 Ribooga Chang
Ribooga Chang Semin Kim
Semin Kim Seungin Lee
Seungin Lee Soyoung Choi
Soyoung Choi Minhee Kim
Minhee Kim Youngjune Park
Youngjune Park- Carbon and Energy Systems, School of Earth Sciences and Environmental Engineering, Gwangju Institute of Science and Technology (GIST), Gwangju, South Korea
The transformation of CO2 into a precipitated mineral carbonate through an ex situ mineral carbonation route is considered a promising option for carbon capture and storage (CCS) since (i) the captured CO2 can be stored permanently and (ii) industrial wastes (i.e., coal fly ash, steel and stainless-steel slags, and cement and lime kiln dusts) can be recycled and converted into value-added carbonate materials by controlling polymorphs and properties of the mineral carbonates. The final products produced by the ex situ mineral carbonation route can be divided into two categories—low-end high-volume and high-end low-volume mineral carbonates—in terms of their market needs as well as their properties (i.e., purity). Therefore, it is expected that this can partially offset the total cost of the CCS processes. Polymorphs and physicochemical properties of CaCO3 strongly rely on the synthesis variables such as temperature, pH of the solution, reaction time, ion concentration and ratio, stirring, and the concentration of additives. Various efforts to control and fabricate polymorphs of CaCO3 have been made to date. In this review, we present a summary of current knowledge and recent investigations entailing mechanistic studies on the formation of the precipitated CaCO3 and the influences of the synthesis factors on the polymorphs.
Introduction
It is generally recognized that global warming is caused by the accumulation of greenhouse gases in the atmosphere, including CO2 in particular. Surprisingly, the atmospheric level of CO2 has reached a significantly symbolic milestone, 400 parts per million (Scripps Institution of Oceanography, 2017), and moreover, a further continuous increase is expected for the foreseeable future in the absence of proper actions (Kim et al., 2013). In the context of global awareness of this issue, in 2015 COP21, also known as the 2015 Paris Climate Conference, proposed an agreement to keep the global average temperature rise below 2°C above preindustrial levels by limiting total carbon emissions in the atmosphere (COP21, 2015). Among the key options to reduce CO2 emissions and finally to meet the aforementioned goal, carbon capture and storage (CCS) technologies are considered to offer the greatest potential for CO2 mitigation from the use of fossil fuels in coal- and gas-fired power plants and in industrial sites [IEA (International Energy Agency), 2010; Smit et al., 2014a], which produce more than 40% of total greenhouse gas emissions [IPCC (Intergovernmental Panel on Climate Change), 2014].
CCS technologies are aimed at mitigating greenhouse gas emissions by capturing CO2 from large point sources, such as fossil fuel power plants and industrial facilities including cement, iron, and steel, chemical and refining facilities, transporting this CO2 to storage sites, and finally storing or sequestering it in geological formations. Among options for CO2 storage, geological CO2 storage is widely accepted as the most viable option for large-scale storage (Leung et al., 2014). In the geological storage scheme, CO2 can be injected into saline aquifers, oil and gas reservoirs, or deep coal beds (Klusman, 2003; White et al., 2003; Fujioka et al., 2010; Garcia et al., 2010; Chiaramonte et al., 2011). The injected CO2 then can be trapped under the ground via a sequence of trapping mechanisms such as stratigraphic, residual, solubility, and mineral trapping (Smit et al., 2014b). In particular, CO2 can be transformed to mineral carbonates by reacting with alkaline minerals present in the geological formation, which is referred to as in situ mineral carbonation. Because mineral carbonates such as CaCO3 or MgCO3 are the thermodynamically most stable form of carbon, long-term storage of CO2 can be achieved once it is transformed to carbonates (Smit et al., 2014b). However, geological CO2 storage poses several uncertainties that must be addressed. For example, potential leakage of injected CO2 is a major concern, and thus accurate quantification of storage potential and constant monitoring of injected CO2 are necessary (Sanna et al., 2014). Finding a storage site having suitable geological formation is also challenging in some regions or countries.
Unlike in situ mineral carbonation, ex situ mineral carbonation carries out a series of chemical processes above ground via reactions between CO2 and alkaline earth metals such as calcium or magnesium that are extracted from naturally occurring silicate minerals, i.e., wollastonite, olivine, serpentine, etc., or industrial by-products or waste materials, i.e., coal fly ash, steel and stainless-steel slags, and cement and lime kiln dusts (Gerdemann et al., 2007). Because this technology involves energy-intensive processes during the preparation of the solid reactants, including mining, transport, grinding and/or activation, as well as the recycling of additives and catalysts, process optimization is required for cost reduction (IPCC, 2005; Oelkers et al., 2008). Despite such shortcomings, ex situ mineral carbonation also has unique advantages. As opposed to in situ methods, this technique allows the utilization of alkaline-metal feedstock extracted from industrial wastes, which are generally recognized to have environmentally hazardous effects, and in this light providing an appropriate method for proper disposal or for recycling is a significant environmental issue. More importantly, the final products, such as CaCO3, can be converted to value-added materials that can be utilized in various applications such as adhesives, sealants, food and pharmaceuticals, paints, coatings, paper, cements, and construction materials (Eloneva et al., 2008a). It was reported that the global calcium carbonate market in 2011 and 2016 was about 81 and 98 million tons, respectively, and further growth is expected. Calcium carbonate is mostly used in the paper industry, followed by plastics, paints, adhesive/sealants, and rubber. Therefore, it can be anticipated that producing value-added mineral carbonates via ex situ mineral carbonation technology may partially reduce the overall expense in CCS as well as in ex situ mineral carbonation processes.
In fact, the precipitated CaCO3 has many industrial applications depending on its physicochemical characteristics such as particle size, shape, density, color, brightness, and other properties, and it is also known that those characteristics are significantly governed by the polymorphs of CaCO3. The precipitated CaCO3 has three polymorphs, such as calcite, aragonite, and vaterite. It has been reported that the formation behavior of each polymorph is affected by synthesis factors including pH, temperature, concentration, and ratio of carbonate and calcium ions, additives, stirring, reaction time, etc. (Zhao et al., 2013; Chang et al., 2017). In this review, we present a summary of current knowledge and recent investigations involving mechanistic studies on the formation of the precipitated CaCO3 and the influences of the synthesis factors on the polymorphs.
Mineral Carbonation Technologies
Since mineral carbonation for CO2 disposal was proposed in the 1990s (Seifritz, 1990), various efforts have been made toward commercialization in connection to CCS schemes (Sanna et al., 2014). The mineral carbonation technologies are based on the spontaneous reaction between CO2 and metal oxide bearing minerals to form insoluble carbonates, and the reactions can be carried out either below (in situ) or above ground (ex situ):
While in situ mineral carbonation involves CO2 injection processes into underground reservoirs where alkali or alkaline earth metals are present in the geological formation, ex situ mineral carbonation technology entails a series of physicochemical processes including mining, grinding, and/or pretreatment processes to secure Ca- or Mg-bearing mineral feedstock. Various naturally occurring mineral silicates or industrial by-products are considered suitable mineral feedstock for ex situ mineral carbonation owing to their large contents of Ca and/or Mg elements. Depending on physical and chemical properties of the mineral feedstock, the reaction pathways for the mineral carbonation must be designed and optimized. In general, the following reactions are anticipated for the representative naturally occurring mineral silicates such as olivine, serpentine, and wollastonite, respectively (Olajire, 2013):
Despite that the natural abundances of oxides and hydroxides of Ca and Mg are relatively low, such silicate minerals including olivine and serpentine, which are relatively abundant over the world, could be an attractive feedstock for CO2 mineralization (Lackner et al., 1997; Styring et al., 2015). Although it is not as abundant as olivine and serpentine, wollastonite also could be a suitable option in limited locations (Lackner et al., 1995), e.g., China, India, United States, Mexico, and Finland (U.S. Geological Survey, 2011).
On the other hand, in some regions or countries where natural silicate minerals of Ca and Mg are not available, industrial by-product or wastes including Ca and/or Mg elements, i.e., coal fly ash, steel and stainless-steel slags, and cement and lime kiln dusts, could be an alternative mineral feedstock to implement CO2 storage via mineral carbonation. The use of industrial by-products or wastes is advantageous over the use of natural silicate minerals, particularly in terms of energy consumption, processing cost, and reuse in products: (i) these materials are often associated with a large point source of CO2 emissions, such as coal fired power plants, cement plants, and the steel and paper industries, and thus combined carbon capture and storage may be achieved with elimination of the CO2 transportation process; (ii) because Ca or Mg elements usually exist in form of a readily reactive states, i.e., CaO or Ca(OH)2, enhanced carbonation yields without significant efforts regarding pre-treatments can be expected (Huijgen and Comans, 2006); (iii) energy-intensive mining and/or grinding processes may be avoided since they are typically in the state of fine-grained particulates (O’Connor et al., 2000a, b); and finally (iv) such industrial by-products or wastes can be accessed easily at low cost, and moreover, they can be transformed into value-added materials of mineral carbonates, which could be applicable to, for example, road base or construction materials, adhesives, sealants, food and pharmaceuticals, paints, coatings, paper, cements, construction materials, etc. (Eloneva et al., 2008a; Huntzinger et al., 2009) and may thereby partially compensate the overall cost of CCS. For these reasons, various types of industrial by-products and wastes have been investigated for ex situ mineral carbonation, including steelmaking slags, cement wastes, and waste ashes (Pan et al., 2012). Table 1 shows the composition of Ca and Mg elements in various feedstocks for ex situ mineral carbonation.
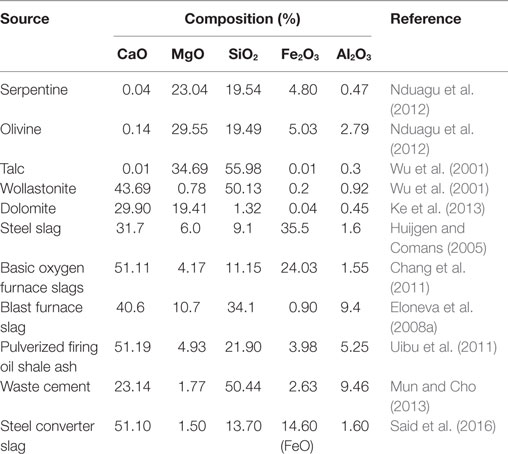
Table 1. Chemical compositions of various sources for mineral carbonation.
The reaction routes for ex situ mineral carbonation can be divided into two processes—direct and indirect mineral carbonations. A direct carbonation is the simplest carbonation method, where Ca or Mg feedstock directly reacts with CO2 in a single step, and further it can be conducted by gas–solid or aqueous route (Eloneva et al., 2007). Direct carbonation offers simplicity and does not require additional chemicals (Bobicki et al., 2012). The direct gas–solid carbonation route was first investigated by Lackner et al. (1997), employing olivine at high CO2 pressure conditions. The slow kinetics of the direct gas–solid carbonation in Ca- or Mg-bearing silicate minerals is the major challenge to be resolved (Lackner et al., 1995). The direct aqueous carbonation route also employs high CO2 pressure at elevated temperature condition for enhanced reaction conversion (O’Connor et al., 2000a, b, 2001). Huijgen et al. (2006) investigated mineral carbonation of finely ground wollastonite via a direct aqueous route introducing CO2 stream into the reactor under continuous stirring to ensure dispersion of the gas. They suggested a two-step reaction: (i) Ca leaching from the silicate mineral and (ii) crystallization of CaCO3. A promising conversion of 75% for 15 min at high temperature and pressure conditions of 200°C and 20 bar CO2 partial pressure, respectively, was reported. However, further efforts are required to enhance the reaction conversion, particularly considering the pretreatment step. The pretreatment process is aimed at promoting the carbonation reaction kinetics by providing larger surface area of the raw materials (Sanna et al., 2014). The process can be conducted through two major processes: (i) mechanical and (ii) thermal pretreatments or hybrid processes. The purpose of mechanical grinding is to destroy the mineral lattice and reduce the particle size, resulting in an increase of the surface area. Various types of mechanical pretreatment methods combined with ultrasonic and wet grinding have been proposed and tested thus far (O’Connor et al., 2001). Nevertheless, the requirement of high energy input is recognized as a critical drawback that must be mitigated. Thermal pretreatment was also investigated by many researchers, particularly focusing on serpentine to remove hydroxyl groups, resulting in a chemical transformation to pseudoforsterite (Sanna et al., 2014). It was revealed that thermotreatment can be a more effective option for Mg-bearing silicate minerals to enhance the carbonation efficiency than mechanical pretreatment, although the energy requirement during the process must be further addressed (Fabian et al., 2010; Sanna et al., 2013).
Indirect mineral carbonation route takes place in more than two steps, typically including (i) extraction of Ca and/or Mg components and (ii) a precipitation reaction step between Ca/Mg and CO2 in either gaseous or aqueous phases. Since the indirect carbonation process separates the precipitation step from dissolution of the raw materials, mineral carbonates with higher purity can be expected compared with the direct carbonation route. In the dissolution step, various additives including strong acids (i.e., HCl, HNO3, and H2SO4) (Teir et al., 2007; Lin et al., 2008; Bobicki et al., 2012), organic acids (i.e., acetic acid, formic acid, succinic acid, oxalic acid, etc.) (Park et al., 2003; Park and Fan, 2004; Bałdyga et al., 2010; Zhao et al., 2013), salts, and alkali solution and ligands (Maroto-Valer et al., 2005; Jarvis et al., 2009; Krevor and Lackner, 2009, 2011) have been investigated to date. Although the extraction efficiencies are promising, the use of such strong acids may provoke significant energy penalties associated with their recovery (Teir et al., 2007; Olajire, 2013; Sanna et al., 2014). Furthermore, some acids such as succinic acid and disodium oxalate often chelate the alkaline metals too strongly, and this does not result in the production of carbonates, but rather the precipitation of succinates or oxalates (Bonfils et al., 2012; Santos et al., 2014).
Utilization of Precipitated Mineral Carbonates
The precipitated mineral carbonates and their derivatives present versatile applications in industrial uses depending on their purity, polymorphism, shape, size and distribution, color, brightness, density, and other many physicochemical properties. Therefore, such a transformation of CO2 into value-added solid carbonates through ex situ mineral carbonation can partially offset the total cost of the carbon capture and storage process, thus making the mineral carbonation process more viable. In addition, the particular use of industrial waste materials (i.e., coal fly ash, steel and stainless-steel slags, and cement and lime kiln dusts) as mineral resources for mineral carbonation can offer an additional benefit providing environmental sustainable option of reuse and recycle of the waste materials.
The final products produced by the ex situ mineral carbonation route can be graded into two categories—(i) low-end high-volume and (ii) high-end low-volume mineral carbonates, regarding market needs, as well as their properties (i.e., purity) (Sanna et al., 2014). Further separation or purification processes are needed before and/or after precipitation steps in the mineral carbonation process for utilization, and thus an indirect process could be more suitable than a direct scheme since the former divides the mineral carbonation cation extraction step, followed by a separated precipitation step (Teir et al., 2005). If necessary, synthesis variables such as temperature, pressure, pH, and concentration of the ingredients in the precipitation step should be manipulated to acquire specific mineral carbonates with targeted properties. It was revealed that high purity calcium or magnesium carbonate can be obtained by the indirect mineral carbonation process separating silica and iron oxide via a pH swing processes (Park and Fan, 2004; Wang and Maroto-Valer, 2011a,b; Sanna et al., 2012).
Recently, a precipitated calcium carbonate (PCC) production technology utilizing steel converter (basic oxygen furnace) slag as a calcium source, referred to as Slag2PCC, has been developed and demonstrated successfully by researchers at Aalto university together with their collaborators (Said et al., 2013, 2016; Mattila et al., 2014). The process includes ammonium salt (e.g., NH4Cl) based calcium extraction and carbonation steps, and it can be operated in both batch and continuous modes. This may be advantageous because the quality of the final product can be controlled by the operation conditions (i.e., concentration of and ).
Molecular Mechanism and Polymorph Formation of CaCO3
Precipitated calcium carbonate exhibits various polymorphs with tunable physicochemical properties, which play a critical role in determining potential markets. Accordingly, more versatile applications of the products of mineral carbonation are anticipated particularly for calcium carbonate or its derivatives. In general, calcium carbonates exist either in the form of amorphous calcium carbonate (ACC) or one of the three polymorphs, namely, calcite, aragonite, and vaterite (Figure 1). Two hydrated phases of calcium carbonate, monohydrate (CaCO3⋅H2O), and hexahydrate of calcium carbonates (CaCO3⋅6H2O) are also possible forms of hydrated calcium carbonates. Recently, it was revealed that ACC normally exists as a monohydrated calcium carbonate (Goodwin et al., 2010). Table 2 briefly presents the properties of the anhydrous crystalline forms of calcium carbonates.
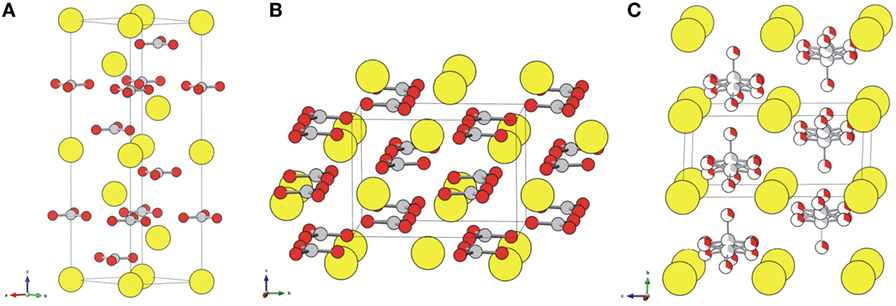
Figure 1. Crystal structures of (A) calcite, (B) aragonite, and (C) vaterite. Ca atoms are displayed as large yellow balls, and carbonate groups are illustrated with gray (carbon) and red (oxygen) balls. Vaterite is depicted with a hexagonal P63/mmc structure that accounts for a partial occupancy of one-third of the carbonate groups. Adapted with permission from Chang et al. (2017). Copyright 2017 American Chemical Society.
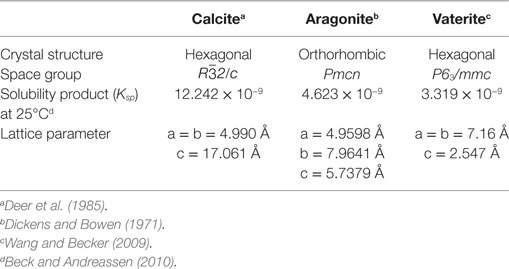
Table 2. Properties of the anhydrous crystalline forms of calcium carbonates.
Among the anhydrous polymorphs of CaCO3, calcite is thermodynamically the most stable at ambient conditions. The order of thermodynamic stability is, from most to least, calcite, aragonite, and vaterite (Declet et al., 2016). Despite lower stabilities from a thermodynamic point of view, aragonite and vaterite can be formed at ambient conditions owing to the kinetic constraints induced by synthesis factors such as temperature and impurities (e.g., Mg) (Ogino et al., 1987; Zhang et al., 2012), which can lead to crystallization of less stable aragonite or the least stable vaterite rather than forming calcite. A number of mechanistic studies have been conducted thus far to reveal the transformation mechanisms among the CaCO3 polymorphs (Kralj et al., 1997; Spanos and Koutsoukos, 1998; Katsifaras and Spanos, 1999; Wei et al., 2003; Rodriguez-Blanco et al., 2011; Zhang et al., 2012; Kabalah-Amitai et al., 2013; Nielsen et al., 2014). Although it is necessary to heat to temperature exceeding 730 K for irreversible transformation of vaterite to calcite (Chang et al., 2017), the least stable vaterite can be stabilized in an aqueous solution at ambient conditions preventing its transformation into calcite or aragonite (Trushina et al., 2014). Despite ongoing debate (Kamhi, 1963; Wang and Becker, 2009; Kabalah-Amitai et al., 2013), the formation of vaterite and its transformation mechanisms among the polymorphs can be explained in terms of sequential dissolution and (re)crystallization processes (Figure 2) (Kralj et al., 1997; Spanos and Koutsoukos, 1998; Katsifaras and Spanos, 1999; Wei et al., 2003): (i) initially formed ACC particles transform to the least stable vaterite and (ii) the most soluble vaterite undergoes dissolution and crystallization finally forming the most stable calcite. The solubility of the CaCO3 polymorphs is in decreasing order of ACC, vaterite, aragonite, and calcite (Beck and Andreassen, 2010). It was revealed that the ACC transformation into vaterite and calcite can be dominantly found below 40°C, while aragonite can be stabilized at elevated temperature above 60°C (Ogino et al., 1987; Chen and Xiang, 2009; Trushina et al., 2014).
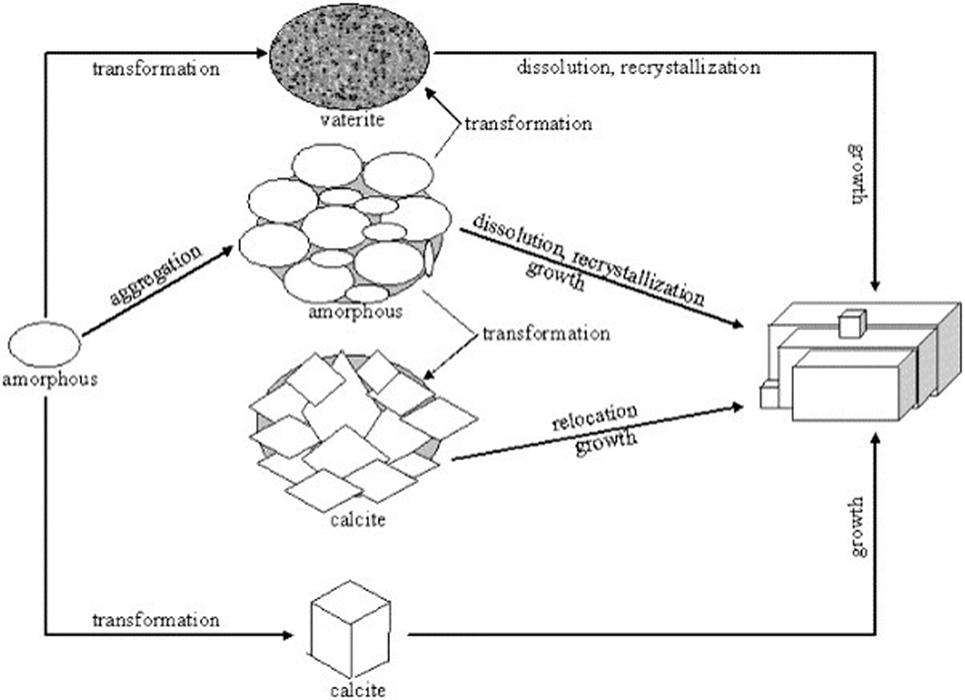
Figure 2. Polymorph formation mechanism of the precipitated calcium carbonate, adapted from Wei et al. (2003). It is being reproduced with permission from the copyright holder.
Zhang et al. (2012) studied the formation mechanisms of aragonite in the presence of Mg2+. They revealed that Mg2+ can promote the formation of aragonite rather than calcite. It was reported that the initially formed ACC by the reaction between Ca2+ and transformed into needle-like aragonite when the Mg2+ concentration was low, while at a high concentration, CaCO3⋅H2O was preferentially formed. Since Mg2+ has smaller ion diameter than Ca2+, the former shows stronger ability to form hydrate. Therefore, when Mg2+ concentration is high enough, it will inhibit the formation of crystalline structures of CaCO3. In contrast, when Mg2+ is incorporated into the calcite lattice, its thermodynamic stability is decreased while aragonite becomes stable in solution. The transformation mechanisms between ACC and aragonite in the presence of Mg2+ have also been investigated in several other studies (Raymond et al., 2007; Munemoto and Fukushi, 2008).
Rodriguez-Blanco et al. (2011) investigated the kinetics of the transformation among the ACC, calcite, and vaterite using various spectroscopic techniques including in situ time-resolved energy dispersive X-ray diffraction in conjunction with high-resolution electron microscopy, ex situ X-ray diffraction, and infrared spectroscopy. They revealed that the second step of transforming metastable vaterite into stable calcite is the rate-determining step, which is controlled by the surface area of calcite via Ostwald ripening (Ostwald, 1897), whereas the first dissolution step of ACC followed by transformation into vaterite occurs rapidly. According to Ostwald ripening, a redeposition of the dissolved small particles (e.g., vaterite) occurs at the large crystal surfaces (e.g., calcite) until they disappear and large particles become even larger. It was found that the solubility of each polymorph (e.g., calcite and vaterite) is the key factor controlling growth of calcite, and thus the solution can be supersaturated with respect to calcite, driving precipitation of the phase. Activation energies for calcite nucleation and crystallization were given as 73 ± 10 and 66 ± 2 kJ/mol, respectively.
The effect of impurities on the transformation of CaCO3 polymorphs was investigated in the presence of Mg ions by Zhang et al. (2012). It was revealed that the presence of Mg2+ in low concentrations promotes the transformation of ACC particles into the thermodynamically less stable aragonite rather than forming calcite, while at a high Mg2+ concentration, monohydrated CaCO3 is favorably formed. They suggested that the presence of Mg ions decreases the thermodynamic stability of calcite through inclusion of the lattice of calcite owing to the Mg ions’ stronger hydrate ability, resulting in preferential formation of aragonite in solution. In contrast, Ca2+ can be substituted by Mg2+ in the calcite lattice since Ca2+ and Mg2+ are interchangeable (Park et al., 2008). As a result, a magnesian calcite ((Ca,Mg)CO3) also can be formed in the presence of Mg2+.
Synthesis Variables and Their Effects on the Formation of CaCO3
The anhydrous crystalline polymorphs of CaCO3 strongly depend on the synthesis variables such as temperature, pressure, pH of the solution, reaction time, degree of supersaturation, ion concentration and ratio, ionic strength, stirring, type and concentration of additives, and feeding order (Tai and Chen, 1998; Jung et al., 2000; García-Carmona et al., 2003a,b; Shen et al., 2006; Meldrum and Cölfen, 2008; Chen and Xiang, 2009; Fuchigami et al., 2009; Ren et al., 2011; Chu et al., 2013; Zhao et al., 2013; Jiang et al., 2014; Ševčík et al., 2015; Chang et al., 2017). Although the formation of CaCO3 can be achieved by a simple precipitation reaction between Ca2+ and ions, the controllable formation of a specific polymorph of CaCO3 is still a practical challenge. Various synthesis factors and their effects on the formation of CaCO3 polymorphs have been investigated thus far using simple model chemicals (e.g., CaCl2 and K2CO3), or via the extraction of Ca2+ from natural silicates (e.g., wollastonite) (Zhao et al., 2013), and industrial wastes (e.g., steel slag, fly ash, waste cement, and blast furnace slag) (Wu et al., 2001; Huijgen and Comans, 2005; Eloneva et al., 2008a; Chang et al., 2011; Nduagu et al., 2012; Mun and Cho, 2013).
Effect of Temperature on the Formation of CaCO3 Polymorphs
While the synthesis factors affect the formation of polymorphs in multiple and interacting ways, temperature is considered the most critical factor affecting the formation of the polymorphs of CaCO3. Ogino et al. (1987) investigated the precipitation of highly supersaturated solutions of Ca2+ and ions and revealed that initially formed ACC transformed into crystalline polymorphs of CaCO3, particularly forming vaterite and calcite at low temperature (14–30°C) and forming aragonite and calcite at high temperature (60–80°C) at pH > 10. The complete transformation of metastable vaterite into calcite was achieved after about 200 min at 25°C, and aragonite was completely transformed into calcite after about 1,000–1,300 min at 60–80°C.
Chen and Xiang (2009) also investigated the effect of temperature on the formation of CaCO3 polymorphs by double injection of model chemicals of CaCl2 and NH4HCO3 solutions (0.125–0.25 mol/l) with a molar ratio of 1:1 at a stirring rate of 450 rpm and temperature ranging from 30 to 80°C. During the precipitations, pH was monitored and the values ranged from 7.03 to 7.48. They found that the vaterite content at 30, 60, 70, and 80°C was 98.6, 74.6, 19.6, and 0%, respectively, whereas the molar content of calcite was lower than 4.4% in this temperature range. The aragonite whiskers formed at 50°C, and the content increased with an increase of temperature. They concluded that the formation of lamellar vaterite at 30–40°C and whisker aragonite at higher temperatures was due to the decrease of values with the increase of temperature, which they determined by thermodynamic calculations.
Chu et al. (2013) examined CO2 mineralization into different polymorphs of CaCO3 using an aqueous CO2 systems employing CaCl2 and sterically hindered 2-amino 2-(hydroxymethly)-1,3-propanediol with or without carbonic anhydrase enzyme. They examined precipitation of CaCO3 for an equimolar system of Ca2+ and ions over 120 min with temperature ranging from 15 to 75°C. At 15°C, vaterite was the major polymorph of CaCO3 showing at 90%. In contrast, aragonite and calcite became dominant phases at 75°C. The composition of calcite increased with temperature up to 40°C, whereas the fraction of vaterite decreased in the same temperature range.
Ševčík et al. (2015) attempted to reveal the optimal synthesis conditions for preparing pure vaterite using CaCl2⋅2H2O and K2CO3 solutions. They performed a quantitative analysis by the Rietveld method employing powder X-ray diffraction (PXRD) with the assumption that vaterite exhibits two crystal structures, the well-established hexagonal structure (space group: P63/mmc) (Meyer, 1959) and triclinic structure (space group C1) proposed by Demichelis et al. (2013). However, the exact crystal structure of vaterite is still under debate due to the difficulties in obtaining large, pure, single crystals of vaterite (Kamhi, 1963; Mugnaioli et al., 2012; Kabalah-Amitai et al., 2013). Although there is uncertainty regarding the structure of vaterite, Ševčík et al. reported that pure vaterite (≥99 wt%), which is composed of 45.7% of hexagonal vaterite and 54.2% triclinic vaterite, could be obtained at 60°C and 600 rpm based on Rietveld analysis results.
Recently, Chang et al. examined the effect of temperatures on CaCO3 polymorphs under controlled pH and ratios using CaCl2 and K2CO3 solutions via ATR FT-IR and PXRD spectroscopies at 25, 50, and 80°C, stirring at 350 rpm (Chang et al., 2017). They confirmed that vaterite is a major component together with calcite at 25°C, and particularly at higher pH (~11) vaterite became the most dominant species. In contrast, at higher temperature of 80°C, aragonite was dominantly observed. Figure 3 illustrates the morphological structures of CaCO3.

Figure 3. SEM images of CaCO3: (A) spherical vaterite, (B) cubic calcite and rosette-shaped aragonite, and (C) needle-like aragonite. (D) Image of structural transformation of vaterite (small spherical particles) into calcite (planar arrays). Adapted with permission from Chang et al. (2017). Copyright 2017 American Chemical Society.
The effect of temperature on CaCO3 polymorphs was also investigated by Zhao et al. (2013). They synthesized PCC by using a calcium-bearing silicate mineral, wollastonite, via two steps of Ca source extraction and carbonation. The Ca source was extracted by using 1 M acetic acid, and the leachate was reacted with 0.25 M K2CO3 solution at 800 rpm for 30 min under two different reaction temperatures of 22 and 82°C for CaCO3 formation. The prepared CaCO3 particles were then quantitatively analyzed by employing PXRD. They revealed that at 22°C the CaCO3 particles were a mixture of calcite (24 wt%) and vaterite (76 wt%). By contrast, the sample synthesized at 82°C contained 87 wt% of aragonite with 13 wt% of calcite.
Said et al. (2013) synthesized PCC through a reaction between CO2 gas and Ca source extracted from steelmaking slag that contained 44.99 wt% of CaO. The Ca source was extracted by various ammonium salts such as ammonium nitrate (NH4NO3), ammonium acetate (CH3COONH4), and ammonium chloride (NH4Cl), and the leachates were reacted with bubbled CO2 gas (1 l/min) at 30°C under a magnetic stirrer operated at 600 rpm.
Although the temperature is the major synthesis factor determining the polymorphs of CaCO3, other factors such as pH, concentration of ions, impurities, and CO2 flow rate also compositively interact in the precipitation and crystallization processes. For instance, aragonite, which is generally formed at elevated temperature, can be obtained at lower temperature in the presence of impurities such as magnesium (Park et al., 2008). Said et al. (2016) also demonstrated aragonite PCC production via the Slag2PCC process. In order to avoid the evaporation of ammonia (NH3) in the NH4Cl solution, the operation was conducted at 45°C. Instead, the CO2 flow rate was optimized to obtain aragonite rather than rhombohedral calcite.
Effect of pH on the Formation of CaCO3 Polymorphs
Solution pH is one of the important factors determining polymorphs of CaCO3, affecting not only the equilibrium concentration of carbonate species (e.g., and ) but also the Ca−CO3 binding strength. In particular, the change in the Ca−CO3 binding strength is considered to play a significant role in the formation of CaCO3 polymorphs, influencing the dissolution behavior of each polymorph including ACC, which later converts into a specific polymorph of CaCO3 (Gebauer et al., 2008).
Gómez-Morales et al. (1996a,b) attempted to compared polymorphs according to different initial pH conditions using 0.0134 M CaCl2⋅2H2O and 0.04 M K2CO3/KHCO3 at 25°C, with stirring at 2,000 rpm. In particular, the pH conditions were adjusted from 7.8 to 10 using KOH and HCl solutions. In their experiments, vaterite was observed under pH lower than 9.3. However, when the pH exceeded 9.3, calcite was started to be produced together with vaterite. Therefore, calcite was expected to form at higher pH. These results partially agree with the work performed by Tai and Chen (1998). They investigated the formation of CaCO3 polymorphs in a pH range of 8.0–12.5 using CaCl2 and Na2CO3 solutions at temperature of 7, 24, and 58°C with stirring at 800 rpm. Their results also indicate that the calcite was produced as the pH increased. However, pure calcite formed in a different pH range depending on the reaction temperature. At 7°C, vaterite and calcite formed at pH ≤ 11 and pure calcite formed at pH > 11. At 24°C, pure calcite, aragonite, and vaterite were obtained at pH > 12, pH = 11, and pH < 10, respectively. At 58°C, aragonite and calcite precipitated at pH ≤ 11 and pure calcite precipitated at pH > 11. They also investigated the effect of ionic strength. It was observed that as the ionic strength was increased, the yield of calcite decreased, whereas the yield of aragonite increased even at conditions where calcite favorably formed.
Recently, Ramakrishna et al. (2016) observed the formation of polymorphs according to the temperature, reaction time, and pH using 0.1 M CaCl2 and 0.1 M Na2CO3 solutions at 25 to 50°C and at pH 10–12. They reported that pH 10 is the most suitable condition for obtaining pure aragonite, whereas calcite became the dominant polymorph when pH exceeded 10. Overall, calcite is favorably formed at high pH conditions, in general, greater than pH 11. However, aragonite is preferentially obtainable in a pH range of 9–11. At low pH conditions, typically below than pH 8, vaterite is favored (Han et al., 2005).
The production of PCC using steel converter slag via the slag2PCC process was investigated by Zevenhoven and coworkers (Eloneva et al., 2008b; Said et al., 2013, 2016; Mattila and Zevenhoven, 2014). The slag2PCC process includes two stages of Ca extraction and carbonation, based on the recirculation of an aqueous ammonium salt solution (Mattila and Zevenhoven, 2014). The ammonium salt solvent extract Ca from CaO or Ca(OH)2, and then it is reacted with CO2 gas. Said et al. (2013) conducted the Ca extraction by using mixtures of ammonium salts including NH4NO3, CH3COONH4, or NH4Cl and the extractant was precipitated in the carbonation process for 60 min with bubbling CO2 gas of 1 l/min at 30°C. The initial pH condition for the carbonation was ~8.5, and it was then stabilized to ~6 (Said et al., 2013). Interestingly, however, the final PCC product was found to be rhombohedral calcite, rather than vaterite or aragonite, which often have been found at similar pH ranges in the previous model chemical studies. They also synthesized calcium carbonates via the slag2PCC process by varying the CO2 flow rate in a pH range of ~9 to ~7 at 45°C, and it was found that both calcite and aragonite formed but the composition ratio depended more strongly on the CO2 flow rate than the pH condition (Said et al., 2016). In conclusion, pH condition affects the formation of polymorphs of calcium carbonate by shifting the equilibrium concentration of coronate species (e.g., , ) or changing the Ca−CO3 binding strength, but other operating factors such as temperature, CO2 flow rate, presence of acid and bases, and carbonation time also appear to more significantly influence the determination of the polymorphs (Saruhashi, 1955; Gómez-Morales et al., 1996a,b).
Effect of Other Factors on the Formation of CaCO3 Polymorphs
Other synthesis factors such as reaction time, stirring rate, impurities, and ultrasound treatment are also known to influence the formation of CaCO3 polymorphs. Reaction (aging) time is another important factor determining the size and shape as well as the polymorphs of CaCO3, by influencing the dissolution and recrystallization of crystals. Rodriguez-Blanco et al. (2011) investigated the kinetics of CaCO3 crystallization focusing on the transformation of ACC to stable calcite, via vaterite using synchrotron-based in situ time-resolved energy dispersive X-ray diffraction (ED-XRD). 1 M Na2CO3 and CaCl2 solutions ( = 1:1 ratio) were reacted at 7.5–25°C for 20 h, and they were monitored through in situ ED-XRD. They were differentiated into two stages, ACC to vaterite and vaterite to calcite transformations. They concluded that the precipitation process occurred in two stages: first, the initially formed ACC particles, which were confirmed as CaCO3⋅H2O, rapidly dehydrated and crystallized to form metastable vaterite; second, the formed vaterite then transformed to stable calcite via dissolution and reprecipitation. The second stage of the reaction was confirmed to be too slow compared with the first stage: the maximum yield of vaterite in the first stage was obtained after ~1 h for the case at 7.5°C, whereas the reaction rate of the second stage was approximately 10 times slower than that of the first stage. Zhao et al. (2015) also investigated the effect of reaction time on the crystallization of CaCO3 using a Ca(OH)2 solution at temperature of 60–120°C. The composition of the polymorphs was analyzed by using PXRD. They observed the formation of vaterite at the initial reaction; however, when the reaction time was increased to 4 h, calcite was found, although vaterite was still the dominant phase. They concluded that the reaction time more strongly affects the size and shape of the CaCO3.
The stirring rate in the precipitation of CaCO3 process affects the particle size and morphologies of the polymorphs. Yan et al. (2009) examined the effect of the stirring rate on the precipitation of the CaCl2 and Na2CO3 solution with varying stirring rate (50–1,000 rpm). At a lower stirring speed (100 rpm), calcite phase was dominantly observed, and a further increase of stirring speed led to the formation of vaterite. Similar results were also observed in a work performed by Ševčík et al. (2015). During the precipitation of the CaCl2⋅2H2O and K2CO3 solutions at a constant temperature of 60°C, lower stirring speed (200 rpm) generated a significant amount of calcite phase (36.3 wt%), whereas vaterite became dominant (≥98 wt%) at higher stirring speeds of 400 and 600 rpm. Although the stirring rate has relatively minor effect on the formation of CaCO3 polymorphs compared to other factors such as temperature, it often influences the hydrodynamics on the particle dynamics (Han et al., 2006). Han et al. observed that at low stirring rate, fine ACC particles were formed and then densely agglomerated. This could be ascribed to the total surface free energy: because the initially formed fine particles were unstable due to their high surface free energy, the fine crystals tended to aggregate to achieve a minimum total surface free energy.
The impurities are also known to significantly influence the formation of CaCO3 particles. Since the ex situ mineral carbonation process utilizes calcium and magnesium sources extracted from naturally occurring silicate minerals or industrial by-products/wastes, various impurities could be included in the carbonation step if a proper separation process is not provided. Therefore, providing an efficient impurity (e.g., magnesium, aluminum, and silica) separation method is an important step to ensure profitable marketability of PCC. De Crom et al. (2015) proposed a three-step process of PCC production using blast furnace slag. The process includes a physicochemical removal step for impurities from the leachate by employing temperature reduction (20 → 1°C) and pH elevation (4.4 → 8.4) with the aim of selectively precipitating Al, Mg, and Si, prior to PCC carbonation. Reportedly, it was possible to obtain a chemically pure PCC (>98% Ca) with a uniform scalenohedral morphology (>88% calcite) and a narrow (1.09 uniformity), small (D50 = 1.1 µm) particle size distribution. Morandeau and White (2015) investigated the effect of MgO content in the mineral carbonation using blast furnace slag. They showed that with a high MgO content, the PCC favorably formed ACC rather than calcite or vaterite.
An ultrasound irradiation technique has been applied to the formation of CaCO3 polymorphs (Price et al., 2011; Stoica-Guzun et al., 2012; Wagterveld et al., 2012; Njegić Džakula et al., 2014). The use of ultrasound can help control the crystallization process, and this is referred to as sonocrystallization (de Castro and Priego-Capote, 2007). Price et al. (2011) investigated the precipitation of calcium carbonate applying ultrasound. Saturated solutions of CaCl2 and NaHCO3 were used for the precipitation, employing ultrasound (20 kHz, intensity ranging from 1.5 to 18.5 W cm−2). It was found that at low ultrasound intensity vaterite was dominantly obtained, but at higher intensities the yields approached 100% calcite. Aragonite only formed at the high-intensity ultrasonic irradiation conditions. Wagterveld et al. (2012) also examined the effect of ultrasonic treatment on the early growth during CaCO3 precipitation. The results revealed that applying ultrasonic treatment helped increase the available surface area for polymorph growth, resulting in a higher volumetric precipitation rate. It was also reported that a more uniform size distribution of the precipitated polymorphs could be anticipated.
Controlling the size and shape of CaCO3 is one of the most important factors when producing a PCC since it determines the quality of the PCC. The particle size is compositively affected by various factors such as temperature, pH, additive types (impurities), concentration of calcium and CO2, solvent ratio, CO2 flow rate, stirring rate, and reaction time (Boyjoo et al., 2014). Bang et al. (2012) investigated the specific surface area and particle size of CaCO3 by bubbling CO2 in Ca(OH)2 solutions. They found that the Ca(OH)2 concentration and the CO2 flow rate significantly influenced the specific surface area, as well as the size of primary and secondary CaCO2 particles. Specifically, as the Ca(OH)2 concentration was increased (from 0.05 to 0.50 M), the BET surface area increased (from ~5 to ~25 m2/g), whereas as the CO2 flow rate was increased, the BET surface area decreased. The primary particle size of the CaCO3 also gradually increased with an increase of the CO2 flow rate at a low Ca(OH)2 concentration, but the tendency became weak at high Ca(OH)2 concentration. They also revealed that the CO2 bubble size also affected the particle size and specific surface area (Bang et al., 2015).
Conclusion
Because ex situ mineral carbonation entails a series of chemical processes above ground via reactions between CO2 and alkaline earth metals such as calcium or magnesium that are extracted not only from naturally occurring silicate minerals, i.e., wollastonite, olivine, and serpentine, but also industrial by-products or waste materials, i.e., coal fly ash, steel and stainless-steel slags, and cement and lime kiln dusts, it offers a number of advantages over the in situ mineral carbonation route. First, this technique allows the utilization of alkaline-metal feedstock extracted from industrial wastes. Therefore, it could provide an appropriate method for proper disposal or for recycling of the industrial wastes. Second, the final products, i.e., CaCO3, can be converted to value-added materials that can be utilized in various applications such as adhesives, sealants, food and pharmaceuticals, paints, coatings, paper, cements, and construction materials. The precipitate calcium carbonate quality is of utmost importance for practical implementation of an ex situ mineral carbonation because it could partly offset the total cost of CCS.
In this review, we addressed the key factors affecting the formation of CaCO3 polymorphs, including temperature, pH, concentration, reaction time, stirring, impurities, and ultrasound, particularly focusing on model chemical studies. Although the precipitation of CaCO3 involves a simple reaction between Ca2+ and , the polymorphs of CaCO3 and its derivatives with various physicochemical properties can be determined by the mutual relations among the synthesis factors. Therefore, a careful review of how each factor affects the polymorphs of CaCO3 could offer insight into the creation of highly value-added CaCO3 and its emerging applications for economically viable deployment. Further efforts to precisely control the morphology, agglomeration, and particle size distribution as well as polymorphs of the PCCs via research on the carbonation kinetics should be made together with designing demonstration processes for commercializing the technology.
Notice
All appropriate permissions have been obtained from the copyright holders of any work that has been reproduced in the manuscript.
Author Contributions
The manuscript was written through contributions of all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the “Climate Technology Development and Application” research project (K07701) through a grant provided by GIST in 2017.
References
Bałdyga, J., Henczka, M., and Sokolnicka, K. (2010). Utilization of carbon dioxide by chemically accelerated mineral carbonation. Mater. Lett. 64, 702–704. doi: 10.1016/j.matlet.2009.12.043
Bang, J.-H., Jang, Y. N., Kim, W., Song, K. S., Jeon, C. W., Chae, S. C., et al. (2012). Specific surface area and particle size of calcium carbonate precipitated by carbon dioxide microbubles. Chem. Eng. J. 198-199, 254–260. doi:10.1016/j.cej.2012.05.081
Bang, J.-H., Song, K., Park, S., Jeon, C. W., Lee, S.-W., and Kim, W. (2015). Effects of CO2 bubble size, CO2 flow rate and calcium source on the size and specific surface area of CaCO3 particles. Energies 8, 12304–12313. doi:10.3390/en81012304
Beck, R., and Andreassen, J.-P. (2010). The onset of spherulitic growth in crystallization of calcium carbonate. J. Cryst. Growth 312, 2226–2238. doi:10.1016/j.jcrysgro.2010.04.037
Bobicki, E. R., Liu, Q., Xu, Z., and Zeng, H. (2012). Carbon capture and storage using alkaline industrial wastes. Prog. Energy Combust. Sci. 38, 302–320. doi:10.1016/j.pecs.2011.11.002
Bonfils, B., Julcour-Lebigue, C., Guyot, F., Bodenan, F., Chiquet, P., and Bourgeois, F. (2012). Comprehensive analysis of direct aqueous mineral carbonation using dissolution enhancing organic additives. Int. J. Greenhouse Gas Control 9, 334–346. doi:10.1016/j.ijggc.2012.05.009
Boyjoo, Y., Pareek, V. K., and Liu, J. (2014). Synthesis of micro and nano-sized calcium carbonate particles and their applications. J. Mater. Chem. A 2, 14270–14288. doi:10.1039/c4ta02070g
Chang, E. E., Chen, C.-H., Chen, Y.-H., Pan, S.-Y., and Chiang, P.-C. (2011). Performance evaluation for carbonation of steel-making slags in a slurry reactor. J. Hazard. Mater. 186, 558–564. doi:10.1016/j.jhazmat.2010.11.038
Chang, R., Choi, D., Kim, M. H., and Park, Y. (2017). Tuning crystal polymorphisms and structural investigation of precipitated calcium carbonates for CO2 mineralization. ACS Sustain. Chem. Eng. 5, 1659–1667. doi:10.1021/acssuschemeng.6b02411
Chen, J., and Xiang, L. (2009). Controllable synthesis of calcium carbonate polymorphs at different temperatures. Powder Technol. 189, 64–69. doi:10.1016/j.powtec.2008.06.004
Chiaramonte, L., Zoback, M., Friedmann, J., Stamp, V., and Zahm, C. (2011). Fracture characterization and fluid flow simulation with geomechanical constraints for a CO2–EOR and sequestration project Teapot Dome Oil Field, Wyoming, USA. Energy Procedia 4, 3973–3980. doi:10.1016/j.egypro.2011.02.337
Chu, D. H., Vinoba, M., Bhagiyalakshmi, M., Hyun Baek, I. I., Nam, S. C., Yoon, Y., et al. (2013). CO2 mineralization into different polymorphs of CaCO3 using an aqueous-CO2 system. RSC Adv. 3, 21722–21729. doi:10.1039/C3RA44007A
COP21. (2015). COP21 Paris France Sustainable Innovation Forum 2015. Available at: http://www.cop21paris.org
de Castro, L., and Priego-Capote, F. (2007). Ultrasound-assisted crystallization (sonocrystallization). Ultrason. Sonochem. 14, 717–724. doi:10.1016/j.ultsonch.2006.12.004
De Crom, K., Chaing, Y. W., Van Gerven, T., and Santos, R. M. (2015). Purification of slag-derived leachate and selective carbonation for high-quality precipitated calcium carbonate synthesis. Chem. Eng. Res. Des. 104, 180–190. doi:10.1016/j.cherd.2015.07.029
Declet, A., Reyes, E., and Suárez, O. M. (2016). Calcium carbonate precipitation: a review of the carbonate crystallization process and applications in bioinspired composites. Rev. Adv. Mater. Sci. 44, 87–107.
Deer, W., Howie, R., and Zussman, J. (1985). An Introduction to the Rock-Forming Minerals, 1st Edn. Harlow, Essex: Longman.
Demichelis, R., Raiteri, P., Gale, J. D., and Dovesi, R. (2013). The multiple structures of vaterite. Cryst. Growth Des. 13, 2247–2251. doi:10.1021/cg4002972
Dickens, B., and Bowen, J. (1971). Refinement of the crystal structure of the aragonite phase of CaCO3. J. Re. Natl. Stand. Sec. A 75A, 27–32. doi:10.6028/jres.075A.004
Eloneva, S., Teir, S., Salminen, J., Fogelholm, C.-J., and Zevenhoven, R. (2008a). Fixation of CO2 by carbonating calcium derived from blast furnace slag. Energy 33, 1461–1467. doi:10.1016/j.energy.2008.05.003
Eloneva, S., Teir, S., Salminen, J., Fogelholm, C., and Zevenhoven, R. (2008b). Steel converter slag as a raw material for precipitation of pure calcium carbonate. Ind. Eng. Chem. Res. 47, 7104–7111. doi:10.1021/ie8004034
Eloneva, S., Teir, S., Savolahti, J., Fogelholm, C.-J., and Zevenhoven, R. (2007). “Co-utilisation of CO2 and calcium silicate-rich slags for precipitated calcium carbonate production,” in ECOS 2007, Padova, 1389–1396.
Fabian, M., Shopska, M., Paneva, D., Kadinov, G., Kostova, N., Turianicová, E., et al. (2010). The influence of attrition milling on carbon dioxide sequestration on magnesium–iron silicate. Miner. Eng. 23, 616–620. doi:10.1016/j.mineng.2010.02.006
Fuchigami, K., Taguchi, Y., and Tanaka, M. (2009). Synthesis of calcium carbonate vaterite crystals and their effect on stabilization of suspension polymerization of MMA. Adv. Powder Technol 20, 74–79. doi:10.1016/j.apt.2008.10.003
Fujioka, M., Yamaguchi, S., and Nako, M. (2010). CO2-ECBM field tests in the Ishikari Coal Basin of Japan. Int. J. Coal Geol. 82, 287–298. doi:10.1016/j.coal.2010.01.004
Garcia, S., Kaminska, S., and Maroto-Valer, M. M. (2010). Underground carbon dioxide storage in saline formations. Proc. Inst. Civil Eng. Waste Resour. Manage. 163, 77–88. doi:10.1680/warm.2010.163.2.77
García-Carmona, J., Gómez-Morales, J., and Rodríguez Clemente, R. (2003a). Rhombohedral-scalenohedral calcite transition produced by adjusting the solution electrical conductivity in the system Ca(OH)2-CO2-H2O. J. Colloid Interface Sci. 261, 434–440. doi:10.1016/S0021-9797(03)00149-8
García-Carmona, J., Morales, J. G., and Clemente, R. R. G. (2003b). Morphological control of precipitated calcite obtained by adjusting the electrical conductivity in the Ca(OH)2–H2O–CO2 system. J. Cryst. Growth 249, 561–571. doi:10.1016/S0022-0248(02)02173-5
Gebauer, D., Völkel, A., and Cölfen, H. (2008). Stable prenucleation calcium carbonate clusters. Science 322, 1819–1822. doi:10.1126/science.1164271
Gerdemann, S. J., O’Connor, W. K., Dahlin, D. C., Penner, L. R., and Rush, H. (2007). Ex situ aqueous mineral carbonation. Environ. Sci. Technol. 41, 2587–2593. doi:10.1021/es0619253
Gómez-Morales, J., Torrent-Burgués, J., López-Macipe, A., and Rodríguez-Clemente, R. (1996a). Precipitation of calcium carbonate from solutions with varying Ca2+ carbonate ratios. J. Cryst. Growth 166, 1020–1026. doi:10.1016/0022-0248(96)00083-8
Gómez-Morales, J., Torrent-Burgués, J., and Rodríguez-Clemente, R. (1996b). Nucleation of calcium carbonate at different initial pH conditions. J. Cryst. Growth 169, 331–338. doi:10.1016/S0022-0248(96)00381-8
Goodwin, A. L., Michel, F. M., Phillips, B. L., Keen, D. A., Dove, M. T., and Reeder, R. J. (2010). Nanoporous structure and medium-range order in synthetic amorphous calcium carbonate. Chem. Mater. 22, 3197–3205. doi:10.1021/cm100294d
Han, Y. S., Hadiko, G., Fuji, M., and Takahashi, M. (2005). Effect of flow rate and CO2 content on the phase and morphology of CaCO3 prepared by bubbling method. J. Cryst. Growth 276, 541–548. doi:10.1016/j.jcrysgro.2004.11.408
Han, Y. S., Hadiko, G., Fuji, M., and Takahashi, M. (2006). Factors affecting the phase and morphology of CaCO3 prepared by a bubbling method. J. Eur. Ceram. Soc. 26, 843–847. doi:10.1016/jeurceramsoc.2005.07.050
Huijgen, W. J. J., and Comans, R. N. J. (2005). Mineral CO2 sequestration by steel slag carbonation. Environ. Sci. Technol. 39, 9676–9682. doi:10.1021/es050795f
Huijgen, W. J. J., and Comans, R. N. J. (2006). Carbonation of steel slag for CO2 sequestration: leaching of products and reaction mechanisms. Environ. Sci. Technol. 40, 2790–2796. doi:10.1021/es052534b
Huijgen, W. J. J., Ruijg, G. J., Comans, R. N. J., and Witkamp, G.-J. (2006). Energy consumption and net CO2 sequestration of aqueous mineral carbonation. Ind. Eng. Chem. Res. 45, 9184–9194. doi:10.1021/ie060636k
Huntzinger, D. N., Gierke, J. S., Sutter, L. L., Kawatra, S. K., and Eisele, T. C. (2009). Mineral carbonation for carbon sequestration in cement kiln dust from waste piles. J. Hazard. Mater. 168, 31–37. doi:10.1016/j.jhazmat.2009.01.122
IEA (International Energy Agency). (2010). Energy Technology Perspectives. Paris: International Energy Agency.
IPCC. (2005). IPCC Special Report on Carbon Dioxide Capture and Storage. New York, NY: Cambridge University Press.
IPCC (Intergovernmental Panel on Climate Change). (2014). Climate Change 2014: Mitigation of Climate Change. New York, NY: Cambridge University Press.
Jarvis, K., Carpenter, R. W., Windman, T., Kim, Y., Nunez, R., and Alawneh, F. (2009). Reaction mechanisms for enhancing mineral sequestration of CO2. Environ. Sci. Technol 43, 6314–6319. doi:10.1021/es8033507
Jiang, J., Zhang, Y., Xu, D., and Liu, J. (2014). Can agitation determine the polymorphs of calcium carbonate during the decomposition of calcium bicarbonate? CrystEngComm 16, 5221–5226. doi:10.1039/C3CE42619J
Jung, W. M., Kang, S. H., Kim, W. S., and Choi, C. K. (2000). Particle morphology of calcium carbonate precipitated by gas-liquid reaction in a Couette-Taylor reactor. Chem. Eng. Sci. 55, 733–747. doi:10.1016/S0009-2509(99)00395-4
Kabalah-Amitai, L., Mayzel, B., Kauffmann, Y., Fitch, A. N., Bloch, L., Gilbert, P. U. P. A., et al. (2013). Vaterite crystals contain two interspersed crystal structures. Science 340, 454–457. doi:10.1126/science.1232139
Kamhi, S. R. (1963). On the structure of vaterite, CaCO3. Acta Crystallogr. 16, 770–772. doi:10.1107/S0365110X63002000
Katsifaras, A., and Spanos, N. (1999). Effect of inorganic phosphate ions on the spontaneous precipitation of vaterite and on the transformation of vaterite to calcite. J. Cryst. Growth 204, 183–190. doi:10.1016/S0022-0248(99)00174-8
Kim, Y. E., Lim, J. A., Jeong, S. K., Yoon, Y. I., Bae, S. T., and Nam, S. C. (2013). Comparison of carbon dioxide absorption in aqueous MEA, DEA, TEA, and AMP solutions. Bull. Korean Chem. Soc. 34, 783–788. doi:10.5012/bkcs.2013.34.3.783
Klusman, R. W. (2003). Evaluation of leakage potential from a carbon dioxide EOR/sequestration project. Energy Convers. Manage. 44, 1921–1940. doi:10.1016/S0196-8904(02)00226-1
Kralj, D., Brečević, L., and Kontrec, J. (1997). Vaterite growth and dissolution in aqueous solution III. Kinetics of transformation. J. Cryst. Growth 177, 248–257. doi:10.1016/S0022-0248(96)01128-1
Krevor, S. C., and Lackner, K. S. (2009). Enhancing process kinetics for mineral carbon sequestration. Energy Procedia 1, 4867–4871. doi:10.1016/j.egypro.2009.02.315
Ke, S., Cheng, X., Wang, Y., Wang, Q., and Wang, H. (2013). Dolomite, wollastonite and calcite as different CaO sources in anorthite-based porcelain. Ceram. Int. 39, 4953–4960. doi:10.1016/j.ceramint.2012.11.091
Krevor, S. C. M., and Lackner, K. S. (2011). Enhancing serpentine dissolution kinetics for mineral carbon dioxide sequestration. Int. J. Greenhouse Gas Control 5, 1073–1080. doi:10.1016/j.ijggc.2011.01.006
Lackner, K. S., Butt, D. P., and Wendt, C. H. (1997). Progress on binding CO2 in mineral substrates. Energy Convers. Manage. 38(Suppl), S259–S264. doi:10.1016/S0196-8904(96)00279-8
Lackner, K. S., Wendt, C. H., Butt, D. P., Joyce, E. L., and Sharp, D. H. (1995). Carbon dioxide disposal in carbonate minerals. Energy 20, 1153–1170. doi:10.1016/0360-5442(95)00071-N
Leung, D. Y. C., Caramanna, G., and Maroto-Valer, M. M. (2014). An overview of current status of carbon dioxide capture and storage technologies. Renew. Sust. Energ. Rev. 39, 426–443. doi:10.1016/j.rser.2014.07.093
Lin, P.-C., Huang, C.-W., Hsiao, C.-T., and Teng, H. (2008). Magnesium hydroxide extracted from a magnesium-rich mineral for CO2 sequestration in a gas–solid system. Environ. Sci. Technol. 42, 2748–2752. doi:10.1021/es072099g
Maroto-Valer, M. M., Fauth, D. J., Kuchta, M. E., Zhang, Y., and Andrésen, J. M. (2005). Activation of magnesium rich minerals as carbonation feedstock materials for CO2 sequestration. Fuel Process. Technol. 86, 1627–1645. doi:10.1016/j.fuproc.2005.01.017
Mattila, H.-P., Hudd, H., and Zevenhoven, R. (2014). Cradle-to-gate life cycle assessment of precipitated calcium carbonate production from steel converter slag. J. Clean. Prod. 84, 611–618. doi:10.1016/j.jclepro.2014.05.064
Mattila, H.-P., and Zevenhoven, R. (2014). Design of a continuous process setup for precipitated calcium carbonate production from steel converter slag. ChemSusChem 7, 903–913. doi:10.1002/cssc.201300516
Meldrum, F. C., and Cölfen, H. (2008). Controlling mineral morphologies and structures in biological and synthetic systems. Chem. Rev. 108, 4332–4432. doi:10.1021/cr8002856
Morandeau, A. E., and White, C. E. (2015). Role of magnesium-stabilized amorphous calcium carbonate in mitigating the extent of carbonation in alkali-activated slag. Chem. Mater. 27, 6625–6634. doi:10.1021/acs.chemmater.5b02382
Mugnaioli, E., Andrusenko, I., Schüler, T., Loges, N., Dinnebier, R. E., Panthöfer, M., et al. (2012). Ab initio structure determination of vaterite by automated electron diffraction. Angew. Chem. Int. Ed. 51, 7041–7045. doi:10.1002/anie.201200845
Mun, M., and Cho, H. (2013). Mineral carbonation for carbon sequestration with industrial waste. Energy Procedia 37, 6999–7005. doi:10.1016/j.egypro.2013.06.633
Munemoto, T., and Fukushi, K. (2008). Transformation kinetics of monohydrocalcite to aragonite in aqueous solutions. J. Mineral. Petrol. Sci. 103, 345–349. doi:10.2465/jmps.080619
Nduagu, E., Björklöf, T., Fagerlund, J., Mäkilä, E., Salonen, J., Geerlings, H., et al. (2012). Production of magnesium hydroxide from magnesium silicate for the purpose of CO2 mineralization – part 2: Mg extraction modeling and application to different Mg silicate rocks. Miner. Eng. 30, 87–94. doi:10.1016/j.mineng.2011.12.002
Nielsen, M. H., Aloni, S., and De Yoreo, J. J. (2014). In situ TEM imaging of CaCO3 nucleation reveals coexistence of direct and indirect pathways. Science 345, 1158–1162. doi:10.1126/science.1254051
Njegić Džakula, B., Kontrec, J., Ukrainczyk, M., Sviben, S., and Kralj, D. (2014). Polymorphic composition and morphology of calcium carbonate as a function of ultrasonic irradiation. Cryst. Res. Technol. 49, 244–256. doi:10.1002/crat.201300388
O’Connor, W. K., Dahlin, D. C., Nilsen, D. N., Rush, G. E., Walters, R. P., and Turner, P. C. (2000a). “CO2 storage in solid form: a study of direct mineral carbonation,” in 5th International Conference on Greenhouse Gas Technologies, Clearwater, FL.
O’Connor, W. K., Dahlin, D. C., Nilsen, D. N., Walters, R. P., and Turner, P. C. (2000b). Proceedings of the 25th International Technical Conference on Coal Utilization & Fuel Systems. Cairns.
O’Connor, W. K., Nielsen, D. N., Gerdemann, S. J., Rush, G. E., Waltera, R. P., and Turner, P. C. (2001). 18th Annual International Pittsburgh Coal Conference. Newcastle.
Oelkers, E. H., Gislason, S. R., and Matter, J. (2008). Mineral carbonation of CO2. Elements 4, 333–337. doi:10.2113/gselements.4.5.333
Ogino, T., Suzuki, T., and Sawada, K. (1987). The formation and transformation mechanism of calcium carbonate in water. Geochim. Cosmochim. Acta 51, 2757–2767. doi:10.1016/0016-7037(87)90155-4
Olajire, A. A. (2013). A review of mineral carbonation technology in sequestration of CO2. J. Pet. Sci. Eng. 109, 364–392. doi:10.1016/j.petrol.2013.03.013
Ostwald, W. Z. (1897). Studien über die Bildung und Umwandlung fester Körper. Z. Phys. Chem. 22, 289–330.
Pan, S. Y., Chang, E. E., and Chiang, P. C. (2012). CO2 capture by accelerated carbonation of alkaline wastes: a review on its principles and applications. Aerosol Air Qual. Res. 12, 770–791. doi:10.4209/aaqr.2012.06.0149
Park, A.-H. A., and Fan, L.-S. (2004). Mineral sequestration: physically activated dissolution of serpentine and pH swing process. Chem. Eng. Sci. 59, 5241–5247. doi:10.1016/j.ces.2004.09.008
Park, A.-H. A., Jadhav, R., and Fan, L.-S. (2003). CO2 mineral sequestration: chemically enhanced aqueous carbonation of serpentine. Can. J. Chem. Eng. 81, 885–890. doi:10.1002/cjce.5450810373
Park, W., Ko, S., Lee, S., Cho, K., Ahn, J., and Han, C. (2008). Effects of magnesium chloride and organic additives on the synthesis of aragonite precipitated calcium carbonate. J. Cryst. Growth 310, 2593–2601. doi:10.1016/j.jcrysgro.2008.01.023
Price, G. J., Mahon, M. F., Shannon, J., and Cooper, C. (2011). Composition of calcium carbonate polymorphs precipitated using ultrasound. Cryst. Growth Des. 11, 39–44. doi:10.1021/cg901240n
Ramakrishna, C., Thenepalli, T., Huh, J.-H., and Ahn, J. W. (2016). Precipitated calcium carbonate synthesis by simultaneous injection to produce nano whisker aragonite. J. Korean Ceram. Soc 53, 222–226. doi:10.4191/kcers.2016.53.2.222
Raymond, S. K. L., Chanock, J. M., Lennie, A., and Meldrum, F. C. (2007). Synthesis-dependent structural variation in amorphous calcium carbonate. CrystEngComm 9, 1144–1152. doi:10.1039/B710895H
Ren, M., Dong, C., and An, C. (2011). Large-scale growth of tubular aragonite whiskers through a MgCl2-assisted hydrothermal process. Materials 4, 1375–1383. doi:10.3390/ma4081375
Rodriguez-Blanco, J. D., Shaw, S., and Benning, L. G. (2011). The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, viavaterite. Nanoscale 3, 265–271. doi:10.1039/C0NR00589D
Said, A., Laukkanen, T., and Järvinen, M. (2016). Pilot-scale experimental work on carbon dioxide sequestration using steelmaking slag. Appl. Energy 177, 602–611. doi:10.1016/j.apenergy.2016.05.136
Said, A., Mattila, H., Järvinen, M., and Zevenhoven, R. (2013). Production of precipitated calcium carbonate (PCC) from steelmaking slag for fixation of CO2. Appl. Energy 112, 765–771. doi:10.1016/j.apenergy.2012.12.042
Sanna, A., Hall, M. R., and Maroto-Valer, M. (2012). Post-processing pathways in carbon capture and storage by mineral carbonation (CCSM) towards the introduction of carbon neutral materials. Energy Envoron. Sci. 5, 7781–7796. doi:10.1039/C2EE03455G
Sanna, A., Uibu, M., Caramanna, G., Kuusik, R., and Maroto-Valer, M. M. (2014). A review of mineral carbonation technologies to sequester CO2. Chem. Soc. Rev. 43, 8049–8080. doi:10.1039/C4CS00035H
Sanna, A., Wang, X., Lacinska, A., Styles, M., Paulson, T., and Maroto-Valer, M. M. (2013). Enhancing Mg extraction from lizardite-rich serpentine for CO2 mineral sequestration. Miner. Eng. 49, 135–144. doi:10.1016/j.mineng.2013.05.018
Santos, R., Chiang, Y., Elsen, J., and Van Gerven, T. (2014). Distinguishing between carbonate and non-carbonate precipitates from the carbonation of calcium-containing organic acid leachates. Hydrometallurgy 14, 90–94. doi:10.1016/j.hydromet.2014.05.001
Saruhashi, K. (1955). On the equilibrium concentration ratio of carbonic acid substances dissolved in natural water. Pap. Meteorol. Geophys. 6, 38–55. doi:10.2467/mripapers1950.6.1_38
Scripps Institution of Oceanography. (2017). The Keeling Curve. Available at: https://scripps.ucsd.edu/programs/keelingcurve
Ševčík, R., Pérez-Estébanez, M., Viani, A., Šašek, P., and Mácová, P. (2015). Characterization of vaterite synthesized at various temperatures and stirring velocities without use of additives. Powder Technol. 284, 265–271. doi:10.1016/j.powtec.2015.06.064
Shen, Q., Wang, L., Huang, Y., Sun, J., Wang, H., Zhou, Y., et al. (2006). Oriented aggregation and novel phase transformation of vaterite controlled by the synergistic effect of calcium dodecyl sulfate and n-pentanol. J. Phy. Chem. B 110, 23148–23153. doi:10.1021/jp064039n
Smit, B., Park, A.-H. A., and Gadikota, G. (2014a). The grand challenges in carbon capture, utilization, and storage. Front. Energy Res. 2:1–3. doi:10.3389/fenrg.2014.00055
Smit, B., Reimer, J. A., Oldenburg, C. M., and Bourg, I. C. (2014b). Introduction to Carbon Capture and Sequestration. London: Imperial College Press.
Spanos, N., and Koutsoukos, P. G. (1998). The transformation of vaterite to calcite: effect of the conditions of the solutions in contact with the mineral phase. J. Cryst. Growth 191, 783–790. doi:10.1016/S0022-0248(98)00385-6
Stoica-Guzun, A., Stroescu, M., Jinga, S., Jipa, I., Dobre, T., and Dobre, L. (2012). Ultrasound influence upon calcium carbonate precipitation on bacterial cellulose membranes. Ultrason. Sonochem. 4, 909–915. doi:10.1016/j.ultsonch.2011.12.002
Styring, P., Quadrelli, E. A., and Armstrong, K. (2015). Carbon Dioxide Utilisation: Closing the Carbon Cycle. Amsterdam: Elsevier.
Tai, C. Y., and Chen, F. B. (1998). Polymorphism of CaCO3, precipitated in a constant-composition environment. AIChE J. 44, 1790–1798. doi:10.1002/aic.690440810
Teir, S., Eloneva, S., and Zevenhoven, R. (2005). Production of precipitated calcium carbonate from calcium silicates and carbon dioxide. Energy Convers. Manage. 46, 2954–2979. doi:10.1016/j.enconman.2005.02.009
Teir, S., Revitzer, H., Eloneva, S., Fogelholm, C.-J., and Zevenhoven, R. (2007). Dissolution of natural serpentinite in mineral and organic acids. Int. J. Miner. Process. 83, 36–46. doi:10.1016/j.minpro.2007.04.001
Trushina, D. B., Bukreeva, T. V., Kovalchuk, M. V., and Antipina, M. N. (2014). CaCO3 vaterite microparticles for biomedical and personal care applications. Mater. Sci. Eng., C 45, 644–658. doi:10.1016/j.msec.2014.04.050
U.S. Geological Survey. (2011). Wollastonite: Mineral Commodity Summaries 2011. Reston, VA: U.S.G. Survey.
Uibu, M., Kuusik, R., Andreas, L., and Kirisimäe, K. (2011). The CO2-binding by Ca-Mg-silicates in direct aqueous carbonation of oil shale ash and steel slag. Energy Procedia 4, 925–932. doi:10.1016/j.egypro.2011.01.138
Wagterveld, R. M., Miedema, H., and Witkamp, G.-J. (2012). Effect of ultrasonic treatment on early growth during CaCO3 precipitation. J. Cryst. Growth Des. 12, 4403–4410. doi:10.1021/cg3005882
Wang, J., and Becker, U. (2009). Structure and carbonate orientation of vaterite (CaCO3). Am. Mineral. 94, 380–386. doi:10.2138/am.2009.293
Wang, X., and Maroto-Valer, M. M. (2011a). Dissolution of serpentine using recyclable ammonium salts for CO2 mineral carbonation. Fuel 90, 1229–1237. doi:10.1016/j.fuel.2010.10.040
Wang, X., and Maroto-Valer, M. M. (2011b). Integration of CO2 capture and mineral carbonation by using recyclable ammonium salts. ChemSusChem 4, 1291–1300. doi:10.1002/cssc.201000441
Wei, H., Shen, Q., Zhao, Y., Wang, D.-J., and Xu, D.-F. (2003). Influence of polyvinylpyrrolidone on the precipitation of calcium carbonate and on the transformation of vaterite to calcite. J. Cryst. Growth Des. 250, 516–524. doi:10.1016/S0022-0248(02)02484-3
White, C. M., Strazisar, B. R., Granite, E. J., Hoffman, J. S., and Pennline, H. W. (2003). Separation and capture of CO2 from large stationary sources and sequestration in geological formations—coalbeds and deep saline aquifers. J. Air Waste Mang. Assoc. 53, 645–715. doi:10.1080/10473289.2003.10466206
Wu, J. C. S., Sheen, J.-D., Chen, S.-Y., and Fan, Y.-C. (2001). Feasibility of CO2 fixation via artificial rock weathering. Ind. Eng. Chem. Res. 40, 3902–3905. doi:10.1021/ie010222l
Yan, F.-W., Zhang, S.-F., Guo, C.-Y., Zhang, X.-H., Chen, G.-C., Yan, F., et al. (2009). Influence of stirring speed on the crystallization of calcium carbonate. Cryst. Res. Technol. 44, 725–728. doi:10.1002/crat.200900190
Zhang, Z., Xie, Y., Xu, X., Pan, H., and Tang, R. (2012). Transformation of amorphous calcium carbonate into aragonite. J. Cryst. Growth 343, 62–67. doi:10.1016/j.jcrysgro.2012.01.025
Zhao, H., Park, Y., Lee, D. H., and Park, A.-H. A. (2013). Tuning the dissolution kinetics of wollastonite via chelating agents for CO2 sequestration with integrated synthesis of precipitated calcium carbonates. Phys. Chem. Chem. Phys. 15, 15185–15192. doi:10.1039/C3CP52459K
Keywords: CO2 storage, CO2 utilization, calcium carbonate, crystallization, polymorphism
Citation: Chang R, Kim S, Lee S, Choi S, Kim M and Park Y (2017) Calcium Carbonate Precipitation for CO2 Storage and Utilization: A Review of the Carbonate Crystallization and Polymorphism. Front. Energy Res. 5:17. doi: 10.3389/fenrg.2017.00017
Received: 11 April 2017; Accepted: 20 June 2017;
Published: 10 July 2017
Edited by:
Renato Baciocchi, University of Rome Tor Vergata, ItalyReviewed by:
Rafael Mattos Dos Santos, Sheridan College, CanadaMai Uibu, Tallinn University of Technology, Estonia
Copyright: © 2017 Chang, Kim, Lee, Choi, Kim and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youngjune Park, eW91bmdAZ2lzdC5hYy5rcg==