 Camille Crouzet1,2,3
Camille Crouzet1,2,3 Fabrice Brunet1*†
Fabrice Brunet1*† German Montes-Hernandez1
German Montes-Hernandez1 Nadir Recham2Nathaniel Findling1
Nadir Recham2Nathaniel Findling1 Jean-Henry Ferrasse3Bruno Goffé4
Jean-Henry Ferrasse3Bruno Goffé4
- 1Univ. Grenoble Alpes, Univ. Savoie Mont Blanc, CNRS, IRD, IFSTTAR, ISTerre, Grenoble, France
- 2LRCS and RS2E, CNRS-UMR7314, Univ. Picardie Jules Verne, Amiens, France
- 3Aix Marseille Univ., CNRS, Centrale Marseille, M2P2, Marseille, France
- 4Aix-Marseille Univ., CNRS, IRD, Coll. de France, CEREGE, Aix-en-Provence, France
A new process route for the valorization of BOF steel slags combining H2 production and CO2 mineral sequestration is investigated at 300°C (HT) under hydrothermal conditions. A BOF steel slag stored several weeks outdoor on the production site was used as starting material. To serve as a reference, room temperature (RT) carbonation of the same BOF steel slag has been monitored with in situ Raman spectroscopy and by measuring pH and PCO2 on a time-resolved basis. CO2 uptake under RT and HT are, respectively, 243 and 327 kg CO2/t of fresh steel slag, which add up with the 63 kg of atmospheric CO2 per ton already uptaken by the starting steel slag on the storage site. The CO2 gained by the sample at HT is bounded to the carbonation of brownmillerite. H2 yield decreased by about 30% in comparison to the same experiment performed without added CO2, due to sequestration of ferrous iron in a Mg-rich siderite phase. Ferric iron, initially present in brownmillerite, is partitioned between an Fe-rich clay mineral of saponite type and metastable hematite. Saponite is likely stabilized by the presence of Al, whereas hematite may represent a metastable product of brownmillerite carbonation. Mg-rich wüstite is involved in at least two competing reactions, i.e., oxidation into magnetite and carbonation into siderite. Results of both water-slag and water-CO2-slag experiments after 72 h are consistent with a kinetics enhancement of the former reaction when a CO2 partial pressure imposes a pH between 5 and 6. Three possible valorization routes, (1) RT carbonation prior to hydrothermal oxidation, (2) RT carbonation after hydrothermal treatment, and (3) combined HT carbonation and oxidation are discussed in light of the present results and literature data.
Introduction
In the context of sustainable development and circular economy growth, alternative sources of raw materials represent an ever-expanding market. In the past, a significant part of industrial wastes and byproducts was considered as ultimate waste. Today, waste regulation is evolving toward more recycling. The steel industry is directly concerned by this mind shift. Steel-making processes, which are mastered for long, produce high amounts of by-products. With a production equivalent to 10–15wt.% of the crude steel output (van Oss, 2013) up to 240 millions of tons of steel slags are annually produced and only partially valorized. In the last decades, several applications using steel slags have been proposed but only a few of them has been developed at an industrial scale. For example, a small proportion of the slag production is spread on agricultural fields for reclamation of acidic lands and soil stabilization (Chand et al., 2015), taking advantage of a high content in reactive calcium hosted in both lime and portlandite, called “free Ca” in the following. The potassium content also motivated its use as fertilizer (Wang and Cai, 2006). Some steel slags are incorporated in binder mix for cement (Shi, 2002; Reddy et al., 2006) or asphalt applications (Ahmedzade and Sengoz, 2009) as already performed with blast furnace slags. Steel slags are most commonly valorized as material for construction and public works. These applications however are limited by the volume instability of steel slags when submitted to weathering and natural aging. Out of the 2.0 Mt steel slag produced in 2014 in France, 54% were transformed as public work materials, whereas 27% were added to the existing stock of 15.2 Mt (http://www.ctpl.info/wp-content/uploads/2015/06/CTPL-Stats-laitiers-acierie-2014-2013.pdf).
It can be seen that current valorization of steel slags is associated with a direct use of limited added value. Among more advanced slag valorization options which require significant processing, Quaghebeur et al. (2010) have developed a new method to achieve, at elevated P and T, an accelerated carbonation of non-hydraulic slags for the manufacture of high-quality construction products. Malvoisin et al. (2013) and Brunet et al. (2013) have proposed to produce high-purity hydrogen from the hydrothermal oxidation of BOF steel slags according to a geoinspired process. In this process, ferrous iron, mainly present in the form of Mg-bearing wüstite, (Fe,Mg)O, is oxidized in the presence of water to form magnetite, Fe3O4, along with H2; the corresponding reaction can be written as follows:
In order to better characterize this RedOx reaction, the pure FeO–water system has been investigated by Crouzet et al. (2017) under hydrothermal conditions; FeO oxidation and H2 production rates were found to be strongly enhanced by replacing pure water by dilute organic acids. For example, Crouzet et al. (2017) showed that FeO is fully oxidized into Fe3O4 within less than 10 h at a temperature as low as 150°C using a 0.05 M acetic acid solution (pH of ca. 4.7). In comparison, less than a quarter of the oxidation reaction is achieved after 144 h at 300°C in pure water. However, the kinetics effect of dilute acid obtained on pure FeO cannot be directly transposed to steel slags. Indeed, due to their high Ca content, steel slags will tend to neutralize mild acids and buffer the pH to high values (Crouzet et al., 2017). Therefore, in the perspective of H2 production from steel slags at an industrial scale, large volumes and/or high concentrations of acid would be required in order to reach significant kinetics enhancement.
In order to meet the objective of enhanced H2/magnetite production kinetics through acidification of the reacting aqueous medium, we propose here to test the effect of maintaining a CO2 partial pressure in the course of the hydrothermal treatment of the slag. Indeed, CO2 partial pressure will buffer the concentration of aqueous carbonic acid and drag the pH to acidic values. In addition to its potential on the enhancement of H2/magnetite production kinetics, CO2 sequestration will occur coincidentally through carbonation of steel slag minerals. Actually, the carbonation capacities of BOF (or LD) steel slags are high and have been, respectively, estimated, on samples of different origin, to 250 and 142 kg CO2/t by Huijgen et al. (2005) and Malvoisin et al. (2013), respectively. Therefore, high-temperature oxidation under CO2 partial pressure can potentially meet a double valorization objective.
Based on the literature data cited above, three valorization routes which combine H2 production and CO2 sequestration can be envisaged. H2 can be produced first (Route A, Figure 1), hydrothermal oxidation Reaction (1) will occur at a pH that is imposed by the steel slag chemistry (basically pH > 10). As no other gas is added, H2 will be produced with high purity and will be compatible with Fuel Cell technologies (Malvoisin et al., 2013) unless the initial slag is significantly carbonated after aging in air. Then, 2.13 g H2/kg FeO can be produced after 72 h reaction at 300°C. A later room temperature (RT) aqueous carbonation step can be easily added to sequester CO2. Aqueous carbonation acts as an accelerated aging and hence improves the residue for its valorization as road engineering product.
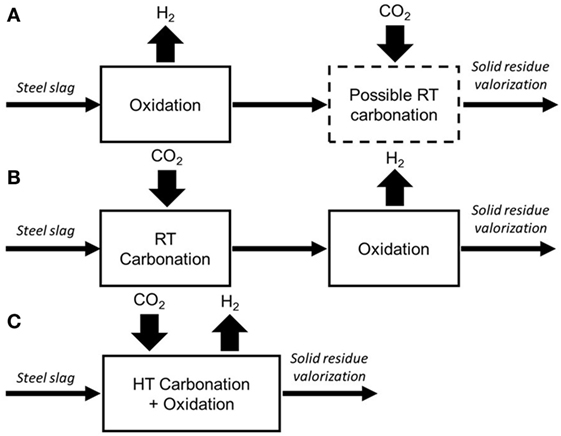
Figure 1. Three possible routes (A–C) for the hydrothermal valorization of steel slag through H2 production and CO2 sequestration. Route c is investigated in the present study.
A second possible path consists in performing RT aqueous carbonation prior to hydrothermal oxidation (Route B, Figure 1). This path was tested by Malvoisin et al. (2013) and led to a modification of the steel slag reactivity by carbonating the “free Ca.” Hydrothermal oxidation is expected to occur at pH closer to neutral values. A minor effect was reported on the H2 production yields with the alteration of H2 purity through the formation of methane for T above 200°C (Malvoisin et al., 2013).
We proposed to investigate here a third route through the application of carbonation and oxidation at once in a single high-temperature step (Route C, Figure 1). Potentially, pH can be dragged to acidic values (pH < 6) through the dissolution of CO2 and the formation of carbonates. A kinetics enhancement is then expected (Crouzet et al., 2017).
Materials and Methods
Starting Materials
BOF steel slag was sampled on a French production site. Samples were collected at three different locations on the slag heap where the slag had been dumped, respectively, a few weeks, several months and several years before. The uppermost 10–20 cm were removed and about 30 kg of steel slag were shoveled from the same spot of the heap. In order to increase both reactive surface area and chemical homogeneity, 5 kg of sample were crushed into powder in a jaw crusher and a planetary ball mill. Laser granulometry showed that particles below 50 µm were obtained with sizes centered on 20 µm.
Ambient Carbonation
Carbonation of steel slag was performed in a 1 L hastelloy (Ni-Cr alloy) PARR® autoclave equipped with pH measuring device (Montes-Hernandez et al., 2009). Ten grams of crushed BOF slag were inserted in 500 mL of deionized water. After dispersion and pH stabilization, carbonation experiment was conducted by injecting 10 bar of CO2 gas. Pressure was read on a 0–137 bar range manometer. Pressure and pH were recorded during 24 h under 400 rpm stirring. CO2 pressure drop due to CO2,gaz − CO2,aq dissolution was determined separately with the same set-up but without the slag sample. The CO2 uptake was estimated from the difference between CO2 pressure drop with and without BOF slags, the steady-state pH in both instances being relatively close, 4 and 3–3.5, respectively. The overall sample carbonation which includes the contribution of carbonates already present in the starting material was determined by thermal gravimetric analysis (TGA). Carbonation extent will be expressed in the form of a CO2 uptake as the mass of uptaken CO2 (kg) divided by the equivalent mass of the CO2-free slag (ton) having sequestered that CO2. It must be noted that the initial slag used here is already partly carbonated. In other words, the slag has already uptaken atmospheric CO2 into an amount that will be referred to as starting material carbonation (SMC) and which is also calculated on the basis of the mass of CO2-free starting slag. After a carbonation experiment, TGA data allow to retrieve the overall slag carbonation (OC), i.e., the total amount of CO2 stored by mass of uncarbonated slag. Note that the difference between OC and SMC yields the effective mass of CO2 that has been uptaken during the experiment by ton of uncarbonated starting slag equivalent.
Phase changes over carbonation were monitored in a 500 mL PARR® autoclave equipped with an in situ Raman device through a sapphire window using a 785 nm laser source following the method proposed by Montes-Hernandez and Renard (2016). Twenty grams of BOF slag were inserted in 300 mL of deionized water at ambient temperature under 400 rpm stirring. Thirty bars of CO2 were injected after 18 min of pressure and pH stabilization. Raman spectra were collected over 24 h, every minute during the first hour and then every 15 min.
Hydrothermal H2 Production
Hydrothermal slag oxidation was conducted in a third 500 mL Hastelloy® autoclave (Crouzet et al., 2017) under 500 rpm stirring. Four grams of steel slag were inserted in 200 mL of deionized water. About 10 bar of CO2 were injected at ambient temperature. After 2 h, PCO2 stabilized after it dropped by about 6 bar, argon was then injected to reach 100 bar of total pressure (Ptotal). Ptotal was then lowered by 50 bar in order to reach 190 bar at 300°C. Gas phase was sampled at regular intervals to monitor hydrogen and other gases (CO2, N2, O2, CO, and CH4). Gas was sampled directly in the hot autoclave, water was condensed and the dried gas readily analyzed with a Clarus 500 gas chromatography (GC). The GC is equipped with a thermal conductivity detector and a Restek ShinCarbon® column, polymer filled and 2 m long. Argon was used as gas carrier. The temperature of the detector, the injection system and the oven were, respectively, set to 250, 100, and 80°C. Each gas sample was analyzed at least three times consecutively. H2 concentration in the sampled gas is converted into absolute quantity (e.g., mass) in the reactor using the ideal gas law. Mass of H2 is normalized to the equivalent mass of FeO included in the solid starting material. Initial steel slag FeO contents were deduced from chemical analysis (Table 1).
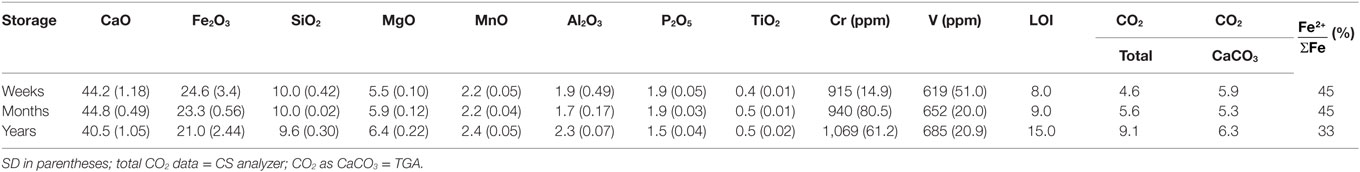
Table 1. Chemical composition (wt.%) of initial BOF slags with different outdoor storage durations.
Solid-Phases Characterization
Chemical composition (major and trace elements) of the starting materials was determined at CRPG-SARM (Nancy, France) following the procedure described in Carignan et al. (2001). Powdered samples were fused in LiBO2, dissolved by acid attack and analyzed using flow injection and low pressure on-line liquid chromatography coupled to ICP-MS. Iron (II) was quantified separately. Samples were first dissolved in a sulfuric, hydrofluoric and boric acid mix. Iron (II) amount was then determined by potassium dichromate in phosphoric acid and potentiometric titration.
Sample carbonation was characterized by TGA (Metler Toledo TGA/DSC 3+). Between 30 and 50 mg of sample were heated from 25 to 1,100°C at a rate of 10°C/min in N2 atmosphere. Calcium carbonates from steel slags decompose in the 500–1,000°C range (e.g., Huijgen et al., 2005). Weight loss observed in the 475–800°C range for all studied samples, was therefore assumed to represent CO2 loss and was used to retrieve carbonation extent. In other words, CO2 contents derived from TGA here assume CaCO3 as sole carbonate phase in the sample. Accuracy on the CO2 content determination is mostly bounded to the fit of the TGA data and is estimated to better than 5%. Total carbon was measured at CRPG-SARM (Nancy) with a CS analyzer (EMIA 320V2 HORIBA) with a precision better than 5%.
Mineral phases were identified by X-ray powder diffraction (XRPD) with a D8 diffractometer (Bruker, CuKα radiation) operated with a 2θ step size of 0.026°. Quantitative analysis of crystalline phases was conducted on the HT-carbonated sample by Rietveld refinement using the BGMN software (Taut et al., 1998). Attempts on the two other samples (starting and RT-carbonated slag) were not conclusive due to the likely presence of amorphous to poorly crystalline phases.
Solid samples were also observed by field-emission scanning electron microscopy (FE-SEM) on a ZEISS Ultra 55 equipped with an energy dispersive X-ray spectroscopy (EDS) detector. Two preparations were employed prior to carbon coating. For chemical analysis, the sample was embedded in epoxy and polished. Crystal morphologies were observed on powder sample dispersed on a double-sided carbon tape.
Results
Starting Steel-Slag Characterization
Chemical Composition
Composition of the three starting steel slags was measured by ICP-AES and -MS analysis. Average results from three replicas are presented in Table 1. Accounting for nearly half of the sample mass, calcium oxide is the main constituent of the steel slag samples followed by iron oxide and silica. Average chemical compositions are in good agreement with literature values (Proctor et al., 2000; Malvoisin et al., 2013; Piatak et al., 2015). Only minor variations in chemical composition were observed between the three samples, this chemical homogeneity among samples reflects the Basic Oxygen Furnace process mastering. Considering that CO2 sequestration occurs through calcium carbonate formation, an overall carbonation potential of 387 kg CO2 can be deduced from the bulk CaO content. SMC was determined to be 63 kg CO2/t steel slag on the freshest sample (weeks, Table 1) by TGA (Figure 2). This SMC figure corresponds to 5.9 wt.% CO2 in the slag (Table 1).
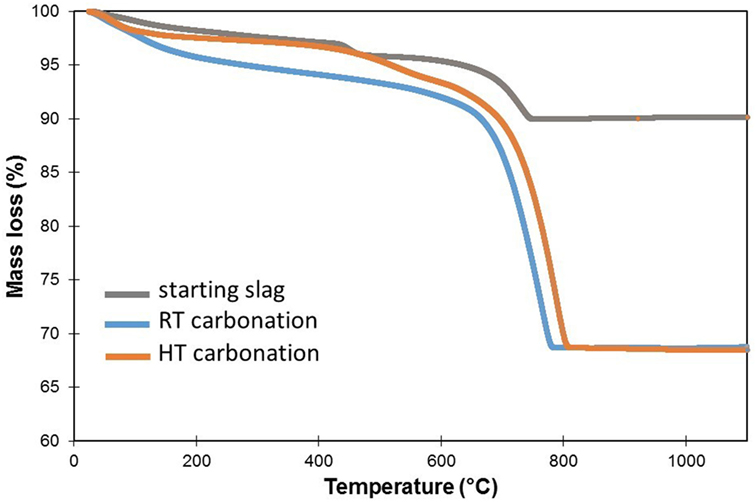
Figure 2. Thermal gravimetric analysis (TGA) data collected on the starting steel slag and the experimentally carbonated samples at room temperature (RT) and at 300°C (HT). The starting steel slag shows two successive mass loss features associated with portlandite and Ca-carbonate decomposition, respectively. Note that the HT-carbonated sample losses more mass, interpreted as due to a higher carbonation degree, than the RT-carbonated one.
Total CO2, LOI, and iron oxidation are the chemical parameters that significantly vary with storage time (Table 1). The longer the steel slag was stored, the higher the total CO2 content. Iron oxidation due to weathering is evidenced by a decrease of the ratio as storage time increases. This ratio drifted from 45% after weeks and months to 33% after years of outdoor storage.
Starting Steel Slag Mineralogy
Mineralogical content of the BOF slag sample aged for several weeks on the storage site (Figure 3) was determined combining bulk composition (Table 1), phase identification by XRPD, FE-SEM imaging, and EDS analysis (Table 2; Figure 4). Three calcium carbonates were identified by XRPD: calcite, aragonite, and vaterite (Table 2). Based on TGA measurement which yielded 5.9 wt.% CO2, these calcium carbonates represent around 15 wt.% of the initial steel slag (Figure 2). Larnite, Ca2SiO4 (C2S), and brownmillerite are the main mineral phases in the starting slag which account together for more than 50% of the slag mass. Brownmillerite and magnetite host the ferric iron contained in the starting steel slag. Ferrous iron is hosted in magnesian wüstite (called wüstite for simplification hereafter).
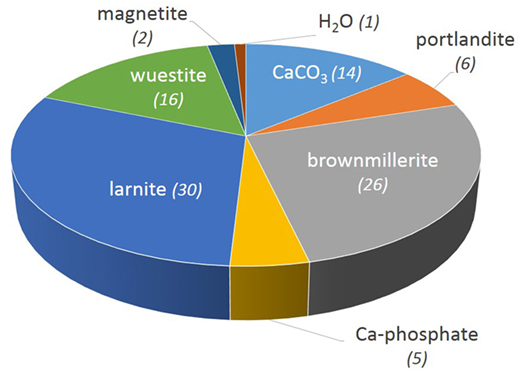
Figure 3. Mineral mass percentage in the starting slag. Mass percentages (in parentheses) are calculated by least square regression through the bulk chemical data (Table 1) on seven mineral components plus water. Both thermal gravimetric analysis (TGA) data (carbonate and portlandite mass proportions, Table 1) and LOI data (CO2 + H2O, Table 1) are used for the regression. The H2O component is the amount of water in the slag that is not accounted for by OH-bearing minerals.
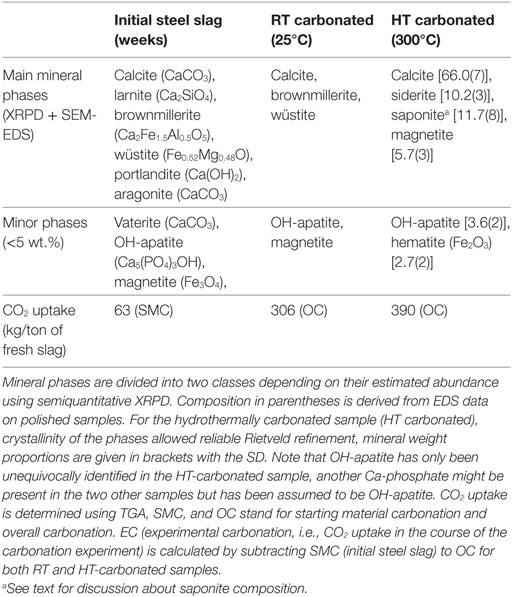
Table 2. Mineralogical characterization of both starting material and experimental products.
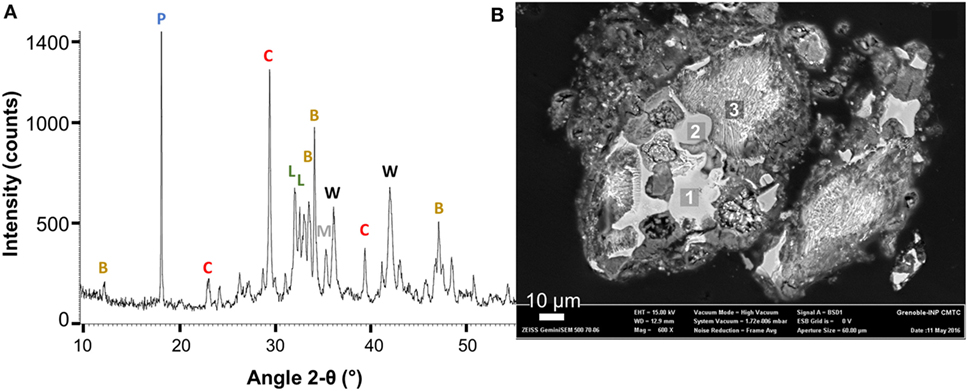
Figure 4. Mineralogical content of the starting steel slag; (A) X-ray powder diffraction (XRPD) data: portlandite (P), calcite (C), larnite (L), brownmillerite (B), and wüstite (W); (B) field-emission scanning electron microscopy (FE-SEM) image in back-scattered electron mode: (1) (Fe0.52Mg0.48)O wüstite, (2) (Fe0.45Mg0.55)O wüstite, and (3) intergrowth: 85 mol.% Ca2SiO4 + 15 mol.% of Ca-phosphate assumed to be Ca5(PO4)3OH.
RT Carbonation
Time-Resolved Carbonation: pH, PCO2 Drop, and In Situ Raman Monitoring
Carbonation was conducted at RT on the freshest slag immerged in water. CO2 pressure drop and pH were first monitored over 24 h. The carbonation experiment started effectively with the CO2 injection (t = 0 min) after pressure and pH stabilization, at 10 bar and a pH value of 10.6, respectively. Temperature rose from 25 to 30°C and the CO2 pressure dropped exponentially before it stabilized at around 4 bar after ca. 85 min (Figure 5A). Similarly, pH decreased exponentially and reached 3.7 in less than 15 min and 3.3 after 40 min and for the rest of the experiment (Figure 5B). The initial steel slag which was already partly carbonated was found to store an equivalent of 1.5 bar CO2.
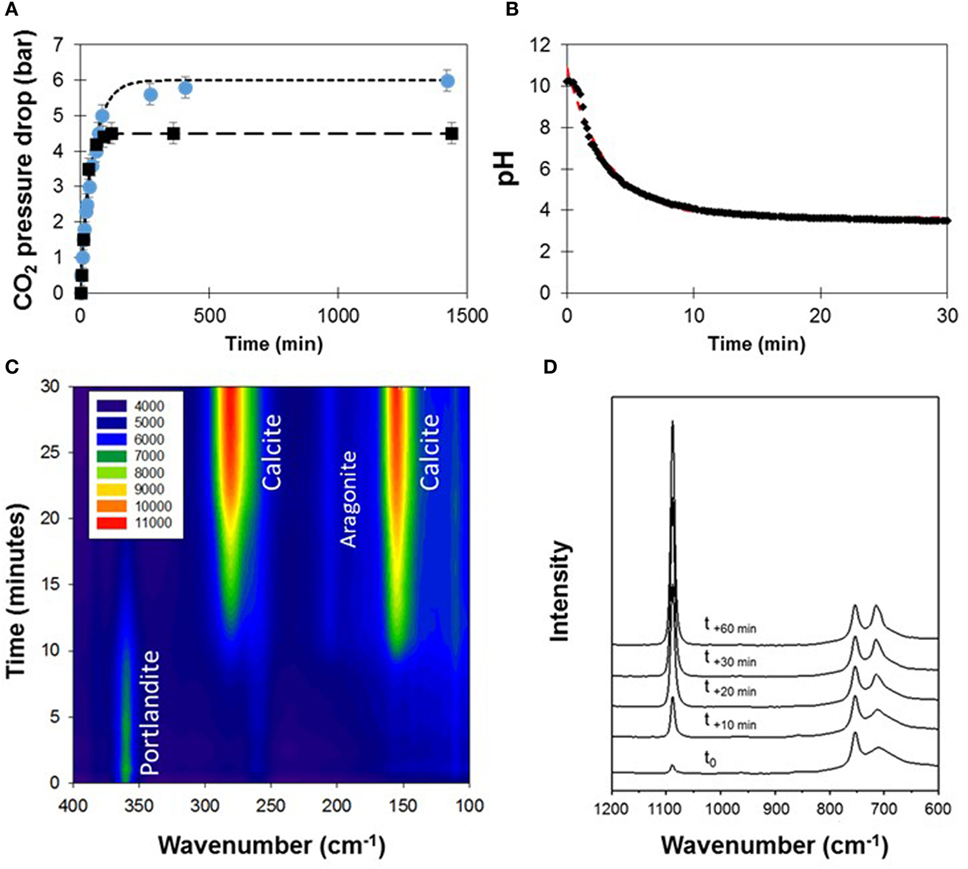
Figure 5. Time-resolved monitoring of steel slag carbonation at ambient temperature. (A) CO2 pressure drop in water (squares) and with the steel slag (circles). Dotted lines correspond to a fit to the equation: . (B) Time-resolved pH data fitted with three parameters . (C,D) In situ Raman spectra presented in two wavenumber ranges.
In parallel to CO2-pressure and pH monitoring, time-resolved phase changes in the aqueous suspension were monitored by in situ Raman spectroscopy (Figures 5C,D). Before CO2 injection, portlandite and calcite were the two main components identified in the suspension. After CO2 was injected, portlandite consumption started and lasted until its total disappearance after 20 min of reaction. Calcite signal started to grow after 10 min of reaction. It kept growing even after portlandite consumption until 720 min of reaction and the observation of signal instabilities. In addition to calcite growth, a minor peak belonging to aragonite was transiently detected at 210 cm−1, which disappeared after 420 min of reaction.
Figure 5D shows spectra in the 1,200–600 cm−1 wavenumber range for five different run durations. Calcite is easily identified with two characteristic bands, one at 1,095 cm−1 and another at 715 cm−1. As reaction proceeds, a narrowing of the Raman band in the 710–680 cm−1 range is observed after more than 30 min of reaction. This signal is attributed to the respective contributions of portlandite and a calcium silicate, possibly larnite. At t0, another small contribution is also detected at 960 cm−1 which quickly disappeared after 20 min of carbonation. This contribution may be related to the partial hydration product of calcium silicate such as afwillite or hibschite. Signal instabilities were observed after 720 min of reaction. As the sapphire peak from the window remains constant, these instabilities are not related to the Raman light source. After opening the autoclave, most of the solid sample was found to have formed a hardened paste, stuck at the bottom of the autoclave, i.e., away from the volume sampled by the Raman probe.
Solid Characterization
X-ray powder diffraction patterns confirmed the full consumption of portlandite and calcium silicates (larnite) as well as the crystallization of calcite (Table 2). Starting aragonite and vaterite all converted into calcite. TGA data indicate a single weight loss event (ca. 23 wt.%) consistent with calcite as the sole Ca-carbonate in the system (Figure 2) and which is characterized by a DTA peak centered around 765°C. An OC of 306 kg CO2/t is deduced which corresponds to an experimental carbonation (EC) of 243 kg CO2/t steel slag. It is calculated that about 79% of the CaO mass in the slag is now combined to CO2 in the form of carbonates. In the starting steel slag (Figure 3), CaCO3, larnite, and portlandite hosted about 70% of the CaO mass contained in the sample. Therefore, the derived OC is broadly consistent with the carbonation of portlandite and larnite although partial carbonation of brownmillerite cannot be excluded. No major mineralogical changes were observed among iron-bearing compounds. Iron was mostly recovered as oxides in the form of brownmillerite, magnetite, and wüstite (Table 2).
Steel Slag Hydrothermal Oxidation under CO2 Pressure
Hydrogen Production
Steel slag hydrothermal oxidation was performed at 300°C according to two experiments, i.e., with and without CO2 (Figure 6). Hydrogen was produced in both cases following a similar kinetic model which is satisfactorily fitted to a simple squared root function of time:
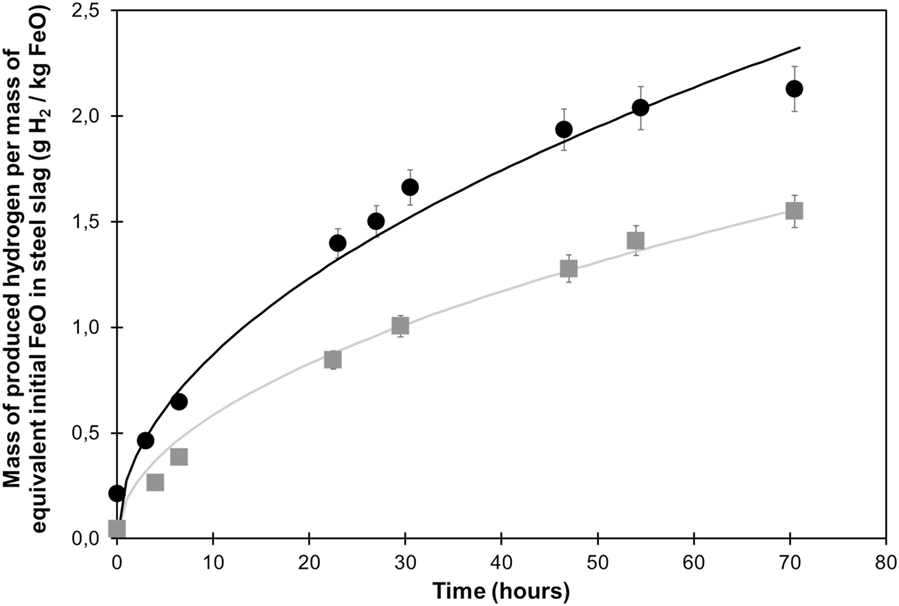
Figure 6. Hydrogen production from steel slag in presence (square) and absence (circle) of added CO2. A square root function of time was used to fit the data.
In the absence of added CO2, hydrogen production rate was found to be roughly constant during the 30 first hours before it progressively slowed down. A total of 2.13 g H2/kg FeO were produced after 72 h and A1 was estimated to 0.276 g H2/h1/2/kg FeO. In the presence of CO2 (initial PCO2 = 2 bar), a total of 1.55 g H2/kg FeO was produced after 72 h; A2 in Eq. 2 is estimated to 0.185 g H2/h1/2/kg FeO. CO2 gas concentration was found roughly constant over the whole reaction. It corresponded to an average CO2 partial pressure of 16 bar at 300°C as calculated assuming CO2 as an ideal gas. No CH4 was detected in the analyzed gas with a CH4 detection limit estimated at 100 ppm.
Characterization of the HT Carbonation Mineral Products
As for RT carbonation, calcite is the dominant mineral phase in the sample carbonated at 300°C (Table 2). XRPD (Figure 7A) and SEM characterization (Figure 7B) revealed another carbonate with a composition (Fe0.56Mg0.36Mn0.06Ca0.02)CO3 as inferred from EDS data. The diffraction signal of this carbonate could be refined using a siderite structure assuming however lattice parameters that significantly exceed those expected along the siderite-magnesite solid solution. XRPD pattern of HT-carbonated products also revealed the crystallization of saponite, a trioctahedral clay mineral which belongs to the smectite group. The retrieved lattice parameter, b, of around 9.22 Å, suggests that the newly formed saponite has octahedral sites dominated by iron (Brigatti, 1983), Fe2+ (Baldermann et al., 2014), and/or Fe3+ (e.g., Treiman et al., 2014). Iron mass balance from Rietveld data along with the constraint that Fe2+/Fetot in the slag must decrease in order to account for measured H2 production, suggesting that Fe in saponite is dominantly ferric.
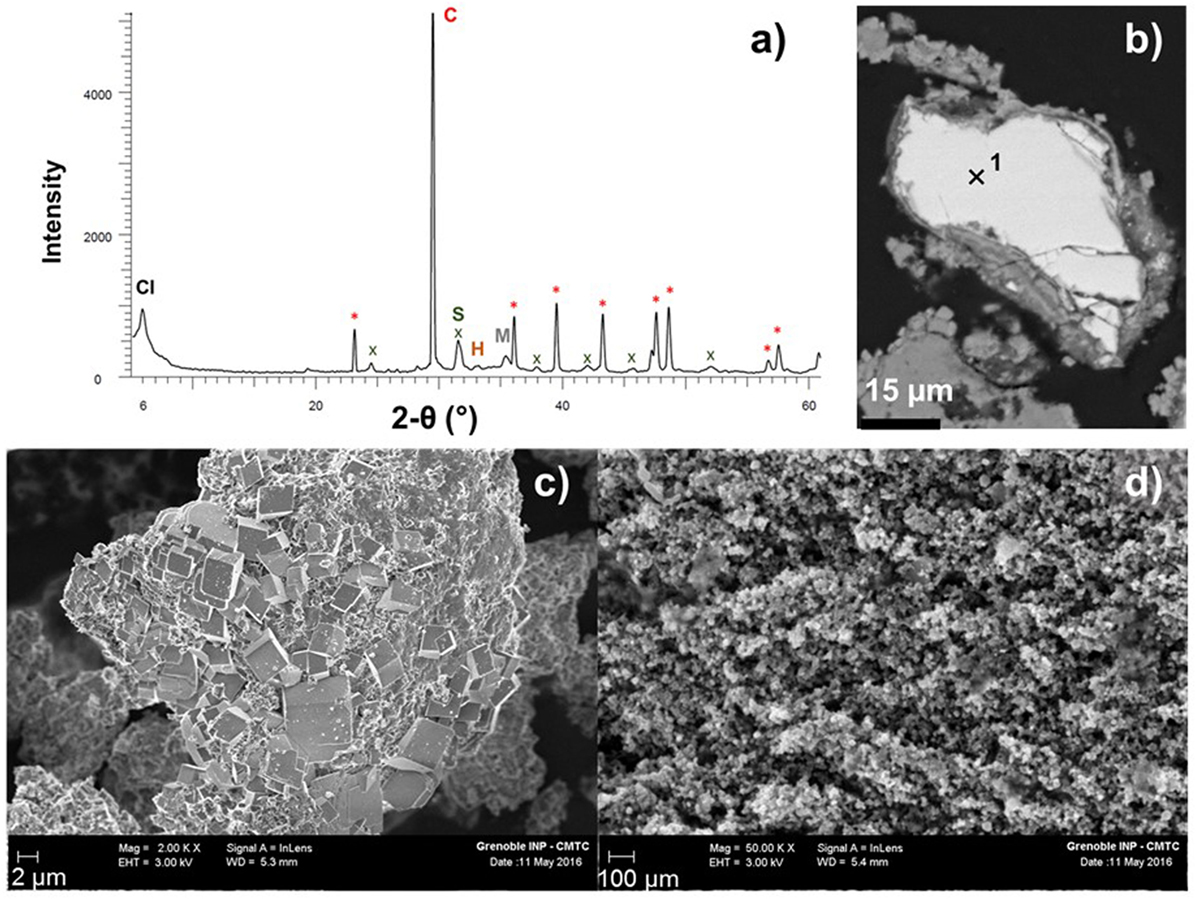
Figure 7. X-ray powder diffraction (XRPD) pattern and field-emission scanning electron microscopy (FE-SEM) imaging of the HT-carbonated steel slag; (A) XRPD data: calcite as (C,*), siderite (S,x), clay minerals (Cl), magnetite (M), and hematite (H); (B) BSE mode imaging on polished sample: EDS chemical analysis (1) of Mg-rich siderite (Fe0.56Mg0.36Mn0.06Ca0.02)CO3; secondary electron mode imaging of (C) micrometric euhedral calcium carbonates and (D) iron oxide nanoparticles in the 10–30 nm size range.
Thermal gravimetric analysis data indicate a two-stage mass loss (Figure 2) with two DTA peaks located at 530 and 789°C. From 120 to 1,000°C, the sample has lost about 30% of its initial mass, whereas Rietveld refinement predicts a (H2O + CO2) content in the HT-carbonated slag of 35.5 wt.%. The main mass loss event (24.2 wt.%) corresponds to the 789°C DTA peak and is very likely correlated with calcite decomposition since calcite is the dominant mineral according to XRPD. The lower temperature mass loss (ca. 4 wt.%) may be correlated with either Fe-rich saponite dehydroxylation (Brigatti, 1983; Földvári, 2011) or Mg-siderite decomposition since 530°C is comprised between magnesite and siderite TGA decomposition temperatures (Földvári, 2011). Based on the amount of Fe-saponite (11 wt.%) obtained from Rietveld refinement, the contribution of saponite dehydroxylation to the sample weight loss should not exceed half a weight percent and therefore the lower temperature TGA peak is more likely related to Mg-siderite decomposition. A 3.5–4% mass loss would correspond to the decomposition of 8–9 wt.% siderite which compares relatively well with the 10 wt.% inferred from Rietveld refinement. EC is estimated to 327 kg CO2/t from TGA. Moreover, no more wüstite nor brownmillerite was found in the XRPD pattern. Iron is now partitioned between magnetite, hematite, saponite and siderite. Carbonates were observed in the form of rhombohedral micrometric particles (Figure 7C). Mineralogical analysis of the sample oxidized at 300°C without added CO2 yielded, by order of abundance, hydrated calcium silicates (katoite–hibschite–jaffeite), calcite, hydroxylapatite, magnetite, wüstite, and portlandite (Crouzet et al., in press).
Further FE-SEM characterization was performed on iron oxides extracted magnetically with a permanent magnet in a water-filled ultrasonic bath. This magnetic separation allowed to emphasize a multitude of iron oxide nanoparticles (Figure 7D).
Discussion
RT Carbonation: Time-resolved Monitoring
As shown by previous studies on steel slags and other Ca-rich materials at RT (e.g., Huijgen et al., 2005; Montes-Hernandez et al., 2009; Fritz et al., 2013), aqueous carbonation leads to a pH shift from basic to acidic conditions. We find for the steel slag studied here a pH decrease from 10.6 to 3.7 in less than 15 min. From 15 to 40 min and until the end of experiment, pH stabilizes from 3.7 to 3.3. In the Ca(OH)2–water subsystem, portlandite consumption is enslaved to the supply of protons from CO2 dissolution; variation in the ionic activity product leads to the very rapid consumption of portlandite and a pH drop by 5–7 units (Fritz et al., 2013). In our experiments with steel slag, a first similar stage was observed but limited to the first minute. This stage may be shortened due to the steel slag chemistry and the availability of ionic charges, supplied by the partial dissolution of other solids either amorphous or crystalline.
In situ Raman spectroscopy was used to follow up the mineralogy of the suspension. Before room-temperature (RT) carbonation, portlandite and calcite were the two main mineral species. The first 20 min showed the crystallization of calcite at the expense of portlandite which fully disappeared according to the overall reaction:
As CO2 is in excess, this reaction only stops when portlandite is consumed. In situ pH measurement shows that the main pH drop occurs during these first 20 min. Despite the persistence of portlandite in the first 20 min of reaction, pH cannot be maintained to high values; it follows that portlandite dissolution is slower than CaCO3 formation. It must also be noted that metastable carbonates (aragonite and vaterite) which likely formed by interaction between the slag and atmospheric CO2 converted into calcite in a CO2-rich aqueous medium at RT (Ogino et al., 1987). Aragonite was even shown to grow metastably in the first hours of reaction before it finally disappeared from Raman spectra after 7 h and was no longer found in the XRPD pattern of the recovered solid sample.
PCO2 has been used as a third proxy of the carbonation kinetics. We showed that 83% of the total PCO2 drop occurred in the first 85 min and almost no further variation was observed after 407 min. CO2 uptake from the gas phase lasted however longer than the single portlandite-carbonation event which is estimated to less than 20 min according to our in situ Raman spectroscopy data. Following the results by Huijgen et al. (2005), it can be argued that calcium silicates and Ca-bearing oxides carbonation may still proceed after portlandite consumption. This assumption is corroborated by further calcite growth after portlandite disappearance as observed by Raman spectroscopy. At the end of RT carbonation experiment, steel slag is recovered in the form of a hardened paste stuck at the bottom of the autoclave. This hardening process may be enhanced by the cementitious properties of calcium carbonates (Johnson et al., 2003) and of the products of dicalcium silicate (C2S) hydration.
Based on the CaO content of 49.3 wt.% in the fresh slag (i.e., CO2-free reference) and on the relationship between CO2 content and carbonation degree (Relation 6 in Huijgen et al., 2005) for a carbonation degree of 100%, the maximum CO2 sequestration (maximum OC) by our steel slag sample is estimated to 387 kg CO2/t of fresh slag. After carbonation at RT, an OC of 306 kg CO2/t of fresh slag is attained corresponding to a carbonation degree of around 80%, i.e., 80% of the Ca contained in the starting material has reacted to form calcium carbonate. The XRPD pattern of the carbonated slag shows that the unreacted Ca is bounded to brownmillerite which appears to be the least reactive Ca-bearing mineral in the starting material.
High-Temperature Steel Slag Carbonation
Influence of CO2 Partial Pressure on Hydrogen Production
In order to investigate the potential of the CO2 injection at high temperature in lowering the pH and enhancing oxidation rate (H2 production), hydrothermal steel slag oxidation was performed in deionized water at 300°C to 150 bar under argon atmosphere with and without added CO2 partial pressure. Hydrogen production was observed in both cases, following similar kinetic models. So apparently, the addition of CO2 does not fundamentally modify the steel slag oxidation process. In absence of a plateau in H2 production, a square root function of time was found to yield a better fit to the H2 production data than a first-order kinetic model (Crouzet et al., 2017). These data indicate a H2 yield by 30% lower when CO2 gas is added. We also show that the addition of CO2 gas leads to the disappearance of wüstite which is otherwise observed as residual phase.
There is a clear discrepancy between recovered H2 and wüstite consumption in the HT carbonation experiments. This apparent H2 deficit can be explained by the formation of an (Fe,Mg)-carbonate, (Fe0.56Mg0.36Mn0.06Ca0.02)CO3, of siderite structure (Rietveld refinement) which actually incorporated ferrous iron and prevented it from further oxidation. The Fe/Mg ratio of this carbonate as well as the presence of manganese traces suggest that it directly formed from wüstite with little fractionation of the divalent cations (Fe, Mg, Mn). Another remarkable feature is the presence of hematite. Using the concentration of aqueous H2 at the magnetite–hematite equilibrium and the Henry constant of H2 as provided by SUPCRT92 (Johnson et al., 1992), it is calculated that, at 300°C and 190 bar, hematite should no longer be stable over 1% reaction progress (Appendix). Hematite might therefore be an intermediate compound of brownmillerite breakdown which persists as a metastable phase indicating a relatively low reduction kinetics into magnetite.
Total H2 produced in the carbonation experiment was found to only represent 37 mol.% of maximum H2 that can be produced based on the initial Fe2+ content of the starting slag. On the other hand, about 50 wt.% of the initial FeO content is sequestered in iron carbonates. Furthermore, the reduction of hematite into magnetite might consume H2 and thereby decrease the overall H2 yield.
Assuming that the pH in the HT carbonation experiments is controlled by the calcite–CO2 equilibrium, an estimated pH of ca. 5.6 is calculated for the investigated experimental conditions. Crouzet et al. (2017) noted a significant kinetic improvement in FeO oxidation in the presence of acetic acid solution at a pH of ca. 4.7. Unfortunately, in the present HT carbonation experiments, slag wüstite is involved in two competing reactions, oxidation on one hand and carbonation on the other. It is therefore not possible to precisely assess the effect of shifting the pH below 7 on wüstite oxidation kinetics alone. However, in the 72-h carbonation/oxidation experiment, 1.55 g H2/kg FeO has been produced. If now, we consider that about half of the FeO has been entrapped into carbonates (additional Fe2+ might also have been incorporated into saponite), it follows that more than 3 g H2/kg of available FeO has been effectively produced in the time span, i.e., 40% more than in the hydrothermal experiment where no CO2 was added. It remains that HT carbonation lowered the H2 yield in comparison to wüstite oxidation in water without added CO2. Moreover, injection of CO2 gas for the sake of kinetic improvements led to the alteration of hydrogen purity. As far as H2 production is concerned slag oxidation under PCO2 has obviously no added value.
In parallel to H2 production, hydrothermal oxidation/carbonation of steel slag led to the formation of magnetite. FE-SEM investigations were conducted on magnetically separated samples. Magnetite was observed as nanoparticle agglomerates. Crouzet et al. (2017) already reported the formation of magnetite nanoparticles as the result of the hydrothermal oxidation of FeO in the presence of acetic acid solution. Magnetite nanoparticles and its economical relevance are discussed in the companion article (Crouzet et al., in press). It would however be interesting to study whether HT carbonation could influence the properties of the produced nanomagnetite (size, morphology, and composition).
Other H2 production routes using CO2 still need to be explored. For example, recently, Michiels et al. (2015) showed that the in situ formation of aqueous has a catalytic effect on the mild hydrothermal oxidation of iron metal powder. The stability of aqueous required these experiments to be conducted at pH = 11 with potassium hydroxide. This catalytic effect was found to be dependent of the CO2 partial pressure. For an initial CO2 overpressure of 15 bar in the reactor at 25°C, the authors reported that siderite formation limits H2/magnetite yield in the 140–260°C range. If this result on Fe metal can be transposed to FeO, slag oxidation under PCO2 at pH > 12 may be worth exploring.
Improvement of Mineral CO2 Sequestration at HT
Although HT carbonation occurred to be detrimental to the H2 yield, it led to higher carbonation degree compared to RT carbonation. Indeed, mineral sequestration (OC) is, respectively, estimated to 306 and 390 kg CO2/t of fresh steel slag at 30 and 300°C by TGA (Table 2). Note that the carbonation degree obtained at HT exceeds 100% as defined on the basis of the Ca-content of the steel slag used in this study. This CO2 sequestration gain at 300°C is explained by (1) the partial carbonation of magnesio-wüstite and (2) the carbonation of the calcium contained in brownmillerite at high temperature. Indeed, no more brownmillerite was detected in the sample recovered from HT carbonation, whereas brownmillerite is still present in the RT-carbonated one. Brownmillerite carbonation liberates Al and Fe3+ which are likely responsible for the formation of saponite and hematite, respectively, as secondary products.
Toward a New Valorization Route?
In addition to valorization routes that would involve room-temperature CO2 sequestration either before or after H2 hydrothermal production, we investigated a third route through the application of carbonation and oxidation, at once, in a single high temperature step (Figure 1C). Obviously, the main drawback of this last route is that the purity of hydrogen will be strongly altered by dilution in a CO2-rich gas. Furthermore, contrary to our expectations and even if acidic conditions were reached, H2 yield decreased by 30% when a CO2 partial pressure (around 15 bar) was maintained at HT. The only tangible advantage is the enhanced CO2 sequestration degree of 390 kg CO2/t of fresh steel slag obtained at 300°C instead of 306 kg CO2/t at RT. If this valorization route is apparently not promising, one should bear in mind that the economic value of the proposed route lies on the nanomagnetite (by)product (Crouzet et al., in press). Despite a magnetite yield which will be lowered as for H2 to which Fe3O4 formation is coupled, the effect of combining HT oxidation and carbonation on the properties of the produced magnetite (size, morphology, surface, etc…) remains to be evaluated.
Author Contributions
CC performed all experiments and wrote most of the manuscript. FB supervised the overall work and significantly contributed to the writing of the manuscript. GM-H designed and supervised the carbonation experiments. NR brought is knowledge of RedOx reaction. NF carried out XRPD and contributed to the interpretation of the diffraction data. J-HF helped contributed to the definition of industrial applications. BG provided the knowledge of magnetite applications and market.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by CNRS through the “Mission Interdisciplinaire: Défi Transition Energétique: Ressources, Société, Environnement—ENRS” program. This work is part of the HyMag’In project funded by the SATT Linksium (Grenoble). Labex OSUG@2020 is also thanked for additional financial support. The authors thank Jérôme Rose and Jérôme Labille (CEREGE—Aix-en-Provence) for laser ganulometry, Ayumi Koishi for TGA measurements and Bruno Lanson for his help with interpreting the X-ray diffraction data. The two reviewers are acknowledged for their thorough review work which substantially improved the manuscript.
References
Ahmedzade, P., and Sengoz, B. (2009). Evaluation of steel slag coarse aggregate in hot mix asphalt concrete. J. Hazard. Mater. 165, 300–305. doi: 10.1016/j.jhazmat.2008.09.105
Baldermann, A., Dohrmann, R., Kaufhold, S., Nickel, C., Letofsky-Papst, I., and Dietzel, M. (2014). The Fe-Mg-saponite solid solution series – a hydrothermal synthesis study. Clay Miner. 49, 391–415. doi:10.1180/claymin.2014.049.3.04
Brigatti, M. F. (1983). Relationships between composition and structure in Fe-Rich Smectites. Clay Miner. 18, 177–186. doi:10.1180/claymin.1983.018.2.06
Brunet, F., Malvoisin, B., Vidal, O. and Goffé, B. (2013). Method for Producing High-purity Hydrogen Gas. Patent WO2014154910 A1.
Carignan, J., Hild, P., Mevelle, G., Morel, J., and Yeghicheyan, D. (2001). Routine analyses of trace elements in geological samples using flow injection and low pressure on-line liquid chromatography coupled to ICP-MS: a study of geochemical reference materials BR, DR-N, UB-N, AN-G and GH. Geostand. Geoanal. Res. 25, 187–198. doi:10.1111/j.1751-908X.2001.tb00595.x
Chand, S., Paul, B., and Kumar, M. (2015). An overview of use of linz-donawitz (LD) steel slag in agriculture. Curr. World Environ. 10, 975–984. doi:10.12944/CWE.10.3.29
Crouzet, C., Brunet, F., Recham, N., Findling, N., Ferrasse, J.-H., Magnin, V., et al. (in press). Hydrothermal steel slag valorization-PartII: Hydrogen and nano-magnetite production, in press. doi:10.3389/feart.2017.00086
Crouzet, C., Brunet, F., Recham, N., Findling, N., Lanson, M., Guyot, F., et al. (2017). Hydrogen production by hydrothermal oxidation of FeO under acidic conditions. Int. J. Hydrog. Energy 42, 795–806. doi:10.1016/j.ijhydene.2016.10.019
Földvári, M. (2011). Handbook of the Thermogravimetric System of Minerals and Its Use in Geological Practice, Vol. 213. Budapest: Occasional Papers of the Geological Institute of Hungary, 180.
Fritz, B., Clement, A., Montes-Hernandez, G., and Noguera, C. (2013). Calcite formation by hydrothermal carbonation of portlandite: complementary insights from experiment and simulation. CrystEngComm 15, 3392–3401. doi:10.1039/c3ce26969h
Huijgen, W. J. J., Witkamp, G.-J., and Comans, R. N. J. (2005). Mineral CO2 sequestration by steel slag carbonation. Environ. Sci. Technol. 39, 9676–9682. doi:10.1021/es050795f
Johnson, D. C., MacLeod, C. L., Carey, P. J., and Hills, C. D. (2003). Solidification of stainless steel slag by accelerated carbonation. Environ. Technol. 24, 671–678. doi:10.1080/09593330309385602
Johnson, J. W., Oelkers, E. H., and Helgeson, H. C. (1992). SUPCRT92: a software package for calculating the standard molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1 to 5000 bar and 0 to 1000°C. Comput. Geosci. 18, 899–947. doi:10.1016/0098-3004(92)90029-Q
Malvoisin, B., Brunet, F., Carlut, J., Montes-Hernandez, G., Findling, N., Lanson, M., et al. (2013). High-purity hydrogen gas from the reaction between BOF steel slag and water in the 473–673 K range. Int. J. Hydrog. Energy 38, 7382–7393. doi:10.1016/j.ijhydene.2013.03.163
Michiels, K., Spooren, J., and Meynen, V. (2015). Production of hydrogen gas from water by the oxidation of metallic iron under mild hydrothermal conditions, assisted by in situ formed carbonate ions. Fuel 160, 205–216. doi:10.1016/j.fuel.2015.07.061
Montes-Hernandez, G., Pérez-López, R., Renard, F., Nieto, J. M., and Charlet, L. (2009). Mineral sequestration of CO2 by aqueous carbonation of coal combustion fly-ash. J. Hazard. Mater. 161, 1347–1354. doi:10.1016/j.jhazmat.2008.04.104
Montes-Hernandez, G., and Renard, F. (2016). Time-resolved in Situ Raman spectroscopy of the nucleation and growth of siderite, magnesite, and calcite and their precursors. Cryst. Growth Des. 16, 7218–7230. doi:10.1021/acs.cgd.6b01406
Ogino, T., Suzuki, T., and Sawada, K. (1987). The formation and transformation mechanism of calcium carbonate in water. Geochim. Cosmochim. Acta 51, 2757–2767. doi:10.1016/0016-7037(87)90155-4
Piatak, N. M., Parsons, M. B., and Seal, R. R. (2015). Characteristics and environmental aspects of slag: a review. Appl. Geochem. 57, 236–266. doi:10.1016/j.apgeochem.2014.04.009
Proctor, D. M., Fehling, K. A., Shay, E. C., Wittenborn, J. L., Green, J. J., Avent, C., et al. (2000). Physical and chemical characteristics of blast furnace, basic oxygen furnace, and electric arc furnace steel industry slags. Environ. Sci. Technol. 34, 1576–1582. doi:10.1021/es9906002
Quaghebeur, M., Nielsen, P., Laenen, B., Nguyen, E., and Van Mechelen, D. (2010). Carbstone: sustainable valorisation technology for fine grained steel slags and CO2. Refract. Worldforum 2, 75–79.
Reddy, A. S., Pradhan, R. K., and Chandra, S. (2006). Utilization of basic oxygen furnace (BOF) slag in the production of a hydraulic cement binder. Int. J. Miner. Process. 79, 98–105. doi:10.1016/j.minpro.2006.01.001
Shi, C. (2002). Characteristics and cementitious properties of ladle slag fines from steel production. Cem. Concr. Res. 32, 459–462. doi:10.1016/S0008-8846(01)00707-4
Taut, T., Kleeber, R., and Bergmann, J. (1998). The new Seifert Rietveld program BGMN and its application to quantitive phase analysis. Mater. Struct. 5, 57–66.
Treiman, A. H., Morris, R. V., Agresti, D. G., Graff, T. G., Achilles, C. N., Rampe, E. B., et al. (2014). Ferrian Saponite from the Santa Monica Mountains (California, U.S.A., Earth): characterization as an analog for clay minerals on mars with application to Yellowknife bay in Gale Crater. Am. Mineral. 99, 2234–2250. doi:10.2138/am-2014-4763
van Oss, H. G. (2013). Slag, iron and steel: U.S. Geological Survey. Miner. Yearb. 2011. 1, 69.1–69.9.
Wang, X., and Cai, Q.-S. (2006). Steel slag as an iron fertilizer for corn growth and soil improvement in a pot experiment. Pedosphere 16, 519–524. doi:10.1016/S1002-0160(06)60083-0
Appendix
Stability limit of hematite is calculated using the two following equilibria and their respective pK calculated with SUPCRT92 (Johnson et al., 1992):
Assuming that both solution and gas volumes in the autoclave remain constant between ambient and 300°C–190 bar, i.e., 200 and 300 mL, respectively, and neglecting the initial presence of O2, it is calculated that ca. 20 μmol of H2 are produced to reach hematite/magnetite equilibrium. Four grams of slag containing 10 wt.% FeO can produce 1.8 10−3 mol H2 at most. Then, the minimum of 20 μmol of H2 which is required to drive hematite reduction will be produced after a reaction progress of ca. 1–1.5%.
Keywords: hydrogen production, CO2 sequestration, BOF steel slag, hydrothermal oxidation, saponite
Citation: Crouzet C, Brunet F, Montes-Hernandez G, Recham N, Findling N, Ferrasse J-H and Goffé B (2017) Hydrothermal Valorization of Steel Slags—Part I: Coupled H2 Production and CO2 Mineral Sequestration. Front. Energy Res. 5:29. doi: 10.3389/fenrg.2017.00029
Received: 08 August 2017; Accepted: 09 October 2017;
Published: 30 October 2017
Edited by:
Renato Baciocchi, Università degli Studi di Roma Tor Vergata, ItalyReviewed by:
Jeroen Spooren, Flemish Institute for Technological Research, BelgiumHannu-Petteri Mattila, finland
Copyright: © 2017 Crouzet, Brunet, Montes-Hernandez, Recham, Findling, Ferrasse and Goffé. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrice Brunet, ZmFicmljZS5icnVuZXRAdW5pdi1ncmVub2JsZS1hbHBlcy5mcg==
†Present address: Fabrice Brunet ISTerre, Maison des Géosciences, BP 35, Grenoble, France