lignin in straw of herbaceous crops. Ind. Crops Prod. 28, 237–259. doi:10.1016/j.indcrop.2008.03.008
Erdocia, X., Toledano, A., Corcuera, M. A., and Labidi, J. (2012). Organosolv black liquor hydrolysis to obtain low molecular weight phenolic compounds. Chem. Eng. Trans. 29, 535–540. doi:10.3303/CET1229090
Faix, O. (1991). Classification of lignins from different botanical origins by FT-IR spectroscopy. Holzforschung 45, 21–27. doi:10.1515/hfsg.1991.45.s1.21
González Alriols, M., Tejado, A., Blanco, M., Mondragon, I., and Labidi, J. (2009). Agricultural palm oil tree residues as raw material for cellulose, lignin and hemicelluloses production by ethylene glycol pulping process. Chem. Eng. J. 148, 106–114. doi:10.1016/j.cej.2008.08.008
Gosselink, R. J. A., van Dam, J. E. G., de Jong, E., Scott, E. L., Sanders, J. P. M., Li, J., et al. (2010). Fractionation, analysis, and PCA modeling of properties of four technical lignins for prediction of their application potential in binders. Holzforschung 64, 193–200. doi:10.1515/hf.2010.023
Kleinert, M., and Barth, T. (2008). Phenols from lignin. Chem. Eng. Technol. 31, 736–745. doi:10.1002/ceat.200800073
Li, J., Henriksson, G., and Gellerstedt, G. (2007). Lignin depolymerization/repolymerization and its critical role for delignification of aspen wood by steam explosion. Bioresour. Technol. 98, 3061–3068. doi:10.1016/j.biortech.2006.10.018
Miller, J. E., Evans, L., Littlewolf, A., and Trudell, D. E. (1999). Batch microreactor studies of lignin and lignin model compound depolymerization by bases in alcohol solvents. Fuel 78, 1363–1366. doi:10.1016/S0016-2361(99)00072-1
Miller, J. E., Evans, L. R., Mudd, J. E., and Brown, K. A. (2002). Batch Microreactor Studies of Lignin Depolymerization by Bases. 2. Aqueous Solvents. Albuquerque, New Mexico: Sandia National Laboratories, Report SAND2002-1318.
Nenkova, S., Vasileva, T., and Stanulov, K. (2008). Production of low molecular phenolic compounds from technical hydrolysis lignin and wood biomass. Chem. Nat. Comp. 44, 182–185. doi:10.1007/s10600-008-9009-z
Roberts, V. M., Stein, V., Reiner, T., Lemonidou, A., Li, X., and Lercher, J. A. (2011). Towards quantitative catalytic lignin depolymerization. Chemistry 17, 5939–5948. doi:10.1002/chem.201002438
Thring, R. W. (1994). Alkaline degradation of ALCELL® lignin. Biomass Bioenergy 7, 125–130. doi:10.1016/0961-9534(94)00051-T
Toledano, A., Serrano, L., and Labidi, J. (2011). Enhancement of lignin production from olive tree pruning integrated in a green biorefinery. Ind. Eng. Chem. Res. 50, 6573–6579. doi:10.1021/ie102142f
Toledano, A., Serrano, L., and Labidi, J. (2012). Organosolv lignin depolymerization with different base catalysts. J. Chem. Technol. Biotechnol. 87, 1593–1599. doi:10.1002/jctb.3799
Toledano, A., Serrano, L., and Labidi, J. (2014). Improving base catalyzed lignin depolymerization by avoiding lignin repolymerization. Fuel 116, 617–624. doi:10.1016/j.fuel.2013.08.071
Valenzuela, M. (2011). Base-Catalyzed Depolymerization of Lignin and Hydrodeoxygenation of Lignin Model Compounds for Alternative Fuel Production. Ph.D. thesis, Atlanta: Georgia Institute of Technology.
Wahyudiono, Kanetake, T., Sasaki, M., and Goto, M. (2007). Decomposition of a lignin model compound under hydrothermal conditions. Chem. Eng. Technol. 30, 1113–1122. doi:10.1002/ceat.200700066
Yokoyama, C., Nishi, K., Nakajima, A., and Seino, K. (1998). Thermolysis of organosolv lignin in supercritical water and supercritical methanol. J. Jpn. Petrol. Inst. 41, 243–250. doi:10.1627/jpi1958.41.243
Yuan, Y., Cheng, S., Leitch, M., and Xu, C. (2010). Hydrolytic degradation of alkaline lignin in hot-compressed water and ethanol. Bioresour. Technol. 101, 9308–9313. doi:10.1016/j.biortech.2010.06.140
Zakzeski, J., Bruijnincx, P. C. A., Jongerius, A. L., and Weckhuysen, B. M. (2010). The catalytic valorization of lignin for the production of renewable. Chem. Rev. 110, 3552–3599. doi:10.1021/cr900354u
Keywords: lignin, depolymerization, homogeneous catalysis, organosolv
Citation: Erdocia X, Prado R, Corcuera MÁ and Labidi J (2014) Influence of reaction conditions on lignin hydrothermal treatment. Front. Energy Res. 2:13. doi: 10.3389/fenrg.2014.00013
Received: 30 December 2013; Accepted: 19 March 2014;
Published online: 01 April 2014.
Edited by:
Arthur Ragauskas, Georgia Institute of Technology, USAReviewed by:
Michelle J. Serapiglia, USDA-ARS Eastern Regional Research Center, USAYunqiao Pu, Georgia Institute of Technology, USA
Copyright: © 2014 Erdocia, Prado, Corcuera and Labidi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jalel Labidi, Chemical and Environmental Engineering Department, University of the Basque Country, Plaza Europa 1, San Sebastián 20018, Spain e-mail:amFsZWwubGFiaWRpQGVodS5lcw==