 John Ozdemir1
John Ozdemir1 Imann Mosleh2†
Imann Mosleh2† Mojtaba Abolhassani2†Lauren F. Greenlee2Robert R. Beitle Jr.2
Mojtaba Abolhassani2†Lauren F. Greenlee2Robert R. Beitle Jr.2 M. Hassan Beyzavi1*
M. Hassan Beyzavi1*- 1Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville, AR, United States
- 2Ralph E. Martin Department of Chemical Engineering, University of Arkansas, Fayetteville, AR, United States
Covalent organic frameworks (COFs) are porous crystalline organic polymers which have been the subject of immense research interest in the past 10 years. COF materials are synthesized by the covalent linkage of organic molecules bonded in a repeating fashion to form a porous crystal that is ideal for gas adsorption and storage. Chemists have strategically designed COFs for the purpose of heterogeneous catalysis of gaseous reactants. Presented in this critical review are efforts toward developing COFs for the sequestration of CO2 from the atmosphere. Researchers have determined the CO2 adsorption capabilities of several COFs is competitive with the highest surface area materials. Engineering the pore environment of COFs with chemical moieties that interact with CO2 have increased the CO2 adsorption performance. The installation of CO2 binding moieties in the COF has made possible the selective adsorption of CO2 over other gases such as N2. The high degree of control of internal pore composition in COFs is coupled with high CO2 adsorption to develop heterogeneous catalysts for the conversion of CO2 to value added products. Two notable examples of this catalysis are the fixation of CO2 to epoxides for the synthesis of cyclic carbonates and the reduction of CO2 to CO. Recent examples of COFs for the capture of CO2 will be discussed followed by COF catalysts which use CO2 as a feedstock for the production of value-added products.
Outlook
The release of CO2 during the combustion of fossil fuels is an environmental detriment linked to global warming and the acidification of the planet's oceans. The concentration of CO2 in the atmosphere is higher than it has been in 20 million years (Tripati et al., 2009) and 2/3 of its release by humankind can be traced back to 90 state or investor-owned enterprises (Boneham et al., 2017). This small conglomerate, and others, could be encouraged to curtail the release of CO2 into the atmosphere quickly if given a financial incentive. One economic motivation for the capture of CO2 at the source of emission is the 110 Mt of CO2 used annually as a chemical reagent (Aresta and Dibenedetto, 2007). Current methods for the separation of CO2 at the point of emission would reduce the energetic output of a powerplant by 25–40% (Haszeldine, 2009) so clearly the economic incentive for CO2 sequestration with current technologies is minimal. New materials must be developed that are capable of facile CO2 capture and subsequent catalytic conversion of CO2 to more valuable products. Two of these value-added products are CO and industrially relevant cyclic carbonates.
Current processes for the capture of CO2 are dependent on amine solutions or chilled ammonia for which the issues of degrading equipment, toxicity, and the high energy cost of amine regeneration are problems (Lu et al., 2015). Inquiries into the gas storage applications of porous materials arose from their low density accompanied by a high internal surface area (Lu and Hao, 2013; Estevez et al., 2018). Inorganic zeolites were the first porous materials to be investigated for the ability to sequester CO2 (Siriwardane et al., 2005; James et al., 2011). Zeolites main attribute however is their use in catalytic cracking of hydrocarbons; an ability that is derived from the high acidity of zeolite frameworks (Zeolite Synthesis, 1989). Compared to COF materials however zeolites will have a smaller pore size and a greater affinity to water, limiting the CO2 capture performance compared to COFs. Other candidates for mitigating the greenhouse effect via CO2 capture include non-crystalline polymers such as covalent microporous polymers (CMPs) (Dawson et al., 2011a; Sun et al., 2015). CMPs have been used successfully in the fixation of CO2 to epoxides and like COFs can also be co-ordinated to metal atoms (Xiong et al., 2017). CMPs though lack crystallinity making their pore size less uniform than COF materials. Due to the amorphous nature of CMPs there is a lack of precise atomic control of the structure and spacing between the molecular building blocks of the polymer, which is something that has been achieved in COFs (Feng et al., 2012a). Polymeric graphitic carbon nitride (g-C3N4) is promising heterogenous catalyst for the metal-free photocatalytic conversion of CO2 to value added products (Shi et al., 2014; Sun and Liang, 2017). Unlike COFs the porosity of g-C3N4 is derived through the use of templating agents and in bulk application g-C3N4 often suffers low solar light absorption. COFs are one of the most recently discovered families of porous materials (Dinga and Wang, 2013; Huang et al., 2016; Lohse and Bein, 2018; Zhao F. et al., 2018). COFs are crystalline polymers synthesized from the covalent linkages of building blocks (Figure 1). COF materials can consist of 2D sheets of linked building blocks held together by π-stacking, or also 3D frameworks. The rapid development of reticular chemistry in recent years was motivated in great part by the demonstrated ability for CO2 capture and conversion by synthesized zeolitic imidazole frameworks (ZIFs) (Wang and Wang, 2016) and metal organic frameworks (MOFs) (Beyzavi et al., 2014, 2015; Ji et al., 2018). The high porosity and low volumetric density of COFs inspired the thorough investigation into the rational design of COF for CO2 sequestration (Jiang et al., 2019). The pore size of COFs can be varied from small to large by the selection of different sized building blocks. COFs with large pores are better suited for CO2 capture at high pressures wherein the interactions between gas molecules plays a larger role in storage than the interactions between CO2 and the pore walls (Jiang et al., 2019). Meanwhile the chemical makeup of the internal pores in small channeled COFs can be tuned to enhance CO2 affinity thus maximizing the ability for low pressure CO2 adsorption in these materials (Morris and Wheatley, 2008; Li B. et al., 2014). Moreover the porosity of COF materials endows a low density; for instance COF-108, which is a 3D COF, achieved a density of 0.17 g/cm3 which at the time was the lowest density of any COF synthesized (El-Kaderi et al., 2007).
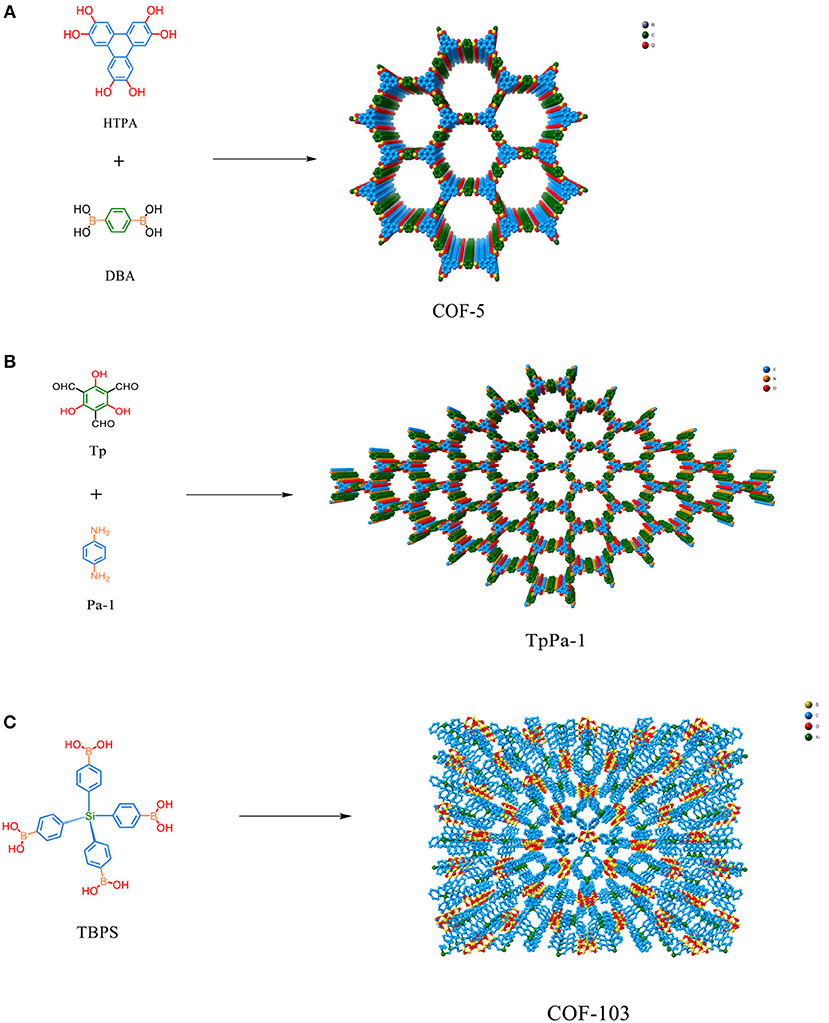
Figure 1. (A) Structure and composition of COF-5 (Table 1 entry 2). (B) Structure and composition of TpPa-1 (Table 1 entry 17). (C) Structure and design of COF-103 (Table 1 entry 7).
Currently COF materials are attracting the attention of an increasing number of scientists as seen by the number of publications focusing on COF materials. With so much scientific focus it would seem that industrialization of COFs is approaching (Zhao W. et al., 2018). As it stands there is an large incentive for the design of resilient materials which can valorize CO2 through a catalytic means (Aresta et al., 2014). Moreover the covalent linkages which comprise COFs could reach a higher stability than the dative interactions between linkers and nodes which hold MOFs together. MOF materials have been used with great success for the fixation of CO2 to epoxides for the synthesis of cyclic carbonates, much of this success is due to the high Lewis acidity of the metal centers (Beyzavi et al., 2015). Metal atoms can however be incorporated into COF frameworks for this catalytic advantage.
Designing COFs for CO2 Adsorption
The synthesis of porous polymers by the use of diverse building blocks of various size is called reticular chemistry (Cordova and Yaghi, 2017). One of the most attractive features of COFs is the large amount of structural diversity that can be obtained by the deliberate selection of the organic monomers used to synthesize the COF (Zhou et al., 2014; Pang et al., 2016; Jin et al., 2017). Crystalline boron-linked COFs were discovered in 2005 by Côté et al. through the condensation of diboronic acid to produce COF-1 with an internal surface area of 711 m2/g and the condensation of diboronic acid with hexahydroxy triphenylene creating COF-5 with internal surface area of 1,590 m2/g (Côté et al., 2005). In 2007 Côté et al. applied co-condensation reactions between 2,3,6,7,10,11-hexa-hydroxytriphenylene (HHTP) and 1,3,5-benzenetriboronic acid (BTBA), 1,3,5-benzenetris(4-phenylboronic acid) (BTPA), and 4,4′- biphenyldiboronic acid (BPDA) to produce COF-6,−8, and−10 (Côté et al., 2007). Next, El-Kaderi et al. synthesized 3-D COFs with high crystallinity by condensation reactions of tetrahedral tetra(4-dihydroxyborylphenyl) methane to create COF-102 with a surface area of 3,472 m2/g or tetra(4-dihydroxyborylphenyl) silane to synthesize COF-103 with a surface area of 4,210 m2/g (El-Kaderi et al., 2007). COFs synthesized for CO2 storage include azo (Ge et al., 2016), azine (Li Z. et al., 2014; Li et al., 2015), imine (Rabbani et al., 2013; Huang et al., 2015a,b; Zhai et al., 2017; Gao et al., 2018), and triazine linkers. The advent of reticular chemistry has inspired computational chemists to simulate COFs for the hope of better optimizing CO2 capture (Zeng et al., 2016). Chemists have been able to incorporate a litany of catalytic centers within COF materials that enable the conversion of CO2 to cyclic carbonates (Roeser et al., 2012; Saptal et al., 2016; Xu et al., 2017; Yu et al., 2018; Zhi et al., 2018) and CO (Lin et al., 2015; Diercks et al., 2018a; Lin and Chen, 2018; Liu et al., 2018; Yang et al., 2018; Yao et al., 2018). Some of the most recently developed and advanced COFs for CO2 adsorption are listed in Table 1.
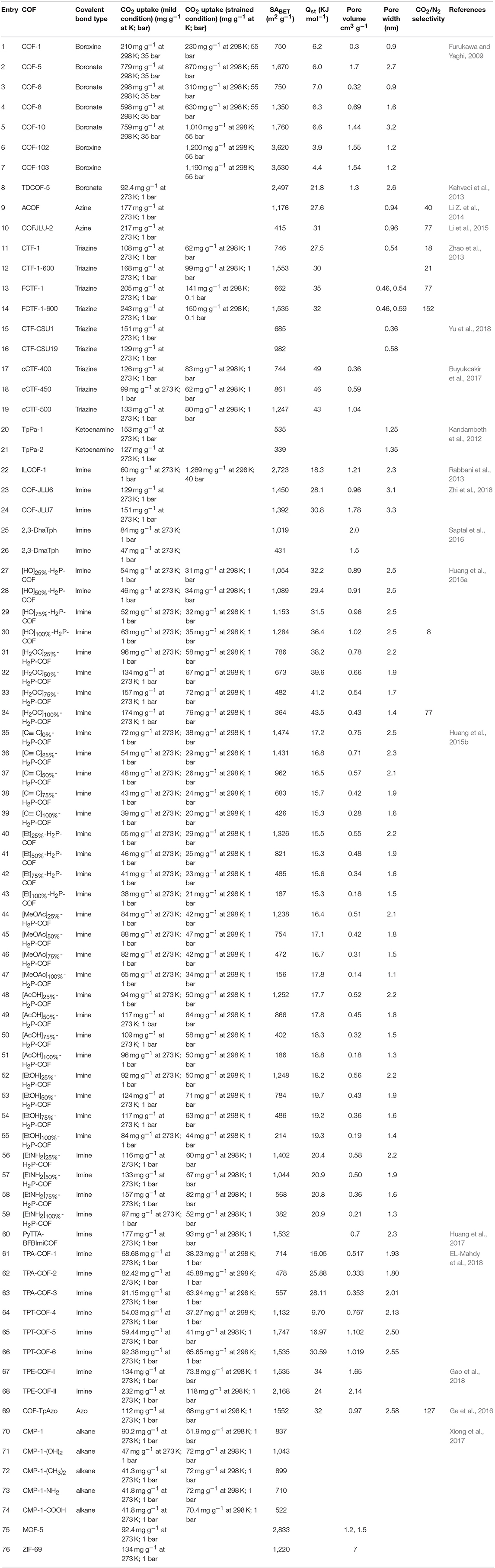
Table 1. COFs for CO2 adsorption.
COFs for the Reduction of CO2 to CO
Various photocatalytic and electrocatalytic systems have been developed for large scale CO2 conversion. However, large scale CO2 conversion to value-added carbon products deals with challenges that need to be addressed. Photocatalytic systems suffer poor selectivity and CO2 adsorption, rapid charge recombination, and unfortunate active site architecture (Inoue et al., 1979; Wang et al., 2013; White et al., 2015; Wang and Wang, 2016). One key challenge in the reduction of CO2 is ensuring specificity because CO2 can be reduced to a broad range of products including carbon monoxide, methanol, formic acid, methane, or even higher order hydrocarbons such as ethanol and ethylene (Qiao et al., 2014). The large-scale CO2 conversion to desired carbon products requires the development of new materials designed through reticular chemistry which improve selectivity toward the desired products and can adsorb exceptionally high amounts of CO2 (Diercks et al., 2018b). In addition, water has been used as a cheap and environmentally friendly solvent to facilitate electron and proton transfer in an electrolytic approach for the CO2 reduction to CO (Hori et al., 1994).
COFs for the Fixation of CO2 to Epoxides for the Synthesis of Cyclic Carbonates
In recent years, COF materials have been widely reported as exceptional heterogeneous catalysts achieving high efficiency with recyclability. In this section, we present recent literature in which COFs are exploited as a heterogeneous catalyst for the coupling of CO2 and epoxides to form cyclic carbonates. These examples can be divided into two groups, metal free COFs for the conversion of epoxides to carbonates (Roeser et al., 2012; Saptal et al., 2016; Yu et al., 2018; Zhi et al., 2018) and COFs with metal centers for the fixation of CO2 to epoxides (Xu et al., 2017). Fixation of CO2 to epoxides by the action of a Lewis acid within the COF framework requires initiation of the epoxide through coordination to some anion, the source of this anion is most often an ammonium salt such as tetrabutylammonium bromide (TBAB) or tetrabutylammonium iodide (TBAI). In the unique case of the charged covalent triazine framework synthesized by Buyukcakir and coworkers the necessary ion is incorporated into the crystal framework de-novo and so no cocatalyst is need for CO2 fixation (Buyukcakir et al., 2017).
The efficiency of CO2 fixation to an epoxide by a catalyst is measured by the pressure and time required for the reaction to proceed (Table 3). High internal surface area could reasonably be assumed to increase the amount of CO2 which is made available to the catalytic centers housed in the pores of the materials. Porous materials are advantaged by a high internal surface area but the pore size will determine which molecules can access the reactive centers. To determine the effect of substrate size on the catalysis substrates of diverse dimensions should be screened. In the case of COF materials, most of which are 2-dimensional sheets; the lower surface area when compared to MOFs could be mitigated by successful exfoliation during the reaction.
COFs for CO2 Capture
First COFs for CO2 Storage: Boron Linked COFs
Furukawa and Yaghi (2009) began the work into COFs for CO2 sequestration by the examining the gas storage capability of boron COFs for the storage of CO2 at both high and low pressures and found that the ability to adsorb CO2 was directly related to the internal surface area. In this study by Furukawa et al. the COFs were divided into three groups: The first two groups were 2D structures that had 1D pores and the third group consisted of 3D structures with 3D pores. The pores of group 1 were 9Å (COF-1 and COF-6), while group 2 materials had larger pores (27,16, and 32 Å for COF-5, COF-8, and COF-10, respectively). The 3D COFs in group 3 had medium-sized pores (12 Å for COF-102 and COF-103 both). The 3D COFs in the third group had the best gas uptake capacities. For instance, COF-102 had gas uptake of 1,180 mg g−1 at 298 K for carbon dioxide measured at 35 bar.
The synthesis of the boron linked COF for the purpose of CO2 capture was continued by Kahveci et al. with the development of TDCOF-5 which was synthesized from a mixture of 1,4-benzene diboronic acid and hexahydroxytriptycene heated in a mesitylene–dioxane solvent mixture for 5 days at 120°C (Kahveci et al., 2013). The calculated Brunauer–Emmett–Teller (BET) surface area was found to be 2,497 m2g−1 while the Langmuir model gave a surface area of 3,832 m2g−1 that is similar to the calculated Connolly surface (4,973 m2g−1). TDCOF-5 can store up to 9.2 wt% of CO2 (2.1 mmol g−1) with a Qst value of 21.8. The use of the triptycene core in TDCOF-5 leads to more accessible boron sites that can facilitate interaction with gas molecules. Greater accessibility to these boron sites increased the CO2 adsorption performance of TDCOF-5 at low pressures.
Synthesis of COFs capable of competitive CO2 adsorption was expanded to the design of imine linked COFs by Rabbani et al. they reported the synthesis of ILCOF-1 with an internal surface area (Braunner-Emmet-Teller) of 2,723 m2 g−1 and a Langmuir surface area of 3,453 m2 g−1 (Rabbani et al., 2013). ILCOF-1 was shown to be exceptionally well suited for high pressure H2, CH4, and CO2 storage. ILCOF-1 was synthesized by the condensation reaction between 1,3,6,8-tetrakis(p-formylphenyl)pyrene and p-phenylenediamine in 1,4-dioxane in the presence of aqueous acetic acid at 120°C for 3 days (Figure 2). The use of expanded pyrene cores in the construction of imine-linked COFs represented a breakthrough in imine COF synthesis as previous work such as the 3D COF-300 had a surface area of SABET = 1,360 m2 g−1 (Uribe-Romo et al., 2009). Previous to this work by Rabbani et al. the use of imine-linked COFs in gas storage had been only investigated theoretically (Mendoza-Cortes et al., 2012). ILCOF-1 was stable up to 400°C as determined by thermogravimetric analysis (TGA). The calculated surface area (SABET) and major pore-size distribution are much larger than other crystalline imine-linked COFs including COF-300 (1,360 m2 g−1, 7.2 Å) (Uribe-Romo et al., 2009); COF-366 (735 m2 g−1, 17.6 Å) (Wan et al., 2011); and COF-LZU1 (410 m2 g−1, 12 Å) (Ding et al., 2011). ILCOF-1 showed significant CO2 uptake at high pressure (29.3 mmol g−1 at 298 K and 40 bar) these values are greater than those for 2D and 3D COFs (5.2–27.3 mmol g−1 at 298 K and 55 bar). ILFOC-1 also outperforms hyper-branched conjugated polymers (HCPs; 10.6–13.3 mmol g−1 at 298 K and 35 bar) (Martín et al., 2011) and PAF-1(29.6 mmol g−1 at 298 K and 40 bar) (Ben et al., 2009). An explanation for the high measured CO2 uptake of ILCOF-1 is the enhancement of CO2 binding created by the N···CO2 interactions in the nitrogen-containing pore walls ILCOF-1 which do not exist in the nitrogen-free PAF and HCP structures.
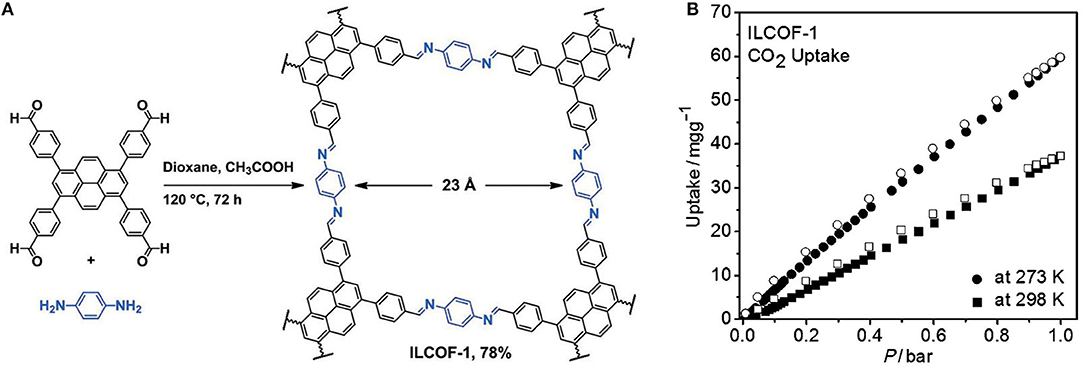
Figure 2. (A) Structure of ILCOF-1 by synthesized by Rabbani et al. (2013) and (B) CO2 uptake capacity of ILCOF-1 as measured by Kaderi et al. Reprinted with permission ©2013 Wiley VCH.
Ge et al. (2016) synthesized an azo (N = N) based covalent organic framework (COF-TpAzo) via a Schiff base condensation reaction. COF-TpAzo was synthesized by the condensation reaction between 1,3,5-triformylphloroglucinol (Chong et al., 2003) (0.3 mmol, 63.0 mg) and 4,4′-azodianiline (Santurri et al., 2003) (0.45 mmol, 95.4 mg) in 1,4-dioxane (3 mL). The maximum CO2 adsorption of COF-TpAzo was 105.6 mg·g−1 (Ge et al., 2016). Yet COF-TpAzo was also 39 times more selective to CO2 over CH4 and 127 times more selective of CO2 over N2 (Table 1 entry 69). At 298 K the CO2/N2 and CO2/CH4 selectivity of COF-TpAzo was 145 and 43, respectively. The increase at in CO2 selectivity at a higher temperature was explained by conventional CO2 affinities and the N2 phobicity of azo group (Patel et al., 2013).
Work by Li Z. et al. toward the development of stable COFs with C-N linkages for CO2 specific adsorption involved the synthesis of a new azine-linked covalent organic framework, ACOF-1, by condensation of hydrazine hydrate and 1,3,5-triformylbenzene under solvothermal conditions (Li Z. et al., 2014). ACOF-1 is with high selectivity toward CO2 over N2 and CH4. ACOF-1 can store up to 177 mg g−1 of CO2 at 1 bar. Dalapati et al. (2013) studied the Py-Azine COF, because it can be engineered with smaller pore sizes due to the short structural length of the azine unit. The CO2 isotherms of ACOF-1 at 273 K (Figure 3), showed 17.7 wt% CO2 uptake at 273 K and 1 bar. This values is greater than CO2 uptake reported for COF-5 (5.9 wt%, SABET = 1,670 m2 g−1) (Furukawa and Yaghi, 2009), COF-103 (7.6 wt%, SABET = 3,530 m2g−1) (Furukawa and Yaghi, 2009), TDCOF-5 (9.2 wt%, SABET = 2,497 m2 g−1) (Kahveci et al., 2013), and ILCOF-1 (6.0 wt%, SABET = 2,723 m2 g−1) (Rabbani et al., 2013). This is despite the fact that the aforementioned COFs have larger internal surface areas. Dalapati et al. account for the higher CO2 uptake capacity of ACOF-1 by considering the great number of nitrogen sites at the pore walls and excellent porosity as well as the high specific surface area, large pore volume and small pore size of the material (Shen et al., 2013).
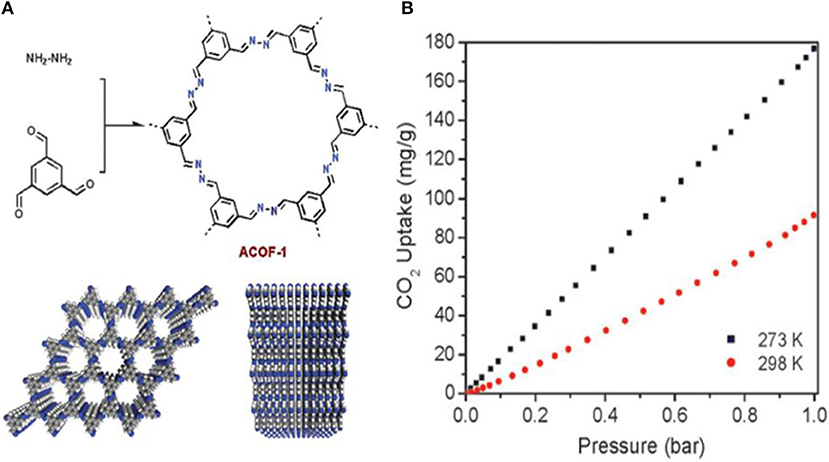
Figure 3. (A) Structure of ACOF-1 and (B) CO2 adsorption isotherms of COF-JLU2 measured up to 1 bar at both 273 and 298 K. Reprinted with permission ©The Royal Society of Chemistry 2014.
Li et al. continued their work on azine linked COFs for the capture of CO2 by synthesizing COF-JLU2 this time utilizing 1,3,5-triformylphloroglucinol and hydrazine hydrate as starting materials (Li et al., 2015). COF-JLU2 (Figure 4A). The CO2 adsorption isotherms were measured as high as 1 bar at 273 and 298 K. COF-JLU2 shows a CO2 uptake of 21.7 wt % at 273 K and 1 bar (Figure 4B, Table 1 entry 10). The isosteric heats of adsorption (Qst) of COF-JLU2 were estimated for CO2 by analyzing adsorption data collected at various temperatures so that better understanding of the CO2 uptake properties could be obtained (Reid et al., 1998). COF- JLU2 shows a Qst of 31 kJ mol−1 (Table 1 entry 10) at lower adsorption values, this is greater than the values reported for COFs (Côté et al., 2007; El-Kaderi et al., 2007; Tilford et al., 2008; Furukawa and Yaghi, 2009; Kahveci et al., 2013), imine-linked organic cages (Jin et al., 2011), and diimide polymer (Farha et al., 2009). Li et al. attribute the excellent CO2 uptake capacity with high adsorption to the inherent microporosity of framework and the strong dipole-quadrupole interactions between the nitrogen and oxygen atoms in COF-JLU2 and CO2.
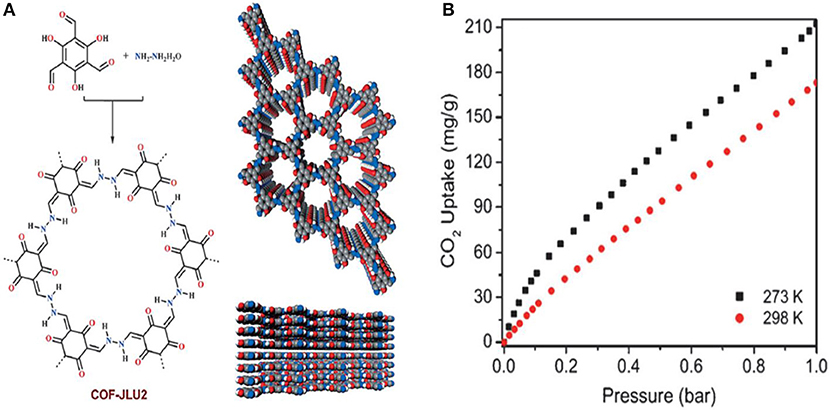
Figure 4. (A) Structure of COF-JLU2 synthesized by Li et al. (2015) and (B) the CO2 adsorption isotherms of COF-JLU2 measured at both 273 and 298 K up to 1 bar pressure. Reprinted with permission ©2015 Wiley VCH.
While several COFs show the ability to capture CO2, industrial applications of the COFs are hampered by their intolerance to harsh conditions especially water (Lanni et al., 2011; Uribe-Romo et al., 2011; Li et al., 2018; Zhang et al., 2018). The crystallinity of COF materials is dependent upon of the reversible reactions that comprise their synthesis; with the kinetic, amorphous products initially being the dominant species but the eventual formation of the lower energy thermodynamic product leading to COF crystals as the major product (Zhu and Zhang, 2017). The four reactions by which most COFs are synthesized, boronic trimerization, boronate ester formation, trimerization of nitriles, and Schiff base condensation are all reversible and so any crystal formed by these reactions will suffer degradation even in mild aqueous environments (Lanni et al., 2011; Spitler et al., 2011). Making COFs from non-reversible reactions is a challenging but effective alternative (Zhang et al., 2018). In a breakthrough work by Kandambeth et al. (2012), a robust COF was synthesized from an irreversible reaction in two steps; the first being a reversible Schiff base reaction and the second being an irreversible keto-enol tautomerization. The new COFs (TpPa-1 and TpPa-2) (Kandambeth et al., 2012) were tested by Kandambet et al. to determine the CO2 uptake capacity. The CO2 uptake of TpPa-1 was found to be 153 mg g−1 at 273 K which makes it as effective as COF-6 (Furukawa and Yaghi, 2009) but with a great increased stability. TpPa-1 and TpPa-2 were soaked in water for 7 days after which pXRD peaks showed no change in position and FTIR analysis gave no new peaks to indicate the COF returning to starting material. Even after treatment in 9N HCl for 3 days the positions and intensities of pXRD peaks of the two COFs remained the same. TpPa-2 was able to withstand 9N NaOH for 7 days without a change in surface area, FTIR peaks, or pXRD pattern. This impressive stability is a result of the irreversible enol-to-keto tautomerism. Later work by Zou et al. used a mechanochemical synthesis of TpPa-COF nanosheet clusters and effectively applied the nanosheet clusters for CO2 capture (Zou et al., 2017). TpPa-COF was also synthesized by use of microwave radiation in 1 h (Wei et al., 2015).
Studies on COF Structures and Composition for Optimizing CO2 Adsorption
Recently EL-Mahdy et al. (2018) reported the synthesis and CO2 adsorption of triphenylamine COFs (TPA-3NH2) and (triphenyltriazine COFs) (TPT-3NH2). TPA-3NH2 was synthesized by the polycondensation of tris(4-aminophenyl)amine with various triarylaldehydes while TPT-3NH2 was synthesized by the polycondensation of 2,4,6-tris(4-aminophenyl)triazine with various triarylaldehydes (Figure 5). The triacrylaldehydes differed in planarity, symmetry, and nitrogen content. By considering the symmetry and planarity of the monomers the highly crystallin COFs can be custom designed for specific applications. El-Mahdy et al. noted that the use of symmetrical monomer units for the synthesis of COFs with hexagonal structure was successfully achieved by other researchers (Haase et al., 2017; Zhai et al., 2017; Gao et al., 2018) but these materials have limited application in gas storage. El-Mahdy et al. sought to control the crystallinity of synthesized COF and elucidate the effect of crystallinity on the CO2 uptake. The pXRD patterns of the COFs confirmed the crystallinity of the synthesized materials and pointed to an increasing degree of crystallinity upon increased planarity of the monomers. El-Mahdy et al. related the surface are of the COF to the symmetry and planarity of their monomers by determining the Brunauer–Emmett–Teller (BET) and Langmuir surface areas of the synthesized COFs (Table 1).
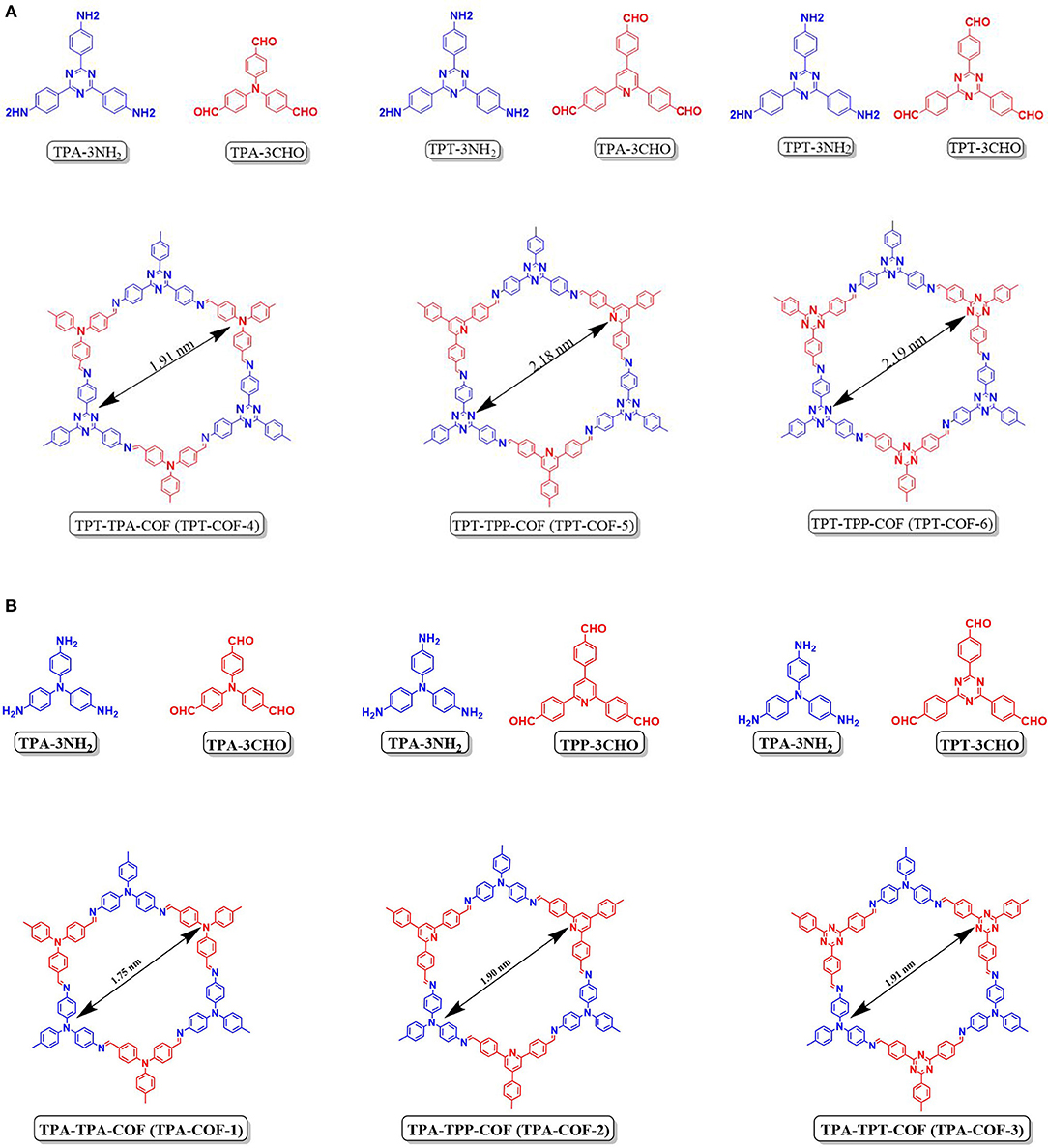
Figure 5. (A,B) Building blocks and structure of TPA and TPT COFs as reported by EL-Mahdy et al. (2018) Reprinted with permission ©2018 Royal Society of Chemistry.
The BET surface area of TPT-COF-6 which was made from the most symmetrical and planar linkers was 1,535 m2 g−1. This value was more than double that of the TPA–TPA-COF (TPA-COF-1) (714 m2 g−1), synthesized from a mixture of the symmetrical and least planar building blocks. This was not the case for unsymmetrical monomers. The TPA–TPP-COF (TPA-COF-2), synthesized from the least planar building blocks (TPA-3CHP), had a BET surface area of 478 m2 g−1 and TPA-COF-3 had a BET surface area of 557 m2 g−1 prepared from the most planar TPT-3CHO monomer (Table 1 entry 63). In the same vein, TPT-COF-4 had a BET surface area of 1,132 m2 g−1 which was markedly less than that of TPT-COF-5 (1,747 m2 g−1) (Table 1 entries 64–65). EL-Mahdy et al. (2018) summarize that the symmetry and planarity are the two main factors that affect the COF's total surface area. Synthesizing COFs with less planar starting materials reduced the surface area for both the symmetric and unsymmetrical COFs.
The CO2 adsorption of TPA-COFs and TPT-COFs at temperatures of 298 and 273 K (Table 1 entries 61–66) were examined by El-Mahdy et al. The CO2 uptake efficiency was heavily influenced by the nitrogen content and nitrogen nucleophilicity of the COFs. The TPA-TRF-COF (TPA-COF-3) had the highest CO2 uptake capacity (63.94 and 91.15 mg g−1 at 298 and 273 K) and had the highest nitrogen content of all the TPA-COFs synthesized in this study. The TPA-TPP-COF (TPA-COF-2) showed CO2 uptake 1.2-fold higher than that of the TPA-TPA-COF (TPA-COF-1), despite the fact that the former had three triarylamine and three triarylpyridine unites in each pore while the latter had 6. It should be added that the nitrogen content for these COFs is similar. El Mahdy et al. deduced that the better uptake efficiency of TPA-COF-2 was caused by the steric hindrance around the nitrogen atoms in the non-planar triarylamine unit. The nucleophilicity of the nitrogen atom decreased as the steric hindrance around the nitrogen increased; which decreases the number of carbon dioxide molecules adsorbed (Kortunov et al., 2016). The same concepts can be applied to the TPT-COF series for which TPT-COF-6 had the highest CO2 uptake capacities 65.65 and 92.38 mg g−1 at 298 and 273 K, respectively, TPT-COF-6 had six triaryltraizine units. TPT-COF-5 showed CO2 uptake capacity 1.1 times higher than TPT-COF-4 (Table 1 entries 64–66).
The aforementioned works make it clear that controlling the stacking and connectivity of COF materials will lead to an increase in CO2 uptake. However, while synthetic procedures for controlling the structure in MOFs (Stock and Biswas, 2012; Furukawa et al., 2013) have been well studied less is known about altering synthetic methods to realize structural differences in a COF. Gao et al. (2018) set out to understand if different structural COF variant could be made from the same flexible building blocks but by using different solvents for synthesis. Gao et al. found the mixture of tetraphenylethane (Ascherl et al., 2016; Pang et al., 2016) building blocks; which contain 4 amine or aldehyde units, heated for 7 days in a o-dichlorobenzene and n-butanol solvent mixture will synthesize [4+4] TPE-COF-I but when the building blocks were heated in 1,4-dioxane the new [2+4]. TPE-COF-II was synthesized (Figure 6). TPE-COF-II had exceptional CO2 adsorption performance of 118.8 cm3g−1 (23.2 wt% CO2 at 1 atm, 273 K). TPE-COF-I was less effective for CO2 uptake with 68.6 cm3 g−1 (13.4 wt%, 1 atm, 273 K) and 37.8 cm3 g−1 (7.38 wt%, 1 atm, 298 K). The BET surface area of TPE-COF-I and TPE-COF-II were found to be 1,535 and 2,168 m2g−1. The pore volume of TPE-COF-II was 2.14 cm3 g−1 (P/P0 = 0.984) this was larger than the pore size of TPE-COF-I (1.65 cm3g−1 P/P0 = 0.983) (Table 1 entries 67, 68).
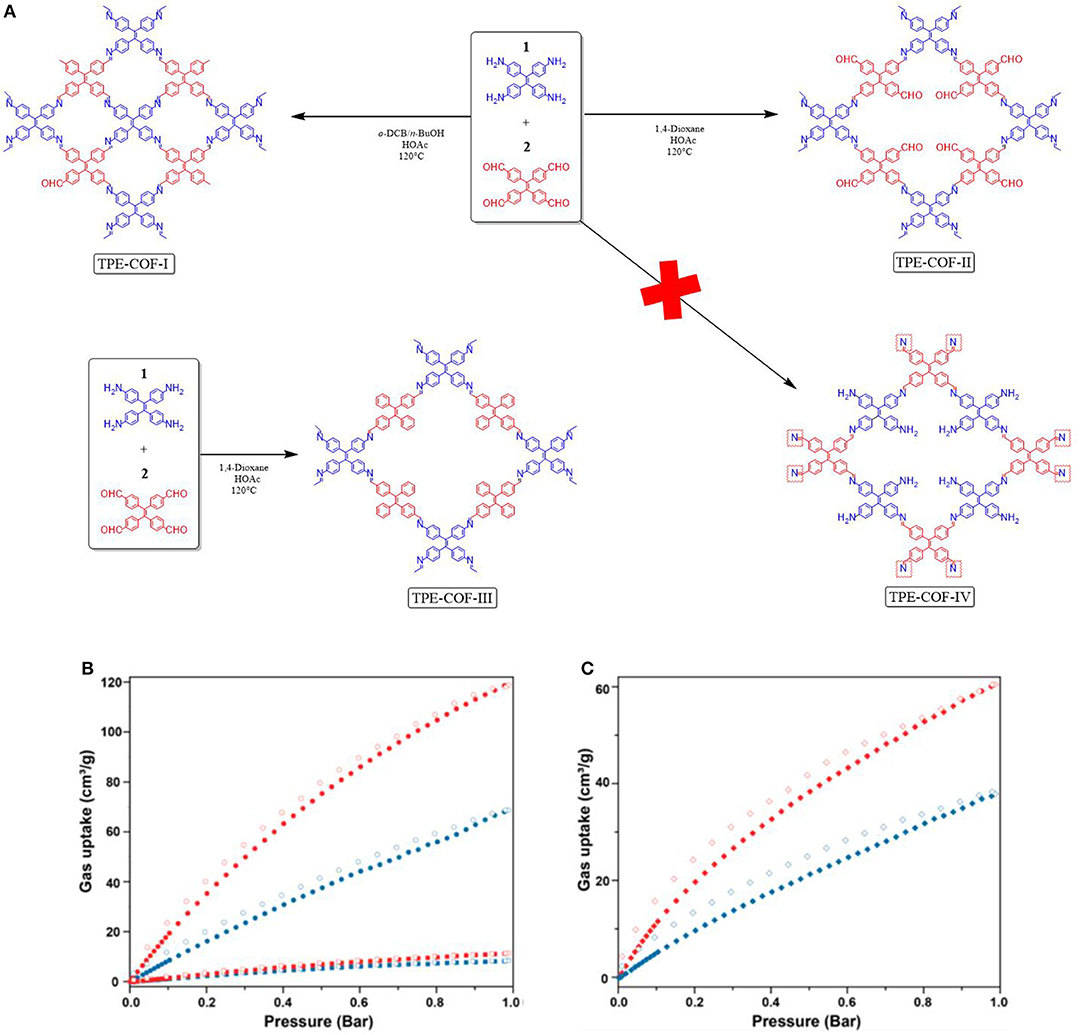
Figure 6. Preparation of TPE-COFs by different reaction conditions yielding different COF structures. (A) Gas sorption isotherms of TPE-COFs. (B) CO2 adsorption isotherm of TPE-COF-II at 273 K (red •) and TPE-COF-I (blue •) at 273 K; N2 adsorption isotherm of TPE-COF-II at 273 K (red ■) and TPECOF-I (blue ■). (C) CO2 adsorption isotherm at 298 K of TPE-COFII (red ♦) and TPE-COF-I (blue ♦) as determined by Loh et al. Reprinted with Permission ©2018 The American Chemical Society.
COFs Which Utilize Channel Wall Functionalization for Increased CO2 Affinity
Molecules with perfluorinated moieties were developed by Zhao et al. as starting materials for COFs capable of effectively adsorbing CO2 in environments with high water levels (Zhao et al., 2013). Zhao et al. devised a perfluorinated covalent triazine-based framework (FCTF-1) that could capture CO2 with high selectivity. The fluorine (F) groups were crucial for increasing FCTF-1′s CO2 adsorption and selectivity because the very polar C–F bonds encourage CO2 adsorption by inducing electrostatic interactions. These electrostatic interactions become more important at low pressures. At 273 K and 0.1 bar, FCTF-1's CO2 uptake was 1.76 mmol g−1 through equilibrium adsorption. It is known that perfluorinated alkenes have a remarkable affinity toward CO2 (DeSimone et al., 1994; Eastoe et al., 2006) motivating the incorporation of perfluorinated alkene linkers into a COF framework. Zhao et al. utilized the known CTF-1 triazine framework as the host for perfluorinated molecules. The combination of trazine frameworks with perfluorinated moieties is beneficial for the following reasons:
(i) It has been shown that the nitrogen-rich CTF-1 framework is ideal for CO2 adsorption (Hao et al., 2010; Sevilla et al., 2011; Zhao et al., 2012), and adsorption could be amplified by electrostatic interactions between CO2 and electronegative fluorine (Cho et al., 2012; Yu et al., 2012).
(ii) The pore size of CTF-1 is decreased to <0.5 nm upon introduction of F atoms. The smaller pores would have greater adsorption potential and be more kinetically selective of CO2 over N2.
(iii) The C–F bonds are hydrophobic and would increase the water tolerance of CTF-1.
(iv) The lack of chemical adsorption allows for regeneration of the sorbent COF.
FCTF-1 was synthesized by Zhao et al. following the same procedure as developed for CTF-1 (Kuhn et al., 2008) except that tetra-fluoroterephthalonitrile was used as the precursor. The incorporation of F groups into CTF not only enhanced the CO2 adsorption capacity but also led to more preferential adsorption of CO2 over N2, i.e., higher CO2-N2 selectivity. FCTF-1, however, lacked the crystallinity of CTF-1.
Later work on the synthesis of COFs for the adsorption of CO2 sought to increase the affinity of the porous materials for CO2 through the introduction of CO2 binding moieties onto the channel walls of the COF while retaining crystallinity (Huang et al., 2015a,b; Ge et al., 2016; Wang et al., 2018). Huang et al. showcased this strategy by fixing COOH moieties into the channel walls of a imine-linked 2D COF. This COF consists of porphyrins linked by phenol containing bridges with phenol groups on the pore walls (Figure 7); this 2D COF has a low carbon dioxide adsorption capacity prior to installation of the COOH functional groups (Huang et al., 2015a). The carboxylic acid content in the COF pores was controlled by altering the ratio of the 2,5-dihydroxyterephthalaldehyde (DHTA) and 1,4-phthalaldehyde (PA) bridging linkers.
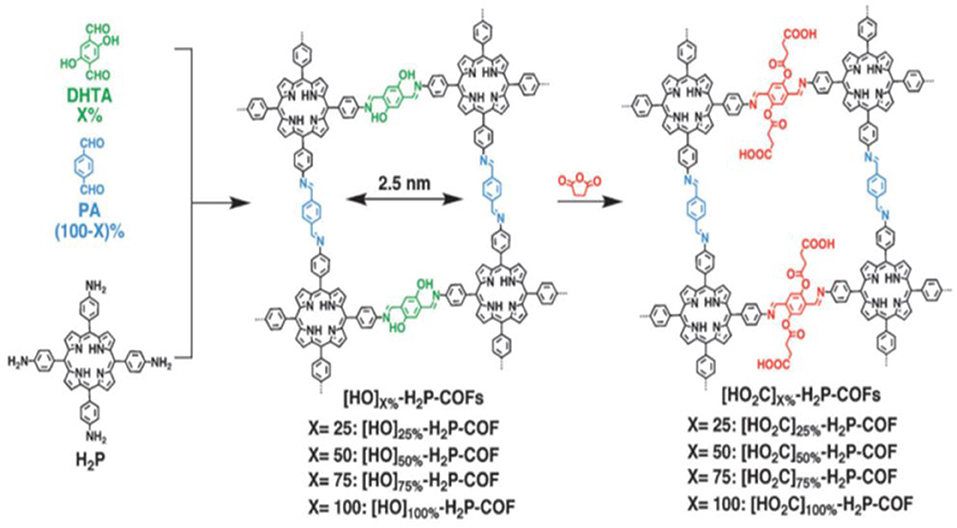
Figure 7. Structure of [HO2C]x%-H2P = COFs synthesized by Huang et al. Reprinted with Permission ©2015 Wiley VCH.
After addition of the COOH moieties in the pores of [HO2C]x%-H2P-COFs, the COF becomes microporous, this is made evident by the typical type I sorption curve in Figure 8. As the percent of bridging linkers containing COOH groups increased from 25 to 100% the BET surface area decreased from 786 to 364 m2 g−1 whereas the pore size decreased from 2.2 to 1.4 nm (Table 1 entries 31–34). It has been previously reported that carboxylic acid groups engage in dipolar interactions with carbon dioxide (Rochelle, 2009; Dawson et al., 2011b). The carboxylic acid functional groups in [HO2C]x%−H2P-COFs are positioned at the termini and have the relatively same pKa value (5.86) as free carboxylic acids. Huang et al. show that [HO]x%-H2P-COFs exhibit low CO2 uptake capacities at 1 bar and 273 K when compared to the [HO2C]x%-H2P-COFs. The HO]x%-H2P-COFs had capacities of 31–35 mg g−1 at 273 K and 46–63 mg g−1 at 298 K whereas the [HO2C]x%-H2P-COFs had capacities of 180 mg g−1 at 273 K and 76 mg g−1 at 298 K. The adsorption capacities of the [HO2C]x%-H2P-COFs was directly correlated to the carboxylic acid content as shown in Table 1, entries 31-34. This is strong evidence for the validity of channel wall COOH functionalization as a method to increase CO2 adsorption.
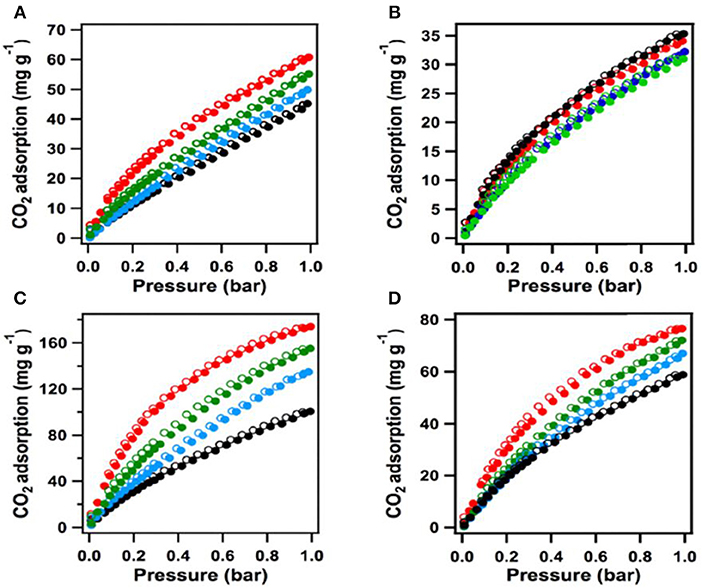
Figure 8. The [HO]x%-H2P-COF CO2 sorption curves measured at (A) 273 K and (B) 298K (black [HO]25%-H2P-COF CO2, blue: [HO]50%-H2P-COF CO2, green: [HO]75%-H2P-COF CO2, red: [HO]100%-H2P-COF CO2. The CO2 sorption curves of [HO2C]x%-H2P-COFs measured at (C) 273K and (D) 298K (black: [HO2C]25%-H2P-COF, blue: [HO2C]50%-H2P-COF, green: [HO2C]75%-H2P-COF, red: [HO2C]100%-H2P-COF). Filled circles represent adsorption and open circles represent desorption. Reprinted with Permission ©2015 Wiley VCH.
Huang et al. (2015b) expanded the study of increasing CO2 uptake in COFs through channel wall optimization by using click reactions to install a litany of functional groups in the pores of an imine linked porphyrin COF. Huang et al. synthesized four novel COFs each with a different percentage of ethynyl functional groups installed in the pore walls. This was accomplished through condensation between 5,10,15,20-tetrakis(p-tetraphenylamino)porphyrin and a variable mixture of 2,5-bis(2-propynyloxy)terephthalaldehyde (BPTA) and 2,5-dihydroxyterephthalaldehyde (DHTA). The resultant [HC≡C]x-H2P- COFs had ethynyl contents of X = 25, 50, 75, and 100 where X = [BPTA]/([BPTA] + [DHTA]) ×100. After isolation of the COFs, Huang et al. used click reactions to produce 20 new COFs with ethyl, acetate, hydroxyl, carboxylic acid, and amino groups; ranging from hydrophobic to hydrophilic and from basic to acidic (Figure 9).
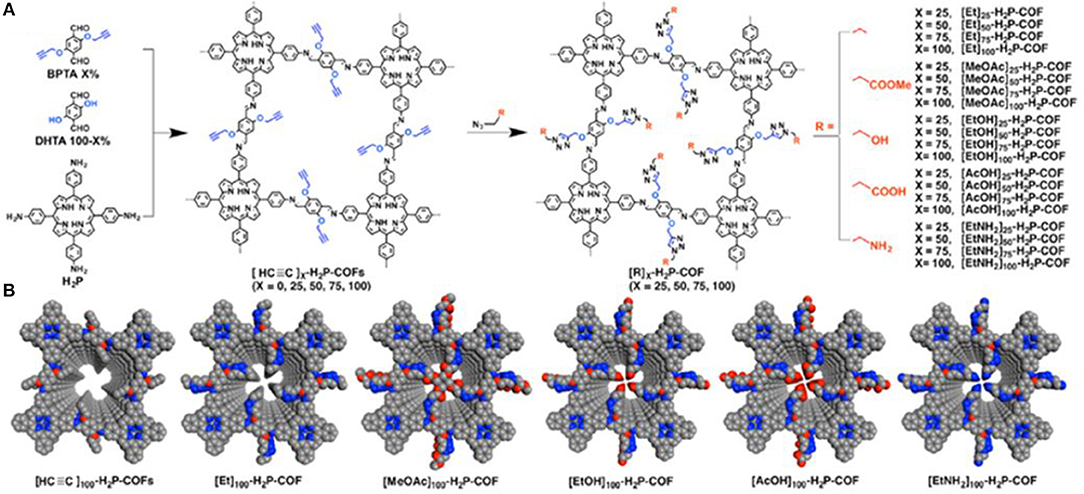
Figure 9. (A) Installation of various functional groups onto the pore surface of an imine linked porphyrin COF as depicted by Huang et al. and (B) Pore structures of the synthesized COFs (Gray, C; Blue, N; Red, O). Reprinted with permission ©2015 The American Chemical Society.
Huang et al. noted that CO2 uptake was influenced by the functional groups that had been installed. The introduction of ester units onto the pore walls of the COF to make ([MeOAc]x-H2P-COFs) resulted in CO2 uptake ability that far surpassed COFs made through the introduction of ethyl units ([Et]x-H2P-COFs) x = 25, 50, 75, 100 (Table 1 entries 39–47). [MeOAc]50-H2P-COFs exhibited CO2 adsorption capacities of 47 and 88 mg g−1 at 298 and 273 K, respectively (Table 1 entries 44–47). This value is 1.6-fold greater than that of the best-performing [Et]x-H2P-COFs (Table 1 entries 40–43). As was determined in the previous work Huang et al. found that the introduction of carboxylic acid groups improved the CO2 uptake ability of the COF greatly. Another well performing functional group was the hydroxyl group (Table 1 entries 52–55). For instance, [EtOH]50-H2P-COF exhibited a capacity of 71 mg g−1at 298 K and 124 mg g−1 at 273 K, these values are 2.3–2.4 times higher than those found for [HC≡C]25-H2P-COF. The CO2 adsorption was markedly improved upon installation of amino groups on the pore surface (Table 1 entries 56–59). The capacities of [EtNH2]25-H2P-COF, [EtNH2]50-H2P-COF, [EtNH2]75- H2P-COF, and [EtNH2]100-H2P-COF were 60, 82, 67, and 52 mg g−1 at 298 K and 116, 157, 133, and 97 mg g−1 at 273 K, respectively. The highest adsorption capacity of [EtNH2]50-H2P-COF showed an increase nearly three times higher than the adsorption capacity of [Et]50-H2P-COF and [HC≡C]50-H2P-COF.
All the COFs mentioned so far possess neutral frameworks. Incorporation of ionic building blocks would introduce a myriad of tunable and novel functions to a COF framework but would interrupt the π-π stacking which is essential to 2D COF crystallinity (Du et al., 2015; Huang et al., 2016; Ma et al., 2016; Diercks and Yaghi, 2017). Huang et al. (2017) were able to design an ionic COF (PyTTA-BFBIm-iCOF) which was able to achieve high crystallinity despite the ionic interfaces on the pore walls due to an atypical reverse AA-stacking mode. PyTTA-BFBIm-iCOF was synthesized by 4,4′,4″,4‴–(pyrene-1,3,6,8-tetrayl) tetraaniline (PyTTA) as the neutral knot and 5,6-bis(4-formylbenzyl)- 1,3-dimethyl-benzimidazolium bromide as the cationic linker; the two moieties were linked by an imine bond. The two building blocks were mixed in o-dichlorobenzene and n-butanol in the presence of acetic acid catalyst and heated to 120°C for 3 days under solvothermal conditions and produced a crystallin powder as shown by the pXRD pattern Figure 10. The pXRD of PyTTA-BFBIm-iCOF after 24-h immersions in 3M HCl, 3M NaOH, or DMF were still crystalline as determined by pXRD.
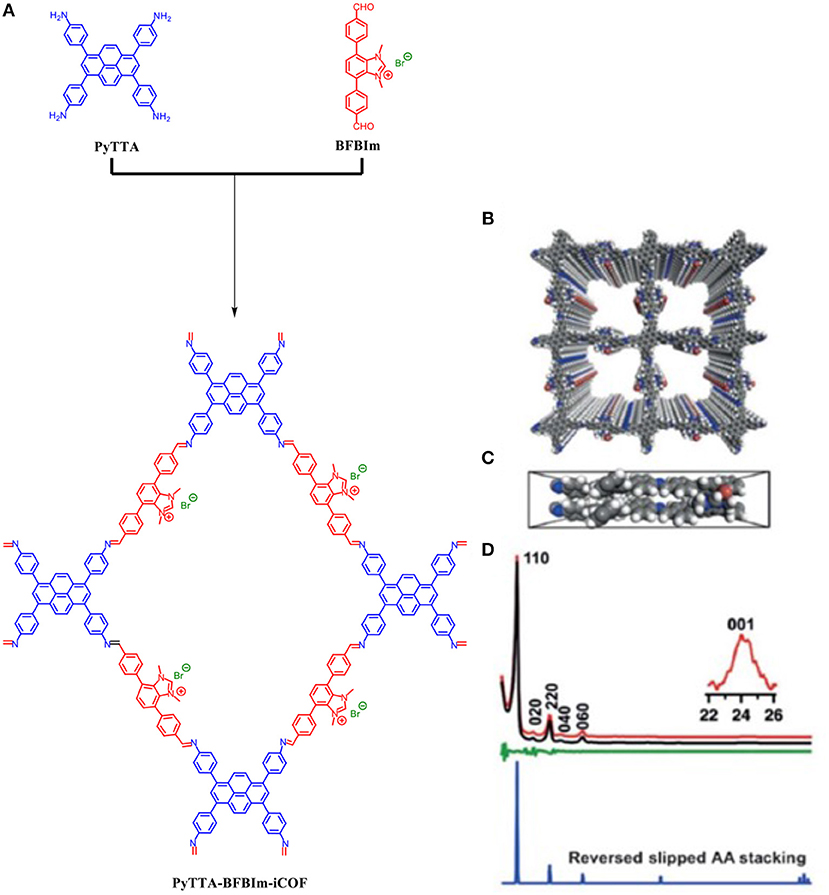
Figure 10. (A) Building blocks and chemical structure of PyTTA-BFBIm-iCOF, (B) Top view of the reversed slipped AA-stacking mode, (C) Bottom view of the reversed slipped AA-stacking mode, and (D) PXRD pattern of PyTTA-BFBIm-iCOF (red) as determined by Huang et al. (2017) Pawley refined pXRD pattern (black), their difference (green) a simulated curve for reversed slipped mode (blue) as determined by Huang et al. (2017) Reprinted with permission ©2017 Wiley VCH.
The BET surface area was determined to be up to 1,532 m2 g−1; only one 2.3 nm pore was determined by DFT with the total pore volume estimated to be 0.7 cm3g−1. PyTTA-BFBIm-iCOF uptakes CO2 with capacities of 93 mg g−1 at 298 K and 177 mg g−1 at 273 K and 1 bar (Table 1 entry 60). This was determined to be almost three times the performance of the analogous neutral COF (PyTTA-TPhA-COF) which was 65 mg g−1 at 273 K and 36 mg g−1 at 298 K and 1bar. PyTTA-BFBIm-iCOF had a Qst value of 30.2 kJ mol−1 as calculated from the isotherms.
Computational Studies on CO2 Adsorption by COF Materials
Babarao and Jiang (2008) performed atomistic simulation studies on 3D, 2D, and 1D COFs. Simulations were performed with a Gibbs ensemble Monte Carlo (GEMC) simulations at set pressure (Jiang and Sandler, 2003, 2006). One simulation box was used for the adsorbent and another for bulk adsorbate. Molecules could move from one box to the other, but the total number of adsorbent molecules remained constant.
In these simulations, the adsorbent volume remained constant, but the bulk phase was allowed to change at a fixed pressure. This method allows the uptake to be determined at the desired bulk pressure, density, and enthalpy. By simulating the aggregation of centers-of-mass for 100 equilibrium CO2 configurations, Babarao and Jiang (2008) were able to illustrate the density distribution contours for COF-106, COF-6, and COF_NT at 1,000 kPa, as seen in Figure 11. This allows the favorable locations of CO2 adsorption could be estimated accurately. Babarao and Jiang (2008) found that CO2 adsorption in COF-108 takes place close to the carbon-oxygen-boron rings, as the pressure increases adsorption will take place in the central cavity. In COF-6 CO2 molecules accumulate at the cramped central pores; this is because the inter-layer distance in COF-6 is small. In the 1D COF_NT, the CO2 molecules are adsorbed in the center of the nanotubes as well as the small spaces amongst the tubes.
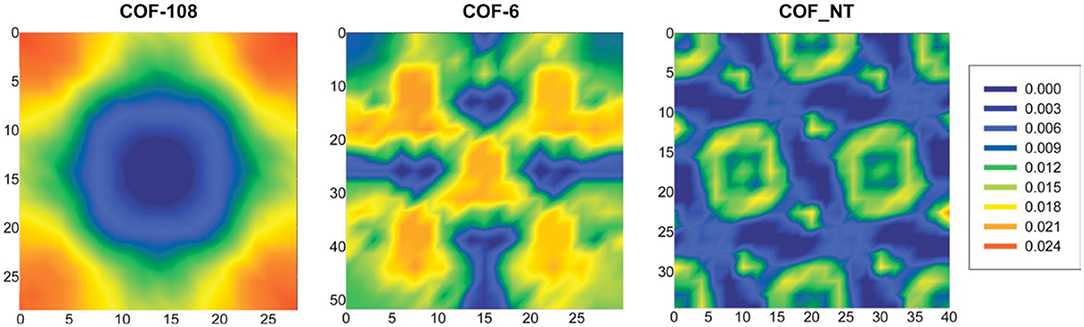
Figure 11. Density distribution contours for the center of mass CO2 created by Babarao and Jiang (2008) depicting COF-108; COF-6; and COF_NT from left to right, each at 1,000 kPa. Reprinted with permission ©2008 The Royal Society of Chemistry.
Babarao and Jiang (2008) developed a function of the framework density ρf, free volume Vfree, porosity ϕ, and accessible surface area Asurf as a method to quantitatively determine the CO2 storage capacity. The framework density (ρf) and gravimetric capacity () are inversely related. The volumetric capacity () decreases linearly. Expressions determined by Babarao and Jiang (2008) for and (1, 2) can be seen below.
As free volume (Vfree) rises, rises as well. has a small drop after an initial small rises. Expressions determined by Babarao and Jiang (2008) for the relation between and with Vfree are shown below (3, 4).
and rise with the porosity guided by the expressions (5, 6) determined by Babarao and Jiang (2008)
and can be expressed as functions of accessible surface area (Asurf) as seen in expressions (7, 8) as determined by Babarao and Jiang (2008) As Asurf increases so do and .
There is a useful interrelationship between and with the framework density, free volume, porosity, and accessible surface area. and would be complemented through a decrease in the framework density or by increasing the free volume, porosity or accessible surface area. These insights elucidated by Babarao and Jiang (2008) are useful for predicting the CO2 adsorption uptake prior to a COF synthesis.
Computational studies aiming to develop new strategies for chemists to optimize the CO2 adsorption of new COF materials utilize simulations of known COFs. Tong et al. (2014) investigated the effects of different π-π stacking motifs as they related to the ability for CO2 capture by simulating 46 COFs. 2D-COFs with hexagonal and tetragonal topologies and 3D-COFs with ctn, bor, and dia topologies. This set of COFs included the majority of main group light elements to yet be synthesized (C, O, B, N, H, F, Si, etc.), porphyrinic COFs which incorporated heavy metals like Zn and Ni were also in studied. The comprehensive analysis of elements provided a solid basis for obtaining relevant and accurate results. Tong et al. calculated charges of the atomic particles for the 46 COFs using the DFT calculations for the purpose of modeling guest–host intermolecular interactions. Subsequently grand canonical Monte Carlo (GCMC) simulations were employed to investigate the adsorption behaviors of the CO2-N2 mixture in COFs at 298 K. This was done using CADSS (Complex Adsorption and Diffusion Simulation Suite); a code developed by Tong et al. The simulation consisted of five types of trials for the molecules: attempts (i) to randomly displace a molecule (translation or rotation), (ii) to re-grow a molecule at a random position, (iii) to create a new molecule, (iv) to delete an existing molecule, and (v) to exchange the molecular identity. Three properties of the COF were studied by GCMC simulations: (i) structure–selectivity relationship, (ii) structure–working capacity relationship, (iii) structure–sorbent selection parameter relationship. In prior work, Wu et al. had it developed a concept dubbed “adsorbility (AD)” for MOFs (Wu et al., 2012a) and found a good correlation between the selectivity and difference in adsorbility of adsorbates (ΔAD = /φ) in MOFs (Wu et al., 2012a,b). The good correlation between selectivity and ΔAD makes ΔAD a valuable indicator for selectivity. This means COFs should be designed with a large ΔAD to improve the separation selectivity. Two broad routines were suggested by Tong et al. to boost ΔAD for adsorbent COFs:
(i) Introduction of polar functional groups which cause , to increase. This can also be accomplished by the incorporation of unsaturated metal sites.
(ii) Structural shift induced by interpenetration which will create new small pores and boost ΔAD.
An important discovery of Tong et al. (2014) was the crucial role that the staggered stacking of COF layers plays in increasing the adsorption of CO2 in 2D COFs. As can be observed from the snapshots shown in Figure 12, CO2 is adsorbed in the pores of eclipsed CTF-2 at random positions. Meanwhile, in staggered CTF-2, the adsorbed CO2 molecules reside at the slit-shaped pores between the staggered layers and most of the CO2 molecules are in the center of these pores. While located at the center of these slit-shaped pores the CO2 molecules are interacting with two COF layers. Tong et al. note that in this simulation more CO2 is adsorbed on the staggered CTF-2 then the eclipsed CTF-2.
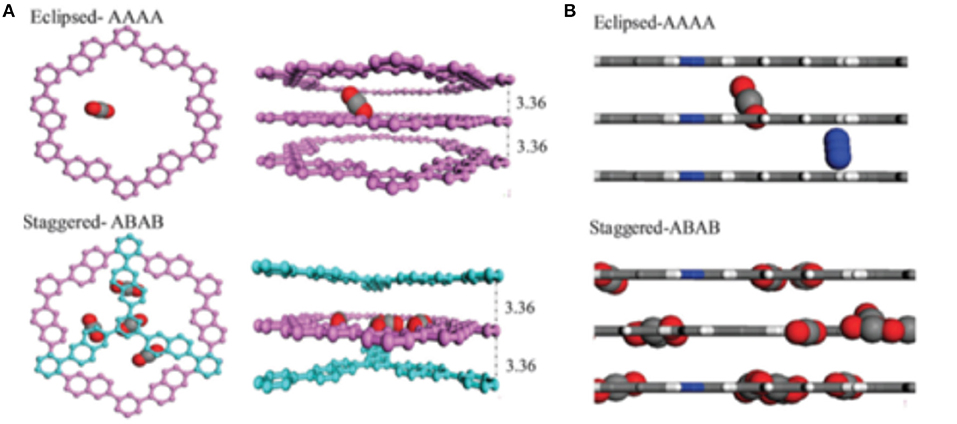
Figure 12. (A) Adsorbed CO2 in eclipsed (top) and staggered (bottom) CTF-2 and (B) CO2-N2 gas mixture in the eclipsed (top) and staggered (bottom) CTF-2. Reprinted with permission ©2014 The Royal Society of Chemistry.
Interlayer slipping between the π-π stacked layers of 2D COFs is a controllable feature that is shown to be exploitable when optimizing a COF material for the increased adsorption of gasses (Uribe-Romo et al., 2011; Feng et al., 2012b; Kandambeth et al., 2012; Biswal et al., 2013; Chandra et al., 2013; Fang et al., 2014). Sharma et al. (2017) studied the effect of interlayer slipping on CO2 adsorption and CO2/N2 separation in microporous TpPa1 and mesoporous TpBD and PI-COFs using quantum mechanical and grand canonical Monte Carlo simulations. In this study, Babarao et al. modeled CO2 bonding sites in the slipped COF structures focusing on bilayer slipping only. Three COFs were simulated in this work: TpPa1 (Kandambeth et al., 2012), TpBD (Biswal et al., 2013), and PI-COFs (Fang et al., 2014). When modeling the slipped structures of TpPa1 and TpBD COFs calculations were simplified by only considering two COF layer supercells with shifting only in the 90 and 60° directions, as shown in Figure 13. Sharma et al. (2017) acknowledge that slipping in other directions are a possibility but base their models on the classification scheme of Koo et al. (2012). Ab initio DFT was carried out in Vienna Ab Initio Simulation Package (VASP) to model the COF structures with at various modes of slipping (Kresse and Furthmüller, 1996). Sharma et al. also calculated binding energy of CO2 by modeling the lone gas molecule in a 2 × 2 × 7 supercell. The position of the CO2 molecule in the supercell was determined by simulated annealing. CO2-CO2 interactions and CO2-COF interactions were simulated by Sharma et al. with the Lennard–Jones (LJ) potential model with Coulombic interactions. Using the RASPA code to implement Monte Carlo (MC) simulation in the grand canonical ensemble, Babarao et al. calculated the single component adsorption isotherms of CO2 and N2 (Dubbeldam et al., 2013). To obtain the mixture (CO2:N2 in a 15:85 ratio) adsorption selectivity, Sharma et al. (2017) performed IAST calculations (Myers and Prausnitz, 1965). Sharma et al. found that changes in the alignment of COF layers during slipping changes the CO2 binding sites.
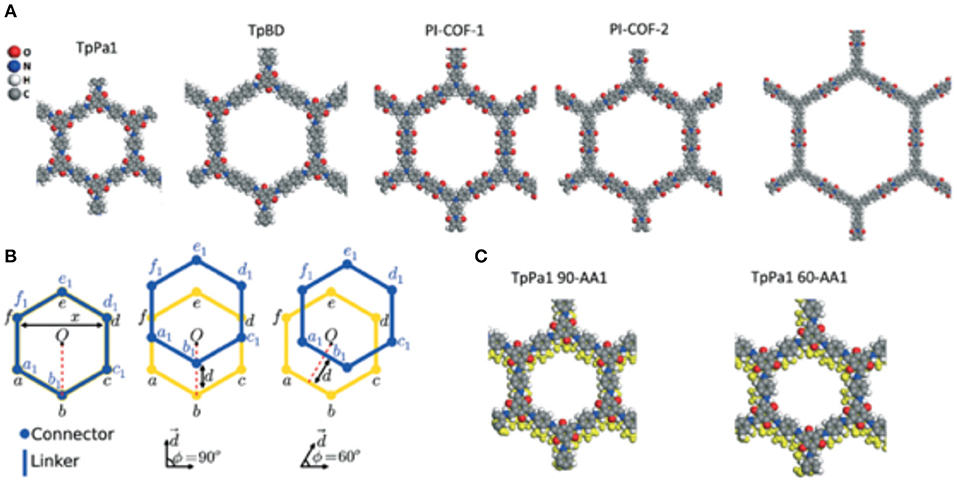
Figure 13. The eclipsed (A) stacked structures of the COFs, (B) illustration of the stacked and slipped structures along 90 and 60° directions a-f (yellow) represents the bottom layers and a1-f1 (blue) represents the top layers and (C) illustration of the TpPa1 COF with 90° slipping and 60° slipping by 1.7 Å in both cases. Reprinted with permission ©The Royal Society of Chemistry.
Sharma et al. (2017) found that CO2 molecules in both AA and slipped COFs show an inclination to absorb close to oxygen atoms between the linkers because the oxygen atoms are more negatively charged than nitrogen and more prevalent in the pores. The CO2 molecules absorbed in the AA will bind at a site between two connector oxygens from two different COF layers, being the same distance from both oxygens, this has been noted in other studies (Dinga and Wang, 2013; Xu et al., 2017). In the slipped COF structures CO2 binds adjoining to a single COF layer and is interacting with three oxygen atoms and with a smaller distance between layers compared to the AA. These two aforementioned factors increase the binding energy to −44.62 kJ mol−1 (Evdw ~ −23.86 kJ mol−1) for 90-AA1 TpPa1 COFs and −43.57 kJ mol−1 (Evdw~ −23.71 kJ mol−1) for 60-AA1 TpPa1 COFs. An important point emphasized by Sharma et al. (2017) is that all oxygen atoms in the AA structure are degenerate. This is not true for the 90-AA1 slipped structure because there is a loss of symmetry. This results in the AA structure having six binding sites per unit cell while the 90-AA1 slipped structure has only one. So even though the slipped structure has higher binding energy the CO2 uptake is reduced. The 60-AB structure of TpPa1 had the greatest CO2 uptake capacity followed by the 90-AA1 structure of the PI-COF-2. Sharma et al. conclude that premeditated design of a slipped COF with exceptional CO2 uptake ability hinges on various factors, such as the chemical structure of building blocks, the energetic interaction between neighboring COF layers and the synthesis protocol.
Computation studies aiming to determine the optimal makeup of CO2 binding functional groups on the channel walls of COF materials have also been undertaken (Dash, 2018; Wang et al., 2018). Work by Lu et al. used grand canonical Monte Carlo (GCMC) to investigate the CO2 adsorption and separation behaviors in four alkyl anime functionalized triphenylamine-based COFs (TPA-xC-NH2, x = 1–4) (Wang et al., 2018). It is reasoned that the CO2 adsorption capacity at 273 K is greater than CO2 adsorption capacity at 298 K due to the exothermic process (Zhou et al., 2017). The reduction of CO2 adsorption capacity when−1C-NH2 is introduced is rationalized by the loss of surface area in TPA-1C-NH2 in relation to the unmodified TPA-COF. This trend does not continue through as the length of the alkylamine chain increases; as evident by the high CO2 adsorption capacities of TPA-4C-NH2 which had the longest alkylamine chain. TPA-4C-NH2 had a CO2 adsorption capacity of 3.41 mmol/g at 298 K and 5.26 mmol/g at 273 K. This represents a 50.2% increase in CO2 adsorption capacity at 298 K and a 30.5% increase at 273 K. TPA-4C-NH2 adsorbs 1.58 mmol/g CO2 at 0.15 bad and 298 K making it viable for CO2 capture at power plants (Hu et al., 2016).
Lu et al. also investigate the selectivity of CO2 over N2 by TPA-COFs. Lu et al. used to define selectivity where qCO2 is the adsorption capacity of CO2 and qN2 is the adsorption capacity of N2 (Wang et al., 2018). The bulk phase ratio of CO2 and N2 (pCO2:pN2) was set to 15:85. Since N2 has very little polarity the addition of longer alkyl amine chains only reduced N2 uptake as internal surface area decreases. This explains why the ability of the TPA-COFs to adsorb CO2 is greatest for the pristine TPA-COF and decreases as the length of the alkylamine chain increases. Since CO2 affinity increases as the alkylamine chain increases, by increasing the length of the alkylamine chains so does the selectivity of CO2/N2. This is affirmed by the analysis of the CO2 selectivity wherein the TPA-COF with−4C-NH2 shows a CO2/N2 selectivity of 11.7 at 298 K and 1 bar, representing a 121% increase in selectivity from the unmodified TPA-COF.
The study of CO2 adsorption into COFs synthesized with reticular N containing molecules was studied through the computational work of Bibek Dash; in this study, the CO2 adsorption capabilities of COFs with N-containing linkers (Dash, 2018) were calculated. Bibek Dash used the Gaussian 09 package to perform the DFT calculations. CO2, N2 molecules on the COF models were optimized with all atoms fully relaxed. The binding affinity of CO2 to the azo group was compared to the affinity for N2 to the azo group using an azobenzene model. Electrostatic interactions between the N of the ring and the CO2 molecule were designated as dipole-quadrupole and dipole-induced dipole interactions (Vogiatzis et al., 2009; Altarawneh et al., 2014).
The synthesis of COF-505 consisting of helical, intertwined threads to make a weaving crystal structure by Liu et al. was an advancement in the COF chemistry (Liu et al., 2016). COF-505 consists of imine linked threads are woven around copper ions; the flexible threads allow for many degrees of freedom so that the structure can stretch and bend without collapsing the general framework. The copper ions can also be removed and subsequently replaced with no loss of crystallinity. Chen et al. performed computational studies to examine the adsorption of CO2 onto COF-505 as well as the potential of COF-505 as a photocatalyst (Chen et al., 2019). Chen et al. used Grimme's DFT-D3 dispersion correction scheme with the Becke–Johnson damping function to perform the dispersion correction to DFT calculations (Grimme, 2011; Grimme et al., 2011). Chen et al. determined that the CO2 molecule could be adsorbed onto both the metal ions and the organic linkers in COF-505. The adsorption energy of CO2 onto COF-505 was found to be −0.16 eV for Cu-COF-505 and −0.18 for Pd-COF-505.
COFs for the Photochemical or Electrochemical Reduction of CO2 to CO
Overview of CO2 Reduction
The first successful use of a COF for the reduction of CO2 to CO was reported by Lin et al. (2015). In this work the researchers synthesized two imine-linked COFs consisting of a cobalt porphyrin, 5,10, 15,20-tetrakis(4-aminophenyl)porphinato] cobalt [Co(TAP)] linked with bidentate aldehyde linkers. The two reticular COFs, COF-366-Co and COF-367-Co differed by the length of the aldehyde linker with the first using 1,4-benzenedicarboxaldehyde (BDA) and then using a 4,4'-Biphenyldicarbaldehyde (BPDA). The COFs were deposited on a conductive and porous carbon fiber for use in electrochemical experiments. Electrolysis of aqueous CO2 saturated solutions with a sodium bicarbonate buffer with applied potentials between −0.57 and −0.97 V produced carbon monoxide.
Lin et al. (2015) hypothesized that due to the low solubility of CO2 in H2O that not all the electroactive cobalt porphyrin sites would fully participate in the electrocatalysis. The researchers elected to separate the active Co sites with catalytically inactive copper porphyrin struts which increased the turn over frequency per cobalt atom. The copper porphyrin was selected by screening the surface area of various COF-367-M (M = metal) compounds and selecting the metal porphyrin which provided the highest surface area. The new bimetallic COFs were denoted COF-366-Co(10%) and COF-366-Co(1%) to denote the percent of Co amongst metals. The bimetallic COFs had a higher TON than the cobalt COFs, as can be seen in Figure 14.
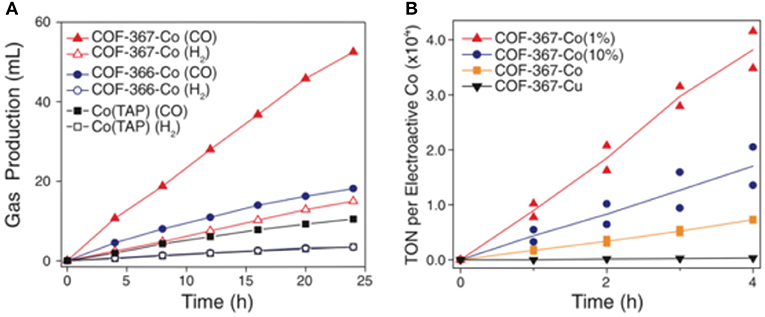
Figure 14. (A) The CO production of various COFs as determined by Lin et al. (2015): (red solid triangles) COF-367-Co; (blue solid circles) COF-366-Co; (black solid squares) Co(TAP); Also shown is the volume of hydrogen produced by various COFs as determined by Lin et al. (2015): (red open triangles) COF-367-Co; (blue open circles) COF-366-Co; (black open squares) Co(TAP) and (B) TON for the CO production of various bimetallic COFs as measured by Lin et al. (2015) at −0.67 V. COF-367-Co(1%) (red solid triangles); COF-367-Co(10%) (blue solid circles); COF-367-Co (orange solid squares) and Co-367-Cu (purple solid inverted triangle; TON in this case is calculated per unit of copper porphyrin). Reprinted with permission ©2015 Science.
Work on Co-porphyrin containing COFs for the electrochemical reduction of CO2 to CO was expanded into the realm of thin films by Diercks et al. (2018a). In this work the Co porphyrins were connected by imine linkages to struts modified with different electron withdrawing groups to synthesize 4 new COFs COF-366-Co, COF-366(OMe)2-Co, COF-366-F-Co and COF-366-(F)4-Co as shown in Figure 15. Yaghi et al. sought to determine the which functional groups provided the optimal electronic effect for the reduction of CO2 to CO. To ensure that performance was a result of only the electrons the adsorption and Qst for all the COFs were measured by Diercks et al. The CO2 adsorption at 295 K was similar for all the four COFs (23.5–27.4 cm3 g−1) and the Qst of all COFs was between 24.6 and 24.1 kJ mol−1. Cyclic voltammograms and X-ray absorbance measurements made clear the differences in electrochemical properties of the COFs as highlighted in Figure 16A.
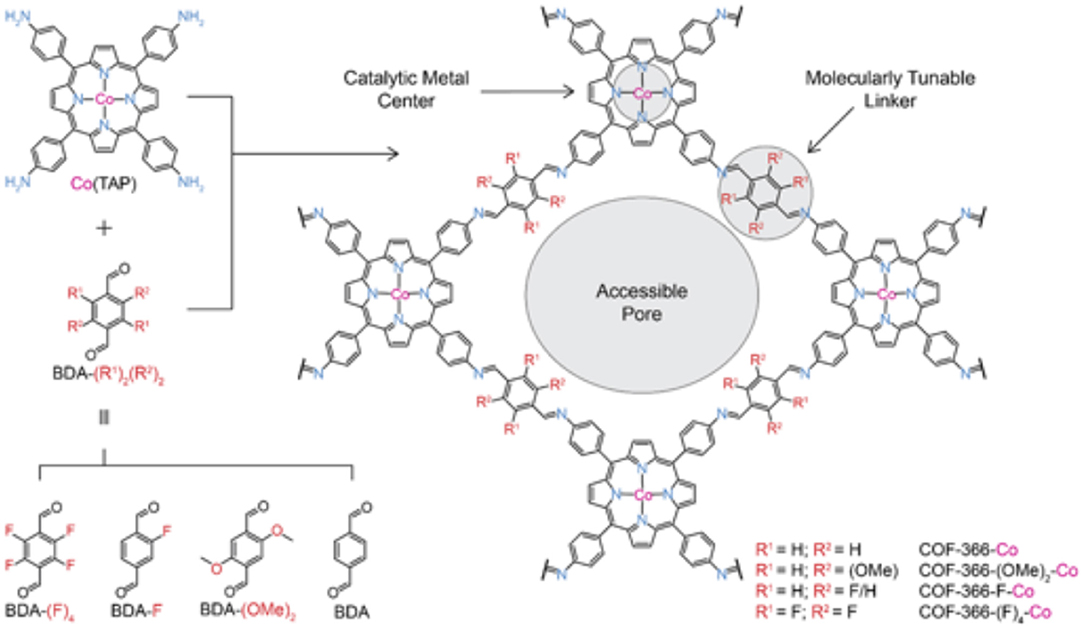
Figure 15. The various inkers used by Diercks et al. (2018a) to produce a series of new Co-COFs. Reprinted with permission ©2018 The American Chemical Society.
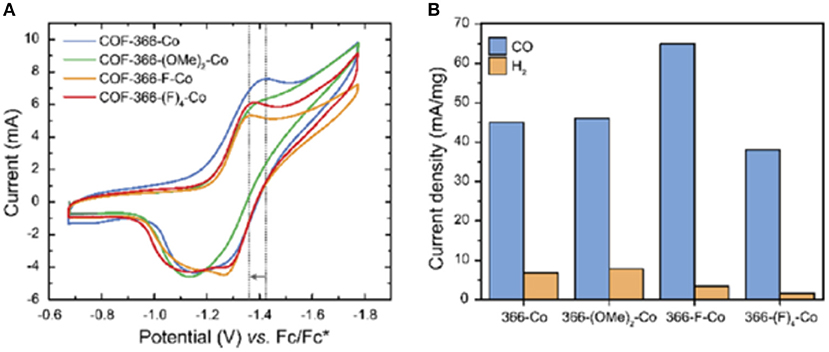
Figure 16. (A) Cyclic voltammograms of four COFs synthesized by Diercks et al. (2018a) in N,N-dimethylformamide. Tetrabutylammonium hexafluorophosphate was used as the electrolyte and (B) the current densities of four COFs synthesized by Diercks et al. (2018a) reported per milligram of cobalt. Measurements were reported by Diercks et al. (2018a) in a 0.5 M aqueous potassium bicarbonate buffer under an applied potential of −0.67 V vs. Reprinted with permission ©2018 The American Chemical Society.
For electrocatalysis, the COF was deposited onto a conductive electrode. Since it has been shown that deposition of a COF onto an electrode as a microcrystalline powder results in minimal electrochemical accessibility (DeBlase et al., 2018) the researches in this study elected to deposit COFs onto the surface of highly ordered pyrolytic graphite (HOPG) so as to increase accessibility of the cobalt centers. These oriented thin films outperformed the traditionally deposited COFs and bulk samples. Electrocatalysis took place in a 0.5 M sodium bicarbonate solution at pH 7.2 and an overpotential of −0.67 V (vs. RHE). Overall higher electron withdrawing character improves the catalytic activity of the COFs. As depicted in Figure 16B COF-366-F-Co has a current density for CO formation of 65 mA mg−1 which is a 20 mA mg−1 increase over COF-366-Co. COF-366-(F)4-Co was an outlier as it had the lowest current density for CO formation but the was the second highest electron withdrawing character. Diercks et al. rationalize this observation by considering the hydrophobicity of the COF-366-(F)4-Co framework which hinders electrolyte availability at the active site (Chen et al., 2013; Deria et al., 2013).
More recent work on cobalt containg COFs for the production of CO consists of three new COFs, Co-Pc-PBBA, Co-PcbPBBA, and Co-Pc-tPBBA; analyzed by Yao et al. (2018). In this computational work the cobalt atoms were held in the center of phthalocyanine linkers. Yao et al. used computational methods to first determine the CO2 storage capacity of the COFs then identified the COFs as promising candidates for the electrochemical reduction of CO2 to CO. The three reticular COFs are linked by boronic acid linkers between the phthalocyanine moieties with the difference being the length of the boronic acid linkers. Through this analysis, the effect of pore size could be investigated. Yao et al. first found that CO2 molecules will be distributed at the corner sites of the COFs at normal pressure. Changing the pore size has little effect on this behavior because the 4 corner sites are equal in all three reticular COFs. This analysis, illustrated in Figure 17, describes the varying performance of the COFs at CO reduction at various pressures. At lower pressures, the COFs with smaller pore size are more economical. COFs with larger pore size can adsorb more CO2 molecules on the surface of the walls at higher pressures while the COFs with smaller pores would begin to hold CO2 in the empty center of their pores. This computational observation dictates that the large-pored COFs would be more efficient at higher pressures.
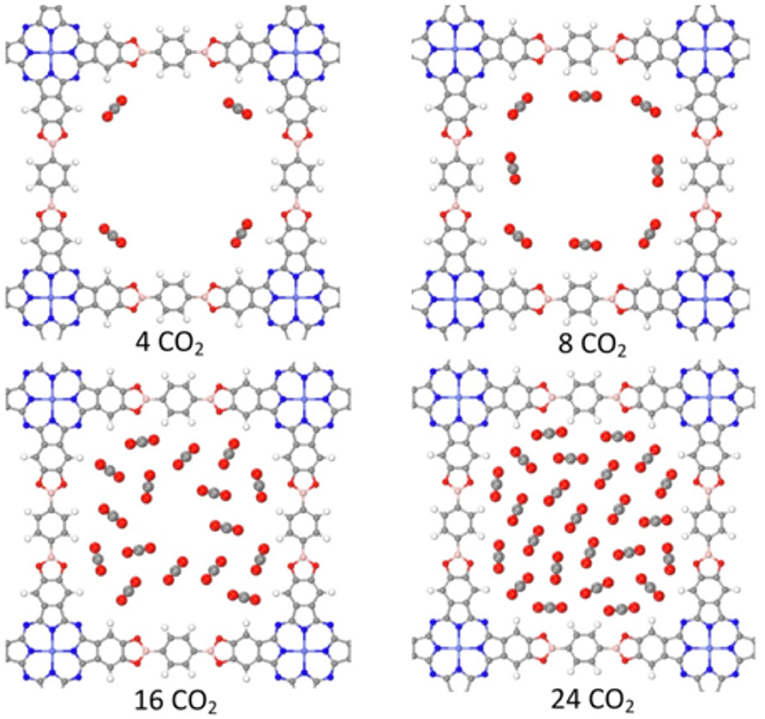
Figure 17. CO2 adsorption into Co-Pc-bPBBA as modeled by Yao et al. (2018). Reprinted with permission copyright 2018 Wiley VCH.
Yao et al. (2018) selected Co-Pc-bPBBA for their computational study. The reduction pathway from CO2 to CO and various other hydrocarbons as elucidated by Yao et al. is depicted in Figure 18 with free energies of the reaction pathways are shown below in Table 2. CO2 reduction begins by the evolution of a carboxyhydroxyl *COOH intermediate (Here the * signifies the adsorbed state); the energy of this *COOH formation is 0.30 eV on Co-Pc-PBBA, this is less than the what has been determined for the Co-porphine (0.46 eV) used in previous works. The *COOH then undergoes electron discharge and proton transfer; followed by loss of the hydroxyl group to form *CO. Yao et al. calculated the adsorption energy (Ead) of CO on Co-Pc-PBBA to be −0.36 eV, which is < -0.26 eV on Co-porphine.
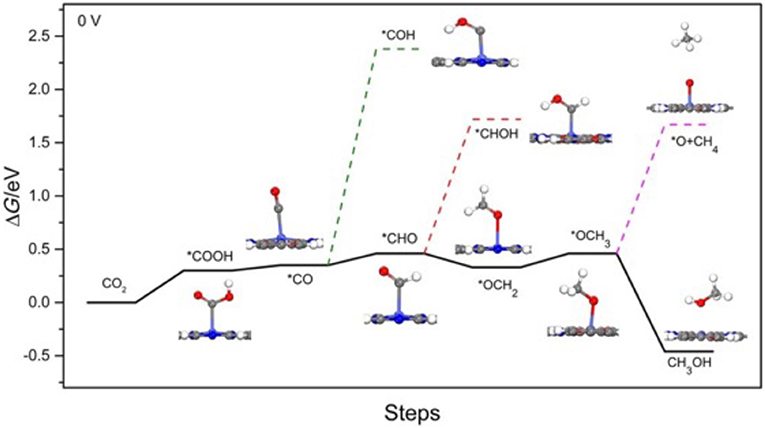
Figure 18. (Solid line) Depiction of thermodynamically favored reaction pathways from CO2 to CO and to CH3OH as calculated by Yao et al. (2018) (Dashed line) depicts the competing pathways. Reprinted with permission ©2018 Wiley VCH.
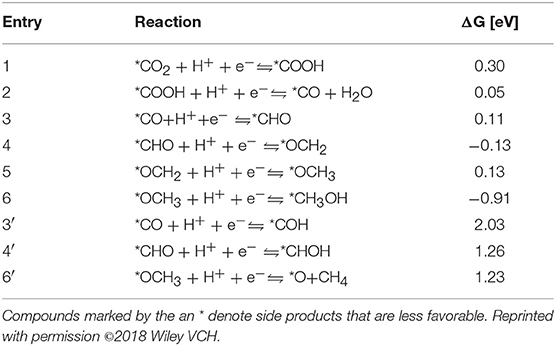
Table 2. The ΔG of CO2 to CO, methane and methanol at U = 0 V vs. the reversible hydrogen electrode (RHE) as determined by Yao et al. (2018).
In 2018 Yang et al. reported the development of a 2D COF photocatalyst with an incorporated Re complex for the photocatalytic reduction of CO2 to CO (Yang et al., 2018). To synthesize the so-named Re-COF, Huang et al. first mixed 2,2-bipyridyl-5,5-dialdehyde (BPDA) and 4,4′,4″-(1,3,5-triazine-2,4,6-triyl) trianiline (TTA) to create the COF scaffold by solvothermal reactions. Upon collection of the COF scaffold, the Re moieties were added by the reaction between bipyridine ligand and Re(CO)3Cl to form Re-COF. The diffuse reflectance UV-vis of Re-COF shows an adsorption band at ~440 nm which is a product of the delocalized intramolecular charge transfer (ICT) band created by the π-conjugation of TTA and BPDA (Pop et al., 2013; Cho et al., 2016; Qian et al., 2017).
The collected gas phase of Re-COF photocatalysis was only 2% H2 and 98% CO showing a 98% selectivity to CO production. The conversion of CO2 to CO by the Re-COF has a TON 48 times greater than homogenous reaction with a Re(bpy)(CO)3Cl catalyst. The mechanism of the CO2 reduction by Re-COF was elucidated by Huang et al. through analysis of diffuse reflectance UV-visible spectra taken during catalysis. The proposed mechanism is shown in Figure 19.
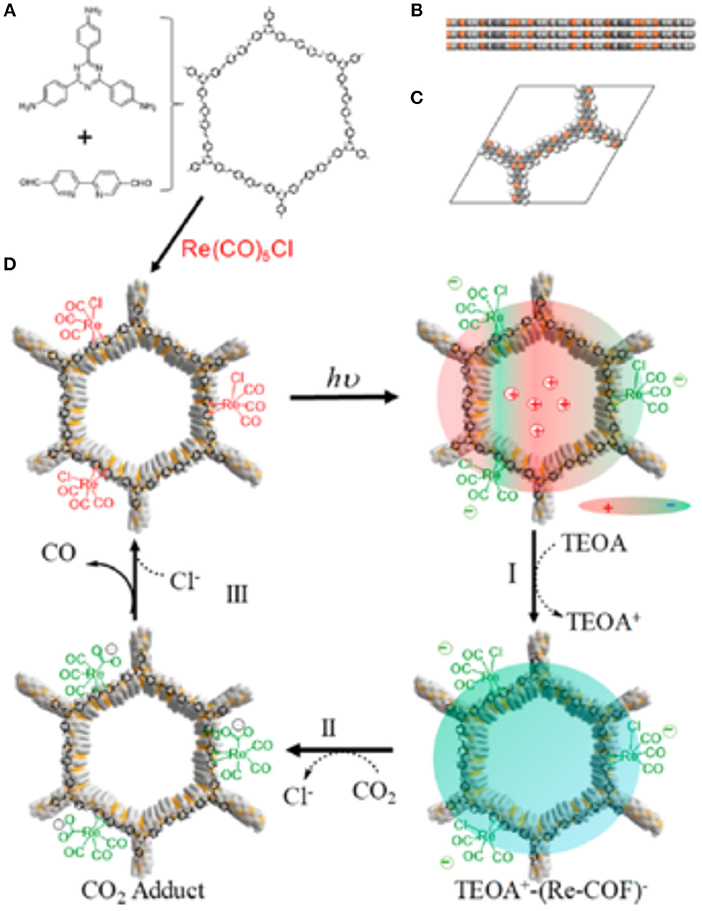
Figure 19. (A) the building blocks and composition of Re-COF (B) View of stacked 2D sheets of Re-COF and (C) the unit cell of Re-COF. (D) Proposed mechanism of CO2 reduction to CO by Re-COF: (Step I) Illumination initiates the catalytic cycle and forms the TEOA+-(COF-Re)− CS state by reducing ICT state via TEOA. (Step II) After 3 h the absorbance from 500 to 800 nm decreases while the absorption at <430 nm and an isosbestic point of 430 nm are observed. (Step III) Consistent CO production and spectral evolution halts. Reprinted with Permission ©2018 The American Chemical Society.
The development of the reduction of CO2 by COFs through the photocatalytic route was also pursued by Fu et al. in their development of the two azine based COFs for the reduction of CO2 to methanol (Fu et al., 2018).
In 2018 work by Liu et al. (2018) used a COF framework to enhance the specificity of silver metal catalyzed reduction of CO2 to CO through the use of COF thin film. Enhancing the selectivity of electrochemical reductions of CO2 by silver is necessary due to the lower potential of the hydrogen evolution reaction (HER). Liu et al. constructed COF-300-AR; a highly stable amine linked COF made by reduction of its imine equivalent (COF-300). The reduction of extended network imine solids to their amine equivalents must overcome a limited diffusion of the reactants and the rigidity of the extended network (Swamy et al., 2010; Liu et al., 2014; Gui et al., 2015). To optimize the reduction of COF-300, Liu et al. studied the reduction of benzylidene aniline to benzyl aniline with NaBH4. Having fine-tuned this procedure Deng et al. quantitively reduced the 3D framework of COF-300 into its amine analog maintaining the crystallinity of the COF. Deposition of COF-300-AR onto the surface of silver improved the selectivity of electrochemical reduction of CO2 to CO from 13 to 53% and from 43 to 80% at −0.70 V and −0.85 V, respectively when compared to just the silver electrode. Addition of the COF thin film also inhibited the HER reaction from 80 to 22% at −0.70 V and 60 to 9% at −0.85V. Concomitantly, silver electrodes coated with a Nafion binder showed no improvement in CO evolution and glassy carbon coated with CO-300-AR showed no CO evolution either. Liu et al. deduced that the increased selectivity for CO production over HER is a result of carbamate formation at the COF-metal interface. The formation of this carbamate intermediate was confirmed through FTIR. Liu et al. summarized the mechanism of the CO reduction into three steps. First, amine functional groups close the surface of the silver electrode attracted CO2 molecules. The second step is the formation of the carbamate close to the surface of the Ag electrode. Close proximity to the silver electrode allows for electron transfer to bypass the COF backbone. In the third step, the carbamate is reduced to CO and OH anions, regenerating the amine moieties of the COF. The third step is facilitated by the aqueous reaction environment.
A comprehensive study of photocatalytic CO2 reduction at the single Ni sites of a COF was recently undertaken by Zhong et al. (2019). The synthesized material, so named Ni-TpBpy is a ketoenamine linked COF resultant from the solvothermal condensation of 1,3,5-triformylphloroglucinol and 5,5′-diamino-2,2′-bipyridin. The resultant COF with honeycomb channels was subsequently treated with Ni(ClO4)2 to make the metalated structure. BET analysis of the COF shows that post-metalation the material retains enough porosity to effectively admit CO2 gas into the reactive channel sites, although the pore volume as determined by the N2 adsorption/desorption measurements decreases from 0.6 cm−1/g prior to metalation to 0.4 cm−1/g. Despite this decrease in pore size Zou et al. discovered that the metalated Ni-TpBpy COF has a higher CO2 adsorption and isosteric heat of adsorption than its unmetallized counterpart. The authors ascribe this observation to the increased Lewis acid-base interactions between the CO2 and Ni sites in the COF. The Ni-TpBpy photocatalyst had an exceptional selectivity toward CO production over H2 or other CO2 reduction products with a 96% CO selectivity over H2.
Reported COFs as Catalysts for the Cycloaddition of CO2 to Epoxides Leading to Organic Carbonates
COFs for the Conversion of Epoxides to Carbonates
The first report of a COF catalyst for the synthesis of cyclic carbonates by CO2 fixation was the 2012 work by Thomas and coworkers in which the researches synthesized three metal-free covalent triazine framework (CTF) cataysts (Roeser et al., 2012). CTFs are a subset of COFs synthesized by the trimerization of dicyanocompounds often at temperatures ≥400 C and with melted zinc chloride solvent (Kuhn et al., 2008; Bojdys et al., 2010). The CTFs prepared by Thomas and coworkers for their 2012 study were crystalline framework (CTF-1), fully amorphous analog of CTF-1, denoted as CTF-1-HSA, and fully amorphous framework (CTF-P-HSA). The synthesis of CTF-1 and the amorphous CTF-1-HSA differ in reaction temperature with CTF-1 being synthesized from 1,4-dicyanobenzene at 400°C and CTF-1-HSA being synthesized from 1,4-dicyanobenzene at 600°C. CTF-P-HSA was synthesized from heating of 2,6-dicyanopyrideine in ZnCl2 at 600°C. Zinc chloride can potentially be catalytic itself so was removed by extensive washing with acidic water.
Thomas and coworkers confirmed the framework with more amorphous structure CTF-P-HSA to be the best choice for the cycloaddition of CO2 to epoxides. The CTFs studied by Thomas and coworkers are highly active under mild conditions and had short reaction times even in the absence of solvent or co-catalyst. The authors explained the high catalytic activity of CTF-P-HSA was due to its amorphous structure resulting in a high surface area, hierarchical porosity, and additional mesopores which facilitated the diffusions to active sites when compared to crystalline frameworks with micropores. Different epoxides including propylene oxide (PO), styrene oxide (SO), cyclohexene oxide (CHO), and epichlorohydrin (CIPO) were used for the cycloaddition of CO2 over the CTF-P-HSA to form cyclic carbonates. The catalysis was carried out at 130°C at times of 4, 8, and 12 h at 6.9 bar of CO2 with high yields (Table 6). In the case of cycloaddition of CO2 over the CTF-P-HSA, it was found that epichlorohydrin is more active than propylene oxide, styrene oxide, and cyclohexene oxide. CTF-P-HSA was recycled 6 times with no loss of activity. Thomas and coworkers also studied the catalytic formation of linear carbonates. Due to thermodynamic limitations, the formation of linear carbonates from CO2 remained as a major issue. One of the synthetic routes starting from CO2 was the one-pot synthesis of dimethyl carbonate from an epoxide, methanol, and CO2. In that route, the cycloaddition to epoxide as well as the transesterification of a cyclic carbonate with methanol were performed in one single batch reaction. The researchers of this work used CTF-P-HSA catalyst for the direct formation of DMC. Between all the epoxides studies by Thomas and coworkers the maximum obtained selectivity of 17.5% was from the propylene oxide reaction under the 20 bar and 160°C.
In 2017 an interesting development was achieved by Buyukcakir et al. in the development of a series of cCTF materials, cCTF-400, CTF-450, and cCTF-500 which consisted of a cationic framework with a Cl− counterion (Buyukcakir et al., 2017). The presence of this anion in the framework allows for the synthesis of cyclic carbonates via fixation of CO2 to an epoxide without the addition of ionic cocatalyst (Table 3 entry 2). cCTF-500 showed an excellent CO2 uptake at low pressure of 133 mg g−1 at 273 K and 80 mg g−1 at 298 K at 1 bar. This makes the charged cCTF-500 framework on par with other CTF's for CO2 adsorption. Synthesis of the charged covalent triazine frameworks (cCTFs) differed only in the temperature of the reaction. Each cCTF was synthesized by placing 1,1′-bis(4-cyanophenyl0-[4,4′-bipyridine]-1,1′-diium dichloride into a sealed glass ampule with ZnCl2 and heating for 48 h at 400, 450, and 500 C to afford the corresponding the cCTF.
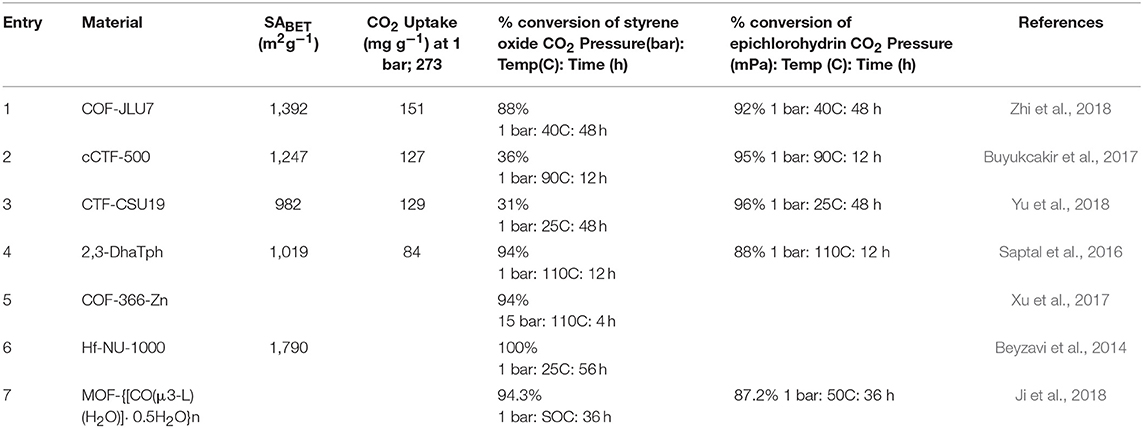
Table 3. Table of COFs and selected MOFs capable of fixation of CO2 to epoxides for the synthesis of cyclic carbonates.
Progress in the development of CTFs for CO2 fixation continued to 2018 when Yu et al. devised a bottom-up approach entailing deliberate linker selection for the synthesis of function-specific CTFs capable of cycloaddition of CO2 to epoxides (Yu et al., 2018). Using precisely selected polymerization conditions, porous crystalline frameworks CTF-CSU1 and CTF-CSU19 with long-range structure were attained. To synthesize the CTFs, Yu et al. heated 3,6-dicyano-9H-carbazole and 3,6-dicyano-9-methylcarbazole in ZnCl2 at 400°C for 48 h with the former producing CTF-CSU1 in 93% yield and the latter producing CTF-CSU19 in 91% yield. These CTFs have an exceptionally high nitrogen content due to the presence of both carbazole and triazine moieties. The high nitrogen content of the of extended polymers is known to be beneficial for the capture and catalytic chemical fixation of CO2 (Talapaneni et al., 2015). Their results showed that in the CO2 cycloaddition, CTF-CSU19 had higher contribution under atmospheric pressure and room temperature (0.1 MPa, 25°C, 48 h) with a TBAB co-catalyst. The elevation in catalytic efficiency was prescribed to the large surface area, pore volume and high nitrogen content. The cycloaddition reaction of CO2 and epoxides having small-sized substituents can be catalyzed using CTF-CSU19 under mild conditions. In addition, CTF-CSU19 can catalyze the fixation of CO2 to epichlorohydrin in high yields although the reacitivty of the larger styrene oxide substrate was more mild (Table 3 entry 3). On the other hand, substrates possessing large substituents like phenyl are relatively slow to react because they cannot easily diffuse into the channels for reactions. The scope of the molecules synthesized by Yu et al. in this study is presented in Table 4. For reusability test, the reaction of epibromohydrin with CO2 catalyzed by CTF-CSUs/TBAB was studied and it was showed that both CTFs can be used for 5 times with only a slight decrease in activity.
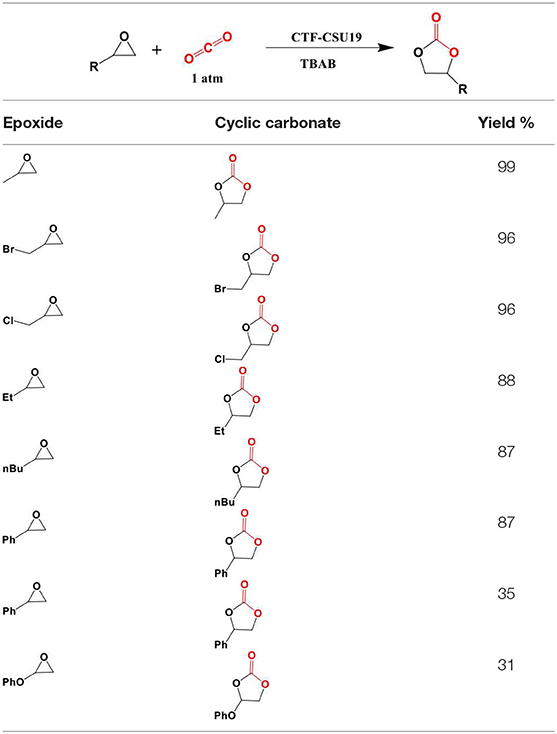
Table 4. Scope of reactions carried out by Yu et al.
In 2016, Saptal et al. synthesized crystalline catechol porphyrin COFs (2,3-DhaTph and 2,3-DmaTph) as a catalyst for the chemical CO2 fixation to epoxides for the synthesis of oxazolidinones and cyclic carbonates (Saptal et al., 2016) 2,3-DmaTph showed remarkable thermal stability and was able to effect CO2 fixation at 100 C (Table 3 entry 4). The molecular structures of COFs 2,3-DhaTph and 2,3-DmaTph are both depicted in Figure 20. The formation of cyclic carbonate under transition metal free and solvent-free conditions was notable.
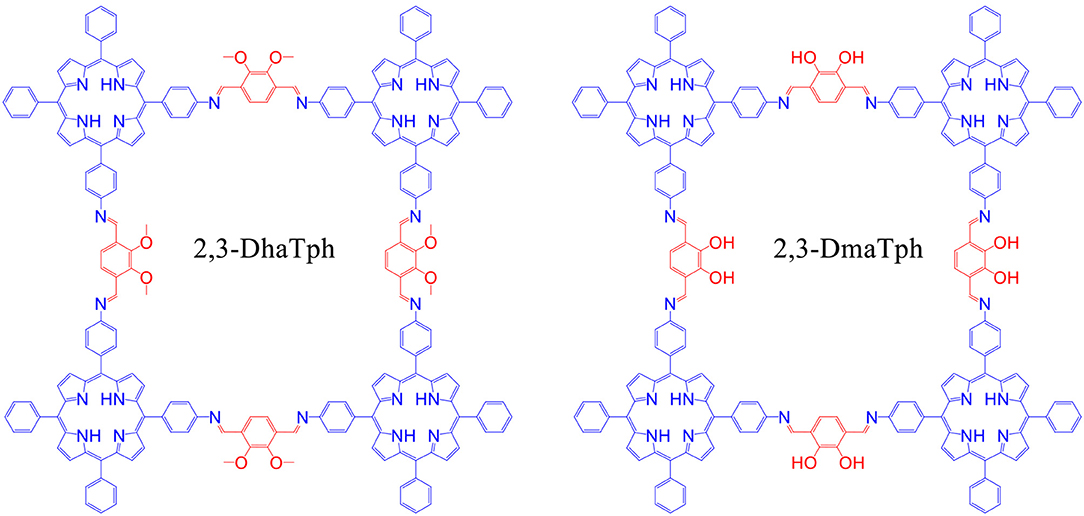
Figure 20. Structure of 2,3-DhaTph and 2,3-DmaTph. Reprinted with Permission ©2016 The Royal Society of Chemistry.
During optimization, Saptal et al. began with the cycloaddition reaction of styrene oxide (SO) in order to synthesize styrene carbonate (SC) as a model reaction. Saptal et al. studied both polar and non-polar solvents and concluded that optimized catalysis could proceed without solvent. Also, Saptal et al. have studied different co-catalysts such as TBAB, TBAI, KI, and NaI, which serve to open the nucleophilic ring of epoxide. Their experiments show that using TBAI as co-catalyst results in a high yield of 94%. Sapatal and coworkers also reported the higher activity of 2,3-DhaTph, the catalyst with hydrogen bond donor capability, comparing to 2,3-DmaTph at 0.1MPa. Furthermore, Saptal et al. investigated the effect of pressure on the yield of the process and found that at high pressure (1MPa), styrene carbonate (SC) concentration was increased. Also, using co-catalyst (TBAI) was very important as a negligible amount of was SC obtained when no co-catalyst was used. Sapatal and coworkers have also found that high surface area of 2,3-DhaTph COF for cycloaddition reaction resulted in a high yield of the process.
Due to the presence of many active sites as well as porphyrin groups and well-oriented hydroxyl functional groups together, 2,3-DhaTph COF showed higher activity compared to 2,3-DmaTph COF for the chemical carbon dioxide fixation. Sapatal et al. state that this feature promoted interactions between active sites of the COF and incoming substrates to effectively transform the product. In the case of catalyst loading, temperature, and time Saptal and coworkers found that the optimum amount of catalyst for the highest conversion of SC is 0.02 mmol, the optimum temperature is 110°C, and optimum time is 12 h. Saptal et al. have also found that the epoxides with aromatic substituents worked better for possessing substituents such as F, Cl, and Br compared to the aliphatic epoxides. The molecules synthesized by Saptal et al. are laid out in Table 5.
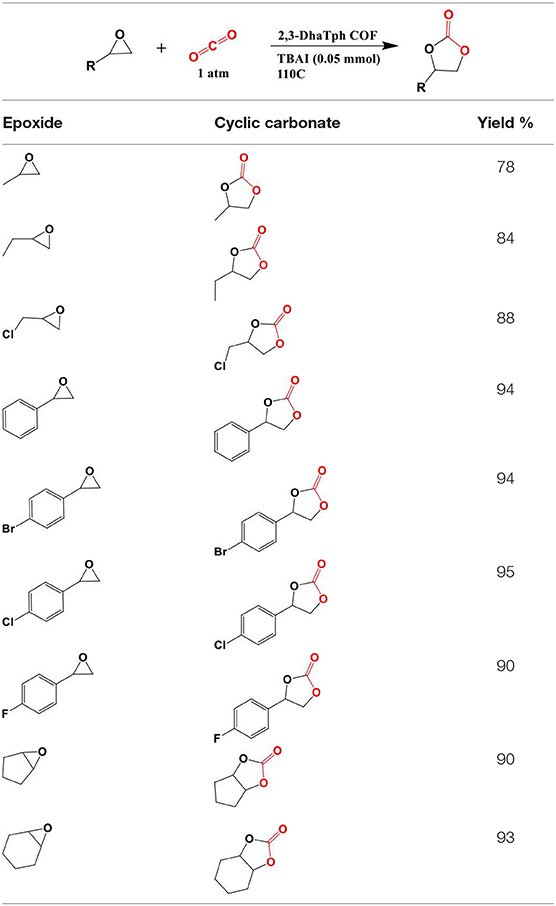
Table 5. Synthesis of various cyclic carbonates using the 2,3-DhaTph COF catalysts.
Using 2,3-DhaTph COF to describe the mechanism, Saptal et al. note that vicinal alcohol groups of the catechol linker increase polarization of the C-O bond through hydrogen bonding. The polar C-O bond is then attacked by the anionic halogen of the co-catalyst facilitating cycloaddition as seen in Figure 21. As porous heterogeneous catalysts can be reused and recycled, cycloaddition reaction of SO to SC was used to investigate the recyclability of the COF catalyst. Sapatal et al. reported that the 2,3-DhaTph COF catalyst could be effectively reused with high catalytic activity.
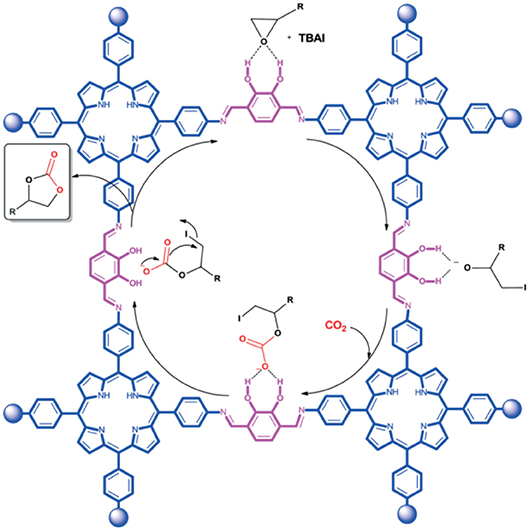
Figure 21. Reaction mechanism proposed by Saptal et al. for the synthesis of cyclic carbonates from carbon dioxide and epoxide catalyzed by 2,3-DhaTph COF as a catalyst system. Reprinted with permission ©The Royal Chemistry Society.
In 2018 Zhi et al. identified two more COFs, so named COF-JLU6 and COF-JLU7; for the efficient synthesis of cyclic carbonates from CO2 (Zhi et al., 2018). Rather than using a triazine linkage to construct the COF framework Zhi et al. used a triazine-based anilines linked with 2,5-dihydroxyterephaladehyde through imine bonds formed during solvothermal synthesis as depicted in Figure 22. In their study, Zhi et al. used the synthesized imine-linked COFs with hydroxy groups as heterogeneous catalyst for cycloaddition of CO2 into cyclic carbonates under mild conditions. Zhi reported the average pore width of 3.1 and 3.3 nm for COF-JLU6 and COF-JLU7, respectively. The cycloaddition of CO2 with epichlorohydrin and tetra-n-butylammonium bromide (TBAB) as co-catalyst was used as a model reaction and resulted in the yield of 86% and 92% for COF-JLU6 and COF-JLU7, respectively, at 40°C and 0.1MPa CO2 pressure. The higher catalytic activity of COF-JLU7 was attributed to its greater CO2 adsorption capacity. The scope of cyclic carbonates synthesized by Zhi et al. is shown in Table 6.
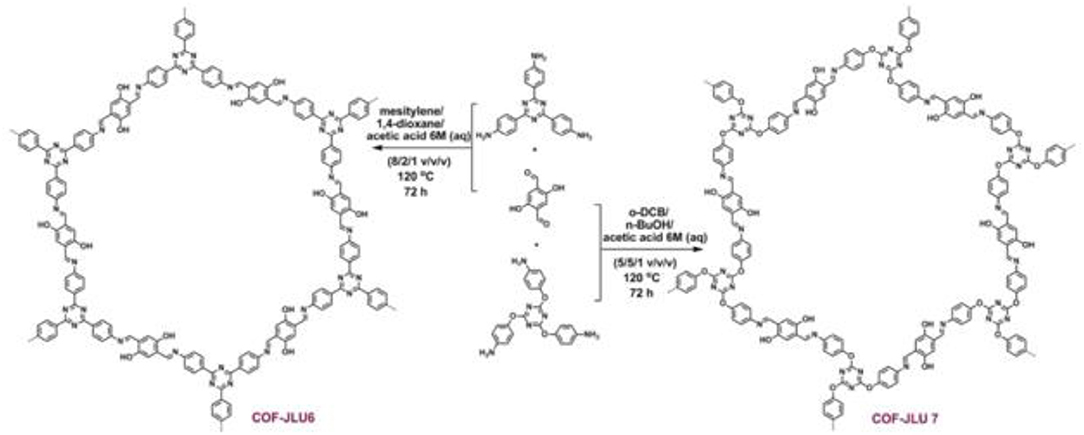
Figure 22. Synthesis and resulting structure of COF-JLU6 and COF-JLU7. Reprinted with permission ©2017 The Royal Society of Chemistry.
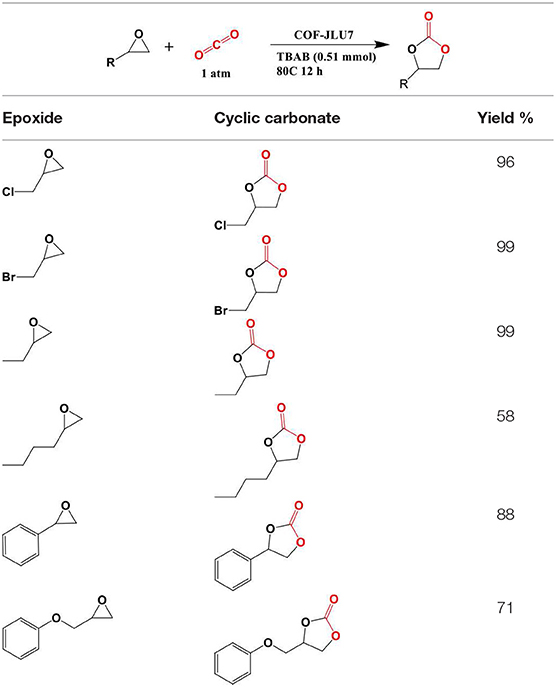
Table 6. Scope of molecules synthesized by Zhi et al. using COF-JLU7 catalyst.
For the recyclability study, Zhi et al. used epichlorohydrin as the substrate and reported that COF-JLU7 could be recycled for five repeating cycles with high catalytic activity and selectivity. The proposed mechanism for CO2 fixation by COF-JLU6 and COF-JLU7 can be seen in Figure 23 and is initiated by hydrogen bonding between the hydroxy groups on the pore walls of the COFs and the C-O bond in the epoxide accompanied by the Br− attacking the epoxide carbon at the less sterically hindered side. This is followed by the formation of an oxyanion stabilized an adjacent hydroxy group from a COF layer. The oxyanion nucleophile then attacks CO2 to form an alkylcarbonate anion which creates the cyclic carbonate upon ring closing while the COF is regenerated.
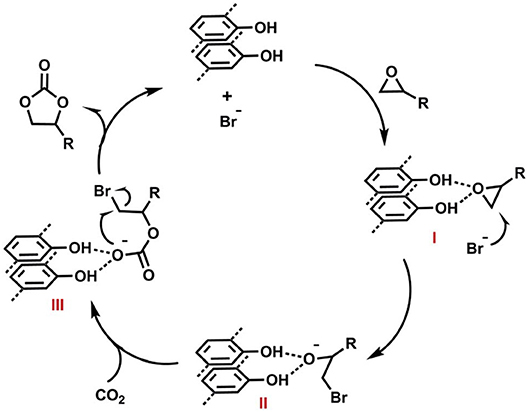
Figure 23. Mechanism proposed by Zhi et al. (2018) for the formation of cyclic carbonates from CO2 and epoxides as catalyzed by COF-JLU6 or COF-JLU7. Reprinted with permission ©2017 The Royal Society of Chemistry.
In 2017, Xu et al. synthesized and employed two dimensional (2D) metalloporphyrin-based covalent organic framework (COF) composites [COF-366-M and COF-SQ-M (M = Zn and Co)] in order to form cyclic carbonates through catalyzing the coupling of CO2 and epoxides (Xu et al., 2017). Under mild conditions, COF-366-Zn was successfully employed as a co-catalyst for the synthesis of 1,2-butylene carbonate and 98.9% selectivity as well as 99.2% activity were obtained. The most pressing advantage of the heterogenous COF catalyst is the comparative ease with which the COF can be recycled when compared to the metalloporphyrin which is soluble in many cyclic carbonates. Xu et al. stated that the metalloporphyrin sites activation improvement was mainly because of the positive role of the microenvironment inside COF-366 frameworks. This could lead to an excellent catalytic performance. Due to their unique 2D COF structure, COF-366-Ms showed high adaptability for various epoxide substrates and solvent choice.
Conclusion
To manufacture a financial incentive for the sequestration of CO2 requires scientific innovation which will make CO2 a valuable commodity. COF materials are one of the most recently developed branches of reticular chemistry with an objectively real potential to motivate the sequestration of emitted CO2. COF materials have been developed that simultaneously achieve the efficient capture of CO2 and the catalytic conversion of CO2 to value-added products including CO and cyclic carbonates. Research that amplifies the ability of COFs to adsorb CO2 will develop COFs with higher internal surface areas. This work will also have to scrutinize the internal chemical environment of COFs to fine-tune the interactions between the COF and CO2. Adjustments toward this end will also include rational pore architecture in regards to the shape, size, and stacking of COF pores. Computational experiments will be essential in predicting the structure and composition of ideal COF materials.
The photocatalytic and electrocatalytic conversion of CO2 to CO as a renewable and sustainable fuel is one avenue in which COFs are showing potential. Present COF electrocatalysts offer high efficiency and selectivity. The stability of COFs in high and low pH can be enhanced through the use of robust chemical bonds. Yet the low solubility of CO2 in water causes inefficient CO2 mass transport and proton-induced catalyst degradation. Another hurdle is the splitting of water to release hydrogen in a competitive off-pathway reduction. However; COF based electrodes provide high selectivity of CO2 reduction reaction against hydrogen evolution reaction; this provides a promising pathway to success for researchers.
Another avenue by which COF materials might serve to incentivize the sequestration of CO2 is the fixation of CO2 to epoxides to form cyclic carbonates. This catalysis has traditionally required the use of high pressures making but advances in COF design now allow for the formation of cyclic carbonates from CO2 and epoxides at atmosphere. The development of COFs for the formation of cyclic carbonates by COFs should focus on enhancing the scope of viable epoxide substrates. The pore size of many COFs could exclude accessibility of catalytic centers to larger epoxides this challenge can be overcome by the use of longer building blocks to develope COFs with larger pores.
Author Contributions
MB directed the project. All authors contributed in writing this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Altarawneh, S., Behera, S., Jenab, P., and El-Kaderi, H. M. (2014). New insights into carbon dioxide interactions with benzimidazole-linked polymers. Chem. Commun. 50, 3571–3574. doi: 10.1039/C3CC45901B
Aresta, M., and Dibenedetto, A. (2007). Utilisation of CO2 as a chemical feedstock: opportunities and challenges. Dalton Trans. 28, 2975–2992. doi: 10.1039/b700658f
Aresta, M., Dibenedetto, A., and Angelini, A. (2014). Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 114, 1709–1742. doi: 10.1021/cr4002758
Ascherl, L., Sick, T., Margraf, J. T., Lapidus, S. H., Calik, M., Hettstedt, C., et al. (2016). Molecular docking sites designed for the generation of highly crystalline covalent organic frameworks. Nat. Chem. 8, 310–316. doi: 10.1038/nchem.2444
Babarao, R., and Jiang, J. (2008). Exceptionally high CO2 storage in covalent-organic frameworks: atomistic simulation study. Energy Environ. Sci. 1, 139–143. doi: 10.1039/b805473h
Ben, T., Ren, H., Ma, S., Cao, D., Lan, J., Jing, X., et al. (2009). Targeted synthesis of a porous aromatic framework with high stability and exceptionally high surface area. Angew. Chem. Int. Ed. 48, 9457–9460. doi: 10.1002/anie.200904637
Beyzavi, M. H., Klet, R. C., Tussupbayev, S., Borycz, J., Vermeulen, N. A., Cramer, C. J., et al. (2014). A hafnium-based metal–organic framework as an efficient and multifunctional catalyst for facile CO2 fixation and regioselective and enantioretentive epoxide activation. J. Am. Chem. Soc. 136, 15861–15864. doi: 10.1021/ja508626n
Beyzavi, M. H., Stephenson, C. J., Liu, Y., Karagiaridi, O., Hupp, J. T., and Farha, O. K. (2015). Metal–organic framework-based catalysts: chemical fixation of CO2 with epoxides leading to cyclic organic carbonates. Front. Energy Res. 2:63. doi: 10.3389/fenrg.2014.00063
Biswal, B. P., Chandra, S., Kandambeth, S., Lukose, B., Heine, T., and Banerjee, R. (2013). Mechanochemical synthesis of chemically Stable isoreticular covalent organic frameworks. J. Am. Chem. Soc. 135, 5328–5331. doi: 10.1021/ja4017842
Bojdys, M. J., Jeromenok, J., Thomas, A., and Antonietti, M. (2010). Rational extension of the family of layered, covalent, triazine-based frameworks with regular porosity. Adv. Mater. 22, 2202–2205. doi: 10.1002/adma.200903436
Boneham, J., Dalton, M. W., Heede, R., Mera, R. J., Allen, M. R., and Frumhoff, P. C. (2017). The rise in global atmospheric CO2, surface temperature, and sea level from emissions traced to major carbon producers. Climate Change 144, 579–590. doi: 10.1007/s10584-017-1978-0
Buyukcakir, O., Je, S. H., Talapaneni, S. N., Kim, D., and Coskun, A. (2017). Charged covalent triazine frameworks for CO2 capture and conversion. ACS Appl. Mater. Interfaces 9, 7209–7216. doi: 10.1021/acsami.6b16769
Côté, A. P., Benin, A. I., Ockwig, N. W., O'Keeffe, M., Matzger, A. J., and Yaghi, O. M. (2005). Porous, crystalline, covalent organic frameworks. Science 310, 1166–1170. doi: 10.1126/science.1120411
Côté, A. P., El-Kaderi, H. M., Furukawa, H., Hunt, J. R., and Yaghi, O. M. (2007). Reticular synthesis of microporous and mesoporous 2D covalent organic frameworks. J. Am. Chem. Soc. 129, 12914–12915. doi: 10.1021/ja0751781
Chandra, S., Kandambeth, S., Biswal, B. P., Lukose, B., Kunjir, S. M., Chaudhary, M., et al. (2013). Chemically stable multilayered covalent organic nanosheets from covalent organic frameworks via mechanical delamination. J. Am. Chem. Soc. 135, 17853–17861. doi: 10.1021/ja408121p
Chen, K., Yang, L., Wu, Z., Chen, C., Jiang, J., and Zhang, G. (2019). A computational study on the tunability of woven covalent organic frameworks for photocatalysis. Phys. Chem. Chem. Phys. 21, 546–553. doi: 10.1039/C8CP04373F
Chen, T.-H., Popov, I., Zenasni, O., Daugulisa, O., and Miljanić, O. Š. (2013). Superhydrophobic perfluorinated metal–organic frameworks. Chem. Commun. 49, 6846–6848. doi: 10.1039/c3cc41564c
Cho, S., Yu, H.-R., Kim, K.-D., Yi, K. B., and Lee, Y.-S. (2012). Surface characteristics and carbon dioxide capture characteristics of oxyfluorinated carbon molecular sieves. Chem. Eng. J. 211, 89–96. doi: 10.1016/j.cej.2012.09.047
Cho, Y.-J., Lee, A.-R., Kim, S.-Y., Cho, M., Han, W.-S., Son, H.-J., et al. (2016). The influence of π-conjugation on competitive pathways: charge transfer or electron transfer in new D–π-A and D–π-Si–π-A dyads. Phys. Chem. Chem. Phys. 18, 22921–22928. doi: 10.1039/C6CP03259A
Chong, J. H., Sauer, M., Patrick, B. O., and MacLachlan, M. J. (2003). Highly stable keto-enamine salicylideneanilines. Org. Lett. 5, 3823–3826. doi: 10.1021/ol0352714
Cordova, K. E., and Yaghi, O. M. (2017). The ‘folklore' and reality of reticular chemistry. Mater. Chem. Front. 1, 1304–1309. doi: 10.1039/C7QM00144D
Dalapati, S., Jin, S., Gao, J., Xu, Y., Nagai, A., and Jiang, D. (2013). An azine-linked covalent organic framework. J. Am. Chem. Soc. 135, 17310–17313. doi: 10.1021/ja4103293
Dash, B. (2018). Carbon dioxide capture using covalent organic frameworks (COFs) type material—a theoretical investigation. J. of Mol. Mod. 24, 120–128. doi: 10.1007/s00894-018-3646-3
Dawson, R., Adamsa, D. J., and Cooper, A. I. (2011b). Chemical tuning of CO2 sorption in robust nanoporous organic polymers. Chem. Sci. 2, 1173–1177. doi: 10.1039/c1sc00100k
Dawson, R., Stöckel, E., Holst, J. R., Adams, D. J., and Cooper, A. I. (2011a). Microporous organic polymers for carbon dioxide capture. Energy Enviorn. Sci. 4, 4239–4245. doi: 10.1039/c1ee01971f
DeBlase, C. R., Silberstein, K. E., Truong, T.-T., Abruña, H. D., and Dichtel, W. R. (2018). β-ketoenamine-linked covalent organic frameworks capable of pseudocapacitive energy storage. J. Am. Chem. Soc. 135, 16821–16824. doi: 10.1021/ja409421d
Deria, P., Mondloch, J. E., Tylianakis, E., Ghosh, P., Bury, W., Snurr, R. Q., et al. (2013). Perfluoroalkane functionalization of NU-1000 via solvent-assisted ligand incorporation: synthesis and CO2 adsorption studies. J. Am. Chem. Soc. 135, 16801–16804. doi: 10.1021/ja408959g
DeSimone, J. M., Maury, E. E., Menceloglu, Y. Z., McClain, J. B., Romack, T. J., and Combes, J. R. (1994). Dispersion polymerizations in supercritical carbon dioxide. Science 265, 356–359. doi: 10.1126/science.265.5170.356
Diercks, C. S., Lin, S., Kornienko, N., Kapustin, E. A., Nichols, E. M., Zhu, C., et al. (2018a). Reticular electronic tuning of porphyrin active sites in covalent organic frameworks for electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 140, 1116–1122. doi: 10.1021/jacs.7b11940
Diercks, C. S., Liu, Y., Cordova, K. E., and Yaghi, O. M. (2018b). The role of reticular chemistry in the design of CO2 reduction catalysts. Nat. Matr. 17, 301–307. doi: 10.1038/s41563-018-0033-5
Diercks, C. S., and Yaghi, O. M. (2017). The atom, the molecule, and the covalent organic framework. Science 355:923. doi: 10.1126/science.aal1585
Ding, S.-Y., Gao, J., Wang, Q., Zhang, Y., Song, W.-G., Su, C.-Y., et al. (2011). Construction of covalent organic framework for catalysis: Pd/COF-LZU1 in suzuki–miyaura coupling reaction. J. Am. Chem. Soc. 133, 19816–19822. doi: 10.1021/ja206846p
Dinga, S.-Y., and Wang, W. (2013). Covalent organic frameworks (COFs): from design to applications. Chem. Soc. Rev. 42, 548–568. doi: 10.1039/C2CS35072F
Du, Y., Yang, H., Whiteley, J. M., Wan, S., Jin, Y., Lee, S. H., et al. (2015). Ionic covalent organic frameworks with spiroborate linkage. Angew. Chem. Int. Ed. 55, 1737–1741. doi: 10.1002/anie.201509014
Dubbeldam, D., Calero, S., Ellis, D. E., and Snurr, R. Q. (2013). RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Chem. Rev. 113, 8261–8323. doi: 10.1080/08927022.2015.1010082
Eastoe, J., Gold, S., and Steytler, D. C. (2006). Surfactants for CO2. Langmuir 22, 9832–9842. doi: 10.1021/la060764d
El-Kaderi, H. M., Hunt, J. R., Mendoza-Cortés, J. L., Côté, A. P., Taylor, R. E., O'Keeffe, M., et al. (2007). Designed synthesis of 3D covalent organic frameworks. Science 316, 286–272. doi: 10.1126/science.1139915
EL-Mahdy, A. F. M., Kuo, C.-H., Alshehri, A., Young, C., Yamauchi, Y., Kim, J., et al. (2018). Strategic design of triphenylamine- and triphenyltriazine-based two-dimensional covalent organic frameworks for CO2 uptake and energy storage. J. Mater. Chem. A. 6, 19532–19541. doi: 10.1039/C8TA04781B
Estevez, L., Barpaga, D., Zheng, J., Sabale, S., Patel, R. L., Zhang, J.-G., et al. (2018). Hierarchically porous carbon materials for CO2 capture: the role of pore structure. Ind. Eng. Chem. Res. 57, 1262–1268. doi: 10.1021/acs.iecr.7b03879
Fang, Q., Zhuang, Z., Gu, S., Kaspar, R. B., Zheng, J., Wang, J., et al. (2014). Designed synthesis of large-pore crystalline polyimide covalent organic frameworks. Nat. Commun. 5, 4503–4511. doi: 10.1038/ncomms5503
Farha, O. K., Spokoyny, A. M., Hauser, B. G., Bae, Y. S., Brown, S. E., Snurr, R. Q., et al. (2009). Synthesis, properties, and gas separation studies of a robust diimide-based microporous organic polymer. Chem. Mater. 21, 3033–3035. doi: 10.1021/cm901280w
Feng, X., Chen, L., Honsho, Y., Saengsawang, O., Liu, L., Wang, L., et al. (2012b). An ambipolar conducting covalent organic framework with self-sorted and periodic electron donor-acceptor ordering. Adv. Mater. 24, 3026–3031. doi: 10.1002/adma.201201185
Feng, X., Dinga, X., and Jiang, D. (2012a). Covalent organic frameworks. Chem. Soc. Rev. 41, 6010–6022. doi: 10.1039/c2cs35157a
Fu, Y., Zhu, X., Huang, L., Zhang, X., Zhang, F., and Zhu, W. (2018). Azine-based covalent organic frameworks as metal-free visible light photocatalysts for CO2 reduction with H2O. Appl. Catal. B Env. 239, 46–51. doi: 10.1016/j.apcatb.2018.08.004
Furukawa, H., Cordova, K. E., O'Keeffe, M., and Yaghi, O. M. (2013). The chemistry and applications of metal-organic frameworks. Science 341:1230444. doi: 10.1126/science.1230444
Furukawa, H., and Yaghi, O. M. (2009). Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J. Am. Chem. Soc. 131, 8875–8883. doi: 10.1021/ja9015765
Gao, Q., Li, X., Ning, G.-H., Xu, H.-S., Liu, C., Tian, B., et al. (2018). Covalent organic framework with frustrated bonding network for enhanced carbon dioxide storage. Chem. Mater. 30, 1762–1768. doi: 10.1021/acs.chemmater.8b00117
Ge, R., Hao, D., Shi, Q., Dong, B., Leng, W., Wang, C., et al. (2016). Target synthesis of an Azo (N = N) based covalent organic framework with high CO2-over-N2 selectivity and benign gas storage capability. J. Chem. Eng. Data 61, 1904–1909. doi: 10.1021/acs.jced.6b00071
Grimme, S. (2011). Density functional theory with London dispersion corrections. WIREs Comput. Mol. Sci. 1, 211–228. doi: 10.1002/wcms.30
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465. doi: 10.1002/jcc.21759
Gui, B., Meng, X., Chen, Y., Tian, J., Liu, G., Shen, C., et al. (2015). Reversible tuning hydroquinone/quinone reaction in metal–organic framework: immobilized molecular switches in solid state. Chem. Mater. 27, 6426–6431. doi: 10.1021/acs.chemmater.5b02648
Haase, F., Gottschling, K., Stegbauer, L., Germann, L. S., Gutzler, R., Duppel, V., et al. (2017). Tuning the stacking behaviour of a 2D covalent organic framework through non-covalent interactions. Mater. Chem. Front. 1, 1354–1361. doi: 10.1039/C6QM00378H
Hao, G. P., Li, W. C., Qian, D., and Lu, A. H. (2010). Rapid synthesis of nitrogen-doped porous carbon monolith for CO2 capture. Adv. Mater. 22, 853–857. doi: 10.1002/adma.200903765
Haszeldine, R. S. (2009). Carbon capture and storage: how green can black be? Science 325, 1647–1652. doi: 10.1126/science.1172246
Hori, Y., Wakebe, H., Tsukamoto, T., and Koga, O. (1994). Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochimica Acta 39, 1833–1839. doi: 10.1016/0013-4686(94)85172-7
Hu, Z., Nalaparaju, A., Peng, Y., Jiang, J., and Zhao, D. (2016). Modulated hydrothermal synthesis of UiO-66(Hf)-type metal–organic frameworks for optimal carbon dioxide separation. Inorg. Chem. 55, 1134–1141. doi: 10.1021/acs.inorgchem.5b02312
Huang, N., Chen, X., Krishna, R., and Jiang, D. (2015a). Two-dimensional covalent organic frameworks for carbon dioxide capture through channel-wall functionalization. Angew. Chem. Int. Ed. 54, 2986–2990. doi: 10.1002/anie.201411262
Huang, N., Krishna, R., and Jiang, D. (2015b). Tailor-made pore surface engineering in covalent organic frameworks: systematic functionalization for performance screening. J. Am. Chem. Soc. 137, 7079–7082. doi: 10.1021/jacs.5b04300
Huang, N., Wang, P., Addicoat, M. A., Heine, T., and Jiang, D. (2017). Ionic covalent organic frameworks: design of a charged interface aligned on 1D channel walls and its unusual electrostatic functions. Angew. Chem. 129, 4982–4986. doi: 10.1002/anie.201611542
Huang, N., Wang, P., and Jiang, D. (2016). Covalent organic frameworks: a materials platform for structural and functional designs. Nat. Rev. Mater. 1:16068. doi: 10.1038/natrevmats.2016.68
Inoue, T., Fujishima, A., Konishi, S., and Honda, K. (1979). Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 277, 637–638. doi: 10.1038/277637a0
James, C., Fisher, I., Siriwardane, R. V., Robert, W., and Stevens, J. (2011). Zeolite-based process for CO2 capture from high-pressure, moderate-temperature gas streams. Ind. Eng. Chem. Res. 50, 13962–13968. doi: 10.1021/ie201666g
Ji, X.-H., Zhu, N.-N., Ma, J.-G., and Chenga, P. (2018). Conversion of CO2 into cyclic carbonates by a Co(II) metal–organic framework and the improvement of catalytic activity via nanocrystallization. Dalton Trans. 47, 1768–1771. doi: 10.1039/C7DT04882C
Jiang, D., Chen, X., Geng, K., Liu, R., Tan, K. T., Gong, Y., et al. (2019). Covalent organic frameworks: chemical approaches to designer structures and built-in functions. Angew. Chem. Int. Ed. doi: 10.1002/ange.201904291
Jiang, J., and Sandler, S. I. (2003). Monte carlo simulation of O2 and N2 mixture adsorption in nanoporous carbon (C168 Schwarzite). Langmuir 19, 5936–5941. doi: 10.1021/la027030v
Jiang, J., and Sandler, S. I. (2006). Shape versus inverse-shape selective adsorption of alkane isomers in carbon nanotubes. J. Chem. Phys. 124:024717. doi: 10.1063/1.2140703
Jin, Y., Hu, Y., and Zhang, W. (2017). Tessellated multiporous two-dimensional covalent organic frameworks. Nat. Rev. Chem. 1:0056. doi: 10.1038/s41570-017-0056
Jin, Y., Voss, B. A., Jin, A., Long, H., Noble, R. D., and Zhang, W. (2011). Highly CO2-selective organic molecular cages: what determines the CO2 selectivity. J. Am. Chem. Soc. 133, 6650–6658. doi: 10.1021/ja110846c
Kahveci, Z., Islamoglu, T., Shar, G. A., Ding, R., and El-Kaderi, H. M. (2013). Targeted synthesis of a mesoporous triptycene-derived covalent organic framework. CrystEngComm 15, 1524–1527. doi: 10.1039/C2CE26487K
Kandambeth, S., Mallick, A., Lukose, B., Mane, M. V., Heine, T., and Banerjee, R. (2012). Construction of crystalline 2D covalent organic frameworks with remarkable chemical (Acid/Base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 134, 19524–19527. doi: 10.1021/ja308278w
Koo, B. T., Dichtelb, W. R., and Clancy, P. (2012). A classification scheme for the stacking of two-dimensional boronate ester-linked covalent organic frameworks. J. Mater. Chem. 22, 17460–17469. doi: 10.1039/c2jm32009f
Kortunov, P. V., Siskin, M., Paccagnini, M., and Thomann, H. (2016). CO2 reaction mechanisms with hindered alkanolamines: control and promotion of reaction pathways. Energy Fuels 30, 1223–1236. doi: 10.1021/acs.energyfuels.5b02582
Kresse, G., and Furthmüller, J. (1996). Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 54, 11169–11186. doi: 10.1103/PhysRevB.54.11169
Kuhn, P., Antonietti, M., and Thomas, A. (2008). Porous, covalent triazine-based frameworks prepared by ionothermal synthesis. Angew. Chem. Int. Ed. 47, 3450–3453. doi: 10.1002/anie.200705710
Lanni, L. M., Tilford, R. W., Bharathy, M., and Lavigne, J. J. (2011). Enhanced hydrolytic stability of self-assembling alkylated two-dimensional covalent organic frameworks. J. Am. Chem. Soc. 133, 13975–13983. doi: 10.1021/ja203807h
Li, B., Wen, H.-M., Zhou, W., and Chen, B. (2014). Porous metal–organic frameworks for gas storage and separation: what, how, and why? J. Phys. Chem. Lett. 5, 3468–3479. doi: 10.1021/jz501586e
Li, H., Li, H., Dai, Q., Li, H., and Brédas, J.-L. (2018). Hydrolytic stability of boronate ester-linked covalent organic frameworks. Adv. Theory Simulations 1:1700015. doi: 10.1002/adts.201700015
Li, Z., Feng, X., Zou, Y., Zhang, Y., Xia, H., Liu, X., et al. (2014). A 2D azine-linked covalent organic framework forgas storage applications. Chem. Commun. 50, 13825–13828. doi: 10.1039/C4CC05665E
Li, Z., Zhi, Y., Feng, X., Ding, X., Zou, Y., Liu, X., et al. (2015). An azine-linked covalent organic framework: synthesis, characterization and efficient gas storage. Chem. Eur. J. 21, 12709–12084. doi: 10.1002/chem.201501206
Lin, R.-B., and Chen, B. (2018). Reducing CO2 with stable covalent organic frameworks. Joule 2, 1028–1038. doi: 10.1016/j.joule.2018.05.017
Lin, S., Diercks, C. S., Zhang, Y.-B., Kornienko, N., Nichols, E. M., Zhao, Y., et al. (2015). Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208–1213. doi: 10.1126/science.aac8343
Liu, H., Chu, J., Yin, Z., Cai, X., Zhuang, L., and Deng, H. (2018). Covalent organic frameworks linked by amine bonding for concerted electrochemical reduction of CO2. Chem 4, 1–14. doi: 10.1016/j.chempr.2018.05.003
Liu, M., Little, M. A., Jelfs, K. E., Jones, J. T., Schmidtmann, M., Chong, S. Y., et al. (2014). Acid- and base-stable porous organic cages: shape persistence and pH stability via post-synthetic “Tying” of a flexible amine cage. J. Am. Chem. Soc. 136, 7583–7586. doi: 10.1021/ja503223j
Liu, Y., Ma, Y., Zhao, Y., Sun, X., Gándara, F., Furukawa, H., et al. (2016). Weaving of organic threads into a crystalline covalent organic framework. Science 351, 365–369. doi: 10.1126/science.aad4011
Lohse, M. S., and Bein, T. (2018). Covalent organic frameworks: structures, synthesis, and applications. Adv. Func. Mater. 28:1705553. doi: 10.1002/adfm.201705553
Lu, A.-H., and Hao, G.-P. (2013). Porous materials for carbon dioxide capture. Annu. Rep. Prog. Chem., Sect. A. Inorg. Chem. 484, 484–503. doi: 10.1039/c3ic90003g
Lu, X., Jin, D., Wei, S., Wang, Z., An, C., and Guo, W. (2015). Strategies to enhance CO2 capture and separation. J. Mater. Chem. A. 3, 12118–12131. doi: 10.1039/C4TA06829G
Ma, H., Liu, B., Li, B., Zhang, L., Li, H. Y, Tan, H.-Q., Zang, H.-Y., et al. (2016). Cationic covalent organic frameworks: a simple platform of anionic exchange for porosity tuning and proton conduction. J. Am. Chem. Soc. 138, 5897–5903. doi: 10.1021/jacs.5b13490
Martín, C. F., Stöckel, E., Clowes, R., Adams, D. J., Cooper, A. I., Pis, J. J., et al. (2011). Hypercrosslinked organic polymer networks as potential adsorbents for pre-combustion CO2 capture. J. Mater. Chem. 21, 5475–5483. doi: 10.1039/c0jm03534c
Mendoza-Cortes, J. L., Goddard, W. A. III., Furukawa, H., and Yaghi, O. M. (2012). A covalent organic framework that exceeds the DOE 2015 volumetric target for H2 uptake at 298 K. J. Phys. Chem. Lett. 18, 2671–2675. doi: 10.1021/jz301000m
Morris, R. E., and Wheatley, P. S. (2008). Gas storage in nanoporous materials. Angew. Chem. Int. Ed. 47, 4966–4981. doi: 10.1002/anie.200703934
Myers, A. L., and Prausnitz, J. M. (1965). Thermodynamics of mixed-gas adsorption. AICHE 11, 121–127. doi: 10.1002/aic.690110125
Pang, Z.-F., Xu, S.-Q., Zhou, T.-Y., Liang, R.-R., Zhan, T.-G., and Zhao, X. (2016). Construction of covalent organic frameworks bearing three different kinds of pores through the heterostructural mixed linker strategy. J. Am. Chem. Soc. 138, 4710–4713. doi: 10.1021/jacs.6b01244
Patel, H. A., Je, S. H., Park, J., Chen, D. P., Jung, Y., Yavuz, C. T., et al. (2013). Unprecedented high-temperature CO2 selectivity in N2-phobic nanoporous covalent organic polymers. Nat. Comm. 4, 1357–1364. doi: 10.1038/ncomms2359
Pop, F., Riobé, F., Seifert, S., Cauchy, T., Ding, J., Dupont, N., et al. (2013). Tetrathiafulvalene-1,3,5-triazines as (Multi)donor–acceptor systems with tunable charge transfer: structural, photophysical, and theoretical investigations. Inorg. Chem. 52, 5023–5034. doi: 10.1021/ic3027336
Qian, H.-L., Dai, C., Yang, C.-X., and Yan, X.-P. (2017). High-crystallinity covalent organic framework with dual fluorescence emissions and its ratiometric sensing application. ACS Appl. Mater. Interfaces 9, 24999–25005. doi: 10.1021/acsami.7b08060
Qiao, J., Liu, Y., Hong, F., and Zhang, J. (2014). A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675. doi: 10.1039/C3CS60323G
Rabbani, M. G., Sekizkardes, A. K., Kahveci, Z., Reich, T. E., Ding, R., and El-Kaderi, H. M. (2013). A 2D mesoporous imine-linked covalent organic framework for high pressure gas storage applications. Chem. Eur. J. 19, 3324–3328. doi: 10.1002/chem.201203753
Reid, C. R., O'koy, I. P., and Thomas, K. M. (1998). Adsorption of gases on carbon molecular sieves used for air separation. Spherical adsorptives as probes for kinetic selectivity. Langmuir 14, 2415–2425. doi: 10.1021/la9709296
Rochelle, G. T. (2009). Amine scrubbing for CO2 capture. Science 325, 1652–1654. doi: 10.1126/science.1176731
Roeser, J., Kailasam, K., and Thomas, A. (2012). Covalent triazine frameworks as heterogeneous catalysts for the synthesis of cyclic and linear carbonates from carbon dioxide and epoxides. ChemSusChem 5, 1793–1799. doi: 10.1002/cssc.201200091
Santurri, P., Robbins, F., and Stubbings, R. (2003). 4,4′-Diaminoazobenzene. Organic Syntheses 40:18. doi: 10.1002/0471264180.os040.07
Saptal, V., Shinde, D. B., Banerjee, R., and Bhanage, B. M. (2016). State-of-the-art catechol porphyrin COF catalyst for chemical fixation of carbon dioxide via cyclic carbonates and oxazolidinones. Catal. Sci. Technol. 6, 6152–6158. doi: 10.1039/C6CY00362A
Sevilla, M., Valle-Vigón, P., and Fuertes, A. B. (2011). N-doped polypyrrole-based porous carbons for CO2 capture. Adv. Mater. 21, 2781–2787. doi: 10.1002/adfm.201100291
Sharma, A., Malani, A., Medhekar, N. V., and Babarao, R. (2017). CO2 adsorption and separation in covalent organic frameworks with interlayer slipping. CrystEngComm 19, 6950–6963. doi: 10.1039/C7CE01647F
Shen, C., Baoa, Y., and Wang, Z. (2013). Tetraphenyladamantane-based microporous polyimide for adsorption of carbon dioxide, hydrogen, organic and water vapors. Chem. Commun. 49, 3321–3323. doi: 10.1039/c3cc41012a
Shi, H., Chen, G., Zhang, C., and Zou, Z. (2014). Polymeric g-C3N4 coupled with NaNbO3 nanowires toward enhanced photocatalytic reduction of CO2 into renewable fuel. ACS Catal. 4, 3637–3643. doi: 10.1021/cs500848f
Siriwardane, R. V., Shen, M.-S., and Fisher, E. P. (2005). Adsorption of CO2 on zeolites at moderate temperatures. Energy Fuels 19, 1153–1159. doi: 10.1021/ef040059h
Spitler, E. L., Giovino, M. R., Whitea, S. L., and Dichtel, W. R. (2011). A mechanistic study of Lewis acid-catalyzed covalent organic framework formation. Chem. Sci. 2, 1588–1593. doi: 10.1039/C1SC00260K
Stock, N., and Biswas, S. (2012). Synthesis of metal-organic frameworks (MOFs): routes to various MOF topologies, morphologies, and composites. Chem. Rev. 122, 933–969. doi: 10.1021/cr200304e
Sun, L.-B., Kang, Y.-H., Shi, Y.-Q., Jiang, Y., and Liu, X.-Q. (2015). Highly selective capture of the greenhouse gas CO2 in polymers. ACS Sust Chem. Eng. 3, 3077–3085. doi: 10.1021/acssuschemeng.5b00544
Sun, S., and Liang, S. (2017). Recent advances in functional mesoporous graphitic carbon nitride (mpg-C3N4) polymers. Nanoscale 9, 10544–10578. doi: 10.1039/C7NR03656F
Swamy, S. I., Bacsa, J., Jones, J. T., Stylianou, K. C., Steiner, A., Ritchie, L. K., et al. (2010). A metal–organic framework with a covalently prefabricated porous organic linker. J. Am. Chem. Soc. 132, 12773–12775. doi: 10.1021/ja104083y
Talapaneni, S. N., Buyukcakir, O., Je, S. H., Srinivasan, S., Seo, Y., Polychronopoulou, K., et al. (2015). Nanoporous polymers incorporating sterically confined N-heterocyclic carbenes for simultaneous CO2 capture and conversion at ambient pressure. Chem. Mater. 27, 6818–6828. doi: 10.1021/acs.chemmater.5b03104
Tilford, R. W III., Mugavero, S. J. III., Pellechia, P. J., and Lavigne, J. J. (2008). Tailoring microporosity in covalent organic frameworks. Adv. Mater. 20, 2741–2746. doi: 10.1002/adma.200800030
Tong, M., Yang, Q., Xiao, Y., and Zhong, C. (2014). Revealing the structure–property relationship of covalent organic frameworks for CO2 capture from post combustion gas: a multi-scale computational study. Phys. Chem. Chem. Phys. 16, 15189–15198. doi: 10.1039/C4CP02047B
Tripati, A. K., Roberts, C. D., and Eagle, R. A. (2009). Coupling of CO2 and ice sheet stability over major climate transitions of the last 20 million years. Science 326, 1394–1397. doi: 10.1126/science.1178296
Uribe-Romo, F. J., Doonan, C. J., Furukawa, H., Oisaki, K., and Yaghi, O. M. (2011). Crystalline covalent organic frameworks with hydrazone linkages. J. Am. Chem. Soc. 133, 11478–11481. doi: 10.1021/ja204728y
Uribe-Romo, F. J., Hunt, J. R., Furukawa, H., Klöck, C., O'Keeffe, M., and Yaghi, O. M. (2009). A crystalline imine-linked 3-D porous covalent organic framework. J. Am. Chem. Soc. 131, 4570–4571. doi: 10.1021/ja8096256
Vogiatzis, K. D., Mavrandonakis, A., Klopper, W., and Froudakis, G. E. (2009). Ab initio study of the interactions between CO2 and N-containing organic heterocycles. ChemPhysChem 10, 374–383. doi: 10.1002/cphc.200800583
Wan, S., Gándara, F., Asano, A., Furukawa, H., Saeki, A., Dey, S. K., et al. (2011). Covalent organic frameworks with high charge carrier mobility. Chem. Mater. 23, 4094–4097. doi: 10.1021/cm201140r
Wang, M., Wei, S., Wu, Z., Zhou, S., Wang, Z., Wang, J., et al. (2018). Alkyl amine functionalized triphenylamine-based covalent organic frameworks for high-efficiency CO2 capture and separation over N2. Mater. Lett. 230, 28–31. doi: 10.1016/j.matlet.2018.07.071
Wang, S., and Wang, X. (2016). Imidazolium ionic liquids, imidazolylidene heterocyclic carbenes, and zeolitic imidazolate frameworks for CO2 capture and photochemical reduction. Angew. Chem. Int. Ed. 55, 2308–2320. doi: 10.1002/anie.201507145
Wang, S., Yao, W., Lin, J., Ding, Z., and Wang, X. (2013). Cobalt imidazolate metal–organic frameworks photosplit CO2 under mild reaction conditions. Angew. Chem. Int. Ed. 23, 1034–1038. doi: 10.1002/anie.201309426
Wei, H., Chai, S., Hu, N., Yang, Z., Wei, L., and Wangb, L. (2015). Microwave-assisted solvothermal synthesis of a crystalline two-dimensional covalent organic framework with high CO2 capacity. Chem. Commun. 51, 12178–12181. doi: 10.1039/C.5C.C.04680G.
White, J. L., Baruch, M. F III, Pander, J. E III., Hu, Y., Fortmeyer, I. C., Park, J. E., Zhang, T., et al. (2015). Light-driven heterogeneous reduction of carbon dioxide: photocatalysts and photoelectrodes. Chem. Rev. 115, 12888–12935. doi: 10.1021/acs.chemrev.5b00370
Wu, D., Wang, C., Liu, B., Liu, D., Yang, Q., and Zhong, C. (2012a). Large-scale computational screening of metal-organic frameworks for CH4/H2 separation. AIChE 58, 2078–2084. doi: 10.1002/aic.12744
Wu, D., Yang, Q., Zhong, C., Liu, D., Huang, H., Zhang, W., et al. (2012b). Revealing the structure–property relationships of metal–organic frameworks for CO2 capture from flue gas. Langmuir 28, 12094–12099. doi: 10.1021/la302223m
Xiong, J., Yang, R. X., Xie, Y., Huang, N. Y., Zou, K., and Deng, W. Q. (2017). Formation of cyclic carbonates from CO2 and epoxides catalyzed by a cobalt-coordinated conjugated microporous polymer. ChemCatChem 9, 2584–2587. doi: 10.1002/cctc.201700386
Xu, K., Dai, Y., Ye, B., and Wang, H. (2017). Two dimensional covalent organic framework materials for chemical fixation of carbon dioxide: excellent repeatability and high selectivity. Dalton Trans. 46, 10780–10785. doi: 10.1039/C7DT02527K
Yang, S., Hu, W., Zhang, X., He, P., Pattengale, B., Liu, C., et al. (2018). 2D covalent organic frameworks as intrinsic photocatalysts for visible light-driven CO2 reduction. J. Am. Chem. Soc. 140, 14614–14619. doi: 10.1021/jacs.8b09705
Yao, C.-L., Li, J.-C., Gao, W., and Jiang, Q. (2018). An integrated design with new metal-functionalized covalent organic frameworks for the effective electroreduction of CO2. Chem. Eur. J. 24, 11051–11058. doi: 10.1002/chem.201800363
Yu, H.-R., Cho, S., Bai, B. C., Yi, K. B., and Lee, Y.-S. (2012). Effects of fluorination on carbon molecular sieves for CH4/CO2 gas separation behavior. Int. J. Greenhouse Gas Control 10, 278–284. doi: 10.1016/j.ijggc.2012.06.013
Yu, W., Gu, S., Fu, Y., Xiong, S., Pan, C., Liu, Y., et al. (2018). Carbazole-decorated covalent triazine frameworks: novel non-metalcatalysts for carbon dioxide fixation and oxygen reduction reaction. J. Catal. 362, 1–9. doi: 10.1016/j.jcat.2018.03.021
Zeng, Y., Zou, R., and Zhao, Y. (2016). Covalent organic frameworks for CO2 capture Adv. Mater. 28, 2855–2873. doi: 10.1002/adma.201505004
Zhai, L., Huang, N., Xu, H., Chena, Q., and Jiang, D. (2017). A backbone design principle for covalent organic frameworks: the impact of weakly interacting units on CO adsorption. Chem. Commun. 53, 4242–4245. doi: 10.1039/C7CC01921A
Zhang, B., Wei, M., Mao, H., Pei, X., Alshmimri, S. A., Reimer, J. A., et al. (2018). Crystalline dioxin-linked covalent organic frameworks from irreversible reactions. J. Am. Chem. Soc. 140, 12715–12719. doi: 10.1021/jacs.8b08374
Zhao, F., Liu, H., Mathe, S. D. R., Dong, A., and Zhang, J. (2018). Covalent organic frameworks: from materials design to biomedical application. Nanomaterials 8:15. doi: 10.3390/nano8010015
Zhao, W., Xia, L., and Liu, X. (2018). Covalent organic frameworks (COFs): perspectives of industrialization. CrystEngComm 20, 1613–1634. doi: 10.1039/C7CE02079A
Zhao, Y., Yao, K. X., Teng, B., Zhang, T., and Han, Y. (2013). A perfluorinated covalent triazine-based framework for highly selective and water–tolerant CO2 capture. Energy Enviorn. Sci. 6, 3684–3692. doi: 10.1039/c3ee42548g
Zhao, Y., Zhao, L., Yao, K. X., Yang, Y., Zhangc, Q., and Han, Y. (2012). Novel porous carbon materials with ultrahigh nitrogen contents for selective CO2 capture. J. Mater. Chem. 22, 19726–19731. doi: 10.1039/c2jm33091a
Zhi, Y., Shao, P., Feng, X., Xi, H., Zhang, Y., Shi, Z., et al. (2018). Covalent organic frameworks: efficient, metal-free, heterogeneous organocatalysts for chemical fixation of CO2 under mild conditions. J. Mater. Chem. A. 6, 374–382. doi: 10.1039/C7TA08629F
Zhong, W., Sa, R., Li, L., He, Y., Li, L., Bi, J., et al. (2019). A covalent organic framework bearing single Ni sites as a synergistic photocatalyst for selective photoreduction of CO2 to CO. J. Am. Chem. Soc. 141, 7615–7621. doi: 10.1021/jacs.9b02997
Zhou, S., Guo, C., Wu, Z., Wang, M., Wang, Z., Wei, S., et al. (2017). Edge-functionalized nanoporous carbons for high adsorption capacity and selectivity of CO2 over N2. Appl. Surf. Sci. 410, 259–266. doi: 10.1016/j.apsusc.2017.03.136
Zhou, T.-Y., Xu, S.-Q., Wen, Q., Pang, Z.-F., and Zhao, X. (2014). One-step construction of two different kinds of pores in a 2D covalent organic framework. J. Am. Chem. Soc. 136, 15885–15888. doi: 10.1021/ja5092936
Zhu, L., and Zhang, Y.-B. (2017). Crystallization of covalent organic frameworks forgas storage applications. Molecules 22:1149. doi: 10.3390/molecules22071149
Keywords: covalent organic framework, carbon dioxide fixation, carbon monoxide, cyclic carbonates, adsorption
Citation: Ozdemir J, Mosleh I, Abolhassani M, Greenlee LF, Beitle RR Jr and Beyzavi MH (2019) Covalent Organic Frameworks for the Capture, Fixation, or Reduction of CO2. Front. Energy Res. 7:77. doi: 10.3389/fenrg.2019.00077
Received: 16 March 2019; Accepted: 24 July 2019;
Published: 13 September 2019.
Edited by:
Youngjune Jason Park, Gwangju Institute of Science and Technology, South KoreaReviewed by:
Pradip Pachfule, Technische Universität Berlin, GermanyXiong Chen, Fuzhou University, China
Copyright © 2019 Ozdemir, Mosleh, Abolhassani, Greenlee, Beitle and Beyzavi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Hassan Beyzavi, YmV5emF2aUB1YXJrLmVkdQ==
†These authors have contributed equally to this work