 Jason M. Lang1
Jason M. Lang1 Umesh M. Shrestha
Umesh M. Shrestha Mark Dadmun
Mark Dadmun- 1Department of Chemistry, University of Tennessee, Knoxville, TN, United States
- 2Chemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, United States
This work increases our understanding of the effect of plant source on the mechanical and morphological properties of lignin-based polyurethanes (PUs). Lignin is a polymer that is synthesized inside the plant cell wall and can be used as a polyol to synthesize PUs. The specific aromatic structure of the lignin is heavily reliant on the plant source from which it is extracted. These results show that the mechanical properties of lignin-based PUs differ based on lignin’s plant source. The morphology of lignin-based PUs was examined using atomic force microscopy and scanning electron microscopy and the mechanical properties of lignin-based PU samples were measured using dynamic mechanical analysis and shore hardness (Type A). The thermal analysis and morphology studies demonstrate that all PUs prepared form a multiphase morphology. In these PUs, better mixing was observed in the wheat straw lignin PU samples leading to higher moduli than in the hardwood lignin and softwood lignin PUs whose morphology was dominated by larger aggregates. Independent of the type of the lignin used, increasing the fraction of lignin increased the rigidity of PU. Among the different types of lignin studied, PU with wheat straw soda lignin exhibited storage moduli ~2-fold higher than those of PUs incorporating other lignins. This study also showed that during synthesis all hydroxyl groups in the lignin are not available to react with isocyanates, which alters the number of cross-links formed within the PU and impacts the mechanical properties of the material.
Introduction
Over the past few decades, polymer materials have become important industrially produced materials due to their wide range of physical and chemical properties. These polymer materials have found use in a vast array of technologies ranging from wiring, coatings, sports, and medical devices to cell phones, houses, automobiles, and planes. However, the demand for new and more elaborate technologies that are more robust than their predecessors will continue to increase. For instance, according to Grand View Research, Inc., the polyurethane market was estimated at ~$54 billion in 2015 and is projected to grow to ~$105 billion by the year 2025 due to recent high demands for applications in furniture, automotive, electronic devices, and footwear (Grand View Research, 2017). Therefore, further research is needed to synthesize and understand new polymer materials that will allow the advancement of modern technology into the future.
Polyurethane (PU)
Polyurethanes were first discovered by Otto Bayer and his colleagues in 1937 (Seymour and Kauffman, 1992). PUs are known for their wide range of properties including high tensile strength, abrasion resistance, and chemical resistance (Saraf and Glasser, 1984, Sarkar and Adhikari, 2001). These properties, among others, make PUs an important commercial polymer for use as flexible or rigid foams, solid elastomers, coatings, and adhesives among other applications (Stevens, 1999, Szycher, 1999, Akindoyo et al., 2016).
Polyurethanes are polymers that are derived from monomer units joined together by carbamate (urethane) linkages. The most common pathway to synthesize a urethane linkage is to react an isocyanate functional group (R-N=C=O) with a hydroxyl functional group (R-OH). For a PU to be successfully synthesized using this method, a polyisocyanate that contains two or more isocyanate groups per molecule, reacts with a polyol, a compound that contains two or more hydroxyl groups per molecule, as shown in Figure 1. In some circumstances, an organic or inorganic catalyst or ultraviolet light is needed for the urethane reaction to take place (Silva and Bordado, 2004, Soto et al., 2014).
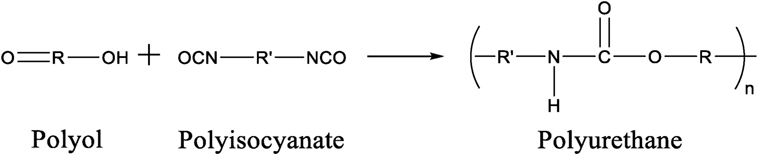
Figure 1. Reaction of a polyol with a polyisocyanate to synthesize a polyurethane.
Isocyanate is the more reactive group than hydroxyl group in PU synthesis. The majority of isocyanates used such as toluene-2,4-diisocyanate, isophorone diisocyanate, and 4,4-methylene diphenyl diisocyanate are aromatic in structure while aliphatic isocyanates, such as hexamethylene diisocyanate (HMDI), are also used (Teo et al., 1997, Chan-Chan et al., 2010, Prisacariu, 2011, Chung and Washburn, 2012, Zhang et al., 2015). Polyols are usually the more flexible of the two components which allows the formation of flexible PU materials and usually consist of polyesters, polyethers, or glycols such as polyethylene glycol (PEG), 1,4-butanediol, and polypropylene glycol (PPG) (Grassie and Zulfiqar, 1978; Wen et al., 2001; Oprea, 2010; Nozaki et al., 2017). The structure of these compounds is presented in Figure 2. The properties of PU depend on the structure and length of polyisocyanate and polyol monomers used during synthesis. Addition of polyisocyanate with aromatic monomer as well as short polyol chains increases the rigidity of the PU backbone which affects the mechanical properties such as Young’s modulus. On the contrary, addition of long aliphatic monomer units, most often as part of the polyol, has the opposite affect and allows the PU to have more rubbery characteristics (Yoshida et al., 1990, Bharadwaj et al., 2002). However, addition of too many aromatic or aliphatic units can produce material that is too brittle or rubbery in character for use. Therefore, a balance between these two traits must be met for a PU to be useful.
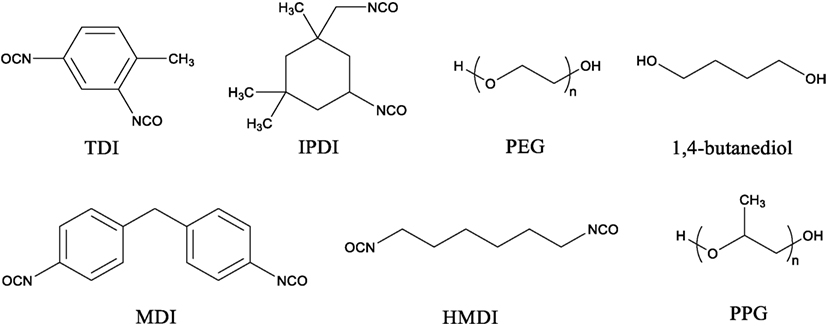
Figure 2. Structures of a select few isocyanate compounds and polyol compounds.
Another way to increase the structural rigidity of a PU is through cross-linking, as seen in Figure 3, which decreases the molecular freedom of the polymer chains. This increases mechanical properties, such as tensile modulus and decreases elongation. PU film that is cross-linked will provide higher chemical and abrasion resistance due to the formation of a tightly packed network. Cross-linking can occur via chemical reactions when at least one of the monomer units contains three or more reactive sites, addition of additives, or can be brought on through the use of radiation (Chen and Rånby, 1989; Basfar et al., 2003; Dong et al., 2012; Iype et al., 2016). PUs have another unique way to cross-link by the reaction of an isocyanate with an already formed urethane linkage to create an allophanate linkage, as shown in Figure 4. Even though the positive impacts that cross-linking has on the properties of PU is attractive, there are some drawbacks as well. One such setback is that cross-links are, for the most part, irreversible and products that have undergone cross-linking are difficult to recycle.
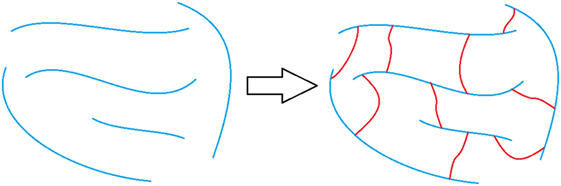
Figure 3. Representation of non-cross-linked polymer chains (left) and polymer chains that are cross-linked (right). The blue lines represent polymer chains and the red lines represent the cross-links.
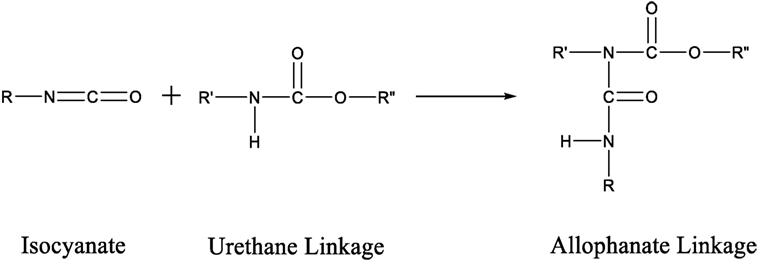
Figure 4. Scheme of the reaction between an isocyanate group and a urethane linkage to form an allophanate linkage.
Over the last decade, there has been an increase in interest in deriving polymers from a renewable resource such as biomass, due to their renewability and biodegradability (Noreen et al., 2016, Gang et al., 2017). Among many renewable resources, one bio-based polymer that is of current interest to scientists and industry is lignin due to its highly aromatic structure, natural abundance, and ability to form lignin-based PU materials.
Lignin
Lignin is a bio-based polyol and the second most naturally occurring polymer on Earth, behind cellulose, and makes up 18–35% of wood (Hon, 1996). Lignin is a highly aromatic polymer found in all plant cell wall alongside cellulose and hemicellulose. It provides structural support as well as a defense mechanism against parasites and diseases (Saito et al., 2013).
Although there is no precise definition of 3D structure of lignin, the structure consists of three-main monomer building blocks. The highly aromatic nature of lignin, shown in Figure 5A, is due to the enzyme initiated dehydrogenation polymerization and radical coupling reactions of three basic monomers or monolignols p-coumaryl alcohol, coniferyl alcohol, and syringyl alcohol shown in Figure 5B (Suhas and Ribeiro, 2007, Chatterjee and Saito, 2015). These three monolignols are very similar in structure consisting of a phenolic ring with an aliphatic side chain that includes an alkene and an alcohol functional group. Structurally they differ in the number of methoxy groups found at each ortho position relative to the phenolic hydroxyl group. These methoxy groups play a key role in the physical characteristics and construction of the lignin molecule.
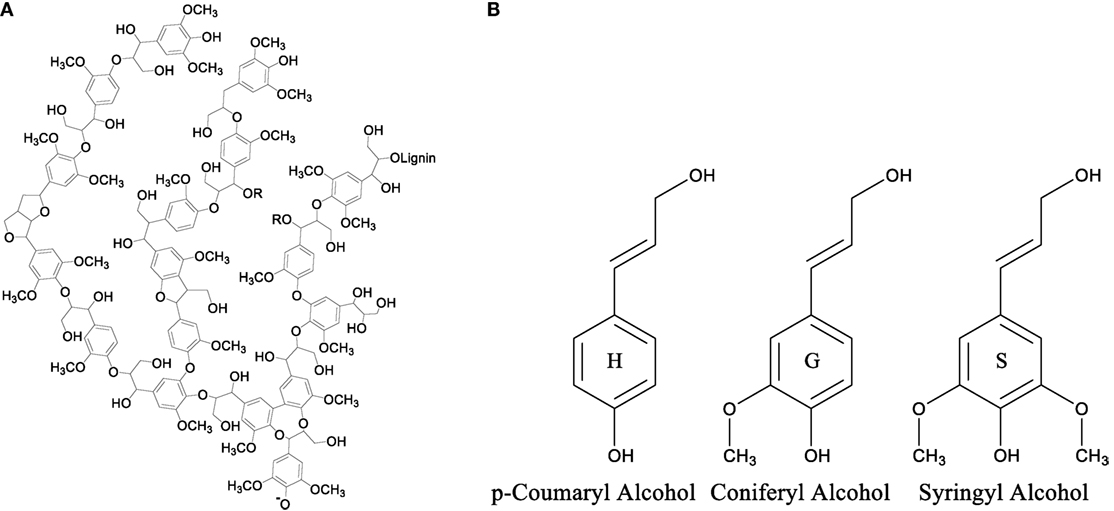
Figure 5. (A) Theoretical representation of the structure of a lignin molecule and (B) the structures of the monolignol units used to polymerize lignin.
The amounts of each monolignol used to form the lignin molecule depend on the type of plants (hardwood, softwood, and grass) from which lignin is extracted. Lignins extracted from hardwood sources contain the highest amounts of syringyl alcohol amongst the three classes of lignins with smaller amounts of coniferyl alcohol monolignols. Softwood lignins, also called coniferous or guaiacyl lignins, are solely made up of coniferyl alcohol monolignols. Lignins that are extracted from grasses, also called non-wood lignin, contain all three monolignols; however, this class of lignins contains the highest amount of p-coumaryl alcohol monolignols. The amount of methoxy groups in the monolignol is important due to the steric hindrance they provide to the phenolic hydroxyl group of the monolignol. This is because the enzyme catalyzed polymerization of the monolignol units heavily targets the two hydroxyl groups connecting the monomers together to form the polymer chain. An increase in steric hindrance leads to a decreased ability for the phenolic site to react with another monomer lowering the amount of cross-linking within the overall lignin structure. From the structure of lignin internal cross-linking, it is understood that hardwood lignin that consists of the highest amount of syringyl alcohol monolignol units possess the least internally cross-linked structure, while grasses that possess the highest amount of p-coumaryl alcohol monolignol have more internally cross-linked structure compared to other classes of lignins. This internal cross-linking affects not only lignin molecule but also the properties of lignin-based materials.
Lignin has gained significant attraction in the scientific community due to its potential use to produce polymer materials. One of the largest processes where lignin is extracted from plants is during the chemical pulping processes at paper mills as by-product (Chakar and Ragauskas, 2004). The main product for this process is cellulose to produce paper. Up until recently, lignin was viewed as waste that was only used as fuel to power the boilers during the paper-making process. In 2010, it was estimated that the paper and pulping industry alone extracted 50 million tons of lignin (Gosselink et al., 2004, Laurichesse and Avérous, 2014). Biorefineries increase that amount making lignin very economical. With lignin being highly abundant, including an estimated 300 billion tons of lignin available in the biosphere with a steady yearly increase of 20 billion tons. The high availability of lignin and its low cost further increases its attraction to be used as a starting material for producing polymer products (Gregorová et al., 2006, Laurichesse and Avérous, 2014).
Other than the plant source they are extracted from, another important classification of lignins is by the extraction process employed. There are several extraction processes that allow the separation of lignin from the lignocellulosic base. The most dominant processes used are the Kraft, sulfite, organosolv, and soda processes. The Kraft process makes up approximately 85% of the total lignin production throughout the world (Tejado et al., 2007). During this process, the wood is dissolved in an aqueous medium of sodium hydroxide and sodium sulfide and is cooked under pressure at 170°C for a couple of hours (Chakar and Ragauskas, 2004, Fatechi and Chen, 2016). During the cooking process, hydroxide and hydrosulfide anions react with the lignin and cleaves α-aryl ether and β-aryl ether bonds producing lignin with higher amounts of phenolic hydroxyl groups and a small amount of sulfur in the form of thiol groups (−SH) (Smook, 1992, Chakar and Ragauskas, 2004, Wang et al., 2016).
In sulfite pulping, lignin is extracted in the form of lignosulfonates due to sulfonic groups being introduced into the structural network of the lignin molecules (Fan and Zhan, 2008). The sulfite process uses sulfurous acid and its alkali salt versions to extract the lignin from wood chips by breaking α-O-4 ether linkages under high pressure at approximately 150°C (Qui et al., 2010, Wang et al., 2016). The attached sulfonic groups also cause lignosulfonate to act as a polyelectrolyte allowing it to be water soluble even though the backbone is composed of a hydrophobic aromatic skeleton. Lignosulfonates contain the highest amount of sulfur among all types of lignins. Although the polydispersity of lignin in general is usually high, lignosulfonates typically have the highest polydispersity among all types of lignins which can be a drawback in material production (Madad et al., 2013).
The organosolv process is a sulfur free pulping process that uses a mixture of solvents such as methanol, ethanol, acetic acid, formic acid, and peroxy-organic acids to cook the wood and separate lignin from the other wood constituents (Xu et al., 2006, Tian et al., 2017). During the organosolv process, lignin becomes solubilized by solvent or solvents used through acidolytic or alkaline cleavage of aryl ether linkages resulting in an increased number of phenolic functionalities. Due to the use of organic solvents and organic acids as the reactants, organosolv lignins have no added functionalities like thiols or sulfonic groups found in Kraft lignins and lignosulfonates (Duval and Lawoko, 2014).
Another pulping process used to extract lignin is the soda process. Like the organosolv process, it is sulfur free and does not include side reactions that add extra functionalities onto the structure. However, in the soda process, alkaline materials without sodium sulfide is used to break down the lignin. The soda process is mainly used for the degradation of grass sources such as wheat straw, hemp, switchgrass, and bagasse and the absence of sulfur content makes it very attractive for polymer applications (Doherty et al., 2011, Laurichesse and Avérous, 2013).
Due to the highly aromatic structure of lignin, different processes have been developed that break down lignin molecules to produce other valuable aromatic chemicals, such as benzene, toluene, xylene, styrene, phenols, cyclohexane, aromatic aldehydes, vanillin, and vanillic acid (da Silva et al., 2009; Varanasi et al., 2013; Yamaguchi et al., 2017). Since lignin is a polyol, different synthesis techniques have also been used to study the effect of lignin incorporation on the properties of biodegradable polymer materials. Due to the heterogeneous nature of lignin and its tendency to aggregate, homogeneous blends of lignin with other polymers are difficult to fabricate and usually results in poor mechanical properties when compared to the pure polymer counterpart (Alexy et al., 2000, Kadla and Kubo, 2004, Chung et al., 2013). However, modification of lignin, such as acetylation and grafting to/from, has been used to disrupt lignin aggregation. Resulting more homogeneous blend of lignin and polymers, as well as an improvement in physical properties (Chung et al., 2013; Sen et al., 2015; Dehne et al., 2016).
The highly aromatic structure of lignin and its ability to act as a polyol have made it a desired building block to produce lignin-based PUs. Some of the more common lignin-based polymeric materials are soft or rigid PU foams, epoxy resins, lignin-grafted-polycaprolactone copolymers, and carbon fiber (Li and Ragauskas, 2012, Baker and Rials, 2013, Laurichesse and Avérous, 2013, Zhao and Abu-Omar, 2015). For most systems, it is desired to employ as much lignin into the polymeric material as possible to further increase targeted properties, use less petroleum-based chemicals, improve economics, and increase the biocompatibility of the material. However, too much lignin incorporated into a system usually leaves the material too brittle to be useful (Saito et al., 2013). Most of the results reported with higher loading of modified lignin-based PUs were too brittle to be studied for their properties; therefore, a compromise must be found between the amount of lignin content and rubbery content used to optimize economics, renewability, and mechanical properties.
Various studies have been carried out to understand the effects on the mechanical and thermal properties of PUs incorporating lignin from hard wood or soft wood or non-wood separately. However, to the best of our knowledge, no study has been done to investigate the effect of lignin from different plant source on the properties of PU. In this study, for the first time, the mechanical, thermal properties and morphology of the PU thermoplastic incorporating lignin extracted from different plant source, hard, soft and wheat straw is studied. Different studies have been carried out to maximize the incorporation of lignin in PU by modifying the lignin with different functionalities. Here, we studied a range of fraction of unmodified lignins from each plant source to understand the balance between the maximum amount of lignin that can be incorporated as polyols to synthesize PU while maintaining flexibility in the material. The results obtained from this study will provide insight into the different parameters that influence the properties of PU. With the understanding of the parameters will allow to develop materials with desired properties.
Materials and Methods
Synthesis of Lignin-Based PU Elastomers
Three different lignins, each from different plant source, were used in this study: a Kraft processed lignin from a softwood source, a lignosulfonate lignin from a hardwood source, and a soda processed lignin from a wheat straw source. The softwood Kraft lignin (SWKL) was purchased from TCI America. The hardwood lignosulfonate lignin (HWLS) was supplied by Borregaard Lignntech and the wheat straw soda lignin (WSSL), also called Protobind 1000, was supplied by GreenValue LLC. The HWLS and the WSSL were used as received while the SWKL was subjected to an acid wash for further purification. The purification was carried out by adding approximately 10 g of SWKL to 300 mL of 2 M hydrochloric acid (HCl) and then SWKL/HCl solution was allowed to stir for 3 h open at ambient temperature and open to air. The mixture solution was then filtered using a fritted funnel with a porosity of 4–5.5 µm to obtain the SWKL particles from the solution. This step was repeated twice and then the SWKL was washed few times with deionized water to remove any leftover HCl. Thus, obtained SWKL was dried overnight in vacuum oven under vacuum strength of 30 mmHg at 60°C. The dried SWKL powder was then stored in a desiccator.
Polypropylene glycol (PPG) (MN = 2,300 g/mol) end-capped on both ends with toluene-2,4-diisocyanate (TDI) (purchased from Sigma-Aldrich) was used as isocyanate cross-linker during PU synthesis. Before synthesis, the lignin being used was heated and dried under full vacuum overnight to remove any residual moisture. All steps for the reaction of lignin with TDI–PPG–TDI to synthesize lignin-based PUs were performed in the nitrogen glove box.
PU Synthesis
2 g of isocyanate was weighed in a 50 mL beaker and allowed to mechanically stir on a hotplate at 100°C for a few minutes. Eight drops of dibutyltin dilaurate (DBTDL) purchased from Sigma-Aldrich was then added as a catalyst and isocyanate/DBTDL mixture was mechanically stirred at 100°C for 10 min to produce a homogeneous mixture. A precise amount of lignin that gave the desired lignin weight percent (wt%) was weighed out and added to isocyanate/DBTDL mixture. The lignin/isocyanate/DBTDL mixture was then allowed to stir on a hotplate at 100°C until the viscosity of the mixture increased due to the progress of the polymerization. The mixture was then poured into a Teflon® mold and allowed to cure at room temperature overnight inside the nitrogen glove box. The dimensions of the mold allowed the formation of samples appropriate as a dual-cantilever sample for the dynamic mechanical analysis (DMA). A range of lignin contents (20, 30, 40, 50, and 60 wt%) were used to synthesize lignin-based PUs. A reference PU sample was also synthesized using the same method as described for the lignin-based PU samples with a hydroxy-terminated PPG (MN = 2,000 g/mol) as the polyol.
Characterization
Dynamic Mechanical Analysis (DMA)
Mechanical properties measurements were performed using a DMA Q800 (TA Instruments) with the dual cantilever geometry and calibrated using the TA Instruments calibration kit. Samples were made by curing in a Teflon® mold with the dimensions 57 mm × 12.5 mm × 3.5 mm (L × W × H). The measurement was carried out at 30°C and 0.1% strain with a frequency of 1 Hz.
Shore Hardness
The shore hardnesses of all lignin-based PU samples were measured according to the ASTM D2240-15 standard procedure with a Type A Model 2000 Durometer made by Rex Gauge Company. Since all lignin-based PU samples did not have a thickness of 6 mm, samples of the same lignin plant source and lignin wt% were piled together to create a sample with the appropriate thickness. Piled samples were measured in three separate areas.
Differential Scanning Calorimeter (DSC)
Thermal properties of the samples were measured using a DSC Q2000 (TA Instruments), where an Indium standard was used for the calibration. Samples weighing between 5 and 10 mg were cut from DMA samples and weighed using a Cahn C-33 microbalance. The samples were then placed in a pre-weighed aluminum standard DSC pan and heated at 10°C per minute over the temperature range of −80–150°C in nitrogen environment.
Phosphorus-31 Nuclear Magnetic Resonance (31P-NMR)
Phosphorus-31 nuclear magnetic resonance spectroscopy was used to determine hydroxyl group content of lignins used. All lignin samples were dried before use to remove any moisture content due to its high reactivity with phospholane reagent. 20 mg of dried lignin sample was added to a 400 µL dimethylformamide/pyridine (1:1, v/v) solution and 200 µL of internal standard (ISTD) solution in a NMR tube. The ISTD was prepared by mixing 75 mg cyclohexanol (CH) (Sigma-Aldrich, 99%) as a standard with 10.084 g deuterated chloroform (CDCl3) and 20 mg chromium (III) acetylacetonate as a relaxation reagent. The sample solution was then derivatized with 100 µL of the phospholane reagent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) (Sigma-Aldrich, 95%). The sample mixtures were analyzed using a JEOL 400 MHz NMR spectrometer over 128 scans with inverse-gated decoupling, a 90° pulse, and a 25 s pulse delay.
A total of five 31P-NMR samples for each lignin source were run to determine an average hydroxyl group concentration and a standard deviation (SD) based on lignin source. Mestrenova software was used to integrate all 31P-NMR data. The following equations were used to calculated the concentrations of each hydroxyl group type (Olarte et al., 2016).
Scanning Electron Microscope (SEM)
The morphologies of lignin-based PU samples were investigated using a Zeiss Auriga 40 field emission SEM. Samples were cut from bulk material and were not coated during sample preparation. Images were captured using a secondary electron detector lens and a voltage level of 1.00 kV.
Atomic Force Microscope (AFM)
Images of the phase morphology of lignin-based PU samples were scanned using an Asylum Research MFP-3D Infinity AFM. Samples were cut from bulk material and were imaged under tapping mode. The uncoated silicon cantilevers with a resonant frequency range or 200–400 kHz and a spring constant of 13–77 N/m from AppNano were used for scanning. The AFM images were analyzed using the WSxM 5.0 software (Horcas et al., 2007).
Results and Discussion
In this study, lignin extracted from a hardwood source, a softwood source, and a non-wood source were used to study the effects that the plant source of lignin has on targeted mechanical properties of lignin-based PUs. As previously discussed, lignin is used as a polyol and reacted with a rubbery cross-linker to synthesize lignin-based PU samples with multiple wt% of lignin loadings. The use of the rubbery cross-linker allows the synthesis of cohesive samples that are not too brittle to be tested. The synthesis of lignin-based PUs with varying lignin loadings provides a pathway to monitor the correlation between targeted mechanical properties and lignin loading and correlate these changes to lignin plant source.
Mechanical Properties
Dynamic mechanical analysis provides information on the influence of lignin source on the mechanical properties of lignin-based PU samples. Figures 6 and 7 show the storage and loss modulus of PUs as a function of lignin content ranging from 10 to 60 wt%. It is evident from Figure 6 that, for all three lignin sources, the storage modulus increases with lignin loading. This can be explained as lignin is the rigid component in PU, while diisocyanate PPG is the rubbery component. Thus, the rigid lignin provides mechanical strength to the system while PPG brings a more viscous response to the sample (Saito et al., 2013). As the ratio between rigid and rubbery component increases, the mechanical stiffness increases, resulting in a higher storage modulus. Figure 6 also shows that the lignin-based PU samples that were synthesized using WSSL provided the highest storage modulus at all lignin loadings, while HWLS provided the lowest storage moduli.
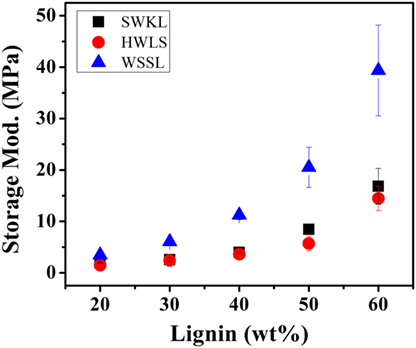
Figure 6. Storage modulus values of the lignin-based polyurethane samples obtained using dynamic mechanical analysis.
The storage moduli of softwood Kraft lignin and hardwood lignosulfonate lignin based PUs exhibited similar storage moduli. At lower lignin loadings (20, 30, and 40 lignin wt%), the difference between the storage moduli of these two samples is minimal. However, the difference between the storage moduli increases at higher lignin loadings (50 and 60 lignin wt%). Even though the storage moduli of SWKL and HWLS samples are similar, SWKL samples consistently exhibited higher moduli than HWLS samples at all lignin loadings. The loss modulus (elastic response) in Figure 7 showed the similar trend as the storage modulus for lignin with different plant source and lignin loading.
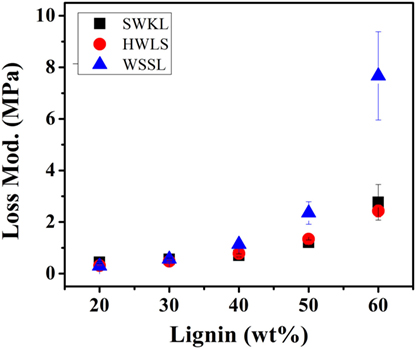
Figure 7. Loss modulus values of the lignin-based polyurethane samples obtained using dynamic mechanical analysis.
Figure 8 shows the shore hardness (Type A) of all lignin-based PU samples, which generally agree with the storage moduli obtained via DMA. All three lignin sources show an increase in hardness as lignin loading increases. This is again due to the increased rigidity that results from the higher lignin content. The increased rigidity resulted from the incorporation of more lignin also increases the resistance to deformation under a compressive force, due to the decrease in molecular mobility with added cross-linking (Saito et al., 2013; Wang et al., 2013; Llovera et al., 2016).
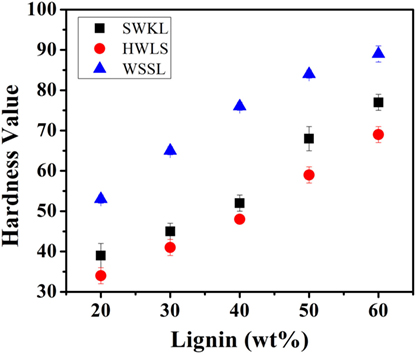
Figure 8. Shore hardness (Type A) results for all lignin-based polyurethane samples.
Lignin-based PU samples synthesized using wheat straw soda lignin show a higher shore hardness at all lignin loadings than softwood Kraft lignin and hardwood lignosulfonate lignin samples, while SWKL samples show a higher shore hardness at all lignin loadings than HWLS samples. This trend is also in agreement with dynamic mechanical analysis results depicting a more cross-linked material for WSSL samples. At higher lignin loading, the hardness values for the WSSL samples deviate from a linear trend due to the hardness value reaching the limits of the Type A durometer used for testing. Values that are close to the limits of the durometer used are considered to have more uncertainty.
To fully understand the mechanical testing results, it is important to determine the nano- and microscale structure of these materials, as these length scales highly influences the mechanical properties of a polymer material. This information will also offer a more thorough explanation on why WSSL samples provided higher storage modulus values relative to the SWKL and HWLS samples. Atomic Force Microscopy and SEM, along with DSC, were used to study the thermal and phase behavior, as well as the morphology of lignin PUs with 20, 40, and 60 wt% lignin loading.
Thermal Properties
The thermal response of the pure lignins and lignin-based PU samples were obtained using DSC. The glass transition temperature (Tg) of the select PUs are shown in Table 1 and the DSC scans are presented in Figures S1–S3 in Supplementary Material in the supplementary material. The thermal properties of the pure lignins show that the WSSL has the highest Tg while HWLS displays the lowest Tg. This is a result of the variation in monolignol content in lignins from various plant sources. Among three types of lignin, WSSL have the highest internal cross-linking due to the presence of the highest amount of p-coumaryl alcohol whereas HWLS have the lowest internal cross-linking. Increase in cross-linking makes molecule less mobile resulting higher Tg. All lignin-based PU samples show multiple Tgs, which is indicative of the presence of multiple phases (Saito et al., 2013, Sen et al., 2015). At low lignin loadings, only one Tg is observed for all lignin sources and is associated with the PPG-dominant domain. As lignin loading increases, a second Tg at a higher temperature is observed for HWLS and WSSL samples and is associated with a lignin-rich phase. Samples synthesized with SWKL did not show an observable second Tg, which we attribute to the faint nature of the Tg of the pure SWKL.
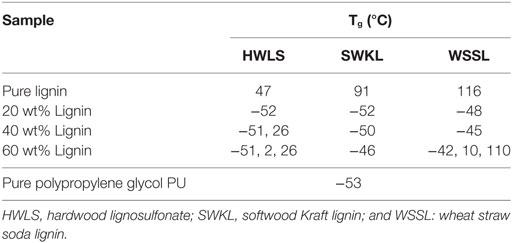
Table 1. Tg values obtained from differential scanning calorimeter scans of lignin-based polyurethane samples of 20, 40, and 60 wt% lignin.
Nano and Microscale Morphology
Phase images obtained via AFM of the various lignin PUs are shown in Figure 9, while the microscopic structure of these samples as determined by SEM are shown in Figure 10. Both of these sets of images show multiphase morphology confirming presence of multi domain in the sample which is clearly visible in HWLS and SWKL samples. Figure S4 in Supplementary Material, in the supplementary material, provides SEM images of pure dry lignins before PU synthesis. Aggregation is observed in both AFM and SEM images for the HWLS-based PU samples at all lignin loadings which become more abundant on both the nanoscale and microscale as lignin loading increases in PUs. The aggregates observed are similar in size to those observed in the SEM images of the pure HWLS, indicating a modest level of mixing between PPG and HWLS during the synthesis. Voids between the aggregates and the surrounding matrix observed in the SEM also indicate that HWLS and PPG do not mix as well as the other lignins do. The size of HWLS particles, along with the minimal level of mixing inhibits the homogeneous dispersion of HWLS in the final PU, detrimentally affecting the mechanical properties. This modest level of mixing is also consistent with the glass transition behavior of the samples, where the Tg of the soft phase (−51 to −53°C) does not vary much from the Tg of the PPG precursor (−53°C).
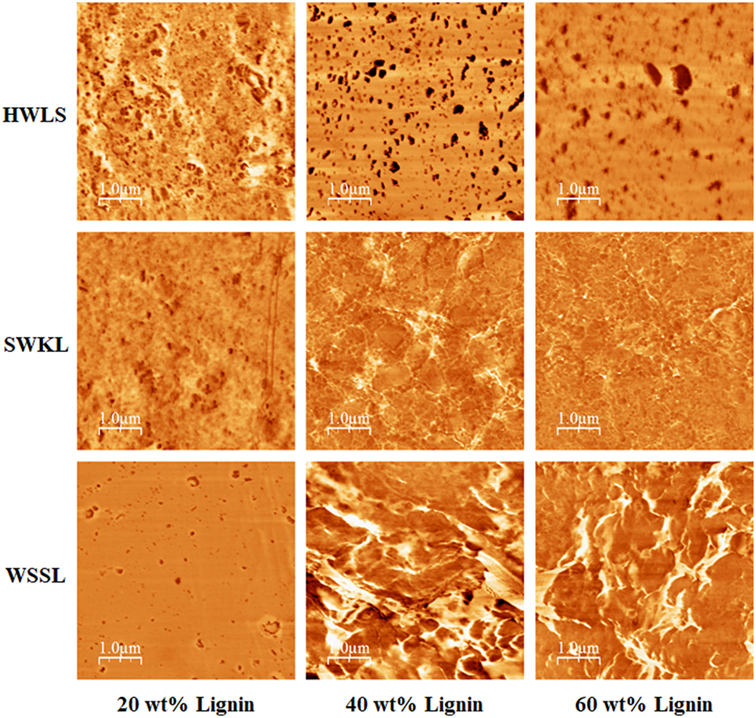
Figure 9. Atomic force microscope phase images of lignin-based polyurethane samples at 20, 40, and 60 wt% lignin loading.

Figure 10. Scanning electron microscope images of lignin-based polyurethane samples with 20, 40, and 60 wt% lignin loading.
The AFM images of SWKL-based PU samples do not show the presence of aggregates indicating better mixing on this length scale. As the result of this improved dispersion of SWKL small distinct domains of a continuous phase are formed, as seen in AFM images, and become more prominent at 60 wt% SWKL loading.
The scanning electron microscopy images show a poor level of mixing on the microscale with the appearance of large particles similar in size to those observed in pure softwood Kraft lignin. Voids are also observed between the particles and the surrounding matrix indicating poor interfacial adhesion between these phases. These particles and voids deteriorate the mechanical properties of the SWKL-based PU samples, similar to hardwood lignosulfonate lignin samples. However, the improved mixing observed at the nanoscale improves the mechanical properties of the material relative to that of the HWLS samples, resulting in higher storage moduli.
For WSSL-based PU samples, the AFM images show improved mixing relative to that of the SWKL and HWLS samples. As WSSL lignin loading increases, the AFM images show the formation of large continuous domains that are considerably larger than those found in SWKL samples. These continuous domains are a result of better mixing of lignin and PPG in WSSL samples. In the SEM images of the pure WSSL lignins, large particles are not observed. The smaller WSSL particles provide a pathway for better dispersion throughout the PU material leading to more enhanced mechanical properties. The SEM images of the WSSL PUs show areas with a smooth surface and some with rough surfaces indicating multiple domains in the sample. However, unlike SWKL samples and HWLS samples, there are no voids between these domains, indicating strong adhesion between domains. Because of the improved mixing, the absence of large lignin particles, and strong adhesion between domains, WSSL samples attain a higher modulus than the other lignin samples at all lignin loadings.
These differing levels of mixing also correlate to the observed thermal behavior of PUs determined from differential scanning calorimeter. The Tg corresponding to the PPG-rich phase increases with lignin loading for all lignins, indicating the incorporation of lignin into these phases. Moreover, the amount of increase in the Tg of this rubbery phase corresponds to the extent of mixing of polypropylene glycol (PPG) and lignin in this phase. For the hardwood lignosulfonate lignin, the Tg increases from −53°C to −51°C, indicating poor mixing between HWLS and PPG. The Tg of the PPG-rich domain is −46°C for the 60 wt% softwood Kraft lignin sample and −42°C for wheat straw soda lignin sample; changes of +7°C and +11°C, respectively. These indicate better mixing between SWKL and WSSL lignins and PPG, in qualitative agreement with the microscopic images, particularly for the WSSL samples. These results also correlate to the mechanical properties of PUs, where the formation of continuous domains in WSSL samples, the better mixing of PPG and lignins in the rubbery phase, and improved adhesion between the domains inhibits the rubbery behavior of the PPG chains enhancing the storage modulus of the material. An opposite effect is evident in HWLS samples where the limited miscibility between the lignin and PPG offer minimal improvement in the storage modulus with addition of lignin.
Correlation to Molecular Level Structure
Correlating the molecular level structure of lignins to the morphology and mechanical properties provides further insight into the effect of plant source on the properties of the synthesized lignin-based PUs. As mentioned earlier, 31P-NMR provides hydroxyl group concentration of each lignin with the ability to distinguish between hydroxyl group types. Therefore, 31P-NMR was used to obtain a better understanding of the structure and reactivity of each lignin by source.
The analysis of the 31P-NMR data is shown in Table 2 and hydroxyl group to isocyanate group ratios (OH:NCO) of each lignin-based PU are given in Table 3. Examples of the 31P-NMR data for all three lignin sources are shown in Figure S5 in Supplementary Material in the supplementary material. Overall, HWLS lignin contains a higher total hydroxyl group content while SWKL and WSSL have approximately the same hydroxyl content. The NMR data also show that HWLS contains approximately four times more aliphatic hydroxyl groups than SWKL and WSSL while the SWKL and WSSL contain more phenolic groups. Based on this hydroxyl content, it would be expected that samples synthesized using HWLS would produce a more cross-linked material with higher mechanical properties than PUs synthesized with SWKL or WSSL. However, according to DMA and hardness testing results, HWLS-based PU has the lowest mechanical properties at all lignin loadings. This apparent discrepancy can be explained by the aggregation of the lignin observed by AFM and SEM, which inhibits the accessibility of these hydroxyl groups to react with isocyanate. The limited number of hydroxyl groups able to react with isocyanate results in a PU with fewer cross-links leading to the inferior storage modulus and shore hardness.
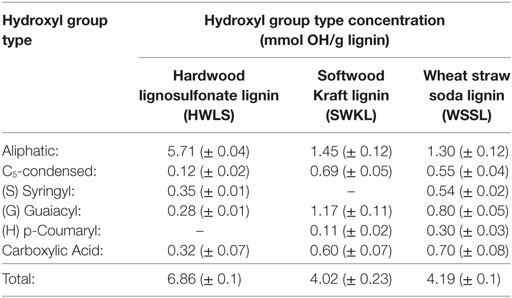
Table 2. Hydroxyl group type concentrations for each lignin source determined by 31P-NMR.
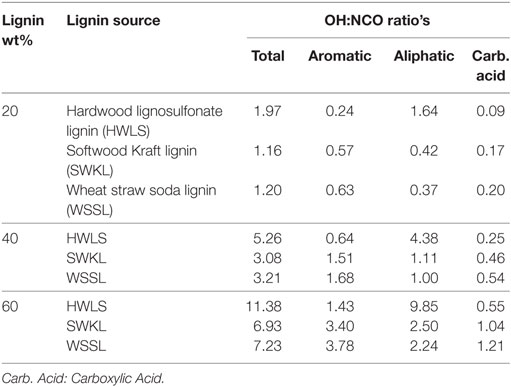
Table 3. Calculated OH:NCO ratios for lignin-based PU samples with 20, 40, and 60 wt% lignin loadings.
Even though SEM images of SWKL PUs showed aggregation within the sample, the AFM images exhibited improved mixing at the nanoscale level compared to HWLS samples. This improved dispersion of SWKL relative to HWLS increases the accessibility of hydroxyl groups of SWKL to react with the isocyanate moieties. This emphasizes that greater hydroxyl group accessibility is more important than the amount of hydroxyl groups present in the formation of cross-links and improvement of the storage modulus of the synthesized PUs. Although there is better dispersion at the nanoscale, SEM images show aggregation throughout SWKL samples, which limits the modulus of the material. The balance of these two factors, therefore, controls the modulus of the SWKL samples, which are higher than that of the HWLS-PU samples, but lower than the WSSL samples.
The large continuous phases observed in the AFM of WSSL PUs and the adhesion between domains in the SEM images indicate a higher level of mixing in WSSL samples. Thus, although WSSL, like SWKL, contains fewer hydroxyl groups than HWLS, even more hydroxyl groups are accessible to react with isocyanate due to the improved mixing. This improved reactivity leads to higher amounts of cross-linking resulting improved adhesion between domains as observed in the SEM images. The greater amount of cross-linking enhances the mechanical properties of the material leading to a higher modulus for WSSL-based PUs compared to the SWKL and HWLS samples.
Therefore, while the amount of hydroxyl groups that exist in each lignin, which is controlled by its plant source, impacts the reaction of the lignin with the isocyanate, the accessibility of hydroxyl groups is more important in controlling the extent of the reaction. The domain adhesion and mixing found in WSSL, brought on by more rigid urethane linkage formation, produces a more rigid framework increasing the mechanical properties of WSSL-based PU beyond those of HWLS and SWKL-based PU. Aggregation of particles and poor domain adhesion in the HWLS and SWKL PUs leaves voids and decreases the continuity of the lignin/PPG framework lowering the mechanical properties of the material. SWKL-based PU show better overall mechanical properties than HWLS samples due to SWKL’s higher level of mixing on the nanoscale, yet are less rigid than the WSSL samples due to the aggregation that is also found throughout the framework albeit to a lesser extent than HWLS.
Conclusion
The lignin plant source clearly affects the mechanical properties of lignin-based PU materials. The storage moduli of lignin-based PUs synthesized with PPG cross-linker increased with lignin loading for all three lignin sources due to lignin acting as the rigid component. PUs synthesized with WSSL lignin exhibited the highest storage modulus at all lignin loadings, while HWLS provided the lowest. The shore hardness of lignin-based PUs agreed with storage moduli (DMA results) showing that the hardness of PU increased with lignin loading and that WSSL samples exhibited the highest hardness at all lignin loadings while HWLS provided the lowest.
Thermal study showed multiple Tgs indicating the presence of multi-phase morphology for all lignin-based PUs. Different levels of mixing for all lignin sources were observed in AFM and SEM images. PUs synthesized with HWLS showed the poorest level of mixing with PPG, drastically lowering the mechanical properties. While distinct domains of a continuous phase formed in their AFM images indicated better mixing of SWKL with PPG leading to enhanced mechanical properties. The smaller particle size of WSSL lignin led to better mixing with PPG and the highest storage modulus at all lignin loadings in WSSL-based PUs. These differing levels of mixing correlated well with the observed thermal behavior of PUs where the Tg corresponding to the PPG-rich phase increased, not only with lignin loading but also with the level of mixing indicating a drop in rubbery behavior of PPG chains and ultimately enhancing the storage modulus.
31P-NMR study revealed that HWLS contained the highest total hydroxyl group content while hydroxyl content of SWKL and WSSL were nearly the same. However, higher storage modulus in PU incorporated with SWKL despite having lower hydroxyl concentration than HWLS showed that not all hydroxyl groups were accessible to isocyanate moieties hindering the number of cross-links that could form. Due to the better dispersion amongst lignins, even more cross-links form in WSSL samples allowing WSSL samples to provide the highest storage modulus and shore hardness.
Author Contributions
MD conceived the experiments, led the interpretation of the results, and helped write the manuscript. US completed some of the experiments, and contributed to both the data interpretation and manuscript writing. JL completed the majority of the experiments and contributed to both the data interpretation and manuscript writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with one of the authors, MD.
Funding
This work was funded by the Soft Materials Research in Tennessee (SMaRT) center, which is supported by the University of Tennessee.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fenrg.2018.00004/full#supplementary-material.
References
Akindoyo, J. O., Beg, M. D. H., Ghazali, S., Islam, M. R., Jeyaratnam, N., and Yuvaraj, A. R. (2016). Polyurethane types, synthesis and applications: a review. RSC Adv. 6, 114453–114482. doi: 10.1039/C6RA14525F
Alexy, P., Kosikova, B., and Podstranska, G. (2000). The effect of blending lignin with polyethylene and polypropylene on physical properties. Polymer 41, 4901–4908. doi:10.1016/S0032-3861(99)00714-4
Baker, D. A., and Rials, T. G. (2013). Recent advances in low-cost carbon fiber manufacture from lignin. J. Appl. Polym. Sci. 130, 713–728. doi:10.1002/app.39273
Basfar, A. A., Ali, K. M. I., and Mofti, S. M. (2003). UV stability and radiation-crosslinking of linear low density polyethylene and low density polyethylene for greenhouse applications. Polym. Degrad. Stab. 82, 229–234. doi:10.1016/S0141-3910(03)00216-7
Bharadwaj, V., Somani, K., and Kansara, S. (2002). The effect of chain length of polyethylene glycol on properties of castor oil based polyurethane elastomers. J. Macromol. Sci. Pure Appl. Chem. 39, 115–127. doi:10.1081/MA-120006522
Chakar, F. S., and Ragauskas, A. J. (2004). Review of current and future softwood Kraft lignin process chemistry. Ind. Crops Prod. 20, 131–141. doi:10.1016/j.indcrop.2004.04.016
Chan-Chan, L. H., Solis-Correa, R., Vargas-Coronado, R. F., Cervantes-Uc, J. M., Cauich-Rodríguez, J. V., Quintana, P., et al. (2010). Degradation studies on segmented polyurethanes prepared with HMDI, PCL and different chain extenders. Acta Biomater. 6, 2035–2044. doi:10.1016/j.actbio.2009.12.010
Chatterjee, S., and Saito, T. (2015). Lignin-derived advanced carbon materials. ChemSusChem 8, 3941–3958. doi:10.1002/cssc.201500692
Chen, Y. L., and Rånby, B. (1989). Photocrosslinking of polyethylene. II. Properties of photocrosslinked polyethylene. J. Poly. Sci. A 27, 4077–4086. doi:10.1002/pola.1989.080271215
Chung, H., and Washburn, N. R. (2012). Improved lignin polyurethane properties with lewis acid treatment. ACS Appl. Mater. Interfaces 4, 2840–2846. doi:10.1021/am300425x
Chung, Y. L., Olsson, J. V., Li, R. J., Frank, C. W., Waymouth, R. M., Billington, S. L., et al. (2013). A renewable lignin-lactide copolymer and application in biobased composites. ACS Sustain. Chem. Eng. 1, 1231–1238. doi:10.1021/sc4000835
da Silva, E. A. B., Zabkova, M., Araujo, J. D., Cateto, C. A., Barreiro, M. F., Belgacem, M. N., et al. (2009). An integrated process to produce vanillin and lignin-based polyurethanes from Kraft lignin. Chem. Eng. Res. Des. 87, 1276–1292. doi:10.1016/j.cherd.2009.05.008
Dehne, L., Babarro, C. V., Saake, B., and Schwarz, K. U. (2016). Influence of lignin source and esterification on properties of lignin-polyethylene blends. Ind. Crops Prod. 86, 320–328. doi:10.1016/j.indcrop.2016.04.005
Doherty, W. O. S., Mousavioun, P., and Fellows, C. M. (2011). Value-adding to cellulosic ethanol: lignin polymers. Ind. Crops Prod. 33, 259–276. doi:10.1016/j.indcrop.2010.10.022
Dong, W., Ren, J., Lin, L., Shi, D., Ni, Z., and Chen, M. (2012). Novel photocrosslinkable and biodegradable polyester from bio-renewable resource. Polym. Degrad. Stab. 97, 578–583. doi:10.1016/j.polymdegradstab.2012.01.008
Duval, A., and Lawoko, M. (2014). A review on lignin-based polymeric, micro- and nano-structured materials. React. Funct. Polym. 85, 78–96. doi:10.1016/j.reactfunctpolym.2014.09.017
Fan, J., and Zhan, H. (2008). Optimization of synthesis of spherical lignosulphonate resin and its structure characterization. Chin. J. Chem. Eng. 16, 407–410. doi:10.1016/S1004-9541(08)60097-X
Fatechi, P., and Chen, J. (2016). Extraction of technical lignins from pulping spent liquors, challenges and opportunities. Product. Biofuels Chem. Lignin Springer Nat. 6, 35–54. doi:10.1007/978-981-10-1965-4_2
Gang, H., Lee, D., Choi, K. Y., Kim, H. N., Ryu, H., Lee, D. S., et al. (2017). Development of high performance polyurethane elastomers using vanillin-based green polyol chain extender originating from lignocellulosic biomass. ACS Sustain. Chem. Eng. 5, 4582–4588. doi:10.1021/acssuschemeng.6b02960
Gosselink, R. J. A., de Jong, E., Guran, B., and Abacherli, A. (2004). Co-ordination network for lignin-standardisation, production and applications adapted to market requirements (EUROLIGNIN). Ind. Crops Prod. 20, 121–129. doi:10.1016/j.indcrop.2004.04.015
Grand View Research. (2017). Polyurethane (PU) Market Analysis by Product (Rigid Foam, Flexible Foam, Coatings, Adhesives & Sealants, Elastomers), by End-Use (Furniture & Interiors, Construction, Electronics & Appliances, Automotive, Footwear, Packaging), & Segment Forecasts, 2014-2025. Available at: http://www.grandviewresearch.com/industry-analysis/polyurethane-pu-market
Grassie, N., and Zulfiqar, M. (1978). Thermal degradation of the polyurethane from 1,4-butanediol and methylene bis(4-phenyl isocyanate). J. Polym. Sci. 16, 1563–1574.
Gregorová, A., Košíková, B., and Moravčík, R. (2006). Stabilization effect of lignin in natural rubber. Polym. Degrad. Stab. 91, 229–233. doi:10.1016/j.polymdegradstab.2005.05.009
Horcas, I., Fernandez, R., Gomez-Rodriguez, J. M., Colchero, J., Gomez-Herrero, J., and Baro, A. M. (2007). WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 78, 013701–013708. doi:10.1063/1.2432410
Iype, E., Esteves, A. C. C., and de With, G. (2016). Mesoscopic simulations of hydrophilic cross-linked polycarbonate polyurethane networks: structure and morphology. Soft Matter 12, 5029–5040. doi:10.1039/c6sm00621c
Kadla, J. F., and Kubo, S. (2004). Lignin-based polymer blends: analysis of intermolecular interactions in lignin-synthetic polymer blends. Compos. Part A. Appl. Sci. Manuf. 35, 395–400. doi:10.1016/j.compositesa.2003.09.019
Laurichesse, S., and Avérous, L. (2013). Synthesis, thermal properties, rheological and mechanical behaviors of lignins-grafted-poly(epsilon-caprolactone). Polymer 54, 3882–3890. doi:10.1016/j.polymer.2013.05.054
Laurichesse, S., and Avérous, L. (2014). Chemical modification of lignins: towards biobased polymers. Prog. Polym. Sci. 39, 1266–1290. doi:10.1016/j.progpolymsci.2013.11.004
Li, Y., and Ragauskas, A. J. (2012). Kraft lignin-based rigid polyurethane foam. J. Wood Chem. Technol. 32, 210–224. doi:10.1080/02773813.2011.652795
Llovera, L., Benjelloun-Mlayah, B., and Delmas, M. (2016). Organic acid lignin-based polyurethane films: synthesis parameter optimization. BioResources 11, 6320–6334. doi:10.15376/biores.11.3.6320-6334
Madad, N., Chebil, L., Charbonnel, C., Ioannou, I., and Ghoul, M. (2013). Enzymatic polymerization of sodium lignosulfonates: effect of catalysts, initial molecular weight, and mediators. Can. J. Chem. 91, 220–225. doi:10.1139/cjc-2012-0036
Noreen, A., Zia, K. M., Zuber, M., Tabasum, S., and Zahoor, A. F. (2016). Bio-based polyurethane: an efficient and environment friendly coating systems: a review. Prog. Org. Coat. 91, 25–32. doi:10.1016/j.porgcoat.2015.11.018
Nozaki, S., Masuda, S., Kamitani, K., Kojio, K., Takahara, A., Kuwarnura, G., et al. (2017). Superior properties of polyurethane elastomers synthesized with aliphatic diisocyanate bearing a symmetric structure. Macromolecules 50, 1008–1015. doi:10.1021/acs.macromol.6b02044
Olarte, M. V., Burton, S. D., Swita, M., Padmaperuma, A. B., Ferrell, J., and Ben, H. (2016). Determination of Hydroxyl Groups in Pyrolysis Bio-Oils Using 31P NMR: Laboratory Analytical Procedure (LAP). Washington, DC; Golden, CO (United States): National Renewable Energy Lab (NREL).
Oprea, S. (2010). Dependence of fungal biodegradation of PEG/castor oil-based polyurethane elastomers on the hard-segment structure. Polym. Degrad. Stab. 95, 2396–2404. doi:10.1016/j.polymdegradstab.2010.08.013
Prisacariu, C. (2011). Polyurethane Elastomers: From Morphology to Mechanical Aspects. Wien, New York: Springer Verlag.
Qui, X., Kong, Q., Zhou, M., and Yang, D. (2010). Aggregation behavior of sodium lignosulfonate in water solution. J. Phys. Chem. B 114, 15857–15861. doi:10.1021/jp107036m
Saito, T., Perkins, J. H., Jackson, D. C., Trammel, N. E., Hunt, M. A., and Naskar, A. K. (2013). Development of lignin-based polyurethane thermoplastics. RSC Adv. 3, 21832–21840. doi:10.1039/c3ra44794d
Saraf, V. P., and Glasser, W. G. (1984). Engineering plastics from lignin. III. Structure property relationship in solution cast polyurethane films. J. Appl. Polym. Sci. 29, 1831–1841. doi:10.1002/app.1984.070290534
Sarkar, S., and Adhikari, B. (2001). Thermal stability of lignin–hydroxy-terminated polybutadiene copolyurethanes. Polym. Degrad. Stab. 73, 169–175. doi:10.1016/S0141-3910(01)00084-2
Sen, S., Patil, S., and Argyropoulos, D. S. (2015). Thermal properties of lignin in copolymers, blends, and composites: a review. Green Chem. 17, 4862–4887. doi:10.1039/C5GC01066G
Seymour, R. B., and Kauffman, G. B. (1992). Polyurethanes: a class of modern versatile materials. J. Chem. Educ. 69, 909–910. doi:10.1021/ed069p909
Silva, A. L., and Bordado, J. C. (2004). Recent developments in polyurethane catalysis: catalytic mechanisms review. Cat. Revi. Sci. Eng. 46, 31–51. doi:10.1081/CR-120027049
Soto, M., Sebastián, R. M., and Marquet, J. (2014). Photochemical activation of extremely weak nucleophiles: highly fluorinated urethanes and polyurethanes from polyfluoro alcohols. J. Org. Chem. 79, 5019–5027. doi:10.1021/jo5005789
Suhas, P. J. M., and Ribeiro, M. M. L. (2007). Lignin – from natural adsorbent to activated carbon: a review. Bioresour. Technol. 98, 2301–2312. doi:10.1016/j.biortech.2006.08.008
Tejado, A., Peña, C., Labidi, J., Echeverria, J. M., and Mondragon, I. (2007). Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 98, 1655–1663. doi:10.1016/j.biortech.2006.05.042
Teo, L. S., Chen, C. Y., and Kuo, J. F. (1997). Fourier transform infrared spectroscopy study on effects of temperature on hydrogen bonding in amine-containing polyurethanes and poly(urethane-urea)s. Macromolecules 30, 1793–1799. doi:10.1021/ma961035f
Tian, D., Chandra, R. P., Lee, J. S., Lu, C., and Saddler, J. N. (2017). A comparison of various lignin-extraction methods to enhance the accessibility and ease of enzymatic hydrolysis of the cellulosic component of steam-pretreated poplar. Biotechnol. Biofuels 10, 1–10. doi:10.1186/s13068-017-0846-5
Varanasi, P., Singh, P., Auer, M., Adams, P. D., Simmons, B. A., and Singh, S. (2013). Survey of renewable chemicals produced from lignocellulosic biomass during ionic liquid pretreatment. Biotechnol. Biofuels 6, 1–9. doi:10.1186/1754-6834-6-14
Wang, C., Kelley, S. S., and Venditti, R. A. (2016). Lignin-based thermoplastic materials. ChemSusChem 9, 770–783. doi:10.1002/cssc.201501531
Wang, Z., Yang, X., Zhou, Y., and Liu, C. (2013). Mechanical and thermal properties of polyurethane films from peroxy-acid wheat straw lignin. Bioresources 8, 3833–3843. doi:10.15376/biores.8.3.3833-3843
Wen, T. C., Fang, J. C., Lin, H. J., and Yang, C. H. (2001). Characteristics of PPG-based thermoplastic polyurethane doped with lithium perchlorate. J. Appl. Polym. Sci. 82, 389–399. doi:10.1002/app.1863
Xu, F., Sun, J.-X., Sun, R., Fowler, P., and Baird, M. S. (2006). Comparative study of organosolv lignins from wheat straw. Ind. Crops Prod. 23, 180–193. doi:10.1016/j.indcrop.2005.05.008
Yamaguchi, A., Mimura, N., Shirai, M., and Sato, O. (2017). Bond cleavage of lignin model compounds into aromatic monomers using supported metal catalysts in supercritical water. Sci. Rep. 7, 1–7. doi:10.1038/srep46172
Yoshida, H., Mörck, R., Kringstad, K. P., and Hatakeyama, H. (1990). Kraft lignin in polyurethanes. II. Effects of the molecular-weight of Kraft lignin on the properties of polyurethanes from a kraft lignin polyether triol polymeric MDI system. J. Appl. Polym. Sci. 40, 1819–1832. doi:10.1002/app.1990.070401102
Zhang, C., Wu, H., and Kessler, M. R. (2015). High bio-content polyurethane composites with urethane modified lignin as filler. Polymer 69, 52–57. doi:10.1016/j.polymer.2015.05.046
Keywords: plant source, lignin, polyol, polyurethane, hardwood, softwood, wheat straw, mechanical properties
Citation: Lang JM, Shrestha UM and Dadmun M (2018) The Effect of Plant Source on the Properties of Lignin-Based Polyurethanes. Front. Energy Res. 6:4. doi: 10.3389/fenrg.2018.00004
Received: 16 October 2017; Accepted: 01 February 2018;
Published: 23 February 2018
Edited by:
Chang Geun Yoo, Oak Ridge National Laboratory (DOE), United StatesReviewed by:
Muhammad Waqas, King Abdulaziz University, Saudi ArabiaMaria Gonzalez Alriols, University of the Basque Country (UPV/EHU), Spain
Copyright: © 2018 Lang, Shrestha and Dadmun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Dadmun, ZGFkQHV0ay5lZHU=