 George R. M. Dowson
George R. M. Dowson Peter Styring
Peter Styring- 1Chemical and Biological Engineering, The University of Sheffield, Sheffield, United Kingdom
- 2UK Centre for Carbon Dioxide Utilisation, Department of Chemistry, The University of Sheffield, Sheffield, United Kingdom
A mixture of 1- and 2-butanol was produced using a stepwise synthesis starting with a methyl halide. The process included a carbon dioxide utilization step to produce an acetate salt which was then converted to the butanol isomers by Claisen condensation of the esterified acetate followed by hydrogenation of the resulting ethyl acetoacetate. Importantly, the CO2 utilization step uses dry, dilute carbon dioxide (12% CO2 in nitrogen) similar to those found in post-combustion flue gases. The work has shown that the Grignard reagent has a slow rate of reaction with oxygen in comparison to carbon dioxide, meaning that the costly purification step usually associated with carbon capture technologies can be omitted using this direct capture-conversion technique. Butanol isomers are useful as direct drop-in replacement fuels for gasoline due to their high octane number, higher energy density, hydrophobicity, and low corrosivity in existing petrol engines. An energy analysis shows the process to be exothermic from methanol to butanol; however, energy is required to regenerate the active magnesium metal from the halide by-product. The methodology is important as it allows electrical energy, which is difficult to store using batteries over long periods of time, to be stored as a liquid fuel that fits entirely with the current liquid fuels infrastructure. This means that renewable, weather-dependent energy can be stored across seasons, for example, production in summer with consumption in winter. It also helps to avoid new fossil carbon entering the supply chain through the utilization of carbon dioxide that would otherwise be emitted. As methanol has also been shown to be commercially produced from CO2, this adds to the prospect of the general decarbonization of the transport fuels sector. Furthermore, as the conversion of CO2 to butanol requires significantly less hydrogen than CO2 to octanes, there is a potentially reduced burden on the so-called hydrogen economy.
Introduction
Carbon capture and storage (CCS) and carbon dioxide utilization (CDU) are two potential approaches to address mitigation of the ever-rising CO2 levels in the atmosphere, which have alarming climate implications (IPCC, 2014). For either approach to be effective in limiting or mitigating emissions, three key criteria have to be achieved. Sufficient amounts of CO2 must be stored or converted, or otherwise prevented from atmospheric release. The CO2 must be stored or converted for a sufficient period of time, and this must be achieved at a reasonable cost. For CCS, the overall storage capacity and intended storage time are potentially sufficient with estimates of total storage capacity exceeding 10 Tt, corresponding to centuries of current-level emissions (IEA, 2009). However, achieving this storage at practical cost is the major challenge facing CCS worldwide, with the exception of enhanced oil recovery (EOR) operations which have questionable total climate impacts given carbon footprint of the CO2 released on use of the additional oil that is produced (Jaramillo et al., 2009). While a handful of geological storage CCS projects without EOR have been initiated, and more are planned, these typically operate where high concentrations of CO2 are available (reducing capture/purification costs) or are reliant on significant government support or subsidy (Global CCS Institute, 2016). In the case of CO2-EOR and CO2 utilization, there is an additional complicating factor that storage is not long term as 92% of the recovered oil will be burned through combustion, with a similar fate for some, but not all of the utilization products.
Therefore, perhaps a better way to look at CO2 utilization is not through the mitigation potential of waste, but in the use of CO2 as a valuable C-1 resource. The latter approach, therefore, considers the utilization as avoiding new fossil carbon entering the supply chain and so indirectly reduces new emissions by mitigation and avoidance (von der Assen and Bardow, 2014). The carbon dioxide is converted for the production of chemicals with higher economic value, which results in a much more favorable immediate economic case than that with CCS. The resulting added-value from the CO2 ideally offsets some or all of the processing costs. This leaves CDU at a decided advantage over CCS in terms of economics, with several commercial processes based on CDU already in operation worldwide (Langanke et al., 2014; Gunning and Hills, 2015; Styring et al., 2015).
However, even with indirect effects included, the maximum amount of CO2 that may be utilized remains very small compared to total emissions. For example, if the entire global annual production of ethylene, the most widely manufactured commodity chemical containing carbon atoms, was carried out using carbon sourced exclusively from CO2, this would result in direct utilization of less than 1.5% of total global CO2 emissions (Stratas Advisors, 2016; Olivier et al., 2017). Even with other commodity chemicals included and assumptions made of indirect mitigation, theoretical utilization could represent only a small proportion of total emissions (Figure 1). Naturally, this further neglects the fact that direct conversion of CO2 into hydrocarbons, and especially aromatics, are not likely to become commercial processes in the near future.
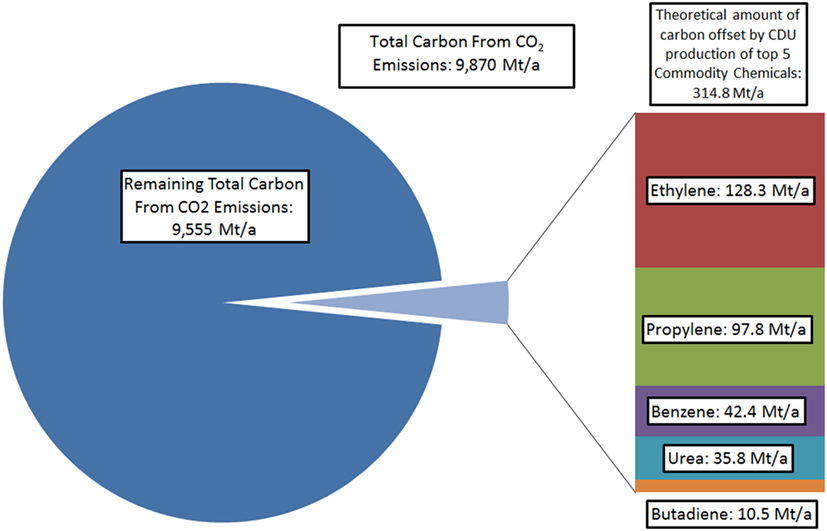
Figure 1. Comparison of total carbon in global CO2 emissions compared to carbon atoms contained in five major carbon-containing commodity chemicals, avoiding derivatives such as ethylene oxide. 2015 figures used (Levdikova, 2014; Yennigallla, 2015; Heffer and Prud’homme, 2015; Coombs, 2016).
It should be noted that while this demonstrates that CDU may only have minor or limited impact on total emissions mitigation, even when imagining a hypothetical sustainable route for the production of the most common commodity chemicals from CO2, profitable CDU processes may provide finance for CCS initiatives, thus offsetting further public or governmental costs (Hendricks et al., 2013). However, if CDU is only considered in isolation from CCS, other potential products in which to sink the emitted carbon must be made to allow for substantial reductions in CO2 emissions. In this context, the only carbon-containing materials that are used on sufficient scale for CDU to impact on total emissions are fuels themselves (Figure 2).
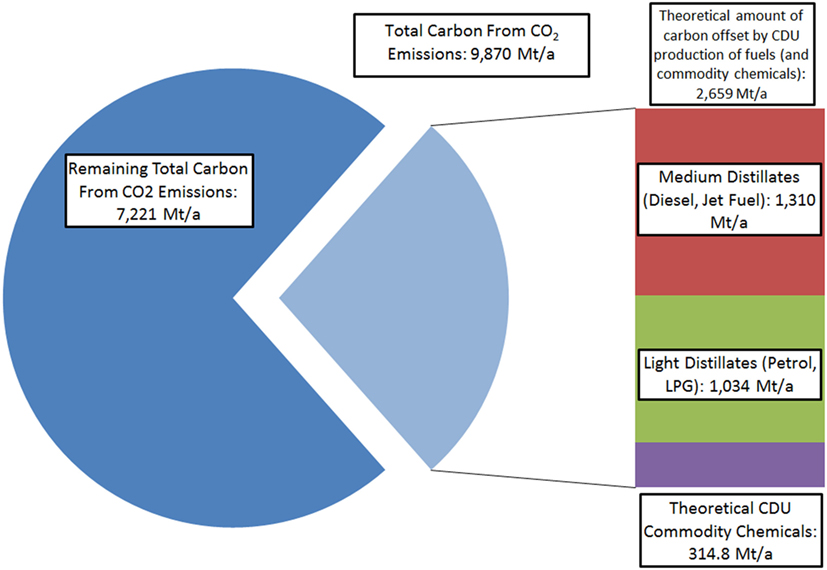
Figure 2. Comparison of total carbon in global CO2 emissions compared to carbon atoms contained in major liquid fuels and the theoretical collective commodity chemicals shown in Figure 1. 2015 Figures used (Dudley, 2015).
Low-Carbon Fuels from Carbon Dioxide
In the case of CDU-derived fuels, which re-release the CO2 that was initially trapped when they are combusted, the efficacy of the process in terms of emissions relies on observing the emissions avoided by the use of such a fuel. Since the CDU fuel itself may be seen as carbon neutral, provided all carbon atoms contained within the liquid fuel come from emission CO2 and no further emissions are generated in production, upon combustion useful work is extracted without the use of new fossil carbon. In the extreme scenario where all major liquid fuels are synthesized from CO2, the result is that road, water, and air vehicles essentially become indirectly powered by renewable electricity. By using synthetic fuels of this kind, it is possible to still use liquid fuel combustion engines and the existing fuels infrastructure. Thus, a 23% reduction/utilization of total emissions could be theoretically achieved (Mobility, 2015). Furthermore, with the large shift in the energy-generation sector toward renewable and lower carbon energy sources, this figure will rise as the total relative share of the emissions from the transport sector grows. It should be noted that other than widespread vehicle electrification, which is strongly limited for long-distance haulage, shipping and air-travel, or dramatic societal change, CDU fuels production represents one of the few potential strategies able to reduce transportation carbon emissions in the short-term future. Furthermore, it is the only strategy that may offer such reductions in transport emissions without significant disruption or onerous infrastructure redevelopment in the short and medium term, provided the CDU fuels are drop-in replacements for existing fossil analogs.
Unlike certain commodity or fine chemicals that can be generated from CO2 without significant energy input, such as polyurethane foams, fuels synthesis by definition requires a large energy input, naturally exceeding the combustion energy of the resulting product. Furthermore, the energy must necessarily be renewable or low carbon and as cheap as possible. In this respect, the conversion of CO2 into fuel may be better considered as an energy-vectoring strategy to balance electricity demand and supply as increasing amounts of renewable energy capacity is installed worldwide. Spot negative energy prices, once very rare, are now becoming more frequent even in large economies like Germany, Australia, UK, and parts of the United States (Martinez-Anido et al., 2016). Furthermore, renewable energy curtailment has grown dramatically. Germany, for example, has had to dramatically increase curtailment of renewable energy supplies in recent years, with over 1.5 GWh of renewable energy curtailed in 2014 (Morris, 2015). This wasted energy alone (not including a much larger amount of ultra-low and negative cost energy generated that year) would have been sufficient energy to produce approximately 50,000–80,000 l of CDU-gasoline, assuming the overall energy efficiency of the conversion of electricity to chemical energy in the fuel to be 30–50% with respect to the combustion energy density of petrol. Although this amount is relatively small in terms of the German liquid fuel market, with nearly 15,000 TWh per year of additional renewable energy expected to be produced worldwide by 2035, production of CDU fuels may be an important tool to balance periods of low demand and high generation in certain areas (BP Statistics, 2015; International Energy Agency, 2016).
It should be noted that even if a high-efficiency pathway to transform CO2 into a liquid fuel can be developed and deployed, the overall use of the energy will have poor efficiency due to the limitations of the combustion engines that use these fuels. Furthermore, since liquid fuels are relatively cheap per liter, if a competitive economic price is desired, the CDU fuel pathway would only be viable when energy prices are very low. Although a simple energy calculation shows that the absolute maximum energy price that allows CDU fuel production at around €1/L is €105/MWh (readily achievable by most low-carbon generation methods), the real energy price will have to be much lower to account for process inefficiencies and further issues such as the cost of capturing and purifying the CO2 from the waste streams to begin with.
The Cost of Carbon Capture
The high cost of capturing carbon dioxide from waste streams is a major challenge for both CDU and CCS strategies. Despite the thermodynamic de-mixing costs being relatively low in flue gas streams (approximately 150–250 MJ/t CO2 depending on concentration), CO2 capture and purification processes have much larger energy costs stemming from the sorption/desorption process. This is evident where an energetic driving force is required for either the sorption of the carbon dioxide, such as in high pressure adsorption and membrane separation, or for the desorption step, such as in amine-based chemisorption or vacuum swing adsorption. This results in a range of energy costs from approximately 1 to 4 GJ/t CO2 with the lower range consisting mainly of immature techniques and the upper range consisting of benchmark amine processes such as monoethanolamine (Dowson et al., 2016). Further issues associated with the capture processes involve the challenges of retrofitting existing plants and the footprint size of the capture process facility, which must be sited near to the point source, as well as the interactions between the sorbent materials and trace gases in the waste stream which are often corrosive or deleterious, especially to the benchmark amine sorbent materials (Uyanga and Idem, 2007; Soosaiprakasam and Veawab, 2008). Furthermore, temperature swing processes such as the benchmark amine-based processes require a substantial amount of waste heat to desorb the captured CO2 which may be available in sufficient quantity for a capture from certain point sources such as power plants although at a high parasitic energy cost (Lin et al., 2016). However, similar waste heat may not be available in industrial manufacturing waste streams, for example.
One potential method to avoid these issues is to directly react the low partial pressure CO2 with a reagent that provides a platform for further product generation. This approach has superficial similarities with amine-based chemisorption processes, in that a new chemical bond is formed with the flue gas CO2. However, such a direct utilization route has the benefit of not requiring large energy expenditure to re-release the CO2 into the gas phase, but instead takes the “capture product” and directly converts it further to form value-added compounds. By necessity, this will require a stoichiometric quantity of the reagent, which must therefore be able to be generated using environmentally benign processes.
Previous work carried out by this group has identified organometallics and particularly Grignard reagents as being suitable for the direct conversion of flue gas concentration of CO2 into value-added products such as acetic, terephthalic, and adipic acids (Dowson et al., 2015). The organomagnesium reagent, which was shown to react readily with CO2, even at low partial pressures, can then be regenerated from the by-product magnesium dihalide (MgX2). Electrolysis of the dihalide produces magnesium metal and the elemental halogen, which can then either directly halogenate certain hydrocarbons or be “burned” in hydrogen and subsequently reacted with an alcohol to produce the organohalide starting precursor (RX) to the initial Grignard reagent (Figure 3). In this way, the Grignard reactant is “looped” making the overall process akin to post-combustion chemical looping, which is typically carried out using calcium oxide as a reactive capture agent (Blamey et al., 2010). Again, the difference in this case is that the looping process produces a value-added compound containing the captured CO2 rather than low-value CO2 gas, combining both capture and utilization in one step.
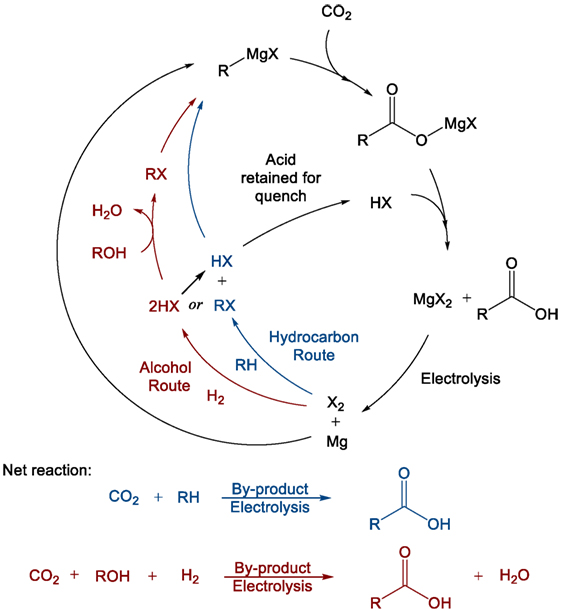
Figure 3. General Grignard CO2 reaction/cycle scheme including Grignard regeneration using alcohols or hydrocarbons as starting reagents.
As a result, the reduced magnesium consumed during the reaction provides the driving force for the overall process, rendering all other steps exothermic, and readily carried out at ambient pressures and temperatures. The hydrogen-halide burning process in particular is extremely exothermic and can also allow for surplus energy recovery (Group, 2014). The crucial magnesium electrolysis step, while highly energy intense, is also highly efficient in industrial magnesium production from sea salts. Faradaic efficiencies approaching 90% have been reported, and energy costs may be further reduced if the expensive magnesium chloride drying process can be avoided by using non-aqueous hydrogen halide for the post-Grignard reaction quench step (Polmear, 1999).
Naturally, it should be noted that Grignard reagents are highly sensitive to water, requiring that any incoming flue gas stream that supplies the CO2 for the first step in Figure 3 must be vigorously dried. While this would normally represent a considerable energy penalty for the process in terms of more standard capture approaches, compared with the electrolysis of one molecular equivalent of magnesium salt per molecule of CO2 captured, the energy cost is relatively small. Furthermore, there is the possibility that the drying process could be carried out using the otherwise non-recoverable portion of the exothermic heat (waste heat) from the reaction process to thermally cycle drying agents. In any case, the flue gas will need to be dried irrespective of the capture process, either post-capture in the case of amines or pre-capture for membranes or direct CDU.
Trace levels of water that are carried through to the Grignard reaction will cause the formation of methane and Mg(OH)X. The methane could potentially be recycled to increase the CO2:O2 ratio in the flue stream or otherwise utilized/reclaimed. The other product, Mg(OH)X, would be converted to the standard MgX2 by-product after the general Grignard quench with HX. Some care would have to be taken to avoid the formation of MgO, which is not typically reduced by electrolysis in the same way as MgX2, although this too is relatively easily converted back to MgX2. Overall, the presence of small quantities of water would primarily serve to only reduce reaction yield. Previous work carried out by our group has indicated that the reaction of Grignard reagents with oxygen is relatively slow compared to its reaction with CO2, although flue gas streams with very high oxygen levels may see trace amounts of the oxygen products, MgO, methanol, and possibly dimethyl ether (Goebel and Marvel, 1933; Dowson et al., 2015).
Without these undesirable reactions, the post-quench products of the reaction of Grignard reagents with CO2 are carboxylic acids, which are not typically used as liquid fuels, although they can be used to generate biodiesel esters. Further conversion steps are therefore required for the generation of a more conventional advanced fuel. One potential advanced fuel target would be butanol. Butanol is well recognized to be a potential drop-in replacement for petrol in road vehicles as well as a suitable blending agent for diesel engines (Yao et al., 2010). Unlike ethanol, which can only be blended with hydrocarbon fuels up to certain limits due to its corrosivity and hydrophilicity, butanol is lipophilic and non-corrosive (New Zealand Ministry of Transport and SGS Industrial, Penrose 2009). Furthermore, it has a slightly higher energy density (36.0 MJ/L) than gasoline (34.2 MJ/L), and similar burn rate and octane number, allowing completely unmodified vehicles to run on both butanol alone and butanol/petrol fuel blends (Szulczyk, 2010; Xu and Avedisian, 2015). Butanol further allows higher ethanol concentrations to be used within blended fuels, as it solubilizes ethanol in the bulk petrol and prevents ethanol volatilization (Yanowitz et al., 2011).
As a CDU fuel, butanol is also attractive as it requires significantly less hydrogen to produce it from CO2 than the proposed synthetic hydrocarbons suitable for petrol engines. This is due to its relatively high density, partial oxidation, and shorter chain length. Simple stoichiometric calculations show that 15% less hydrogen per liter and 26% less hydrogen per kilogram is required in the product fuel than for synthetic octanes. Given the limitations in energy efficiency of hydrogen generation from water electrolysis, this presents a potentially significant energy advantage for butanol production (Dincer and Acar, 2015). Furthermore, as a single-component fuel, it does not encounter issues with the absence of branched and aromatic compounds in synthetic hydrocarbons that reduce the octane numbers of methanol-to-gasoline and Fisher–Tropsch products to unsuitable levels (Dry, 2002). The fact that it can be used as a single-component fuel potentially allows for higher general engine efficiency to be achieved even than conventional petrol as the combustion characteristics are entirely uniform and not disrupted by variations in low-octane components, when the butanol is being used alone (Irimescu, 2012). The other advantage is that mixtures of butanol isomers can be directly used as drop-in fuels, meaning that isomer mixtures do not need to be separated prior to use.
However, the production of butanol from carbon dioxide, other than via biological processes or syngas, has previously only been reported as trace production in the high temperature/pressure hydrogenation of CO2 in water by platinum/cobalt catalysts (He et al., 2016). Here, we report a route to produce butanol-based fuel that is suitable for petrol-based applications in a multi-step-moderate-yield process. This has the potential to become a moderate-to-high yield process from CO2 once fully optimized. The reported process is demonstrated under near-ambient conditions for the major synthesis steps.
Experimental Methods
All reagents and deuterated solvents were purchased from Sigma-Aldrich at the highest available purity and were used without further purification. Schlenk-line techniques, used where indicated, were carried out using furnace-dried glassware under a dry nitrogen atmosphere. The apparatus was purged at least three times prior to handling of air-sensitive Grignard reagents. All solvents were purchased from VWR at HPLC grade or higher and were used without further purification. H2, CO2, N2, O2, and dry synthetic flue gas (12% CO2, 88% N2) were supplied by BOC-Linde. Catalyst pre-treatment and hydrogenation experiments were carried out in a 250-mL stainless steel Parr autoclave with heating mantle and glass insert and were stirred magnetically using an IKE stirrer hot-plate.
1H NMR spectra were recorded on a Bruker Avance 400 spectrometer operating at 400 MHz. All samples were dissolved in deuterochloroform and TMS added as the internal standard. Quantitative analysis of the butanol mixtures was performed by 1H NMR using mesitylene (10 μL) as an internal standard against 0.2 mL of reaction products diluted with deuterochloroform.
Butanol Synthesis: Grignard Reaction and Esterification
Magnesium Acetate Chloride (CH3COOMgCl)
A 250-mL two-necked, round bottom flask with magnetic stirrer bar attached to a reflux condenser and Schlenk-line apparatus with large silicone oil bubbler, under an atmosphere of dry nitrogen was cooled in an ice-water bath. Methylmagnesium chloride (50 mL, 3.0 M solution in THF, 150 mmol) was added under a flow of dry nitrogen. A further 100 mL of dry THF was added to maintain solubility of Grignard reagent throughout the reaction. After liquid additions were complete and the reaction vessel sealed, the nitrogen flow was stopped. Dry synthetic flue gas (12% CO2, 88% N2) was admitted at a slow flow rate that maintained positive overall pressure through a needle adapter and sparging needle inserted as deeply as possible into the liquid layer. Flow was monitored using the oil bubbler. Maintaining low gas flow rates and the use of a reflux condenser was important to limit the evaporative loss of solvent and formation of a gel-like solid within the reaction vessel, indicative of poor initial solubility or inverse solubility of the magnesium chloride acetate salt intermediate product in THF. The latter effect can become pronounced, particularly if temperatures become elevated. Complete reaction was indicated by a stable internal pressure when gas flow was paused (monitored by oil bubbler) or by the reaction remaining cold or cooling even when the ice bath was temporarily removed. The resulting product, magnesium acetate chloride, was used directly in solution without isolation.
Ethyl Acetate by Direct Esterification of Magnesium Acetate Chloride
The Grignard reaction mixture was quenched by the very slow addition of a small excess of sulfuric acid (30 mL, 166 mmol) dissolved in ethanol (100 mL). The resulting mixture was then fractionally distilled, using a long fractionating column due to the relatively close boiling points of THF and the ethyl acetate/ethanol azeotrope (66 and 71.8°C). The resulting product was dried using anhydrous magnesium sulfate and stored over molecular sieves. Crude product yield was 13.62 g, indicating an ethyl acetate yield of approximately 9.41 g (71% overall yield from the starting Grignard agent) with an azeotropic composition of 71.8% ethyl acetate. Separation of the azeotrope was not attempted as neither ethanol nor trace THF would adversely affect subsequent reaction stages.
Ethyl Acetoacetate
A Claisen condensation was carried out using a previously published procedure which emphasized the importance of ensuring the alkoxide salt was of highest purity possible to achieve maximum yield (Fisher and McElvain, 1934). Therefore, freshly prepared sodium ethoxide (1.3 g, 19 mmol) was added to the azeotropic mixture from the previous reaction and heated under reflux for 24 h. The mixture was then cooled and aqueous acetic acid added slowly to quench the base. The product, ethyl acetoacetate was extracted using diethylether, with additional distilled water added to the reaction mixture to limit ethanol contamination of product yield. 4.80 g of very pale yellow ethyl acetoacetate (69% yield from ethyl acetate) was isolated and characterized by 1H NMR spectrometry, comparing the resulting spectrum to reference spectra: δH (ppm) 400 MHz, CDCl3: 1.30 (t, 3 H), 2.28 (S, 3 H), 3.45 (S, 2 H), and 4.20 (q, 2 H).
Hydrogenation Catalyst (Cu/ZnO) Preparation
ZnO (32.55 g, 400 mmol) was added to a 1 M aqueous solution of Cu(NO3)2 (100 mL, 100 mmol) and stirred to form a well-dispersed slurry. The suspension was dried under vacuum using a rotary evaporator and the moist solid further dried using the same method with the addition of 2-propanol to drive off moisture by azeotropic evaporation. The resulting solid was placed in a furnace at 500°C for 16 h to yield a gray-black powder of CuO supported on ZnO. 2 g of the resulting powder was placed in a 250-mL Parr autoclave and heated to 200°C under 10 bar H2 for 4 h. During cooling, the autoclave was evacuated to remove any generated water, yielding a copper-orange powder of Cu on ZnO with a loading of 20 wt% Cu determined by the initial molar ratio of the reactants.
Hydrogenation and Hydrogenolysis
Ethyl acetoacetate (4.80 g, 36.9 mmol) and ethanol (5 mL) were added to the catalyst contained in the 250-mL Parr autoclave which was then sealed and purged using H2 to a pressure of 20 bar. Reaction was continued for 16 h. The reaction mixture was cooled and filtered, then dried over anhydrous magnesium sulfate to yield 10.5 mL of mixed alcohols, which were analyzed by 1H NMR spectroscopy (Table 1) with quantitative analyses carried out using mesitylene [δH(ppm) 400 MHz, CDCl3: 2.25 (s, 9 H), 6.75 (m, 3 H)] as an internal standard. The branched methyl group on the 2-butanol appears as a doublet at a shift of 1.12 ppm was cleanly away from the bulk of the signals. The combined integral of the terminal CH3 groups (at δH 0.7–0.8 ppm) where the 1- and 2-butanol signals overlap was determined and the amount of 1-butanol was determined by subtracting the amount of 2-butanol based on the amount determined from the 1.12 ppm doublet. A small doublet was observed next to the 2-butanol doublet which did not fit with being a carbon satellite and fitted the reference spectrum for 1,3-butane diol (the terminal CH3), which was used to calculate the butanediol concentration. The combined butanol isomers, butanediol and acetone were isolated in 98% conversion from ethyl acetoacetate with 95% selectivity to the butanol isomers. The overall four-step conversion from methyl magnesium chloride (chloromethane) to butanol isomers is 44%.

Table 1. Composition of total product mixture, determined by 1H NMR, 400 MHz, CDCl3.
Results and Discussion
Producing butanol entirely and directly from CO2 using the Grignard process would not be possible without additional steps and starting reagents. First, the Grignard process requires a hydrocarbon or alcohol to form the initial Grignard reagent and the product of the Grignard reaction would have to be converted from a carboxylic acid to the final alcohol product. With CDU processes in mind, the two obvious candidates for the starting hydrocarbon and alcohol shown in Figure 3 would be methane and methanol, both readily synthesized by the hydrogenation of CO2 (Figure 4) (Mikkelsen et al., 2010; Saeidi et al., 2014). The selective formation of methyl halide from the reaction of methane and the elemental halogen is somewhat challenging, due to the higher reactivity of the products than starting materials, although appropriate choice of rare-earth or precious metal catalyst can yield the desired product (Olah et al., 1985; Podkolzin et al., 2007). By comparison, the conversion of methanol into any of the methyl halides is rather trivial using the hydrogen halide to carry out nucleophilic substitution. The former pathway remains potentially interesting as it does not require the addition of hydrogen to the process shown in Figure 3, although additional hydrogen would be needed in the CDU production of methane over methanol. With either of these compounds used as starting materials, the product of the Grignard process, after quenching, is acetic acid. This would have to then be condensed and hydrogenated to form the desired product, butanol.
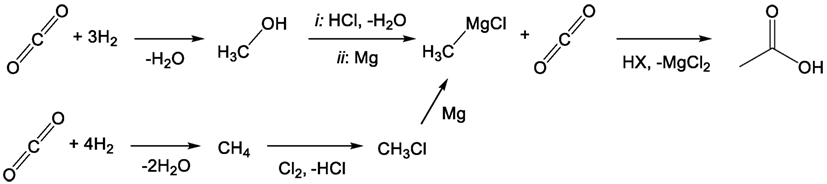
Figure 4. Overall preparation of acetic acid via CO2 hydrogenation followed by Grignard reaction.
Condensing two equivalents of smaller molecules such as ethanol, to produce butanol has previously been reported by Guerbet chemistry (Koda et al., 2009; Dowson et al., 2013; Ho et al., 2016). However, production of butanol from acetic acid has not previously been reported. Although butanol has previously been made directly from CO2, the yield was limited (Irimescu, 2012). Acetic acid can be condensed in the Claisen self-condensation via ethyl acetate to produce the desired four-carbon chain product, as shown in Figure 5.

Figure 5. Esterification of acetic acid and Claisen condensation of ethyl acetate.
While this reaction is readily carried out in the presence of a strong base, the final alkyl acetoacetate (ethyl acetoacetate in the case shown in Figure 5) product cannot be isolated without an acid quench, requiring a stoichiometric equivalent of base to be consumed to drive the otherwise mildly endergonic reaction. Care must also be taken that the acid quench does not hydrolyze the alkyl ester to produce the oxobutanoic acid (acetoacetic acid), as this is not stable, particularly at room temperature, decomposing to produce acetone and carbon dioxide (Hay and Bond, 1967).
Hydrogenation of the produced acetoacetate ester to directly yield butanol had not been previously reported in the scientific literature but similar hydrogenation processes using supported Cu(0) catalysts have been reported, particularly in the reduction of highly oxidized species such as low-molecular weight esters, glycerol, dimethyl maleate, and carbon dioxide (Figure 6) (van de Scheur and Staal, 1994; Brands et al., 1999; Schlander and Turek, 1999; Bienholz et al., 2011).
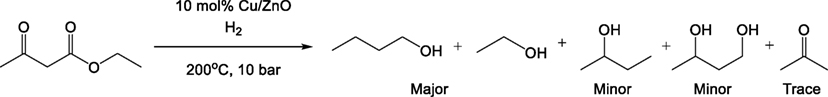
Figure 6. Hydrogenation of ethyl acetoacetate to produce butanol, ethanol, and minor by-products.
By including all compounds generated and consumed during the overall reaction process, a basic energetic pathway of the generation of butanol from the reaction of methanol with CO2 and hydrogen, as described in the experimental section can be constructed using the available reaction enthalpy data (Figure 7).
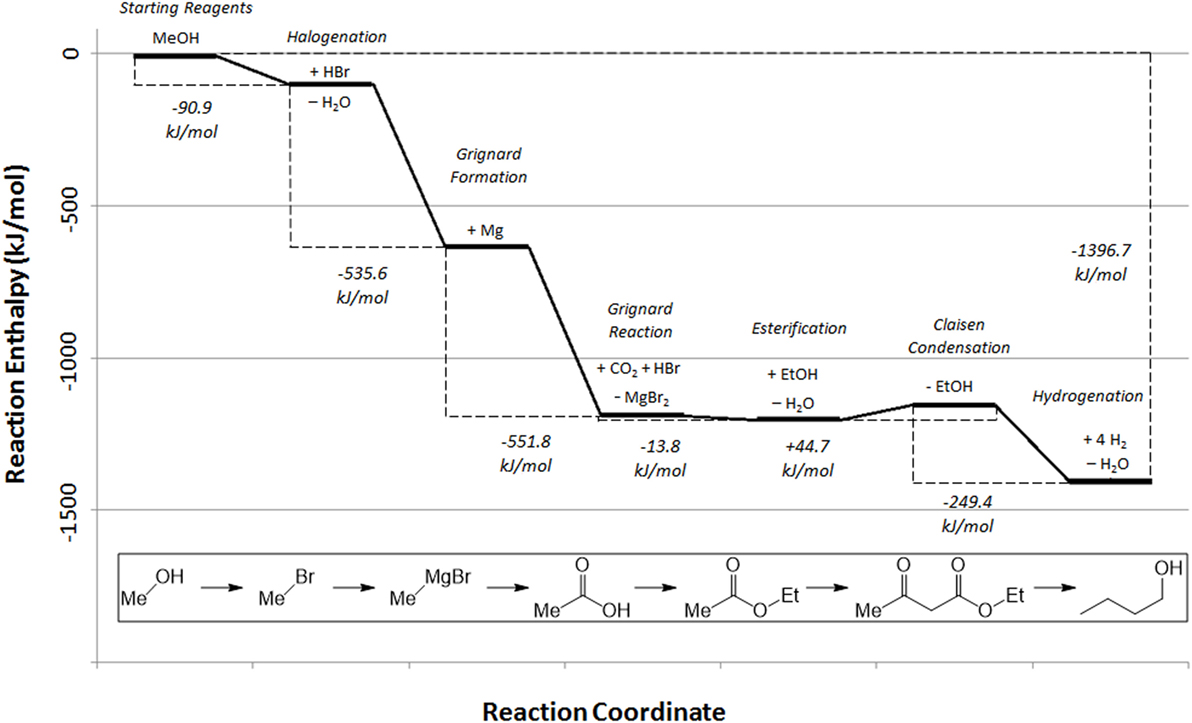
Figure 7. Reaction enthalpy profile for the synthesis of butanol from methanol in a carbon dioxide utilization process (NIST, 2017).
Note that this energy calculation in Figure 7 excludes the initial formation of the methanol (or methane) starting reagent, previously illustrated in Figure 4, for clarity.
From the starting methanol, the reaction profile clearly demonstrates the strongly exothermic nature of the overall reaction pathway, but naturally does not show the energy required to regenerate the Grignard agent, which provides the majority of the driving force for the overall process, with the remaining steps driven by the by-production of water in the esterification and hydrogenation steps. Furthermore, it should be noted that the Claisen condensation has a slightly positive overall enthalpy of reaction. This, in combination with the reaction entropy, renders this step of the reaction non-spontaneous without consumption of one equivalent of base by use of acid work-up (Davis and Garratt, 1991). However, given the exothermic nature of the overall reaction pathway, a broad estimation of the overall energy cost of the reaction, starting from methanol or methane, can be calculated from the energy requirements of magnesium regeneration, hydrogen production, and Claisen base/acid. Note that this calculation does not include energy costs associated with the drying of the carbon dioxide source and assumes 100% reaction yield. Simultaneously, potential energy recovery of the 15.26 MJ/L exotherm from the reaction process shown in Figure 7, has not been included, which would make the overall process more favorable. While the drying costs will be small in comparison with the other energy requirements such as magnesium electrolysis, reaction yields may be limited. In fact, magnesium electrolysis costs can be minimized by realizing that the process operates commercially at large scale and so it would be economically more favorable to batch process the regeneration of magnesium as part of that process rather than using dedicated electrolysers. At this stage, energy costs associated with the process alone are considered. A full life-cycle analysis is being carried out to account for all impacts, including the impact of methanol production and transport of the by-product to the electrolysis site. However, a full scope three LCA falls outside the scope of this paper due to the complexity of the analysis.
By comparison of the specific energy of the starting materials (methanol and methane, respectively) with the energy density of the butanol product, and then comparing this difference with a reasonable minimum overall energy cost of conversion (including magnesium regeneration, hydrogen generation, and the stoichiometric acid and base required to complete the Claisen condensation) a calculation of the efficiency of the transformation via this route, with the assumptions listed above, can be made (Table 2).
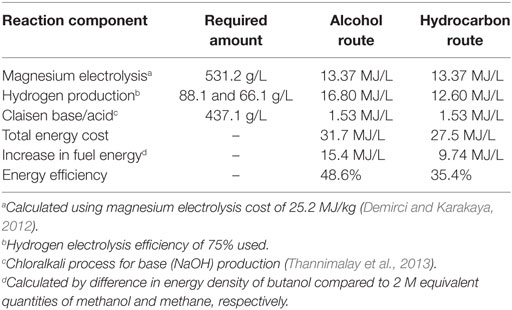
Table 2. Minimum energy cost per liter of butanol fuel produced from methanol and methane.
It should be emphasized that Table 2 represents the minimum energy cost of butanol production calculated with the assumptions listed below the table and assumes 100% chemical yields for all steps but with realistic energy requirements for the reagent costs. While such maximal yields are implausible, near-quantitative yields for each step should be possible in principle.
Note also that this calculation omits the production of methanol/methane from CO2 as with Figure 7. A calculation including the energy costs of the additional hydrogen required to form the methanol and methane required for the Grignard route are shown in Table 3. However, it should be noted that these figures omit any costs of capture and purification of CO2 from a waste gas stream, which would be required for the initial step of this route.

Table 3. Minimum energy cost per liter of butanol fuel produced from CO2 and hydrogen only.
These two tables illustrate that the energy costs associated with chemical looping of magnesium to capture CO2 and upgrade methanol or methane to drop-in petrol replacement does not significantly outstrip the energy gain in the content of the produced fuel. In addition, it can be seen in the two tables that the conversion of CO2, methanol or methane into butanol by a CDU approach, and without any additional energy recovery but with optimistic reaction yields, could in principle convert nearly half of a given quantity of electrical energy into a storable fuel that is directly compatible with existing infrastructure. While it is tempting to therefore make comparisons with the efficiencies of existing energy storage methods such as pumped hydroelectric storage and compressed air energy storage (which are higher), these approaches would have a completely different role in energy storage than a CDU-based fuel.
Overall, we believe that it is not unreasonable to suggest that CDU fuel production could realistically represent a new tool in balancing electricity demand and supply and could, with sufficient installation of renewable energy, make a tangible impact in reducing the carbon footprint of liquid fuel transportation.
Conclusion
Butanol can be produced using a multi-step synthetic approach from methane or methanol via a Grignard reagent that reacts with dilute carbon dioxide in nitrogen to give an acetate intermediate. Classic synthetic organic chemistry then allows homologation to give a four-carbon backbone that is partially hydrogenated to yield a mixture of 1- and 2-butanol. Each isomer has a higher octane number and energy density than octane itself and can be used as a direct drop-in fuel for gasoline-based combustion engines. This means the fuel can be used directly in existing automobile engines without blending or modification of those engines. The transformation, which is overall exothermic from the Grignard reagent, is facilitated by the high energy of the organometallic starting material. Energy is therefore required to regenerate the magnesium from the stoichiometric amount of magnesium halide by-product. If this is reprocessed using standard high-efficiency industry electrolysis techniques using renewable energy then the cost of this step is minimized.
The methodology allows liquid fuels to be produced from renewable, weather-dependent electricity at times where it would otherwise be curtailed, or in situations where dedicated generation is installed. This provides a potentially excellent method for chemical energy storage across seasons, otherwise not possible using other storage methods. The process also adds value to the system by removing carbon dioxide from primary emissions sources, storing otherwise curtailed excess renewable energy and by avoiding new fossil carbon from entering the supply chain when the fuel is used. While current projected utilization quantities are expected to fall short of the vast quantities of CO2 anthropogenically emitted, it provides a tool in the arsenal for the production of low-carbon fuels and to balance energy demands in an increasingly intermittently powered world. It is recognized that the fuel will eventually lead to CO2 emissions, but this will not be new carbon but second generation, upcycled carbon.
The fact that flue gas concentration carbon dioxide is used means that the costly carbon capture step can be omitted, although it would be required in some form for total synthesis of fuel from CO2 and hydrogen alone. In the original case where methanol or methane are used as starting reagents, this is an example of reactive capture of CO2 where the gas is chemically removed from the flue gas stream, thereby purifying the waste stream while at the same time producing a value-added product. The process has a number of steps including the esterification and Claisen condensation where improvements can be made through more precise definition of the reaction parameters. The overall carbon avoidance can also be improved by using methanol sources from CO2 to prepare the Grignard reagent through methanol bromination and by preparing the methyl rather than the ethyl ester. We will also look at using real flue gases from industrial emissions to test the robustness of the methodology and product purity.
Author Contributions
GRMD was the Postdoctoral Research Associate who carried out the laboratory work. PS was the academic supervisor and Principal Investigator. There were equal contributions to the production of this paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
We thank EPSRC for funding the 4CU Programme Grant (EP/K001329/1) at the University of Sheffield which has supported this work and GRMD. We also thank EPSRC for funding of the CO2Chem Grand Challenge Network to PS (EP/K007947/1 and EP/H035702/1).
References
Bienholz, A., Blume, R., Knop-Gericke, A., Girgsdies, F., Behrens, M., and Claus, P. (2011). Prevention of catalyst deactivation in the hydrogenolysis of glycerol by Ga2O3-modified copper/zinc oxide catalysts. J. Phys. Chem. C 115, 999–1005. doi: 10.1021/jp104925k
Blamey, J., Anthony, E. J., Wang, J., and Fennell, P. S. (2010). The calcium looping cycle for large-scale CO2 capture. Prog. Energy Combust. Sci. 36, 260–279. doi:10.1016/j.pecs.2009.10.001
Brands, D. S., Poels, E. K., and Bliek, A. (1999). Ester hydrogenolysis over promoted Cu/SiO2 catalysts. Appl. Catal. A Gen. 184, 279–289. doi:10.1016/S0926-860X(99)00106-4
Coombs, D. (2016). Propylene: The “Other” Olefin, Goldman Sachs Chemicals Intensity Conference, March 2016. Houston, TX, USA.
Davis, B. R., and Garratt, P. J. (1991). “Acylation of esters, ketones and nitriles,” in Comprehensive Organic Synthesis, eds B. M. Trost, and I. Fleming. 2nd Edn, (Pergamon, Oxford, UK), 795–863.
Demirci, G., and Karakaya, I. (2012). “Molten salt hydrolysis of MgCl2 in a cell with rapid chlorine removal feature,” in 2012 Magnesium Technology, eds S. N. Mathaudhu, W. H. Sillekens, N. R. Neelameggham, and N. Hort (Hoboken, NJ, USA: John Wiley & Sons), 59–62.
Dincer, I., and Acar, C. (2015). Review and evaluation of hydrogen production methods for better sustainability. Int. J. Hydrogen Energy 40, 11094–11111. doi:10.1016/j.ijhydene.2014.12.035
Dowson, G. R. M., Dimitriou, I., Owen, R. E., Reed, D. G., Allen, R. W. K., and Styring, P. (2015). Kinetic and economic analysis of reactive capture of dilute carbon dioxide with Grignard reagents. Faraday Discuss. 183, 47–65. doi:10.1039/c5fd00049a
Dowson, G. R. M., Haddow, M. F., Lee, J., Wingad, R. L., and Wass, D. F. (2013). Catalytic conversion of ethanol into an advanced biofuel: unprecendented selectivity for n-butanol. Angew. Chem. Int. Ed. 52, 9005–9008. doi:10.1002/anie.201303723
Dowson, G. R. M., Reed, D. G., Bellas, J.-M., Charalambous, C., and Styring, P. (2016). Fast and selective separation of carbon dioxide from dilute streams by pressure swing adsorption using solid ionic liquids. Faraday Discuss. 2016, 511–527. doi:10.1039/C6FD00035E
Dry, M. E. (2002). The Fischer-Tropsh process: 1950–2000. Catal. Today 71, 227–241. doi:10.1016/S0920-5861(01)00453-9
Fisher, N., and McElvain, S. M. (1934). The acetoacetic ester condensation. VII. The condensation of various alkyl acetates. J. Am. Chem. Soc. 56, 1766–1769. doi:10.1021/ja01323a034
Global CCS Institute. (2016). in Global Status of CCS: 2016, Projects, Policy and Markets, eds P. Grubnic, I. Havercroft, L. Irlam, N. James, P. Tomski, and S. Vaghi (Brussels, Belgium: Global CCS Institute).
Goebel, M., and Marvel, C. (1933). The oxidation of Grignard reagents. J. Am. Chem. Soc. 55, 1693–1696. doi:10.1021/ja01331a065
Gunning, P., and Hills, C. (2015). 9th International Conference on the Environmental and Technical Implications of Construction with Alternative Materials. Santander, Spain.
Hay, R. W., and Bond, M. A. (1967). Kinetics of the decarboxylation of acetoacetic acid. Aust. J. Chem. 20, 1823–1828. doi:10.1071/CH9671823
He, Z., Qian, Q., Ma, J., Meng, Q., Zhou, H., Song, J., et al. (2016). Water-enhanced synthesis of higher alcohols from CO2 hydrogenation over a Pt/Co3O4 catalyst under milder conditions. Angew. Chem. Int. Ed. 55, 737–741. doi:10.1002/anie.201507585
Heffer, P., and Prud’homme, M. (2015). Short-Term Fertilizer Outlook 2015–2016. Paris, France: International Fertilizer Industry Association.
Hendricks, C., Noothout, P., Zakkour, P., and Cook, G. (2013). Implications of the Reuse of Captured CO2 for European Climate Action Policies. Netherlands: ECOFYS.
Ho, C. R., Shylesh, S., and Bell, A. T. (2016). Mechanism and kinetics of ethanol coupling to butanol over hydroxyapatite. ACS Catal. 6, 939–948. doi:10.1021/acscatal.5b02672
IEA. (2009). in Technology Roadmap: Carbon Capture and Storage, eds T. Kerr, and B. Beck (Paris, France: IEA).
IPCC. (2014). in Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, eds R. K. Pachauri, and L. A. Meyer (Geneva, Switzerland: IPCC).
Irimescu, A. (2012). Performance and fuel conversion efficiency of a spark ignition engine fuelled with iso-butanol. Appl. Energy 96, 477–483. doi:10.1016/j.apenergy.2012.03.012
Jaramillo, P., Griffin, W. M., and McCoy, S. T. (2009). Life cycle inventory of CO2 in an enhanced oil recovery system. Environ. Sci. Technol. 43, 8027–8032. doi:10.1021/es902006h
Koda, K., Matsu-ura, T., Obora, Y., and Ishii, Y. (2009). Guerbet reaction of ethanol to n-butanol catalyzed by iridium complexes. Chem. Lett. 38, 838–839. doi:10.1246/cl.2009.838
Langanke, J., Wolf, A., Hofmann, J., Böhm, K., Subhani, M. A., Müller, T. E., et al. (2014). Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 16, 1865–1870. doi:10.1039/C3GC41788C
Levdikova, T. (2014). Butadiene: 2014 World Market Outlook and Forecast up to 2018. Birmingham, UK: The Market Publishers.
Lin, Y.-J., Chen, E., and Rochelle, G. T. (2016). Pilot plant test of the advanced flash stripper for CO2 capture. Faraday Discuss. 192, 37–58; discussion 567. doi:10.1039/C6FD00029K
Martinez-Anido, C. B., Brinkman, G., and Hodge, B.-M. (2016). The impact of wind power on electricity prices. Renew. Energy 94, 474–487. doi:10.1016/j.renene.2016.03.053
Mikkelsen, M., Jørgensen, M., and Krebs, F. (2010). The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 3, 43–81. doi:10.1039/B912904A
Mobility, F. I. A. (2015). Global Reduction in CO2 Emissions from Cars: A Consumer Perspective. Paris, France: FIA Mobility.
Morris, C. (2015). Renewable Power Curtailment Skyrockets in Germany. Energy Transition. Available at: https://energytransition.org/2015/11/renewable-power-curtailment-in-germany/ (Last accessed September 23, 2017).
New Zealand Ministry of Transport and SGS Industrial, Penrose. (2009). Bioethanol Corrosion Testing: Results of Bioethanol Corrosion Testing. Auckland, New Zealand.
NIST. (2017). Thermodynamic Data for All Reactions, Intermediates and Products. US National Institutes of Standards and Technology (NIST), Chemistry Webbook. Available at: http://webbook.nist.gov/chemistry/ (Last accessed September 26, 2017).
Olah, G. A., Gupta, B., Farina, M., Felber, J. D., Ip, W. M., Husain, A., et al. (1985). Electrophilic reactions at single bonds. 20. Selective monohalogenation of methane over supported acidic or platinum metal catalysts and hydrolysis of methyl halides over.gamma.-alumina-supported metal oxide/hydroxide catalysts. A feasible path for the oxidative conversion of methane into methyl alcohol/dimethyl ether. J. Am. Chem. Soc. 107, 7097–7105.
Olivier, J. G. J., Janssens-Maenhout, G., Muntean, M., and Peters, J. A. H. W. (2017). Trends in Global CO2 Emissions. 2016 Report. The Hague, Ispra: PBL Netherlands Environmental Assessment Agency, European Commission, Joint Research Centre.
Podkolzin, S. G., Stangland, E. E., Jones, M. E., Peringer, E., and Lercher, J. A. (2007). Methyl chloride production from methane over lanthanum-based catalysts. J. Am. Chem. Soc. 129, 2569–2576. doi:10.1021/ja066913w
Polmear, I. J. (1999). “Introduction,” in Magnesium and Magnesium Alloys, eds M. M. Avedesian, and H. Baker (OH, USA: ASM International), 3–6.
Saeidi, S., Amin, N. A. S., and Rahimpour, M. R. (2014). Hydrogenation of CO2 to value-added productions – a review and potential future developments. J. CO2 Util. 5, 66–81. doi:10.1016/j.jcou.2013.12.005
Schlander, J. H., and Turek, T. (1999). Gas-phase hydrogenolysis of dimethyl maleate to 1,4-butanediol and y-butyrolactone over copper/zinc oxide catalysis. Ind. Eng. Chem. Res. 38, 1264–1270. doi:10.1021/ie980606k
Soosaiprakasam, I. R., and Veawab, A. (2008). Corrosion and polarization behaviour of carbon steel in MEA-based CO2 capture process. Int. J. Greenhouse Gas Control 2, 553–562. doi:10.1016/j.ijggc.2008.02.009
Styring, P., Quadrelli, A., and Armstrong, K. (2015). Carbon Dioxide Utilisation: Closing the Carbon Cycle. Oxford, UK: Elsevier.
Szulczyk, K. R. (2010). Which is a better transportation fuel – butanol or ethanol? Int. J. Energy Environ. 1, 501–512.
Thannimalay, L., Yusoff, S., and Zawawi, N. Z. (2013). Life cycle assessment of sodium hydroxide. Aust. J. Basic Appl. Sci. 7, 421–431.
Uyanga, I. J., and Idem, R. O. (2007). Studies of SO2- and O2-induced degradation of aqueous MEA during CO2 capture from power plant flue gas streams. Ind. Eng. Chem. Res. 46, 2558–2566. doi:10.1021/ie0614024
van de Scheur, F. T., and Staal, L. H. (1994). Effects of zinc addition to silica supported copper catalysts for the hydrogenolysis of esters. Appl. Catal. A Gen. 108, 63–83. doi:10.1016/0926-860X(94)85181-6
von der Assen, N., and Bardow, A. (2014). Life cycle assessment of polyols for polyurethane production using CO2 as feedstock: insights from an industrial case study. Green Chem. 16, 3272–3280. doi:10.1039/C4GC00513A
Xu, Y., and Avedisian, C. T. (2015). Combustion of n-butanol, gasoline, and n-butanol/gasoline mixture droplets. Energy Fuels 29, 3467–3475. doi:10.1021/acs.energyfuels.5b00158
Yanowitz, J., Christensen, E., McCormick, R., US National Renewable Energy Laboratory (NREL). (2011). Utilization of Renewable Oxygenates as Gasoline Blending Components. CO, USA: Golden, 31.
Yao, M., Wang, H., Zheng, Z., and Yue, Y. (2010). Experimental study of n-butanol additive and multi-injection on HD diesel engine performance and emissions. Fuel 89, 2191–2201. doi:10.1016/j.fuel.2010.04.008
Keywords: carbon dioxide utilization, butanol, energy storage, carbon avoided, transport fuel, Grignard reagent
Citation: Dowson GRM and Styring P (2017) Demonstration of CO2 Conversion to Synthetic Transport Fuel at Flue Gas Concentrations. Front. Energy Res. 5:26. doi: 10.3389/fenrg.2017.00026
Received: 31 May 2017; Accepted: 19 September 2017;
Published: 12 October 2017
Edited by:
Robert J. Farrauto, Columbia University, United StatesReviewed by:
Edward J. Rode, DNV GL, NorwayXuezhong He, Norwegian University of Science and Technology, Norway
Copyright: © 2017 Dowson and Styring. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Styring, cC5zdHlyaW5nQHNoZWZmaWVsZC5hYy51aw==