 Murugesan Vijayakumar1*
Murugesan Vijayakumar1* Niranjan Govind2
Niranjan Govind2 Bin Li1
Bin Li1 Xiaoliang Wei1
Xiaoliang Wei1 Zimin Nie1Suntharampillai Thevuthasan2Vince Sprenkle1
Zimin Nie1Suntharampillai Thevuthasan2Vince Sprenkle1 Wei Wang1*
Wei Wang1*- 1Energy and Environmental Directorate, Pacific Northwest National Laboratory, Richland, WA, USA
- 2Environmental and Molecular Science Laboratory, Pacific Northwest National Laboratory, Richland, WA, USA
Lack of comprehensive understanding about the interactions between Nafion membrane and battery electrolytes prevents the straightforward tailoring of optimal materials for redox flow battery applications. In this work, we analyzed the interaction between aqua-vanadyl cation and sulfonic sites within the pores of Nafion membranes using combined theoretical and experimental X-ray spectroscopic methods. Molecular level interactions, namely, solvent share and contact pair mechanisms are discussed based on vanadium and sulfur K-edge spectroscopic analysis.
Introduction
Nafion®, a perfluorinated cation exchange membrane, has wide technological applications ranging from fuel cells, batteries, to sensors. The excellent chemical stability combined with high proton conductivity and exceptionally wide functionality arises from the stable tetrafluoroethylene (TFE) backbone and acidic sulfonic groups in the pore structure of Nafion. In particular, its applicability in strong acidic and oxidative chemical environments has made it a central component in vanadium redox flow batteries (VRFB) (Zhang et al., 2010; Schwenzer et al., 2011). As a promising stationary energy storage system, VRFB requires cost effective long-term operation capabilities under a wide temperature range. Although, the chemically stable Nafion provides the necessary proton conductivity, it is also permeable for vanadium ion diffusion through its pore structure leading to substantial capacity decay during VRFB operation (Luo et al., 2013). Hence, designing a highly ion-selective membrane is the main challenge in realizing wide range commercial deployment of VRFB technology. However, a molecular level analysis of how the solvated vanadium ions diffuse through the spatially confined Nafion pores must precede any rational design approach of a highly ion-selective membrane.
Typically, cation permeation through Nafion is believed to be a result of its electrostatic interaction with negatively charged sulfonic acid sites within their pore structure (Hsu and Gierke, 1983). Inside the Nafion pore structure, the cations can interact with sulfonic sites though various mechanisms. For example, if the cations are fully solvated within these spatially encapsulated solutions, then it is expected that their solvation structure could effectively screen the electrostatic interaction with sulfonic sites. In this scenario, the solvent molecule as part of solvation sphere can interact with the sulfonic site, i.e., solvent share type interaction, and experience charge driven diffusion behavior. In addition, higher cation concentration, charge, temperature along with pore geometry can lead to contact pair formation where the cation can directly bind to sulfonic acid sites (Komoroski and Mauritz, 1982). The interaction between cations and sulfonic sites often can significantly impact the overall membrane performance. For instance, if the contact pair is formed with covalent type bonding, it could lead to fouling process and decrease the overall cation permeability including the proton conductivity. Hence, it is essential to gain a molecular level understanding of vanadium ion interaction with the Nafion membrane in order for the rational design of membranes tailored for high performance VRB operation. Unfortunately, such study focused on the interactions of vanadium ions within the Nafion membrane is still elusive. Our previous analysis of the Nafion–vanadyl ion interaction is based on nuclear magnetic resonance (NMR) and x-ray photoelectric spectroscopy (XPS) methods (Vijayakumar et al., 2011, 2012), which has inherent limitations in terms of internal solvent dynamics and surface sensitivity, respectively. To overcome these experimental limitations, we employed the vanadium and sulfur K-edge x-ray absorption (XAS) analysis in combination with density functional theory (DFT) based computational methods. This combined approach can help us shed light on the aqua-vanadyl ion interaction with the sulfonic sites.
Materials and Methods
All the measurements described herein were done using the commercial Nafion® 115 membrane. To probe the vanadyl ion interaction, the membrane was soaked in 1 M vanadium (IV) sulfate aqueous solution for 24 h at room temperature. The soaked membrane is washed with distilled water and dried in open air to remove any surface adsorbed vanadium species. Both vanadium and sulfur K-edge x-ray near edge spectroscopic (XANES) data were recorded in fluorescence mode at Stanford synchrotron radiation light source (SSRL), on beam line 4–3. The vanadium and sulfur K-edge XANES peak positions are calibrated using absorption edge of sulfur powder and vanadium metal foil. Ground state quantum chemistry calculations were performed with the ADF 2013 software package. Both geometry optimization and energy barrier analysis carried out using the in-build PBE exchange-correlation functional with the D3 dispersion and triple-zeta double polarization (TZ2P) basis set on all atoms. The Gibbs free energy (G) of each structure was calculated using bond energy, solvation energy, entropy, and internal energy terms, which are computed with inbuilt program in ADF package (te Velde et al., 2001). For geometry optimization and energy calculation, only the side chain with terminal sulfonic sites is considered as it is crucial part of the Nafion pore structure. The TFE backbone is severely hydrophobic and not expected to interact with aqual-vanadyl ion and subsequently not analyzed in this study. The XANES spectra are calculated using time dependent density functional theory (TDDFT) with the NWChem quantum chemistry suite (Valiev et al., 2010). Averaged spectra were generated by sampling 20 geometries near the optimized structure and 1.0 eV Lorentzian broadened. The absorbing centers V and S were represented with the Sapporo-QZP-2012 (Noro et al., 2012) all electron basis set, while the remaining atoms were treated with the 6-311G** basis set (Krishnan et al., 1980). The BHLYP exchange-correlation functional (Lee et al., 1988; Becke, 1993) was used for all the TDDFT XANES calculations.
Results and Discussion
The vanadyl ion normally exists as a distorted octahedral structured cation [VO⋅5(H2O)]2+ in aqueous solution. The molecular structure of vanadyl cation with its solvation phenomena in redox flow battery electrolyte has previously been reported (Vijayakumar et al., 2010, 2014). This distorted octahedral cation has five liable water molecules in octahedral coordination along with one vanadyl oxygen (V =O), which constitutes the first coordination sphere. This aqua-vanadyl ion can diffuse into the Nafion membranes forming electrostatic interactions with negatively charged sulfonic acid sites within the membrane pore structure. The diffusing aqua-cation can interact through the solvent share mechanism where the water molecule from first solvation can interact with sulfonic site through hydrogen bonding interaction. Alternatively, the sulfonic site can directly bond with the central vanadium atom via V–O bonding. This direct bonding represents a contact pair mechanism where the symmetry of the bond with respect to the native vanadyl oxygen (V =O) can result in either cis or trans positions. Schematic view of optimized geometry for solvent share, cis and trans contact pair mechanisms are shown in Figure 1 (optimized molecular structures are available in Supplementary Material). To evaluate these three different possible vanadyl ion specific interactions and possibly identify the dominant mechanism within Nafion pores, we employed the ground state DFT calculations. All three possible structures were constructed from prior optimized vanadyl and Nafion side chain molecular structure and subsequently optimized without any geometry restrictions. From these optimized structure, respective room temperature (295 K) Gibbs energy were calculated. Surprisingly all three structures shows similar energy (±0.6 eV) indicating nearly equal probability of formation inside the Nafion pores. It should be noted here that, our DFT calculations only involves a single side chain molecule and vanadyl ion whereas the experimental conditions contain complex cluster of sulfonic sites, bulk water, and multiple vanadyl ions engaging in highly correlated interactions. Nevertheless, the main focus of this article is to explore the molecular interaction between sulfonic sites and vanadyl ion. To verify the possibility of these different types of sulfonic site – vanadyl ion interactions within Nafion membranes, we carried out XANES measurements on pristine Nafion, vanadyl ion solution, and soaked Nafion membrane.
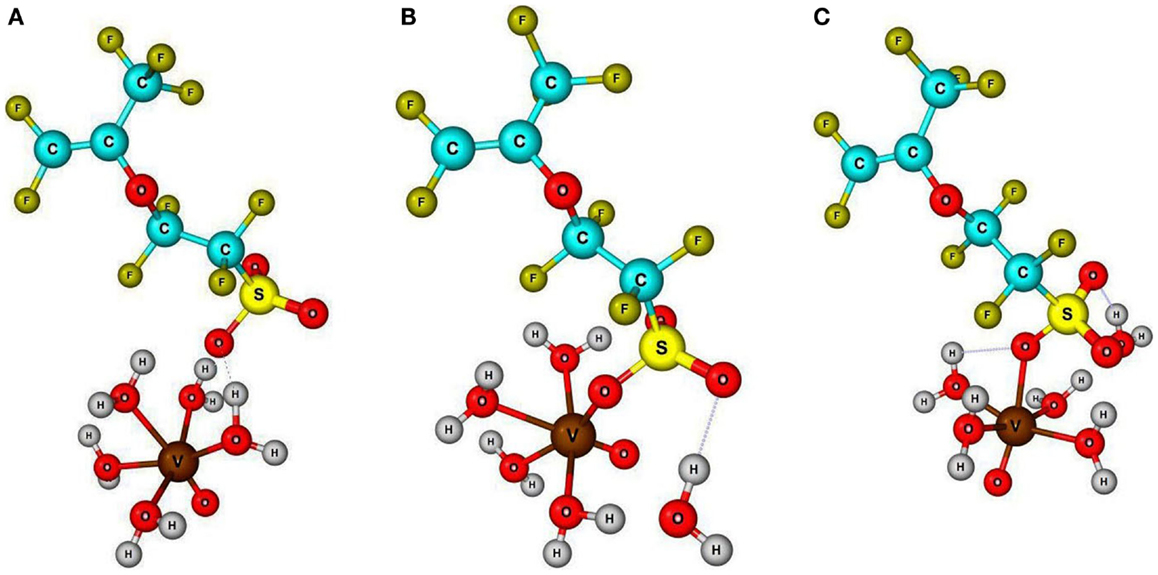
Figure 1. Schematic view three possible interaction mechanisms, namely (A) solvent share, (B) contact pair through cis position, (C) contact pair through trans position, between solvated vanadyl ion with sulfonic acid sites inside the Nafion pores based on DFT analysis. Fully optimized molecular structures are available in Supplementary Material.
The vanadium K-edge spectra of pure vanadyl ion electrolyte solution and soaked Nafion membrane show very similar spectrum (see Figure 2A) with pre-edge absorption peaks (A and B) followed by shoulder peak (C) on strong absorption curve (peak D). While the strong absorption peak (D) and shoulder peak (C) represents dipole allowed 1s → 4p transition and 1s → 4p shakedown transition respectively, the pre-edge peaks (A and B) represents dipole forbidden 1s → 3d transition indicating distorted octahedral symmetry of vanadyl molecular structure (Wong et al., 1984). The intensity of the pre-edge peaks is known to arise due to deviation of centrosymmetry around the absorbing V center allowing for stronge 3d → 4p mixing. Empirically, the intensity of pre-edge absorption peaks will give direct indication of the distortion in vanadium molecular structure, especially deviation from centrosymmetry, provided other factors such as oxidation state and ligand field strength remain comparable. For the soaked Nafion membrane, the dominant pre-edge peak (B) grows about 20% relative to the pure electrolyte solution, indicating the average ligand bond lengths are contracted and vanadyl molecule experiences more disorder inside Nafion pores. Now we can correlate this observation to DFT calculated molecular structure of vanadyl ion interacting with sulfonic sites of Nafion membranes (see Figure 1). The molecular size parameter can be defined as an average ligand bond length, , where r is each individual ligand bond length and n is the total number of ligands (n = 6 for vanadyl molecules). The DFT optimized structure of vanadyl ion interacting with Nafion through solvent share and contact pair mechanism shows similar molecular size parameter (R = 2.03 ± 0.02 Å), but lower than the pure vanadyl molecule (R = 2.07Å). This is in good agreement with increased intensity of pre-edge absorption in vanadium K-edge peaks of soaked Nafion membrane. Now we can focus on the main absorption peaks (C and D), where the soaked Nafion membrane register slight blue shift (~0.5eV) relative to the pure vanadyl solution (see Figure 2A). Typically, the shift in main absorption peaks is due to changes in any combination of valence, electronegativity of the bonding ligands, coordination number and specific structural features of vanadium molecule. Due to its interaction with Nafion the valence state of vanadyl (VO2+) and coordination numbers are not affected whereas overall structure is affected due to the difference in electronegativity of oxygen as part of water and sulfonic molecules. Hence, this slight blue shift is similar to the pre-edge peak intensity and merely represents a structural disorder in vanadyl structure.
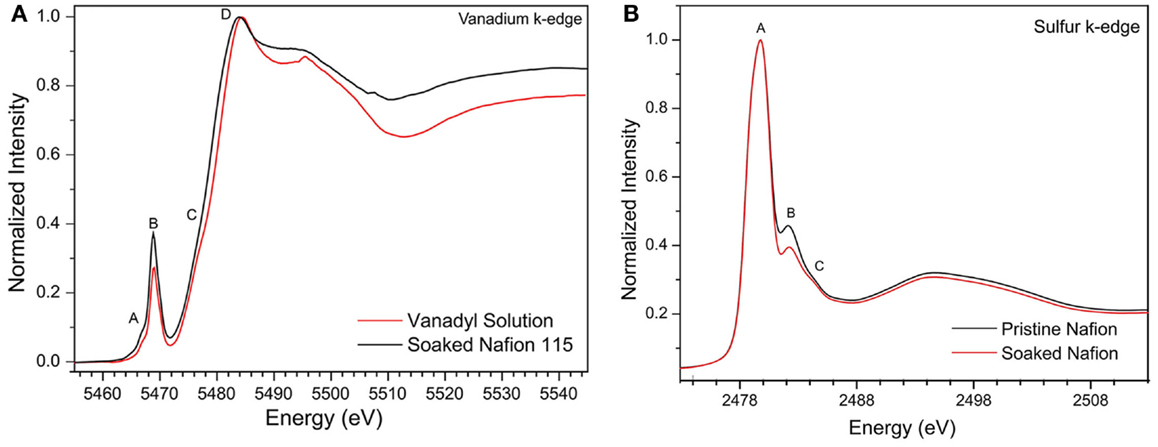
Figure 2. (A) Experimental sulfur K-edge spectra of pristine and soaked Nafion 115 membrane and (B) Experimental vanadium K-edge spectra of 2 M vanadyl ion solution and soaked Nafion 115 membrane. (Nafion membranes are soaked in vanadyl ion solution for 24 h and subsequently washed with DI water and dried).
The sulfur K-edge XANES spectrum of both pristine Nafion and soaked Nafion shows very similar features with one broad asymmetric peak centered at 2480.2 eV (peak A) followed by shoulder type resonances around 2482 and 2483 eV (peak B and C). The high intense broad peak A can be ascribed to an antibonding (C–S) σ* MO of A1 symmetry, whereas the shoulder type peak B and C represents (S–O) π bonding (Damian Risberg et al., 2007). It is evident that, the soaked Nafion shows slightly reduced intensity for peak B and C (see Figure 2B) indicating the possible change in S–O bond length of sulfonic sites due to interaction with vanadyl ions. Our DFT optimized geometry reveals that the typical S–O bond distance (1.46 ÅA) of sulfonic site is elongated due to its solvent share type interaction (1.57 ÅA) and contact pair interaction (~1.51 ÅA) with vanadyl ion. However, the cumulative effect of this bond elongation would be minimal as only one S–O bond is influenced by vanadyl ion interaction with sulfonic site [i.e., ]. Meanwhile, the C–S bonds mostly remain uninterrupted (±0.01 ÅA) due to vanadyl ion interaction (Damian Risberg et al., 2007). Overall, unlike the vanadyl ion structural distortion, the sulfonic site of Nafion side chain retains its chemical structure, which is reflected in similar sulfur K-edge spectra of pristine and soaked Nafion membrane. Despite these observations, it is still unclear about the preferred type of interaction between sulfonic sites and vanadyl ion inside Nafion pores. To further explore the possible interactions, we calculated the XANES spectra for all three interaction models and compare with experimental spectra (see Figure 3). Our ensemble averaged TDDFT XANES calculations for the S and V K-edges are in fair agreement with experiment. As can be seen Figure 3A, there are no significant differences between the trans contact, cis-contact, and solvent share, respectively. This suggests that the experiment can be a combination of all three configurations. For the V K-edge spectra, the pre-edge features A and B are captured well while features C and D are in reasonable agreement. As in the S K-edge spectra, there are no significant differences in the V K-edge spectra of the three configurations. It is clear that, the experimental spectra could be summation of contributions from all three bonding environments as shown in Figure 1. This is in good agreement with our DFT based Gibbs energy calculation, where the energy requirement for these different configurations are similar and represent possibilities of such bonding interaction between Nafion and solvated vanadyl ion.
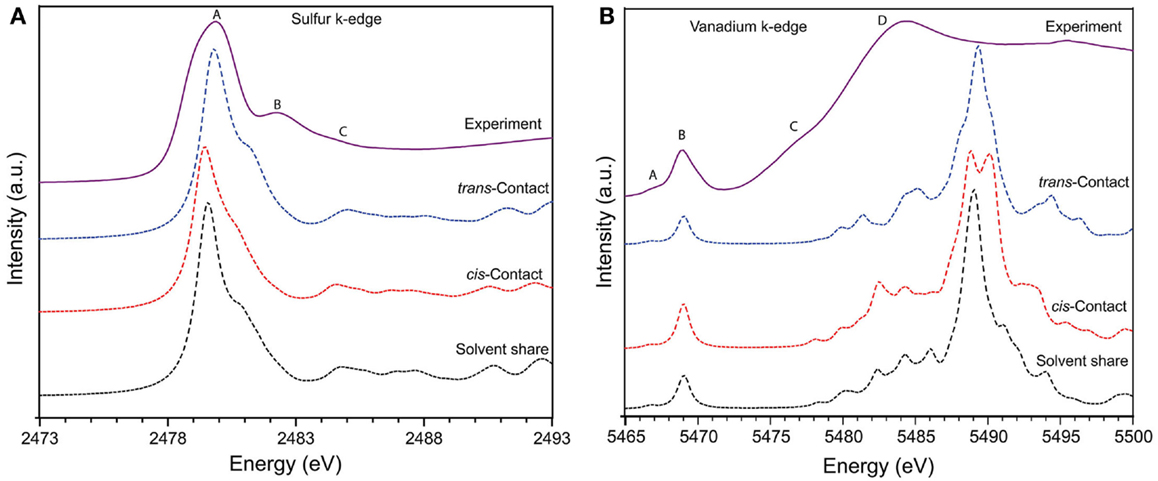
Figure 3. The solid line represents experimental (A) sulfur K-edge and (B) vanadium K-edge X-ray absorption spectra of Nafion 115 membrane soaked in mixed-acid based VO2+ electrolyte solution. Dotted lines represent theoretically calculated spectra of three possible interaction mechanisms, namely, solvent share, contact pair through cis and trans position between solvated vanadyl ion with sulfonic acid sites inside the Nafion pores based on TDDFT XANES calculations.
Conclusion
Predictive level understanding about chemical interaction between solvated cation and Nafion membranes is imperative for designing optimal membranes for all vanadium redox flow battery technology. However, the challenge is to employ the efficient tools to probe the molecular level interaction inside the nano size pores of Nafion membranes. Herein, we employed a combined experimental and theoretical analysis of core-level vanadium and sulfur K-edge spectroscopy to analyze the possible chemical interactions such as, solvent share, cis- and trans-contact pair formation between solvated vanadyl ion and sulfonic sites of Nafion membranes. The DFT-based energy calculation predicted both solvent share and contact pair interactions are probable. The TD-DFT calculated core-level vanadium and sulfur K-edge excitations for these chemical interactions were quite similar, and matches well with experimental spectra. This reveals that both contact pair and solvent share interactions between diffusing vanadyl ion and sulfonic sites of Nafion are probable as predicted by DFT calculations. However, the pore size, vanadyl ion concentration, electric field, and temperature can greatly influence the energy barrier for contact pair and solvent share interactions and result in preference over other interaction. Apparently, more detailed studies are warranted to gain quantitative view about these interactions and subsequently move toward designing of optimal membranes for redox flow batteries.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Ritimukta Sarangi at Stanford Synchrotron Radiation Lightsource (SSRL) for her help in XANES measurements. The sample preparation work is supported by Office of Electricity (OE) Delivery & Energy Reliability (project manager: Dr. Imre Gyuk), U.S. Department of Energy (DOE) under Contract #57558. The synchrotron and computational work is part of Chemical Imaging Initiative, under LDRD program at Pacific Northwest National Laboratory (PNNL). PNNL is a multiprogram laboratory operated for DOE by Battelle under Contract DE-AC05-76RL01830. The computational work is carried out at the Environmental and Molecular Sciences Laboratory, a national scientific user facility sponsored by DOE’s Office of Biological and Environmental Research. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fenrg.2015.00010/abstract
References
Becke, A. D. (1993). A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377. doi: 10.1063/1.464304
Damian Risberg, E., Eriksson, L., Mink, J., Pettersson, L. G. M., Skripkin, M. Y., and Sandström, M. (2007). Sulfur X-ray absorption and vibrational spectroscopic study of sulfur dioxide, sulfite, and sulfonate solutions and of the substituted sulfonate ions X3CSO3- (X = H, Cl, F). Inorg. Chem. 46, 8332–8348. doi:10.1021/ic062440i
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsu, W. Y., and Gierke, T. D. (1983). Ion transport and clustering in nafion perfluorinated membranes. J. Memb. Sci. 13, 307–326. doi:10.1016/S0376-7388(00)81563-X
Komoroski, R. A., and Mauritz, K. A. (1982). “Nuclear magnetic resonance studies and the theory of ion pairing in perfluorosulfonate ionomers,” in Perfluorinated Ionomer Membranes, eds A. Eisenberg and H. L. Yeage (Washington, DC: American Chemical Society), 113–138.
Krishnan, R., Binkley, J. S., Seeger, R., and Pople, J. A. (1980). Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654. doi:10.1063/1.438955
Lee, C., Yang, W., and Parr, R. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 37, 785–789. doi:10.1103/PhysRevB.37.785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, Q., Li, L., Wang, W., Nie, Z., Wei, X., Li, B., et al. (2013). Capacity decay and remediation of nafion-based all-vanadium redox flow batteries. ChemSusChem 6, 268–274. doi:10.1002/cssc.201200730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Noro, T., Sekiya, M., and Koga, T. (2012). Segmented contracted basis sets for atoms H through Xe: Sapporo-(DK)-nZP sets (n D, T, Q). Theor. Chem. Acc. 131, 1–8. doi:10.1007/s00214-012-1124-z
Schwenzer, B., Zhang, J., Kim, S., Li, L., Liu, J., and Yang, Z. (2011). Membrane development for vanadium redox flow batteries. ChemSusChem 4, 1388–1406. doi:10.1002/cssc.201100068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
te Velde, G., Bickelhaupt, F. M., Baerends, E. J., Fonseca Guerra, C., Van Gisbergen, S. J. A., Snijders, J. G., et al. (2001). Chemistry with ADF. J. Comput. Chem. 22, 931–967. doi:10.1002/jcc.1056
Valiev, M., Bylaska, E. J., Govind, N., Kowalski, K., Straatsma, T. P., Van Dam, H. J. J., et al. (2010). NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477–1489. doi:10.1016/j.cpc.2010.04.018
Vijayakumar, M., Bhuvaneswari, M. S., Nachimuthu, P., Schwenzer, B., Kim, S., Yang, Z., et al. (2011). Spectroscopic investigations of the fouling process on nafion membranes in vanadium redox flow batteries. J. Memb. Sci. 366, 325–334. doi:10.1016/j.memsci.2010.10.018
Vijayakumar, M., Burton, S. D., Huang, C., Li, L., Yang, Z., Graff, G. L., et al. (2010). Nuclear magnetic resonance studies on vanadium(IV) electrolyte solutions for vanadium redox flow battery. J. Power Sources 195, 7709–7717. doi:10.1016/j.jpowsour.2010.05.008
Vijayakumar, M., Nie, Z., Walter, E., Hu, J., Liu, J., Sprenkle, V., et al. (2014). Understanding aqueous electrolyte stability through combined computational and magnetic resonance spectroscopy: a case study on vanadium redox flow battery electrolytes. Chempluschem 80, 428–437. doi:10.1002/cplu.201402139
Vijayakumar, M., Schwenzer, B., Kim, S., Yang, Z., Thevuthasan, S., Liu, J., et al. (2012). Investigation of local environments in nafion-SiO2 composite membranes used in vanadium redox flow batteries. Solid State Nucl. Magn. Reson. 42, 71–80. doi:10.1016/j.ssnmr.2011.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, J., Lytle, F., Messmer, R., and Maylotte, D. (1984). K-edge absorption spectra of selected vanadium compounds. Phys. Rev. B Condens. Matter. 30, 5596–5610. doi:10.1103/PhysRevB.30.5596
Zhang, Y. P., Chen, Y., Zhou, Y. L., and He, P. (2010). Membranes for all-vanadium redox flow battery. Prog. Chem. 22, 384–387. doi:10.3390/membranes2020275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ion exchange membrane, redox flow battery, X-ray absorption spectroscopy, TD-DFT
Citation: Vijayakumar M, Govind N, Li B, Wei X, Nie Z, Thevuthasan S, Sprenkle V and Wang W (2015) Aqua-vanadyl ion interaction with Nafion® membranes. Front. Energy Res. 3:10. doi: 10.3389/fenrg.2015.00010
Received: 05 February 2015; Accepted: 19 February 2015;
Published online: 23 March 2015.
Edited by:
Linda F. Nazar, University of Waterloo, CanadaReviewed by:
Yuliang Cao, Chinese Ministry of Education, ChinaGuihua Yu, The University of Texas at Austin, USA
Copyright: © 2015 Vijayakumar, Govind, Li, Wei, Nie, Thevuthasan, Sprenkle and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Murugesan Vijayakumar and Wei Wang, Energy and Environmental Directorate, Pacific Northwest National Laboratory, 902 Battelle Blvd, P.O. Box 999, Richland, WA 99352, USA e-mail:dmlqYXlAcG5ubC5nb3Y=;d2VpLndhbmdAcG5ubC5nb3Y=