 Chen Ling*
Chen Ling* Ruigang Zhang
Ruigang Zhang
- Toyota Research Institute of North America, Ann Arbor, MI, United States
Rechargeable magnesium battery (rMB) has received increased attention as a promising alternative to current Li-ion technology. However, the lack of appropriate cathode that provides high-energy density and good sustainability greatly hinders the development of practical rMBs. To date, the successful Mg2+-intercalation was only achieved in only a few cathode hosts, one of which is manganese dioxide. This review summarizes the research activity of studying MnO2 in magnesium cells. In recent years, the cathodic performance of MnO2 was impressively improved to the capacity of >150–200 mAh g−1 at voltage of 2.6–2.8 V with cyclability to hundreds or more cycles. In addition to reviewing electrochemical performance, we sketch a mechanistic picture to show how the fundamental understanding about MnO2 cathode has been changed and how it paved the road to the improvement of cathode performance.
Introduction
Since the first commercialization in 1991, rechargeable Li-ion battery (LIB) has dominated the market of secondary battery industry and now it is the primary energy source in portable electronic devices. Without any doubt, their dominance will continue and expand to other fields such as electronic vehicles and stationary stations where the large-scale output and storage of energy is essential. At the same time, concerns are also raised by the availability of lithium, the inherent element in LIBs with the abundance of only 20 ppm in the Earth’s crust, and the steeply increased material cost. Consequently, batteries utilizing more earth-abundant element have received more and more attention. Sodium-ion battery, for example, has gained considerable interest recently as potentially cheaper technology especially for large-scale stationary energy storage (Yabuuchi et al., 2014). Another interesting alternative is rechargeable magnesium batteries (rMBs), which shuttles bivalent Mg2+ between metal magnesium anode and a cathode to store and transport the energy (Muldoon et al., 2014). Magnesium is the eighth abundant elements in the Earth’s crust with worldwide availability and much less cost risk. Metal magnesium anode has nearly doubled volumetric capacity (3,833 mAh cc−1) of lithium (2,046 mAh cc−1). In addition, the operation of metal Mg anode seems not to be plagued by the dendritic deposition in recharge as which hampers the usage of metal Li anode in LIB (Matsui, 2010; Ling et al., 2012; Muldoon et al., 2014). Because of these merits, rMBs are now considered as an attractive post-LIB candidate with potentially higher volumetric energy density.
The first prototype rMB was reported by Aurbach’s group in 2000 (Aubach et al., 2000), using metal magnesium anode, Mg(AlCl2BuEt)2 electrolyte, and Chevrel phase Mo6S8 cathode. Coin cells constructed with this configuration delivered a stable performance to >2,000 cycles. After 15 years of research, the development of practical rMB has seen lights from several breakthroughs such as the discovery of electrolytes with wider operation window and less corrosive capability (Muldoon et al., 2012; Yoo et al., 2013; Tutusaus et al., 2015). Meanwhile, the Chevrel phase used in Aurbach’s work is still by far the most successful cathode with remarkable reversibility and cyclability for rMB. However, low-operation voltage (1.2 V) and capacity (theoretically 120 mAh g−1) limit the energy density of Chevrel phase to at most ~20% of typical LIB cathode such as LiCoO2 (Aubach et al., 2000). A crucial hurdle toward the development of practical rMB is therefore to find suitable cathodes meeting the requirements of energy density, cyclability, and rate capability (Yoo et al., 2013; Mohtadi and Mizuno, 2014; Muldoon et al., 2014; Bucur et al., 2015; Huie et al., 2015; Song et al., 2016; Zhang and Ling, 2016a).
The challenge to find suitable rMB cathode is widely believed to be a consequence of bivalency of Mg2+ (Levi et al., 2009, 2010; Yoo et al., 2013; Huie et al., 2015; Ling and Suto, 2017), which not only dramatically reduces the mobility of Mg2+ but also generates other undesirable effects (Ling et al., 2015; Zhang and Ling, 2016b; Ling and Suto, 2017). To date, reversible electrochemical Mg2+-intercalation was only achieved in a few cathode hosts. Chevrel phase and TiS2 showed good cyclability for Mg2+-intercalation but with limited energy densities due to the low voltages. Oxide cathodes generally have higher voltages but the reversible intercalation has only been achieved in V2O5, MoO3, and MnO2 at specific conditions (Gregory et al., 1990; Aubach et al., 2000; Gershinsky et al., 2013; Kim et al., 2015a; Nam et al., 2015; Sa et al., 2016; Sun et al., 2016). Among these materials, MnO2 had received perhaps the best attention not only due to the easiness to synthesis but also because the plentiful polymorphism and easily tuned electrochemical properties provide a good platform to gather fundamental knowledge about cathode chemistry. This review summarizes research activities on using manganese dioxide as rMB cathodes. In addition to reviewing the improvement of electrochemical performance in recent years, we sketch a mechanistic picture to show how the fundamental understanding about MnO2 cathode has been changed and how it resulted in the improvement of cathode performance.
Performance in Dry Non-Aqueous Cells
Owning to the flexibility of MnO6 octahedra to interconnect through edge-shared and corner-shared oxygen, manganese dioxide has a diverse set of distinguished polymorphic structures, providing plentiful chemistry for different applications such as batteries, catalyst, and pigmentation (Post, 1999). The first MnO2 cathode for rMB was reported by our group in 2012 (Zhang et al., 2012). The α-MnO2, also called Hollandite phase after the name of mineral, is constructed of double chains of edge-sharing MnO6 and each chain is interconnected through vortex-shared oxygen in a way to form tunnels of quasi-square sections with two octahedra on each side (Post, 1999). The oxygen anionic framework in α-MnO2 is deficient to form closed packed lattice, hence creating a large open cavity along a one-dimensional channel. Various types of cations such as Li+, Na+, K+, Ag+, Ba2+, and even small molecules such as H3O+ and can occupy the cavity and some of them may also diffuse along the channel. The performance of α-MnO2 in LIB was summarized in Thackeray, (1997). It has a large discharge capacity (~300 mAh g−1) compared to other MnO2 polymorphs. However, it was revealed that the cycling of α-MnO2 in LIB is not stable due to asymmetric distortion of lattice, resulting in poor capacity retention (Thackeray, 1997; Ling and Mizuno, 2012; Yuan et al., 2015).
Because the interstitial site in α-MnO2 is sufficiently large to accommodate the occupation of much larger Ba2+, it was naturally thought that the intercalation of smaller Mg2+ could be feasible. Figure 1A shows the galvanostatic cycling of the electrode in a voltage range between 0.8 and 3.0 V (Zhang et al., 2012). Several apparent characteristics were immediately noticed. First, no clear plateau was observed in the voltage profile and the voltage slope was increased from that of Li+-intercalation (Thackeray, 1997; Ling and Mizuno, 2012; Yuan et al., 2015). The average discharge voltage was typically around 1.5–1.6 V, far below that for Li+-intercalation even if the difference of anionic potential between Li/Li+ and Mg/Mg2+ (0.7 V in aqueous solution and may around 1 V in non-aqueous solution) was taken into account. Second, the charge and discharge displayed a large voltage hysteresis of >0.5 V, indicating the existence of kinetic barrier or asymmetric reaction pathways (Cabana et al., 2010; Yu et al., 2014). Third, despite very high-initial discharge capacity (280 mAh g−1) the cycling always showed very poor capacity retention. In some severe cases, less than 50% capacity remained after only five cycles (Rasul et al., 2012a,b; Kim et al., 2015b).
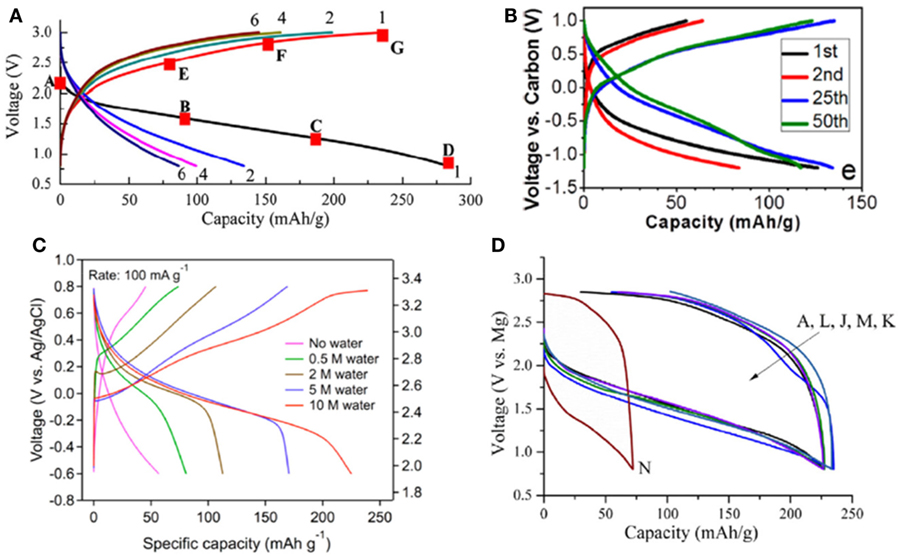
Figure 1. Performance of MnO2 in non-aqueous magnesium battery. (A) α-MnO2 in 0.2 M Mg-HMDS/tetrahydrofuran. Reprinted with permission from Zhang et al. (2012). Copyright 2012 Elsevier. (B) Birnessite-Mg0.15MnO2⋅0.9H2O in 0.25 M Mg(TFSI)2/diglyme. Reprinted with permission from Sun et al. (2016). Copyright 2016 American Chemical Society. (C) Birnessite-MnO2⋅0.94H2O in 0.5 M Mg(ClO4)2/AN with different water concentrations. Reprinted with permission from Nam et al. (2015). Copyright 2016 American Chemical Society. (D) Different polymorphs of MnO2 in 0.2 M APC/THF. A: α-; L: γ-; J: δ-; M: β-; K: γ; N: α-mixed with Mn2O3. Reprinted with permission from Zhang et al. (2015). Copyright 2015 Elsevier.
Because the preparation of α-MnO2 typically involved the participation of secondary cation to stabilize the open channel, it was thought that the cathode performance can be improved by designing stabilizing species (Thackeray, 1997). For example, Li2O stabilized α-MnO2 significantly improved the cyclability in LIB due to the minimized structural damage during Li+-intercalation (Thackeray, 1997). Surprisingly, in rMB, the discharge capacity of K+-stabilized α-MnO2 increased with the amount of K+ (Zhang et al., 2015), although it seemed that by introducing K+ in the channel the diffusion of Mg2+ should be negatively affected. The discharge capacity was also affected by ionic size of stabilizing ion (Zhang et al., 2015). The optimized capacity was reached in the cathode with the radius of stabilizing ions in the range of 1.4–1.5 Å (H3O+ and K+). However, no obvious difference of cycling behavior was observed when K+-stabilized and Li2O-stabilized α-MnO2 was compared in rMB, in contrast to that in LIB.
Interestingly and surprisingly, the characteristics observed in the cycling of α-MnO2 were not the fingerprint for this specific polymorph. Reports of todorokite phase (Kumagai et al., 2001), manganese oxide octahedral molecular sieves (OMS-5 MnO2) cathode (Rasul et al., 2013), Birnessite phase (B-MnO2) (Rasul et al., 2012a), and spinel phase (Kim et al., 2015b) all exhibited similar behaviors in dry non-aqueous electrolyte. We compared the performance of a series of MnO2 with different polymorphs (α-, β-, γ-, and δ-phases) and found that the polymorphic structure of MnO2 did not show great impact for the cathode activity in non-aqueous Mg cells (Figure 1D) (Zhang et al., 2015). It was in sharp contrast with that in LIB, where Li+-intercalation was strongly affected by the crystal structure of the host lattice (Thackeray, 1997; Goodenough and Kim, 2010). On the other side, the surface area of cathode strongly affected the discharge capacity (Zhang et al., 2015). Cathodes with surface area larger than 70 m2 g−1 constantly showed high discharge capacity (~250 mAh g−1).
These results disfavored an intercalation mechanism. Particularly, the observation that the capacity of α-MnO2 increased with surface area until getting stabilized for cathodes with surface area above 70 m2 g−1 (Figure 2A) indicated that the activity was related to a surface process instead of bulk insertion (Zhang et al., 2015). As shown in Figure 2B, a non-crystalline layer apparently appeared on the surface of the α-MnO2 crystalline nanorods after electrochemical magnesiation (Arthur et al., 2014). The majority of magnesium was located in the shell, whereas potassium as stabilizing ion is located primarily in the core. In addition, the shell contained higher concentration of Mn2+ while and the core was rich in unreduced Mn4+. Combining these evidence, the reaction was deciphered as a conversion mechanism with several possible electrochemical and chemical reactions (Arthur et al., 2014):
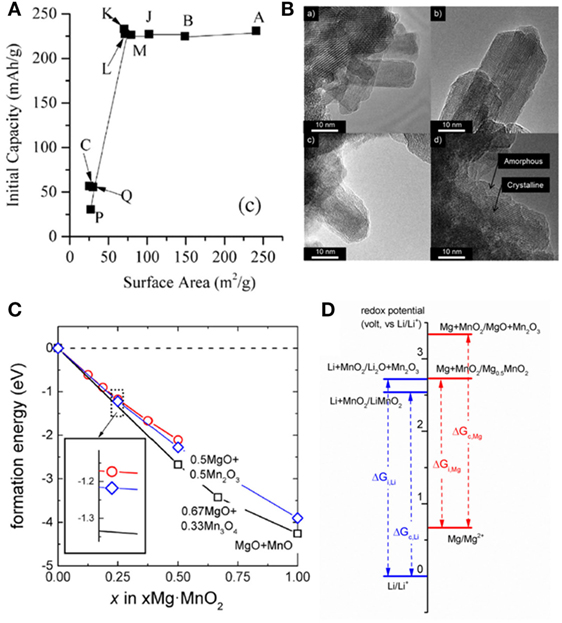
Figure 2. Reaction mechanism of MnO2 in non-aqueous magnesium battery at super dry condition. (A) Capacity of different MnO2 cathodes versus surface area of active material. Reprinted with permission from Zhang et al. (2015). Copyright 2015 Elsevier. (B) Surface amorphourization of α-MnO2 cathode. Reprinted with permission from Arthur et al. (2014). Copyright 2014 American Chemical Society. (C) Reaction energy with the formation of intercalated compound (red), amorphous magnesium and manganese oxides (blue), and crystalline magnesium oxide and manganese oxides (black). (D) Energy diagram (unit: volt) for the magnesiation and lithiation of α-MnO2. Reprinted with permission from Ling et al. (2015). Copyright 2015 American Chemical Society.
Although it was still debated that the conversion of α-MnO2 may be a result of poor compatibility between Grignard-based electrolyte and oxide (Shterenberg et al., 2015), the choice of electrolyte did not cause apparent difference as long as it supported the conduction of Mg2+ and the anode reaction. In a recent work, the discharge voltage curve looked extremely similar in magnesium monocarborane and complex electrolyte (APC—all phenyl complex) (Tutusaus et al., 2015). Wang et al. tested the chemical magnesiation of α-MnO2 using di-n-butylmagnesium/heptane and diphenylmagnesium/THF and failed to observe any intercalated product. Instead, only amorphous MgO was detected in 25Mg NMR (Wang et al., 2015). These results suggested that the conversion is an intrinsic result of MnO2 cathode with little dependence on the choice of electrolyte.
To understand the reason that the classical intercalation did not occur in Mg-MnO2 system, we explored density functional theory to compare the intercalation and conversion path for the magnesiation (Ling et al., 2015). In Mg-Mn-O system, the most stable phase at the composition of MgMn2O4 is spinel (Ling et al., 2016). If the complete structural transformation from α-MnO2 to spinel phase is kinetically hindered in really operations, the intercalated α-MgxMnO2 was revealed to be thermodynamically less stable than amorphous xMg⋅MnO2 or mixed MgO and MnO, as shown in Figure 2C (Ling et al., 2015). As a result, the conversion reaction that generates amorphous magnesium and manganese oxide is thermodynamically more preferable than the intercalation reaction. Even if a direct conversion is presumably hindered by possible kinetic barriers, the intercalation could only occur to a concentration below α-Mg0.125MnO2, beyond which the integrity of the crystalline lattice becomes questionable due to the tetragonal to orthorhombic distortion.
The conversion reaction is thermodynamically driven by the high affinity between magnesium and oxygen (Ling et al., 2015). Figure 2D showed that the redox potential for Li-intercalation is higher than Li-conversion, resulting in thermodynamically stable intercalation. The insertion of Li into α-MnO2 was directly observed in a recent report (Yuan et al., 2015). The redox potential of Mg intercalation is nearly the same as Li-intercalation. However, the stronger affinity between Mg and O greatly stabilizes MgO. Consequently, Mg-conversion, which forms highly stable MgO, occurs at much higher potential than that for Li-conversion. It increases the thermodynamic preference on Mg-conversion reactions. Consistent with our prediction, the conversion reaction was found to be responsible to the electrochemical activity and the poor cyclability of several other oxide-based cathodes such as Mg2Mo3O8 and FePO4 (Gautaum et al., 2016; Zhang and Ling, 2016b).
Performance in Water-Containing Non-Aqueous Cells
In dry non-aqueous cells, the electrochemical reaction occurred between active MnO2 and magnesium. It was proposed to introduce species to screen the polarization of Mg2+ ions (Levi et al., 2009). One of the screening agents was water. The introduction of water in the rMB had significant effect on the electrochemical behavior, as revealed in the study of the Birnessite phase (B-MnO2). The layered structure of B-MnO2 consists of stacked MnO2 plains either with or without crystalline water in the interlayer space. The usage of B-MnO2 in rMB was first attempted by Rasul et al. (2012a). For pristine B-MnO2, the initial discharge capacity was only 65 mAh g−1, much less than that of α-MnO2. Sun et al. synthesized B-Mg0.15MnO2⋅0.9H2O using hydrothermal method and reported a capacity of 80 mAh g−1 in three-electrode cell test (Sun et al., 2016), close to the value reported by Rasul et al. (2012a). However, they observed a flat voltage plateau at ~1.4 V during discharge and ~1.7 V during charge. In the coin cell test, a capacity of about 135 mAh g−1 was achieved after a conditioning of 20 cycles, as shown in Figure 1B. After that the capacity gradually decreased. By measuring the water content in the electrolyte, they found that the crystalline water was steadily released in the electrolyte during the conditioning cycles. Coincidentally, the number of cycles required to release all of the structural water into the electrolyte corresponds to the number of cycles of the conditioning process. The ex situ XPS and EELS analysis revealed the formation of Mg(OH)2, MnO, and MnOOH in the discharge. Similar to the mechanism of α-MnO2, the magnesiation of B-MnO2 was a conversion reaction instead of Mg2+-intercalation. The formation of hydroxide and oxyhydroxide species suggested that the water also participated in the reaction (Sun, 2014):
When more water was added in the electrolyte, the electrochemical performance of B-MnO2 changed dramatically. Nam et al. (2015) observed that the capacity of B-MnO2⋅0.94H2O, synthesized by aqueous electrochemical transformation of the spinel manganese oxide, increased significantly with water concentration, from 56.8 mAh g−1 to 80.7, 112.7, 170.4, and 227.6 mAh g−1 for solutions containing 0 (no water), 0.5, 2, 5, and 10 M of water, respectively (Figure 1C). In addition, the discharge voltage increased and the voltage hysteresis decreased with water content. For 10 M water solution, the operation voltage was around 2.8 V versus Mg/Mg2+, appreciably higher than other reports. After 30 cycles in 10 M water solution, the cathode still retains a capacity of ~200 mAh g−1 (Nam et al., 2015).
The effect of water was also analyzed by Song et al. (2015) in the study of highly porous and amorphous MnO2 nanowires cathode. They controlled water concentration by varying the ratio between hydrated Mg(ClO4)2⋅6H2O and anhydrous Mg(ClO4)2 in PC solution. In electrolyte containing no water, no redox peaks are observed in cyclic voltammetry, and the capacity was low. When the water content increased, two redox peaks at around 0.1 and 0.9 V versus Ag/AgCl were observed. The galvanostatic charge and discharge displayed an initial capacity of 160 mAh g−1 and retained 67% after 200 cycles when 0.1 M hydrated Mg(ClO4)2⋅6H2O was used. Interestingly, they found that after activated in water-containing electrolyte, the cathode behaved significantly better than the pristine phase. After 100 cycles, the activated MnO2 still retained about 70% of the initial capacity in dry electrolyte.
Based on the experimental observations, two functions of water were proposed. The first function is to decrease the desolvation energy of Mg2+ ion (Nam et al., 2015). Because of the bivalence nature and its strong affinity to , the desolvation of Mg2+ is much more difficult than that for monovalent ions such as Li+ and Na+, resulting a strong bonding between Mg2+ and electrolyte molecules. Solvating Mg2+ by water effectively reduced the desolvation cost. As shown by Song et al.’s study (Song et al., 2015), the important factor in the desolvation process was the H2O/Mg ratio instead of total amount of water in the electrolyte. The highest capacity was reached at the H2O/Mg of 6, probably corresponding to the state when Mg2+ was fully or almost fully solvated by H2O molecules.
Another function of water is to shield the charge of bare Mg2+ by forming hydrated species. In the case of Mg2+-intercalation in B-MnO2, the water content in the cathode became higher when water concentration in the solution increased, which confirmed the insertion of Mg2+ in hydrated state. The maximal number of water molecules that were coinserted into Mg-B cathodes together with each Mg ion was estimated to be 3 in two studies (Nam et al., 2015; Song et al., 2015), indicating out of six coordinated water molecules three were removed during the insertion.
As a result of these effects, the intercalation of hydrated Mg2+(H2O)n species instead of bare Mg2+ can occur in water-containing cells (Nam et al., 2015). By avoiding the formation of conversion species, the electrochemical performance was improved. However, the coinsertion of water molecule requires sufficient size of interstitial space in the cathode host, and sometimes induces the modification of framework to accommodate the insertion of hydrated species. Figure 3 illustrated the structure evolution of B-MnO2 cycled in water-containing electrolyte (Nam et al., 2015). After the insertion of hydrated Mg2+, the discharge product was composed of a layered structure whose interlayer spacing was half of the (001), consistent with the insertion of guest cations between slabs. The ABF-STEM revealed random mixed Mg and Mn in each layer, indicating necessary interlayer Mn migration, which may trigger the layer-to-spinel transformation as observed in aqueous cells (Sun et al., 2016). Additional atomic layers were observed between the mixed Mg/Mn layers, which was assigned to the water arrangement and proved the effective shielding between Mg2+ and the host.
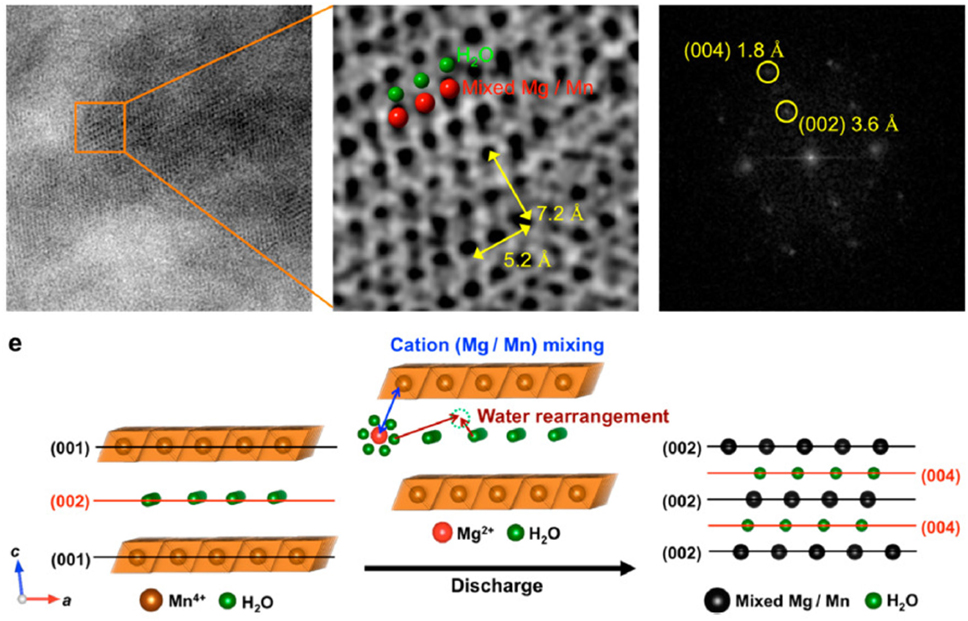
Figure 3. (A) ABF-STEM images of the discharged Mg-B cathode at low and high magnifications and FFT pattern of (B). (B) A graphical illustration of insertion of hydrated Mg2+ and Mg/Mn mixing during discharge process. Reprinted with permission from Nam et al. (2015). Copyright 2016 American Chemical Society.
MnO2 in Aqueous Magnesium Cells
The water improved performance promoted the interest of applying MnO2 cathode in fully aqueous cells. In aqueous cells, B-MnO2 exhibited sloping voltage curves with an average discharge voltage at around 2.8 V (Nam et al., 2015; Sun et al., 2016), significantly higher than that in dry electrolyte and even in wet electrolyte, suggesting the aqueous solution greatly improves the kinetics for Mg2+-transportation. In Sun et al.’s work (Sun et al., 2016), the initial capacity of B-MnO2 was 150 mAh g−1 (Figure 4A). Capacity fading was observed over the first 20 cycles, which was attributed to the enhancement of Mn2+ dissolution from the nanosized material. The stable capacity was ~50 mAh g−1 after ~100 cycles (Figure 4B).
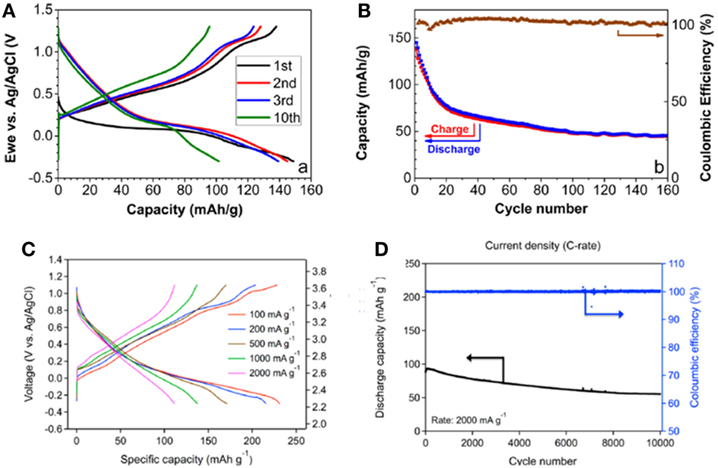
Figure 4. Performance of Birnessite-MnO2 in aqueous magnesium battery. (A) Birnessite-Mg0.15MnO2⋅0.9H2O in 0.5 M Mg(ClO4)2 and (B) capacity and Coulombic efficiency evolution at 2 C. Reprinted with permission from Sun et al. (2016). Copyright 2016 American Chemical Society. (C) Birnessite-MnO2⋅0.94H2O in 0.5 M Mg(ClO4)2 at various current densities and (D) cycling performance at 2000 mA g−1. Reprinted with permission from Nam et al. (2015). Copyright 2016 American Chemical Society.
A good performance was reported by Nam et al. using B-MnO2 cathode in aqueous cells (Nam et al., 2015). The cell exhibited a reversible capacity of 231.1 mAh g−1 (Figure 4C) and excellent rate capability (Figure 4D). When the current density was increased from 100 to 2,000 mA g−1, the capacity retention was still 48.6%. More importantly, at 500 and 2,000 mA g−1, 78.3 and 62.5% of the initial capacities were retained after 500 and 10,000 cycles, respectively. This outstanding cycling behavior was among the best for reported Mg battery cathodes.
The structural evolution of B-MnO2 cycled in aqueous solution was analyzed by Sun et al. (2016). They observed a contraction of the interlayer spacing from 7 to 4.86 Å after discharge. The latter value coincided with the distance between the octahedral slabs in spinel MgMn2O4. Based on this, they proposed Mg2+-intercalation triggered phase transformation: the insertion of Mg2+ at the tetrahedral interlayer site generates a very short (1.84 Å) Mn−Mg distance, repelling Mn out of the triangular lattice and into the interlayer space. The local structure of the discharged phase had the same arrangement of spinel MgMn2O4 as partially occupied triangular slabs of MnO6 octahedra interconnected by tetrahedral MgO4 and octahedral MnO6 moieties.
Direct evidence for Mg2+-intercalation in spinel MnO2 in aqueous cell was obtained by Kim et al. (2015a). They discharged spinel MnO2 (acid-treated LiMn2O4) in 1 M Mg(NO3)2 solution (Figure 5A). The discharge was interpreted as the formation of spinel MgMn2O4. The ABF-STEM image identified clear positions of Mn, Mg, and O in a spinel structure as shown in Figure 5B. Interestingly, the insertion of Mg into the spinel hosts strongly depended on the size of cathode particle. Nanoflakes with short the diffusion lengths showed appreciable insertion while micrometric particles was less intercalated, indicating the intercalation was limited by bulk diffusion.
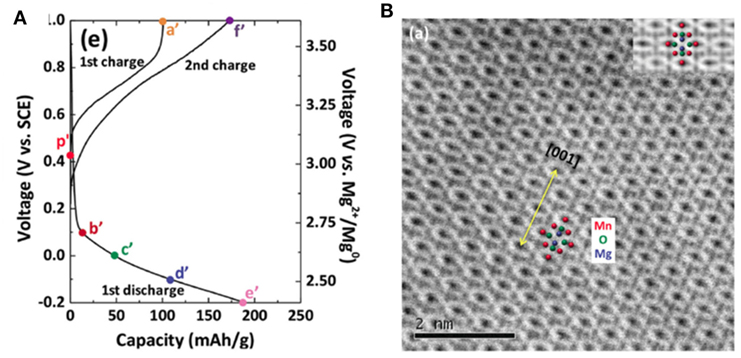
Figure 5. (A) Electrochemical responses of the Pt/1 M Mg(NO3)2/acid-treated spinel LixMn2O4 cell. (B) ABF-STEM image of the acid-treated spinel. Atomic positions are marked as Mn (red), O (green), and Mg (blue). Reprinted with permission from Kim et al. (2015a). Copyright 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Spinel MgMn2O4 has a Jahn-Teller distorted tetragonal lattice with oxygen anion forming a close-packed framework and Mg2+ and Mn3+ occupying tetrahedral and octahedral sites, respectively. Compared to that in B-MnO2, the interlayer spacing in spinel phase is halved. It is unlikely that water molecules can be accommodated in the compact interstitial space of spinel (Sun et al., 2016). Indeed, Kim et al. (2015a) did not find any sign of water insertion in their study, indicating water may not be necessary for Mg2+-intercalation. This conclusion raised an interesting yet unanswered question: why in wet non-aqueous electrolyte water molecules were coinserted with Mg2+ in the form of hydrated species while in aqueous solution only Mg2+ was intercalated? More work is still necessary to understand the mechanistic difference between Mg2+-intercalation in water-containing non-aqueous and aqueous solutions.
Computational Studies
Recently, first principles based computational studies have been applied to study Mg-MnO2 system. In addition to unveiling complicated mechanism in the magnesiation of rMB cathode (Gautam et al., 2015, 2016; Ling et al., 2015), computational work helps find suitable cathode candidate for experimental studies. Liu et al. (2015) found that among various spinel compounds MnO2 is particularly interesting due to the stability especially at charged state. The diffusion of Mg2+ has the barrier in the range of 600–800 meV, suggesting potentially feasible mobility particularly at small particle sizes. High Mg mobility was predicted in an unusual form of MgMn2O4 (Ling and Mizuno, 2013). Its crystalline structure is analog to that of CaFe2O4, with Mg replacing Ca and Mn replacing Fe. The diffusion barrier for Mg migration along the one-dimensional channel was ~0.40 eV, comparable to Li diffusion in many LIB cathodes. If the full capacity of this compound can be utilized, the energy density of this cathode is ~1.3 times to current LIB cathode. However, this unusual polymorph of MgMn2O4 is only thermodynamically stable at high pressures, which creates challenge for experimental synthesis and evaluation.
The prediction of Mg battery cathode performance requires careful examination of both thermodynamics and kinetics. For this purpose, we developed a triangular radar-type graph to quantitatively visualize the thermodynamics, kinetics, and structural information in the same figure (Ling et al., 2016). As shown in Figure 6, the triangular graph contains the calculated free energy difference between conversion and intercalation reaction, the deformation scored defined by the change of key structural parameters such as bond length and bond angles, and the diffusion barrier. The predictions from Figure 6 agreed well with experiments. For example, we predicted that α-MnO2 and Ramsdellite-MnO2 are both prone to conversion reaction due to high ΔG and deformation score, which was consistent with experimental results (Zhang et al., 2015). The most promising candidate to achieve Mg2+-intercalation is the spinel phase, which also agreed well with Kim et al.’s observation and Liu et al.’s prediction (Kim et al., 2015a; Liu et al., 2015).
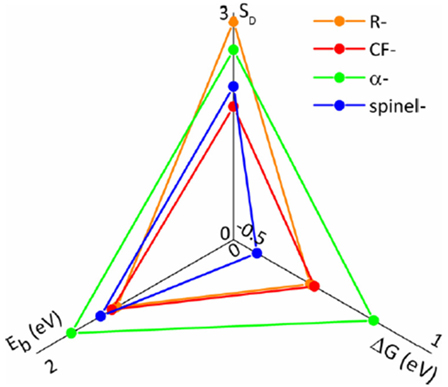
Figure 6. Triangular radar graph to quantitatively assess the free energy change (ΔG), deformation score (SD), and the diffusion barrier (Eb) for the magnesiation of MnO2 polymorphs. Reprinted with permission from Ling et al. (2016). Copyright 2016 American Chemical Society.
Summary and Future Perspective
Table 1 summarizes the reported performance of MnO2 cathode. Currently, the cathodic performance of MnO2 was impressively recorded at the capacity of >150–200 mAh g−1 at voltage of 2.6–2.8 V with cyclability to hundreds or more cycles. If continuous improvement is achieved to realize one-electron transfer in MnO2 cathode, the cathode capacity will be boosted to 308 mAh g−1. We note that the study of the modification of MnO2 cathode materials through the structural control or other optimization methods has not been seriously carried out in most reported work. Thus, it is optimistic to assume that the performance can be further improved from current level. Coupled with the dendrite free metal magnesium anode, the capacity of the active electrode reaches 271 mAh g−1 and the energy density achieves 700 mWh g−1, exceeding to the theoretical energy density of graphite/spinel LiMn2O4 (~440 mWh g−1) and even Si/spinel LiMn2O4 systems (~570 mWh g−1, assuming a theoretical capacity of Si anode is 4,200 mAh g−1). It will greatly help rMB compete with LIB technology. However, it should be noted that additional challenges such as strong Jahn-Teller distortion and Mn-dissolution will appear with the fully reduction from Mn4+O2 to Mn3+ (Mizuno et al., 2014).
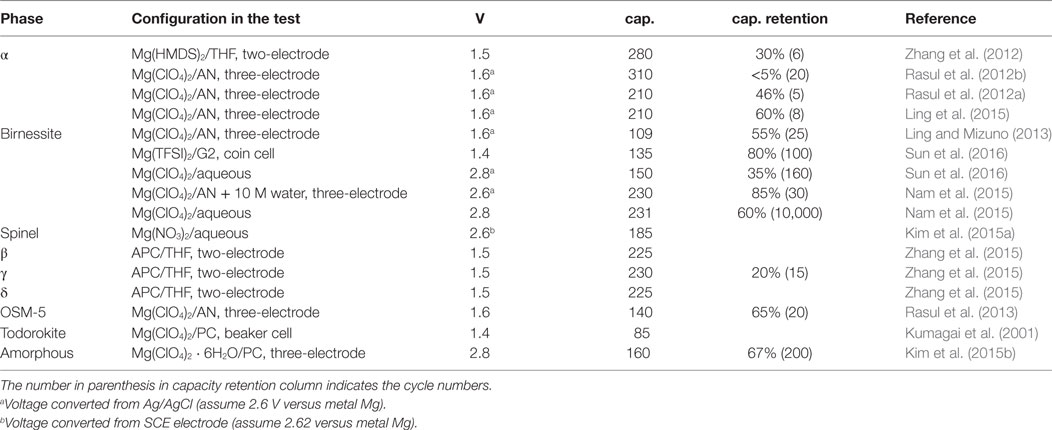
Table 1. Summary of average discharge voltage, highest discharge capacity (cap.), and capacity retention for MnO2 in different magnesium cell configurations.
Despite large capacity recorded for MnO2 cathode, it suffered from rapid capacity decline during the cycling in early reports. Improvement of sustainable cyclability was achieved by adding H2O in the electrolyte solution, or using aqueous cells. The role of water was complicated in magnesium batteries. It can participate as solvation shell, fascinate coinsertion, shield the charge of Mg2+, trigger the phase transformation, and insert as proton (Mizuno et al., 2014). For MnO2 cathode, the electrochemical reaction occurred through conversion reaction, H2O-participated conversion reaction, hydrated Mg2+(H2O)n-intercalation, and Mg2+-intercalation, depending on the concentration and source of H2O, as summarized in Table 2. Further studies are still necessary to provide more details to the presented knowledge, such as the thermodynamics of MnO2-Mg-water phase, the kinetics of Mg2+(H2O)n-intercalation and the effect of water on MnO2 structure. At this moment, the difference between aqueous cells and water-containing non-aqueous cells is still unclear: while Mg2+-intercalation in wet solution was interpreted as the insertion of hydrated species, in aqueous cell where abundant water molecules are available the insertion does not involve the participation of water (Kim et al., 2015a; Nam et al., 2015; Sun et al., 2016). Future DFT-based computational studies are especially welcome to address these questions.
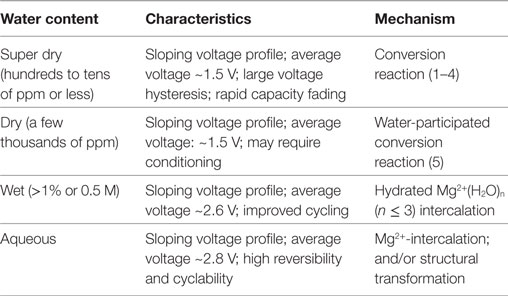
Table 2. Summary of characteristics and reaction mechanism of MnO2 cathode in different magnesium cell configurations.
While the water-containing non-aqueous and aqueous cells seem to be prominent compared to non-aqueous systems in terms of cathode performance, it must be noted that Mg battery containing water is not a practical choice currently because of the incomparability between water and metal Mg anode. The discovery of an additive which solvates Mg2+ similar to H2O but does not have the incompatibility issue is highly welcome to improve the cathode performance, especially the cyclability, in practical rechargeable Mg cells. One of such species might be Cl−, which could form MgCl+ and be inserted in layered sulfides (Yoo et al., 2017). On the other side, a reversible aqueous Mg cell can be achievable if metal Mg anode can be effectively protected from water while allowing stable Mg deposition and dissolution. These directions should deserve some consideration toward developing practical rMBs.
In summary, while rechargeable magnesium battery has received increased attention as a promising alternative to current Li-ion technology, significant effort must be devoted to the discovery of cathode candidate to make it really compelling. Undoubtedly, the exploration of novel cathode cannot be succeeded without sufficient knowledge at fundamental level. From this review, we showed how the mechanistic understanding about the electrochemical activity of MnO2 has been changed and how it paved the road to the improvement of cathode performance. We believe that with continuation of such efforts the research community will finally welcome new breakthroughs toward development of practical rechargeable magnesium batteries.
Author Contributions
CL conceived the idea and wrote the draft. Both authors participated into analyzing the results and finalizing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arthur, T. S., Zhang, R., Ling, C., Glans, P.-A., Fan, X., Guo, J., et al. (2014). Understanding the electrochemical mechanism of K-αMnO2 for magnesium battery cathodes. ACS Appl. Mater. Interfaces 6, 7004–7008. doi: 10.1021/am5015327
Aubach, D., Lu, Z., Schechter, A., Gofer, Y., Gizbar, H., Turgeman, R., et al. (2000). Prototype systems for rechargeable magnesium batteries. Nature 407, 734–737. doi:10.1038/35037566
Bucur, C. B., Gregory, T., Olliver, A. G., and Muldoon, J. (2015). Confession of a magnesium battery. J. Phys. Chem. Lett. 6, 3578–3591. doi:10.1021/acs.jpclett.5b01219
Cabana, J., Monconduit, L., Larcher, D., and Palacín, M. R. (2010). Beyond intercalation-based Li-ion batteries: the state of the art and challenges of electrode materials reacting through conversion reactions. Adv. Energy Mater. 22, E170–E192. doi:10.1002/adma.201000717
Gautam, G. S., Canepa, P., Abdellahi, A., Urban, A., Malik, R., and Ceder, G. (2015). The intercalation phase diagram of Mg in V2O5 from first-principles. Chem. Mater. 27, 3733–3742. doi:10.1021/acs.chemmater.5b00957
Gautam, G. S., Canepa, P., Richards, W. D., Malik, R., and Ceder, G. (2016). Role of structural H2O in intercalation electrodes: the case of Mg in nanocrystalline xerogel-V2O5. Nano Lett. 16, 2426–2431. doi:10.1021/acs.nanolett.5b05273
Gautaum, G. S., Sun, X., Duffort, V., Nazar, L. F., and Ceder, G. (2016). Impact of intermediate sites on bulk diffusion barriers: Mg intercalation in Mg2Mo3O8. J. Mater. Chem. 4, 17643–17648. doi:10.1039/C6TA07804D
Gershinsky, G., Yoo, H. D., Gofer, Y., and Aurbach, D. (2013). Electrochemical and spectroscopic analysis of Mg2+ intercalation into thin film electrodes of layered oxides: V2O5 and MoO3. Langmuir 29, 10964–10972. doi:10.1021/la402391f
Goodenough, J. B., and Kim, Y. (2010). Challenges for rechargeable Li batteries. Chem. Mater. 22, 587–603. doi:10.1021/cm901452z
Gregory, T. D., Hoffman, R. J., and Winterton, R. C. (1990). Nonaqueous electrochemistry of magnesium applications to energy storage. J. Electrochem. Soc. 137, 775–780. doi:10.1149/1.2086553
Huie, M. M., Bock, D. C., Takeuchi, E. S., Marschilok, A. C., and Takeuchi, K. J. (2015). Cathode materials for magnesium and magnesium-ion based batteries. Coordination Chem. Rev. 287, 15–27. doi:10.1016/j.ccr.2014.11.005
Kim, C., Philips, P. J., Key, B., Yi, T., Nordlund, D., Yu, Y.-S., et al. (2015a). Direct observation of reversible magnesium ion intercalation into a spinel oxide host. Adv. Mater. 27, 3377–3384. doi:10.1002/adma.201500083
Kim, J.-S., Chang, W.-S., Kim, R.-H., Kim, D.-Y., Han, D.-W., Lee, K.-H., et al. (2015b). High-capacity nanostructured manganese dioxide cathode for rechargeable magnesium ion batteries. J. Power Sources 273, 210–215. doi:10.1016/j.jpowsour.2014.07.162
Kumagai, N., Komaba, S., Sakai, H., and Kumagai, N. (2001). Preparation of todorokite-type manganese-based oxide and its application as lithium and magnesium rechargeable battery cathode. J. Power Sources 97-98, 515–517. doi:10.1016/S0378-7753(01)00726-1
Levi, E., Gofer, Y., and Aurbach, D. (2010). On the way to rechargeable Mg batteries: the challenge of new cathode materials. Chem. Mater. 22, 860–868. doi:10.1021/cm9016497
Levi, E., Levi, M. D., Chasid, O., and Aurbach, D. (2009). A review on the problems of the solid state ions diffusion in cathodes for rechargeable Mg batteries. J. Electroceram. 22, 13–19. doi:10.1007/s10832-007-9370-5
Ling, C., Banerjee, D., and Matsui, M. (2012). Study of the electrochemical deposition of Mg in the atomic level: why it prefers the non-dendritic morphology. Electrochim. Acta 76, 270–274. doi:10.1016/j.electacta.2012.05.001
Ling, C., and Mizuno, F. (2012). Capture lithium in αMnO2: insights from first principles. Chem. Mater. 24, 3943–3951. doi:10.1021/cm302347j
Ling, C., and Mizuno, F. (2013). Phase stability of post-spinel compound AMn2O4 (A= Li, Na, or Mg) and its application as a rechargeable battery cathode. Chem. Mater. 25, 3062–3071. doi:10.1021/cm401250c
Ling, C., and Suto, K. (2017). Thermodynamic origin of irreversible magnesium trapping in chevrel phase Mo6S8: importance of magnesium and vacancy ordering. Chem. Mater. 29, 3731–3739. doi:10.1021/acs.chemmater.7b00772
Ling, C., Zhang, R., Arthur, T. S., and Mizuno, F. (2015). How general is the conversion reaction in Mg battery cathode: a case study of the magnesiation of α-MnO2. Chem. Mater. 27, 5799–5807. doi:10.1021/acs.chemmater.5b02488
Ling, C., Zhang, R., and Mizuno, F. (2016). Quantitatively predict the potential of MnO2 polymorphs as magnesium battery cathodes. ACS Appl. Mater. Interfaces 8, 4508–4515. doi:10.1021/acsami.5b11460
Liu, M., Rong, Z., Malik, R., Canepa, P., Jain, A., Ceder, G., et al. (2015). Spinel compounds as multivalent battery cathodes: a systematic evaluation based on ab initio calculations. Energy Environ. Sci. 8, 964–974. doi:10.1039/C4EE03389B
Matsui, M. (2010). Study on electrochemically deposited Mg metal. J. Power Sources 196, 7048–7055. doi:10.1016/j.jpowsour.2010.11.141
Mizuno, F., Singh, N., Arthur, T. S., Fanson, P. T., Ramanathan, M., Benmayza, A., et al. (2014). Understanding and overcoming the challenges posed by electrode/electrolyte interfaces in rechargeable magnesium batteries. Front. Energy Res. 2:46–56. doi:10.3389/fenrg.2014.00046
Mohtadi, R., and Mizuno, F. (2014). Magnesium batteries: current state of the art, issues and future perspectives. Beilstein J. Nanotech. 5, 1291–1311. doi:10.3762/bjnano.5.143
Muldoon, J., Bucur, C. B., and Gregory, T. (2014). Quest for nonaqueous multivalent secondary batteries: magnesium and beyond. Chem. Rev. 114, 11683–11720. doi:10.1021/cr500049y
Muldoon, J., Bucur, C. B., Oliver, A. G., Sugimoto, T., Matsui, M., Kim, H. S., et al. (2012). Electrolyte roadblocks to a magnesium rechargeable battery. Energy Environ. Sci. 5, 5941–5950. doi:10.1039/c2ee03029b
Nam, K. W., Kim, S., Lee, S., Salama, M., Shterenberg, I., Gofer, Y., et al. (2015). The high performance of crystal water containing manganese birnessite cathodes for magnesium batteries. Nano. Lett. 6, 4071–4079. doi:10.1021/acs.nanolett.5b01109
Post, J. E. (1999). Manganese oxide minerals: crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. U.S.A. 96, 3447–3454. doi:10.1073/pnas.96.7.3447
Rasul, S., Suzuki, S., Yamaguchi, S., and Miyayama, M. (2012a). High capacity positive electrodes for secondary Mg-ion batteries. Electrochim. Acta 82, 243–249. doi:10.1016/j.electacta.2012.03.095
Rasul, S., Suzuki, S., Yamaguchi, S., and Miyayama, M. (2012b). Synthesis and electrochemical behavior of hollandite MnO2/acetylene black composite cathode for secondary Mg-ion batteries. Solid State Ionics. 225, 542–546. doi:10.1016/j.ssi.2012.01.019
Rasul, S., Suzuki, S., Yamaguchi, S., and Miyayama, M. (2013). Manganese oxide octahedral molecular sieves as insertion electrodes for rechargeable Mg batteries. Electrochim. Acta 110, 247–252. doi:10.1016/j.electacta.2013.06.094
Sa, N., Kinnibrugh, T. L., Wang, H., Gautam, G. S., Chapman, K. W., Vaughey, J. T., et al. (2016). Structural evolution of reversible Mg insertion into a bilayer structure of V2O5·nH2O xerogel material. Chem. Mater. 28, 2962–2969. doi:10.1021/acs.chemmater.6b00026
Shterenberg, I., Salama, M., Yoo, H. D., Gofer, Y., Park, J.-B., Sun, Y.-K., et al. (2015). Evaluation of (CF3SO2)2N-(TFSI) based electrolyte solutions for Mg batteries. J. Electrochem. Soc. 162, A7118–A7128. doi:10.1149/2.0161513jes
Song, J., Noked, M., Gillette, E., Duay, J., Rubloff, G., and Lee, S. B. (2015). Activation of a MnO2 cathode by water-stimulated Mg2+ insertion for a magnesium ion battery. Phys. Chem. Chem. Phys. 17, 5256–5264. doi:10.1039/C4CP05591H
Song, J., Shadeo, E., Noked, M., and Lee, S. B. (2016). Mapping the challenges of magnesium battery. J. Phys. Chem. Lett. 7, 1736–1749. doi:10.1021/acs.jpclett.6b00384
Sun, J. Z. (2014). Preparation and characterization of novel positive electrode material for magnesium cells. Monatsh. Chem. 145, 103–106. doi:10.1007/s00706-013-0977-8
Sun, X., Duffort, V., Mehdi, B. L., Browning, N. D., and Nazar, L. F. (2016). Investigation of the mechanism of Mg insertion in birnessite in nonaqueous and aqueous rechargeable Mg-ion batteries. Chem. Mater. 28, 534–542. doi:10.1021/acs.chemmater.5b03983
Thackeray, M. M. (1997). Manganese oxides for lithium batteries. Prog. Solid St. Chem. 25, 1–71. doi:10.1016/S0079-6786(97)81003-5
Tutusaus, O., Mohtadi, R., Arthur, T. S., Mizuno, F., Nelson, E. G., and Sevryugina, Y. V. (2015). An efficient halogen-free electrolyte for use in rechargeable magnesium batteries. Angew. Chem. Int. Ed. 54, 7900. doi:10.1002/anie.201412202
Wang, H., Senguttuvan, P., Proffit, D. L., Pan, B., Liao, C., Burrell, A. K., et al. (2015). Formation of MgO during chemical magnesiation of Mg-ion battery materials. ECS Electrochem. Lett. 4, A90–A93. doi:10.1149/2.0051508eel
Yabuuchi, N., Kubota, K., Dahbi, M., and Komaba, S. (2014). Research development on sodium-ion batteries. Chem. Rev. 114, 11636–11682. doi:10.1021/cr500192f
Yoo, H. D., Liang, Y., Dong, H., Lin, J., Wang, H., Liu, Y., et al. (2017). Fast kinetics of magnesium monochloride cations in interlayer-expanded titanium disulfide for magnesium rechargeable batteries. Nature Commun. 8, 339. doi:10.1038/s41467-017-00431-9
Yoo, H. D., Shterenberg, I., Gofer, Y., Gershinsky, G., Pour, N., and Aurbach, D. (2013). Mg rechargeable batteries: an on-going challenge. Energy Environ. Sci. 6, 2265–2279. doi:10.1039/c3ee40871j
Yu, H.-C., Ling, C., Bhattacharya, J., Thomas, J. C., Thornton, K., and Van Der Ven, A. (2014). Designing the next generation high capacity battery electrodes. Energy Environ. Sci. 7, 1760–1768. doi:10.1039/c3ee43154a
Yuan, Y., Nie, A., Odegard, G. M., Xu, R., Zhou, D., Santhanagopalan, S., et al. (2015). Asynchronous crystal cell expansion during lithiation of K+-stabilized α-MnO2. Nano Lett. 15, 2998–3007. doi:10.1021/nl5048913
Zhang, R., Arthur, T. S., Ling, C., and Mizuno, F. (2015). Manganese dioxides as rechargeable magnesium battery cathode: synthetic approach to understand magnesiation process. J. Power Sources 282, 630–638. doi:10.1016/j.jpowsour.2015.02.067
Zhang, R., and Ling, C. (2016a). Status and challenge of Mg battery cathode. MRS Energy Sustainbility. 3, E1. doi:10.1557/mre.2016.2
Zhang, R., and Ling, C. (2016b). Unveil the chemistry of olivine FePO4 as magnesium battery cathode. ACS Appl. Mater. Interfaces. 8, 18018–18026. doi:10.1021/acsami.6b03297
Keywords: MnO2, magnesium battery, intercalation, conversion, beyond Li-ion battery
Citation: Ling C and Zhang R (2017) Manganese Dioxide As Rechargeable Magnesium Battery Cathode. Front. Energy Res. 5:30. doi: 10.3389/fenrg.2017.00030
Received: 17 July 2017; Accepted: 11 October 2017;
Published: 03 November 2017
Edited by:
Jian Liu, University of British Columbia Okanagan, CanadaReviewed by:
Yingwen Cheng, Pacific Northwest National Laboratory (DOE), United StatesReza Younesi, Uppsala University, Sweden
Yufeng Zhao, Yanshan University, China
Copyright: © 2017 Ling and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Ling, Y2hlbi5saW5nQHRveW90YS5jb20=