 Brandon Johnston
Brandon Johnston Hakeem Henry
Hakeem Henry Nam Kim
Nam Kim Sang Bok Lee*
Sang Bok Lee*- Department of Chemistry and Biochemistry, University of Maryland, College Park, MD, United States
As lithium-ion batteries approach their theoretical limits for energy density, magnesium-ion batteries are emerging as a promising next-generation energy storage technology. However, progress in magnesium-ion battery research has been stymied by a lack of available high capacity cathode materials that can reversibly insert magnesium ions. Vanadium Oxide (V2O5) has emerged as one of the more promising candidate cathode materials, owing to its high theoretical capacity, facile synthesis methods, and relatively high operating voltage. This review focuses on the outlook of hydrated V2O5 structures as a high capacity cathode material for magnesium-ion batteries. In general, V2O5 structures exhibit poor experimental capacity for magnesium-ion insertion due to sluggish magnesium-ion insertion kinetics and poor electronic conductivity. However, several decades ago, it was discovered that the addition of water to organic electrolytes significantly improves magnesium-ion insertion into V2O5. This review clarifies the various mechanisms that have been used to explain this observation, from charge shielding to proton insertion, and offers an alternative explanation that examines the possible role of structural hydroxyl groups on the V2O5 surface. While the mechanism still needs to be further studied, this discovery fueled new research into V2O5 electrodes that incorporate water directly as a structural element. The most promising of these hydrated V2O5 materials, many of which incorporate conductive additives, nanostructured architectures, and thin film morphologies, are discussed. Ultimately, however, these hydrated V2O5 structures still face a significant barrier to potential applications in magnesium-ion batteries. During full cell electrochemical cycling, these hydrated structures tend to leach water into the electrolyte and passivate the surface of the magnesium anode, leading to poor cycle life and low capacity retention. Recently, some promising strides have been made to remedy this problem, including the use of artificial solid electrolyte interphase layers as an anode protection scheme, but a call to action for more anode protection strategies that are compatible with trace water and magnesium metal is required.
Introduction
For the past several decades, lithium-ion batteries have dominated the area of energy storage devices. As the current state of the art technology, they have been used extensively in a variety of applications, including portable electronics and electric vehicles. However, as energy demands continue to increase, lithium-ion batteries are soon expected to reach their theoretical limit. Lithium metal anodes can be used to increase the energy density. However, lithium metal anodes tend to form dendrites during successive charge and discharge cycles (Bieker et al., 2015). These, and other safety issues, have made it necessary to replace lithium metal anodes with low capacity graphite anodes in nearly all commercialized lithium-ion batteries. Thus, in recent years, significant research efforts have been made to develop new battery chemistries. In particular, magnesium-ion batteries have emerged as a promising alternative to lithium-ion batteries because of their high energy density, low cost, and improved environmental safety (Novák et al., 1999; Deog, 2013). The divalent nature of the Mg2+ ion enables magnesium batteries to attain high volumetric capacities of up to 3,833 mAh/cm3 (Novák et al., 1999). This is significantly higher than the volumetric capacity of the graphite (760 mAh/cm3) anodes currently used in commercialized lithium-ion batteries (Muldoon et al., 2014; Zhang et al., 2019). Moreover, magnesium metal has a very low standard reduction potential of −2.37 V vs S.H.E., making it possible to achieve high energy densities. Additionally, since magnesium carbonate (MgCO3) is abundant in the earth’s crust, magnesium is widely available and less expensive to source than lithium (Aurbach et al., 2001). Unlike lithium anodes, magnesium metal anodes cycled in certain electrolytes have been shown to be non-dendritic in nature, making them an intrinsically safer option. Despite the potential benefits, magnesium batteries are still hindered by three main challenges, as shown in Figure 1 (Levi et al., 2010).
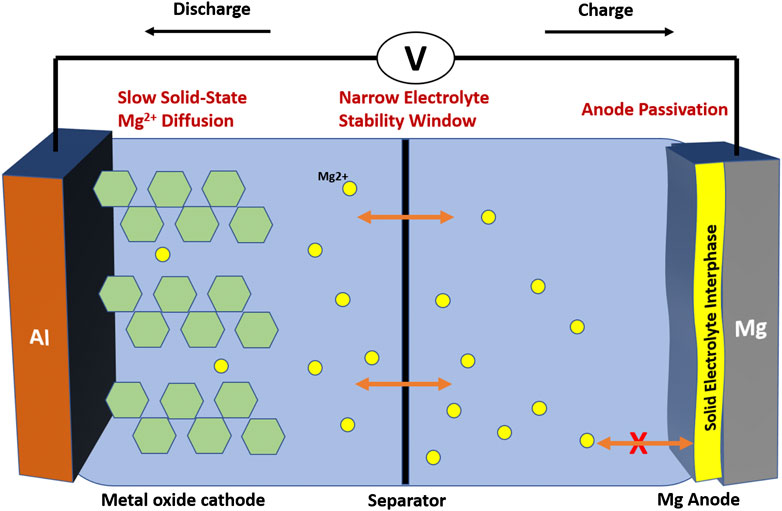
FIGURE 1. Schematic of standard Mg-ion battery and the central three challenges facing researchers. 1) Slow solid-state diffusion 2) Narrow electrolyte stability window. 3) Anode passivation.
First, magnesium metal’s low standard reduction potential makes it relatively reactive. Water and oxygen contamination in the electrolyte are particularly problematic, as parasitic side reactions at the anode-electrolyte interface can result in the formation of solid electrolyte interphase (SEI) layers that inhibit magnesium ion transport (Deog et al., 2013; Attias et al., 2019). Even ppm levels of contamination in the electrolyte can form insulating surface layers on the magnesium metal anode (Attias et al., 2019). Although lithium and magnesium metal both react with water and oxygen to form passivating surface layers, this interfacial reaction is not as limiting for lithium systems because the SEI formed remains lithium-ion conductive (Shterenberg et al., 2014). In addition to reacting with water and oxygen, magnesium metal will degrade reactive electrolyte species and solvent molecules to form thick electronically and ionically insulating surface layers (Attias et al., 2019). This challenge significantly limits the type of electrolytes that may be used in magnesium batteries, as the magnesium analogs of conventional electrolytes used in lithium-ion batteries fall into this category. As a result, researchers have worked to develop new electrolytes that avoid anode passivation and are both chemically and electrochemically compatible with magnesium metal. Early formulations of magnesium electrolytes, such as Grignard-based electrolyte solutions, were expensive and hazardous, making them poor candidates for use in commercial magnesium batteries. Furthermore, they suffered from low oxidative stability, making them incompatible with many high voltage cathode materials (above 2.2 V vs Mg/Mg2+) (Gaddum and French, 1927; Aurbach et al., 2001; Aurbach et al., 2009; Saha et al., 2014). More recently, new electrolytes have been designed to achieve higher anodic stability voltages. All phenyl complex (APC), organoborate, borohydride, and nitrogen containing electrolytes have been shown to have electrochemical stability windows of >3 V vs Mg/Mg2+ (Attias et al., 2019; Deivanayagam et al., 2019). However, although these newer electrolyte solutions can function at higher voltages, finding systems in which the cathode, electrolyte, and anode are compatible with each other remains a significant challenge. Finally, it has been shown that magnesium ions do not readily insert into many of the metal oxide cathodes conventionally used in lithium-ion batteries. Strong electrostatic interactions between the Mg2+ ion and the cathode host material hinder the insertion reaction mechanism. Often times, metal oxide cathodes consist of layered structures. During discharge, magnesium ions migrate between the layers of the reduced metal oxide structure. Since magnesium is divalent, it experiences strong electrostatic interactions with the electronegative oxygen atoms in the metal oxide structure.
These interactions lead to slow solid-state diffusion of the Mg2+ ion, resulting in low specific capacities, low power capabilities, and irreversible Mg-ion insertion (Aurbach et al., 2009). The Chevrel phases (MxMo6T8, M = metal, T = Se or S) were one of the first classes of cathode materials shown to effectively host magnesium ions. They were used in one of the first magnesium-ion battery prototypes developed by Aurbach et al. (2000). While the prototype MgxMo3S4 cathode had a relatively low operating voltage of 1.2 V vs Mg/Mg2+ and a low specific capacity of 130 mAh/g, it was stable for more than 2000 cycles. Ultimately, the impact of this work re-ignited the battery community’s interest in rechargeable magnesium batteries. Since then, many other classes of cathode materials; such as oxides, sulfides, and polyanions have been developed and tailored for magnesium ion storage (Levi et al., 2002; Zhang and Ling, 2016; Canepa et al., 2017). Despite these advancements, there still remains a lack of viable cathodes for magnesium batteries, and research in this area is an ongoing priority.
Of the many materials that have been investigated for applications in magnesium-ion batteries, Vanadium (V) oxide has emerged as one of the more promising candidate cathode materials. First introduced by Whittingham et al. in 1976, V2O5 is safe, has a relatively low cost, and may be easily synthesized in a wide variety of morphologies (Shao et al., 2013; Liu et al., 2018; Whittingham, 1976). Layered orthorhombic (alpha phase) V2O5 has been reported as one of the few metal oxide cathodes capable of successfully hosting Ca2+, Mg2+, and Y3+ ions (Gautam et al., 2015b; Canepa et al., 2017). Additionally, V2O5 has a high cell voltage of 2.66 V vs Mg/Mg2+, as well as a theoretical specific capacity of 294 mAh/g when 1 mol of Mg ion is inserted (Gregory, 1990; Canepa et al., 2017). However, initial studies of electrochemical Mg-ion insertion into V2O5 by Novák et al. (1999) demonstrated that intercalation of Mg2+ ions into bulk V2O5 materials is extremely slow and incomplete at room temperature. Furthermore, most of the Mg ions become adsorbed onto the surface of the cathode (Saha et al., 2014; Shklover et al., 1996). The poor practical magnesium insertion performance of V2O5 can be more easily understood by carefully examining the structural transitions that occur during insertion. In its most thermodynamically stable phase, α-V2O5 is a layered structure consisting of square pyramidal units. These pyramidal units form double chain sheets that lie parallel to the a-axis, as shown in Figure 2A. During insertion, the oxidation state of vanadium decreases from (V) to (IV), and magnesium ions become coordinated between the sheets of the V2O5 pyramids. The strong electrostatic interactions between the electronegative atoms cause the sheets to pucker, as shown in Figure 2C. In the alpha phase, the calculated barrier for magnesium-ion insertion is quite high (∼975 meV), and the insertion voltage is significantly less than the theoretical insertion voltage (∼2.3 V vs. Mg/Mg2+). As a result, the practical energy density of α-V2O5 is limited. Furthermore, because of the large migration barrier, Mg-ion insertion is highly irreversible, leading to poor long-term cycle life during electrochemical testing. Lastly, insertion of greater than 0.5 mol of magnesium is impractical because the V2O5 undergoes a structural transition and becomes ordered in the ε phase, which has a lower potential and weaker driving force for magnesium-ion insertion. Thus, V2O5 capacity for magnesium-ion insertion is usually limited to 147 mAh/g (corresponds to 0.5 mol of Mg inserted per V2O5) (Gautam et al., 2015a).
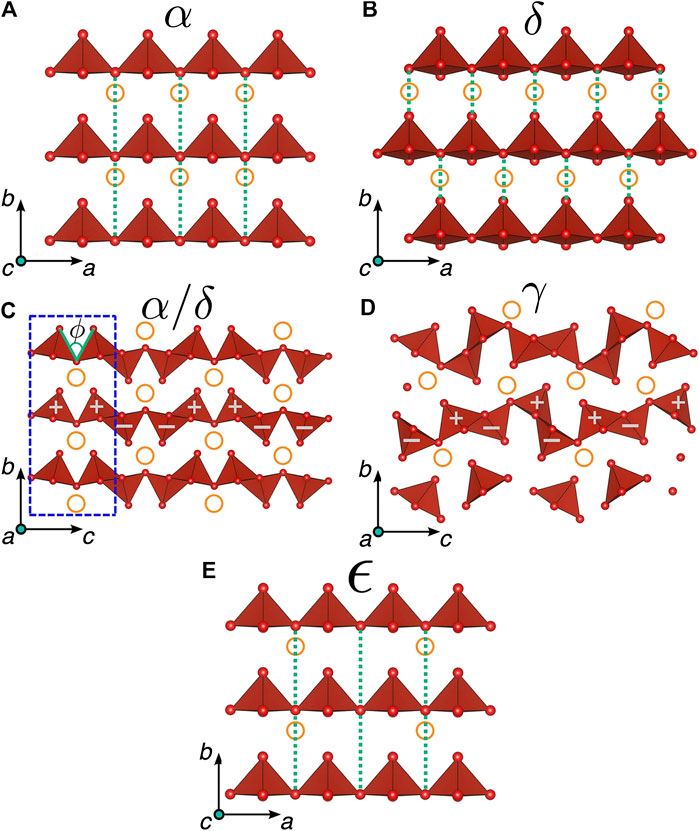
FIGURE 2. (A) α and (B) δ polymorphs of orthorhombic V2O5 are shown along the c-axis (shown to a depth of c/2 for viewing clarity) and (C) along the a-axis, which, compared to the (D) γ polymorph, has a different orientation of VO5 pyramids as denoted by the “+” and “−” symbols along the c-axis. Hollow orange circles correspond to the intercalation sites, the green dotted lines show the differences in layer stacking, and the dashed blue rectangle in panel (C) indicates a distance of c/2. Panel (E) illustrates the ϵ phase corresponding to a specific ordering of Mg atoms in α-V2O5 at half magnesiation, where alternate intercalant sites are occupied in the a-axis, as indicated by the orange circles. The schematics here correspond to “supercells” of the respective polymorph unit cells. Reprinted (adapted) with permission from Chem. Mater. 2015, 27, 10, 3733–3742. Copyright 2015 American Chemical Society.
Consequently, V2O5 cathodes tend to suffer from low specific capacities, poor capacity retention, and short cycle life. However, a few landmark studies in the 1980s and 1990s demonstrated promising electrochemical performance of V2O5 cathodes in magnesium-containing electrolytes. In 1987, Pereira-Ramos et al. (1987) successfully demonstrated improved electrochemical Mg-ion insertion into V2O5 from molten sulfone-based electrolytes at high temperature. The insertion reaction results in the formation of vanadium bronzes according to the reaction mechanism below (Novák et al., 1999):
When x = 0.5 mol of Mg ions are inserted into the bronze, a specific capacity of up to 130 mAh/g can be achieved at low current density (Canepa et al., 2017). In 1998, a V2O5 aerogel was explored as a promising cathode material for Mg batteries, with preliminary results suggesting that Mg2+ insertion into V2O5 aerogel occurs at higher levels than does Li+ intercalation (Le et al., 1998). Furthermore, as early as 2001, Morita et al. developed a prototype solid-state Mg battery utilizing a V2O5 slurry cathode, a novel polymeric solid electrolyte, and a Mg metal anode. The test cell showed an initial discharge capacity of 100 mAh/g (Morita et al., 2001). Unfortunately, the cell displayed low charge capacity, likely due to the degradation of the polymeric electrolyte and subsequent passivation of the Mg metal anode. Despite the success of a few early studies, electrochemical insertion of Mg into V2O5 is generally less effective, more complex, and non-uniform than chemical insertion (Novák et al., 1994).
These preliminary studies demonstrated many of the issues that limit the potential performance of V2O5 as a cathode for Mg batteries. As mentioned earlier, V2O5 cathodes exhibit low capacity and poor reversibility of magnesium-ion insertion. This is due in large part to the high migration barrier (975 meV for α-V2O5) for Mg-ion transport in α-V2O5 (Guatam et al., 2015a). In order to bypass limitations of Mg2+ diffusion in V2O5 structures, the electrochemical performance has been improved by several electrode and electrolyte design changes: nanostructured cathodes, conductive carbon substrates, and most significantly, the presence of water in non-aqueous electrolytes.
Modifying electrode structures by nanostructuring has been recently employed as an effective strategy to enhance the electrochemical performance of V2O5 in magnesium electrolytes. In 2015, Tepavcevic et al. (2015) synthesized a nanostructured bilayered V2O5 cathode that was electrochemically deposited onto a carbon nanofoam substrate. This composite cathode was able to achieve a specific capacity of 240 mAh/g, which is significantly higher than previously reported capacities for pristine V2O5 cathodes. The enhanced performance is due, in large part, to the nanostructured architecture of the electrode, which decreases electron and ion transport distances. As a result, the impact of the sluggish solid-state diffusion kinetics of Mg2+ is mitigated.
In addition to adopting a nanostructured architecture, Tepavcevic et al. (2015) used a carbon nanofoam substrate to improve the capacity of their bilayered V2O5 cathode. The high electrical conductivity of the carbon substrate allows it to function as an integrated current collector, enhancing the electron transport throughout the electrode. Other studies have also made use of carbon additives (carbon black, carbon nanotubes, etc.) to increase the electronic conductivity of V2O5 electrodes (Liu et al., 2010; Zhang et al., 2011; Wang et al., 2014).
Discovery: Effect of Electrolyte Water Content on Mg-Ion Insertion Capacity of V2O5 Cathodes
While V2O5 nanostructuring and the addition of conductive additives improved the capacity of these electrodes for reversible Mg ion insertion, one of the most important discoveries for realizing high capacity V2O5 cathodes in magnesium batteries was the discovery of the role of water molecules in electrolytes in facilitating magnesium-ion insertion. This discovery fueled much of the current research regarding the synthesis of hydrated V2O5 structures, which will be discussed further in later sections of this review. The exact mechanism and role which water plays in the electrolyte and how it improves Mg ion insertion is still not fully understood. Better understanding the role water plays in facilitating Mg-ion insertion will allow researchers to better design high performance hydrated V2O5 structures. Thus, the authors of this review discuss several mechanisms proposed in the literature and propose a new mechanism as well.
In 1993, Novák showed that the capacity of V2O5 could be enhanced when cycled in an electrolyte solution of Mg(ClO4)2 * nH2O in acetonitrile (Novák, 1993). By adjusting the water content, they were able to achieve capacities upwards of 170 mAh/g for n = 1 mol of H2O per mole of magnesium perchlorate. It was proposed that water molecules may act to solvate the magnesium ions and shield their divalent charge in order to facilitate insertion into the interlayer spaces of the V2O5 structure. This mechanism is consistent with the idea that water molecules can more readily solvate magnesium ions than an organic solvent, as the resulting energy of solvation is much lower. However, as the author notes, there is another potential explanation for the improved performance. On most voltage windows used for the electrochemical insertion of magnesium, the electrolysis of water may occur (2H2O → O2 + 4H+ + 4e−) (Ni et al., 2019). As protons are generated, it is possible that they might be reduced or intercalated at the cathode-electrolyte interface, resulting in higher capacity values being observed.
In 2004, Yu et al. found that Novák et al.’s findings translated to systems using different electrolyte solvents. In their study, they showed that the cyclic voltammetry redox behavior of V2O5 is improved when cycled in Mg(ClO4)2 * nH2O in propylene carbonate. Capacities upwards of 158 mAh/g were achieved using galvanostatic charge/discharge cycling(Yu and Zhang, 2004). However, just as in the study performed by Novák (1993) the capacity faded quickly over the first few cycles. This was attributed to parasitic reactions between the magnesium ribbon counter electrode and the water present in the electrolyte. Alternatively, the authors introduced the idea that capacity fade could be attributable to irreversible intercalation transformations, but no specific mechanism was presented.
In 2016, another significant advance in understanding the role of electrolyte water content in improving the magnesium-ion insertion capacity of V2O5 was made by Sa et al. (2016). For the first time, evidence of proton intercalation into V2O5 was presented. As in previous studies, a significant difference between V2O5 electrodes cycled in dry vs. water containing electrolyte was observed. In this particular study, greater than 250 mAh/g was achieved for V2O5 half-cells cycled in Mg (TFSI)2/diglyme with 2,600 ppm H2O. By comparison, V2O5 half cells cycled in dry electrolyte showed much lower capacity, reaching a maximum of less than 65 mAh/g. These cells were then analyzed using proton NMR, with samples cycled in water containing electrolyte showing chemical shifts indicating proton insertion. Ultimately, the authors of this study suggested that proton insertion, rather than water/magnesium-ion co-intercalation was responsible for the improved capacity of V2O5 electrode materials cycled in water containing organic electrolytes.
In order to more rigorously determine which species might be inserting into the interlayer spaces of V2O5 cathodes in water containing electrolytes, Lim et al. (2017) used X-ray diffraction data and DFT calculations to scrutinize proposed insertion sites in the crystal structure. Interestingly, the authors found that the cyclic voltammetry redox behavior indicated a second irreversible reaction, which was attributed to the electrolysis of water. This assessment was supported by the increasing pH of the electrolyte solution due to hydrogen gas evolution during cycling and by the emergence of new V2O5 phases in XRD data during electrochemical reduction. Further XRD analysis of V2O5 samples reduced in water containing electrolyte showed some structural distortions which were more closely examined using a first principles approach. Ultimately, it was determined that only three cavities in each crystalline unit are large enough to accommodate species larger than protons, as shown in Figure 3. However, each of the sites have center to oxygen distances of about 2.5 Å, leaving enough space for ions of ∼1.0 Å in radius. This is large enough to accommodate magnesium ions and protons, but too small to accommodate larger molecules like water, Mg(H2O)n2+, and H3O+. Furthermore, ICP-MS analysis of the Mg-inserted V2O5 sample showed that only 0.17 mol of Mg ions per mole of V2O5 was intercalated. In order to quantify the level of proton insertion into the Mg-inserted V2O5 structure, a DFT approach was used. Although the calculations were unable to exactly quantify the level of proton insertion, it was determined that between 0.66 and 1.17 mol of H+ may be inserted for each V2O5 molecule. Ultimately, these results suggest that the increased capacity of V2O5 electrodes cycled in water containing organic electrolytes is due in large part to an increase in proton insertion, although further study detailing the mechanism and sequence of intercalation is required.
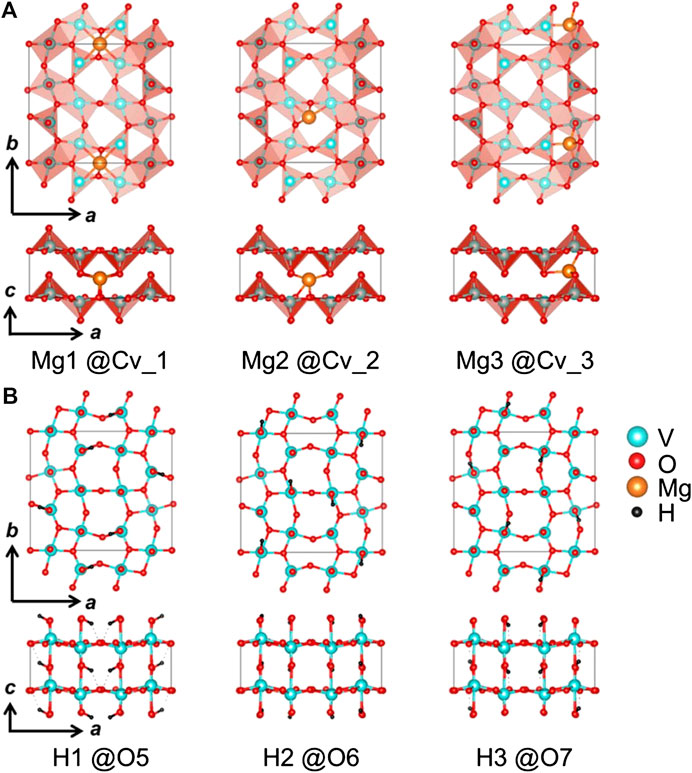
FIGURE 3. Optimized positions of magnesium and proton ions suggested by DFT calculations for Mg0.17HxV2O5 (x = 1): (A) Mg ions in three cavity sites (Cv_1, Cv_2, and Cv_3), and (B) protons around the three apical oxygen atoms (5O, O6, and O7). Reprinted (adapted) with permission from Inorg. Chem. 2017, 56, 14, 7668–7678. Copyright 2017 American Chemical Society.
Previous studies discussing the impact of hydrogenation on V2O5 performance may provide information about the mechanism by which protons are inserted into sites within the V2O5 lattice in wet electrolytes. A 2016 study by Peng et al. (2016) showed that hydrogen atoms may reversibly adsorb onto oxygen atoms in the V2O5 structure to form OH bonds. Similar structural changes following hydrogenation were observed using XPS in a 2016 study by Ma et al. (2016). Furthermore, a recent 2019 study by Ni et al. (2019) showed that increasing the hydrogen content of V2O5 electrodes by hydrogenation substantially lowers the migration barrier for magnesium-ion insertion. In fact, the migration barrier for α-V2O5 may drop from 1.28 eV without hydrogenation to 0.56 eV when 2 mol of hydrogen are inserted per mole of V2O5. These studies show clear evidence that the electronegative oxygen atoms in the V2O5 structure are capable of forming surface hydroxyl groups, a process which may be possible under water containing electrolyte cycling conditions. Additionally, they suggest that increased proton insertion might promote magnesium-ion insertion into V2O5, as increased hydrogen content is associated with a lower diffusion barrier and expanded magnesium-ion insertion pathways.
As a result, there is a new possible mechanism for the improved Mg-ion insertion capacity of V2O5 in wet electrolytes. Protons generated from the electrolysis of water may react with the electronegative oxygen atoms in the V2O5 structure to form structural hydroxyl groups. In turn, these structural hydroxyl groups promote additional Mg-ion insertion by lowering the migratory insertion barrier. This assertion is supported from a structural standpoint by the increased density of proton isosurfaces surrounding oxygen elements of the V2O5 polyhedra, as shown in the 3D bond valence difference maps in Figure 4 below.
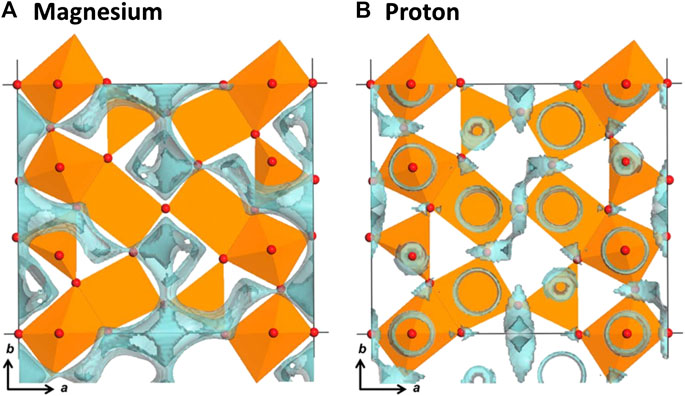
FIGURE 4. 3D bond valence difference map isosurfaces (sky-blue color) for Mg-inserted V2O5 with the isosurfaces of |0.1| vu for (A) Mg ions and (B) protons. Oxygen atoms are denoted as red balls, and vanadium atoms, not seen in this figure, are in the centers of the orange-colored polyhedra. Reprinted (adapted) with permission from Inorg. Chem. 2017, 56, 14, 7668–7678. Copyright 2017 American Chemical Society.
The idea that water in the electrolyte may react with the surface of a transition metal oxide to form stable structural hydroxyl groups has been demonstrated in previous studies (Song et. al., 2015; Sahadeo et al., 2018). Most notably, Sahadeo et al. (2018) presented a rigorous surface study of MnO2 electrodes cycled in hydrated organic electrolyte using surface, angle-resolved, and depth profiling XPS analysis. Their study elucidated the mechanism of MnO2 water activation by showing that a stable surface hydroxide layer consisting of Mg(OH)2, Mn(OH)2, and MnOOH formed during the discharge phase. It should be noted that V2O5 does not exhibit exactly the same water activation capabilities. Unlike MnO2, which retains improved capacity when cycled in dry electrolyte following water activation, it has only been shown that V2O5 demonstrates improved capacity when cycled in wet electrolyte. Still, the role of structural hydroxyl groups in improving the magnesium-ion insertion capacity of V2O5 is well documented. A 2018 study by Tepavcevic et al. (2018) showed that structural hydroxyl groups are an intrinsic feature of electrochemically deposited V2O5 cathodes. In dry electrolytes, these hydroxyl groups were found to provide a solvating environment that cooperatively promotes magnesium-ion insertion and distorts the vanadium polyhedra in a way that increases accessibility of cation insertion sites.
In conclusion, the presence of water in magnesium salt organic electrolytes improves the capacity of V2O5 electrodes, but the mechanism by which this occurs is still unclear. Four primary mechanisms which explain this improvement in capacity have been proposed: water solvation, proton insertion, hydrogenation to reduce magnesium insertion migration barrier, and the coordinating effect of structural hydroxyl groups. There is little evidence that Mg(H2O)n2+ is inserted into the V2O5 structure directly, but the possibility that magnesium ions transporting in the electrolyte are preferentially solvated by water molecules should not be discounted. That being said, the majority of evidence points to the idea that the primary reasons for which V2O5 exhibits increased capacity in water containing electrolytes is due to an increase in proton insertion. It is well documented that protons are generated by the electrolysis of water and that a significant number of protons can be inserted into small cavities present in the crystalline structure of V2O5. As a result, the observed increase in capacity when cycled in wet electrolytes may be attributable to increased insertion of protons. Furthermore, these protons may work to further increase the capacity of V2O5 by promoting the insertion of magnesium-ions by lowering the migration barrier and expanding magnesium-ion diffusion pathways. Finally, the protons generated from the electrolysis of water may react with the electronegative oxygen atoms in V2O5 to form semi-stable structural hydroxyl groups. These structural hydroxyl groups may alter the structural environment in order to promote further magnesium ion insertion. In order to narrow down the possibilities, better spectroscopic studies, especially using surface sensitive XPS analysis, of V2O5 electrode materials cycled in water containing organic electrolytes of magnesium salts is required. Of course, it should be noted that water molecules cannot yet be practically used in magnesium full cells, as they react parasitically with magnesium metal. As discussed previously, magnesium metal’s reactivity makes it incompatible with a suite of electrolyte solvents due to the formation of insulating surface interphase layers (SEI) (Deog et al., 2013). In a similar way, magnesium anodes may react with water molecules to form surface passivation layers of magnesium hydroxide or magnesium oxide, neither of which freely conduct magnesium ions. As a result, water containing electrolytes, despite their promise, cannot be used until schemes which protect the magnesium anode from reacting with water are designed. This is discussed in greater detail later in this review.
In any case, the observed increase in Mg-ion insertion into V2O5 during cycling in water containing organic electrolytes led researchers to pursue hydrated (with crystalline structural water) V2O5 cathode materials. Essentially, hydrated V2O5 structures incorporate water directly into the layers of the V2O5 structure. Ideally, hydrated V2O5 structures will reduce the Mg-ion insertion barrier and exhibit the same Mg-ion insertion capabilities observed in water-containing electrolytes. This is preferable to adding water directly to the electrolyte, as it could prevent passivating surface reactions between a magnesium anode and water molecules.
Efforts to Construct High Capacity Hydrated V2O5 Morphologies for Mg-Ion Batteries
With the general understanding of how water in the electrolyte promotes the insertion of magnesium ions into V2O5, researchers began to look for ways to incorporate water directly into the V2O5 structure and synthesize hydrated V2O5 structures. In theory, by integrating water into the crystal structure, magnesium-ion insertion into V2O5 can be enhanced, as the lattice water molecules can reduce electrostatic interactions between the electronegative lattice oxygen atoms and the positive charge of the magnesium ion through a solvation-type mechanism while eliminating water in electrolytes and avoiding anode passivation. In 1995, Novák (1995) first demonstrated the promise of hydrated V2O5 structures when they investigated the electrochemical insertion of magnesium into chemically deposited hydrated vanadium bronzes. In their study, high capacities of 200 mAh/g were attained, but rapid capacity fade was observed with an increasing number of cycles. Ultimately, it was determined that the level of Mg-ion insertion was directly correlated with the lattice water content in each bronze, and the observed fade in capacity was attributed to structural water loss during cycling. More recently, researchers have worked to develop aqueous electrochemical deposition methods and sol gel techniques for the fabrication of hydrated V2O5 electrodes. In both synthesis routes, crystal (lattice) water is integrated into the V2O5 structure, giving way to a hydrated structure with significantly enhanced Mg-ion insertion capabilities.
In the past several years, aqueous electrodeposition techniques have emerged as one of the more facile methods for producing hydrated V2O5 structures. In particular, it has been shown that electrodeposition of V2O5 from water-containing electrolytes leaves crystal water and adsorbed water in the structure, both of which enhance the electrochemical performance of the cathode (Henry et al., 2020). In 2015, Tepavcevic et al. (2015) demonstrated that the electrochemical deposition of V2O5 from an aqueous solution of VOSO4 produced a bilayered V2O5 film with incorporated structural water and hydroxyl groups. The crystal water worked to solvate the Mg ions during insertion via a charge shielding effect, similar to what was initially proposed for water-containing organic electrolytes. Meanwhile, the structural hydroxyl groups were responsible for expanding and maintaining the interlayer spacing of the V2O5 crystal lattice, allowing for more rapid Mg-ion diffusion within the structure. Second, they altered the symmetry of the V2O5 so as to promote electron transfer and magnesium intercalation (Tepavcevic et al., 2015). Unlike the lattice water, these structural hydroxyl groups were found to be covalently bonded to the V2O5 structure, making them structurally stable during electrochemical cycling. Additionally, the structural hydroxyl groups demonstrated thermal stability up to 500°C during post-deposition annealing. Other more recent investigations of the role of the structural hydroxyl groups have shown that they are an intrinsic component of V2O5 structures synthesized via electrochemical deposition techniques (Tepavcevic et al., 2018).
In addition to aqueous electrochemical deposition techniques, hydrated V2O5 structures have been made in the form of hydrated gels. Sol gel methods are popular, as they can be tailored to produce a wide variety of hydrated structures, and they easily translate to large-scale production. In 1998, Le et al. (1998) in one of the earliest reported sol gel V2O5 studies, investigated the insertion of polyvalent cations into V2O5 aerogels. Aerogels are synthesized by removing liquid under supercritical conditions so that the solid framework present in the gel-state is preserved. As a result, aerogels maintain high surface area and short ion diffusion distances through solid materials. These properties simultaneously enhance the intercalation of guest ions and the overall rate performance of the insertion reaction. If the liquid is removed below the critical point under air or vacuum conditions, the porous three-dimensional network collapses, and a more compact structure, known as a xerogel, forms (Gautam et al., 2016). The structure of the hydrated V2O5 in xerogel is shown in Figure 5. The extremely high capacity and excellent rate performance was attributed to the high surface area of the xerogel structure, the boost in electronic conductivity afforded by the carbon additive, and crystal water charge shielding effects.
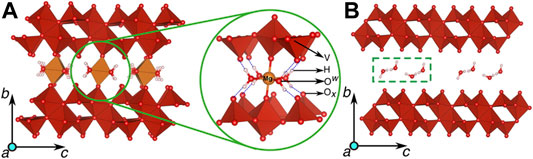
FIGURE 5. Structures of the fully magnesiated (xMg = 0.5) and the fully demagnesiated Xerogel, with 1 H2O per formula unit of V2O5 are displayed in (A) and (B) respectively. The coordination of each Mg by 4 Ow can be observed in the enlarged image in the green circle, with the dashed blue lines indicating hydrogen-bonding between the water molecules and the lattice oxygen. The atomic species in the Xerogel structure are labelled in the enlarged image with Ow and Ox indicating the water and lattice oxygen, respectively. Reprinted (adapted) with permission from Nano Lett. 2016, 16, 4, 2426–2431. Copyright 2016 American Chemical Society.
High performance hydrated V2O5 structures have also been successfully synthesized via hydrothermal sol gel methods. The sol gel synthesis method, coupled with thermal annealing post-processing, results in the formation of crystalline V2O5 structures with lattice bound water molecules. For example, An et al. (2015) demonstrated a graphene-decorated vanadium oxide nanowire aerogel that exhibited a high specific capacity of 330 mAh/g. The exceptional performance of the cathode was attributed to the charge shielding effect caused by the crystal water present in the aerogel, along with graphene’s high electronic conductivity and the nanostructured architecture (An et al., 2015).
Recently, the rational design of hydrated structures of vanadium oxide has become an even more popular method for enhancing the electrochemical performance for Mg systems. In 2016, Sa et al. were one of the first to show direct evidence for Mg2+ intercalation into a bilayer-structured V2O5 • n H2O (n∼ 0.6) xerogel structure (Sa et al., 2016). The cathode consisted of a layered ribbon-like structure, with H2O molecules present between the layers. This type of layered structure is beneficial for Mg2+ intercalation due to its structural flexibility, allowing it to transform its shape to effectively host the inserting cation by modifying the interlayer spacing. A schematic of the ‘pristine’ and ‘discharged’ crystal structures are shown in Figure 6. In this work, the authors found that the interlayer spacing tends to decrease during Mg2+ intercalation and increase during de-intercalation, likely due to strong electrostatic interactions between the Mg cation and the V2O5 cathode. The authors also observed the co-intercalation of solvent molecules along with Mg2+ ions; this insertion mechanism is likely attributable to the expanded interlayer spacing caused by H2O molecules present within the crystal structure. The co-intercalation of solvated Mg2+ ions serves to further shield the divalent charge and promote fast intercalation and diffusion reactions.
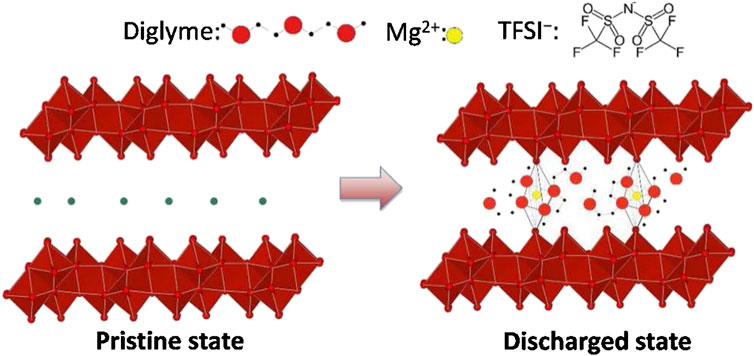
FIGURE 6. Schematic of the V2O5• nH2O crystal structure in the pristine (left) and discharged (right) states. Reprinted with permission from Chem. Mater. 2016, 28, 2962–2969. Copyright 2016 American Chemical Society.
In 2019, Xu et al. (2019) also developed a bilayer-structured vanadium oxide (Mg0.3V2O5 • 1.1 H2O) cathode with a synergistic co-operation between Mg2+ ion and lattice H2O. The lattice H2O promotes fast and reversible Mg2+ ion intercalation while the pre-intercalated Mg2+ ions enhance the electronic conductivity of the cathode while simultaneously improving the structural stability. A schematic of the cathode synthesis is depicted below in Figure 7. The unique electrode structure and nanowire morphology allowed for excellent electrochemical performance and 80% capacity retention after 10,000 cycles at a high current density of 2 A/g. The H2O molecules not only serve to increase the interlayer spacing, they also provide a charge shielding effect by coordinating the inserting Mg2+ ions, thereby decreasing the electrostatic interactions and increasing the efficiency of the intercalation reaction. Moreover, the pre-intercalated Mg2+ ions function as structural pillars in between the V2O5 layers, ensuring that the crystal structure remains stable during Mg2+ intercalation/de-intercalation. This allows for long-term cycling with high coulombic efficiency. At the same time, Deng et al. investigated the first manganese-ion pre-intercalated vanadium oxide (Mn0.04V2O5 • 1.17 H2O) structure as a cathode for Mg batteries (Deng et al., 2019). According to the authors, the water molecules leftover from the deposition process increase the interlayer spacing, and the Mn2+ ions function as interlayer pillars that stabilize the resulting structure, similar to the synergistic effect previously observed in Xu’s work. Consequently, the cathode structure exhibits fast insertion kinetics, leading to good rate performance, and excellent cycle life (82% capacity retention after 10,000 cycles) (Deng et al., 2019).
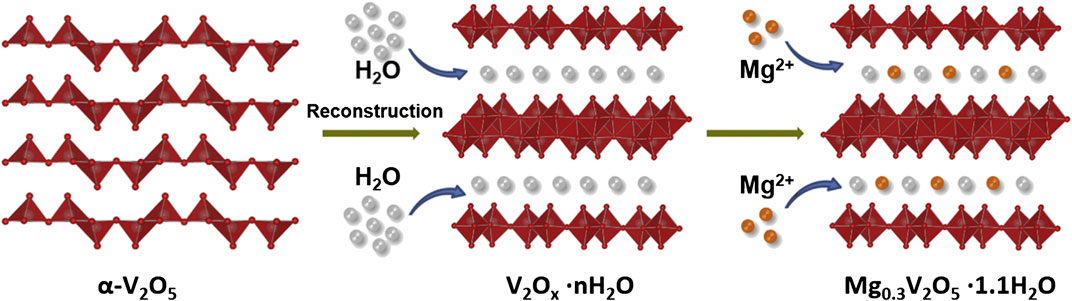
FIGURE 7. Schematic Diagrams of the Formation of the Bilayer-Structured Mg0.3V2O5• 1.1 H2O. Reprinted with permission from Chem 5, 1194–1209, May 9, 2019. Copyright Elsevier 2019.
While V2O5 structures that incorporate structural (lattice) water represent a promising design choice for the future of Mg batteries, several drawbacks must be addressed before they can be practically applied. The most severe challenge facing hydrated V2O5 structures is the loss of structural H2O during electrochemical cycling (Novák, 1995). Despite their improved insertion kinetics and high specific capacities, some hydrated V2O5 structures still suffer from poor cycle life (Imamura et al., 2003; Tepavcevic et al., 2015; Perera et al., 2017). The limited cycle life of these structures is largely attributed to their structural instability. The loss of structural water during electrochemical cycling has two major consequences. First, significant leaching of water during cycling would make hydrated V2O5 structures largely incompatible with Mg metal anodes in full cell applications. The low reduction potential of Mg metal causes it to react strongly with even ppm levels of H2O present in the electrolyte, leading to the formation of passivating solid electrolyte interphase (SEI) surface layers (Lu et al., 1999). These SEI layers on the Mg anode surface are ionically insulating and prevent further Mg2+ ion transport. Second, the presence of structural water is an integral part of the V2O5 layered structure. Without the structural water to shield the charge of the Mg2+ ion and maintain the interlayer spacing, the Mg-ion insertion reaction would be slow, irreversible, and less thermodynamically favorable. This effect has been seen in early studies of hydrated V2O5 structures, many of which observed drastic capacity fade with extended cycling attributable to the loss of water molecules from the cathode into electrolyte during electrochemical cycling (Novák, 1993; Novák, 1995; Tepavcevic et al., 2015). In order for hydrated V2O5 cathodes to have real world applications in batteries, the water loss issue must be remedied. In practice, there are several of different methods that can be used to alleviate this problem, ranging from anode protection to the use of solid electrolytes.
Progress to Realize Hydrated Vanadium Oxide for Mg Battery, Focus on Anode Protection and Solid Electrolytes
As stated previously, despite utilizing crystalline water in these hydrated V2O5 structures, water still tends to slowly leach out of these structures into electrolytes. In order to realize hydrated V2O5 structures as a cathode material for magnesium batteries, solutions which prevent parasitic side reactions between water and magnesium must be more rigorously explored. One of these solutions is to use solid electrolytes for magnesium batteries. By acting as a barrier between the magnesium anode and a hydrated V2O5 structure, such as the V2O5 xerogel, solid electrolytes could help to mitigate anode passivation.
Solid electrolytes are attractive for other reasons as well; they offer improved safety, thermal stability, and electrochemical stability (Jaschin et al., 2020). However, solid electrolytes often suffer from poor ionic conductivity when compared with liquid electrolytes. In most cases, the ionic conductivity of solid electrolytes is several orders of magnitude lower than that of liquid electrolytes (Jaschin et al., 2020). Furthermore, solid electrolytes need high coulombic efficiency, good oxidative stability, low electronic conductivity, and favorable mechanical properties (Jaschin et al., 2020). In particular, solid electrolytes must be sufficiently pliable to conform to the surfaces of the anode and cathode in order to provide a high surface area interface for ionic transport. Unlike lithium-ion batteries, very few solid electrolytes have been successfully demonstrated in full cell magnesium battery applications. Rather, the majority of research has focused on analyzing the conductivity and mechanical properties of candidate polymers, ceramics, and metal-organic frameworks in magnesium symmetric cells (Jaschin et al., 2020). Many of the more promising polymer electrolytes are made from polyethylene oxide (PEO) with magnesium salts (Roedern et al., 2017). For example, a recent study demonstrated a nanocomposite polymer consisting of PEO, magnesium borohydride, and magnesium oxide nanoparticles with 98% coulombic efficiency for magnesium plating and stripping (Shao et al., 2015). However, PEO is incompatible with water, and the majority of PEO based solid electrolytes have low ionic conductivities and demonstrate large overpotentials when cycled in magnesium symmetric cells. Higher ionic conductivities have been observed in blend polymer electrolytes, such as PEO-PVP(polyvinylpyrrolidone) (Anilkumar et al., 2017).
Perhaps the most interesting avenue of research that could facilitate the use of pure magnesium metal with hydrated V2O5 structures is the development of anode protection schemes, where a thin layer is deposited at either the surface of the magnesium or on the V2O5 cathode. The use of anode protection has already been extensively demonstrated in lithium-ion batteries. For example, in 2015, Kozen et al. demonstrated improved capacity retention in lithium-sulfur batteries by using a thin aluminum oxide protection layer on a bare lithium anode deposited via atomic layer deposition (ALD) (Kozen et al., 2015). Atomic layer deposition techniques are able to finely control the thickness of the materials with conformal deposition, as well as the surface chemistry which can enable good interface between the anode and the electrolyte. Recently in 2019, Su et al. used ALD to deposit magnesium phosphate thin films on platinum substrate to measure its electrochemical properties (Su et al., 2019). They found that this magnesium phosphate film had an ionic conductivity of 1.6 × 10–7 S cm−1 at 500°C with an activation energy of 1.37 eV. While the ionic conductivity had to be measured at elevated temperatures due to the low ionic conductivity at room temperature, a nanometer scale layer could act as an artificial interface layer that allows for good Mg ion conductivity.
Electrochemical deposition of artificial interface layers are also possible. In Li-S systems a thin elastomer from the electrolyte can be formed on the Li via electrochemical pretreatment to prevent unwanted parasitic reactions (Wang et al., 2018). Recently, Tang et al. used lithium containing salt, lithium tetrakis (hexafluoroisopropyloxy)borate (LBhfip), in Mg full cell (Tang et al., 2020). The solid electrolyte interface that formed from the reduction of LBhifp was stable and Mg ion conductive which prevented parasitic reactions between the Mg anode and the bulk of the electrolyte, and allowed good Mg depositions and cycling. When paired with Mo6S8, this full cell was stable up to 6,000 cycles with the capacity of 101.3 mAh g−1.
Although investigations of anode protection in magnesium-ion batteries are limited, some landmark studies have been recently published. For example, Son et al. (2018) recently reported the development of an artificial interphase that permits the reversible cycling of a Mg/V2O5 full cell in hydrated organic electrolyte (Son et al., 2018). The elastic interphase, which was crafted from cyclic polyaniline, enabled reversible plating/stripping of magnesium and prevented electrochemical reduction of the electrolyte. When cycled in a full cell with 3M water in propylene carbonate-based electrolyte, capacities upwards of 140 mAh/g were reached, along with greater than 65% capacity retention over 40 cycles. Ultimately, this work represents one of the first reported instances of successful pairing of a V2O5 cathode and a magnesium anode in the presence of water. By improving the ionic conductivity of the interphase (1.19 × 10–6 S/cm), greater magnesium-ion storage capability and capacity retention might be achieved. In this sense, the development of artificial solid electrolyte interphases is one of the most promising avenues for developing compatible magnesium full cells that incorporate hydrated V2O5 structures.
Conclusion and Outlook
For the past several decades, the commercialization of magnesium-ion batteries has been seriously constrained by the lack of energy dense cathode materials that are compatible with conventional electrolytes and a magnesium metal anode. V2O5, owing to its safety, high theoretical capacity for magnesium-ion insertion, and facile synthesis; is perhaps one of the most promising potential cathode materials. In practice, V2O5 exhibits poor electrochemical performance and slow, irreversible insertion of Mg2+ when cycled in conventional organic electrolytes. However, when water is added to the electrolyte, magnesium-ion insertion can be bolstered significantly. Although the mechanism is not entirely clear, the improved performance is likely attributable to increased levels of proton insertion, which, by decreasing the migratory insertion barrier, leads to increased magnesium ion intercalation. Additionally, a new hypothesis is proposed that protons may react with the electronegative oxygen atoms in V2O5 to form structural hydroxyl groups. In turn, these structural hydroxyl groups may alter the structural environment in order to promote further magnesium ion insertion. Ultimately, the mechanism is not entirely clear, and the authors of this work suggest that more studies, especially those which focus on XPS and other surface sensitive analysis techniques, be performed.
Irrespective of the mechanism, the improved magnesium-ion insertion capacity of V2O5 in wet organic electrolyte has led to the development of high capacity hydrated V2O5 structures. In particular, researchers have been able to demonstrate high capacity V2O5 structures prepared from aqueous electrodeposition and sol gel techniques. However, these hydrated structures are not yet completely compatible with magnesium anodes. Over the course of many cycles, water tends to leach from the structure into the electrolyte and passivate the surface of the magnesium metal anode, resulting in rapid capacity fade. This central issue remains the most significant challenge to realizing high capacity hydrated V2O5 structures for magnesium batteries. In order to remediate this problem, researchers have looked into the development of solid electrolytes. However, the one of the most promising avenues for reconciling hydrated V2O5 structures with magnesium metal anodes appears to be the development of magnesium anode protection layers (artificial SEI), as they have been successfully demonstrated in Mg/V2O5 full cells with a hydrated electrolyte (Son et al., 2018). Ultimately, hydrated V2O5 electrodes are a promising candidate cathode material for high capacity magnesium-ion batteries. However, a call to action for the development of anode protection schemes that are compatible with both trace water and magnesium metal is required.
Author Contributions
BJ and HH conceptualized and wrote the manuscript. BJ, HH, and NK edited and prepared the manuscript for publication. SL supervised and edited the manuscript for publication.
Funding
This work was supported as part of the Nanostructures for Electrical Energy Storage (NEES), an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DESC0001160, and continued in analysis, manuscript writing and discussions supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021070.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
An, Q., Li, Y., Deog Yoo, H., Chen, S., Ru, Q., Mai, L., et al. (2015). Graphene decorated vanadium oxide nanowire aerogel for long-cycle-life magnesium battery cathodes. Nano Energy 18, 265–272.
Anilkumar, K. M., Jinisha, B., Manoj, M., and Jayalekshmi, S. (2017). Poly(ethylene oxide) (PEO) – poly(vinyl pyrrolidone) (PVP) blend polymer based solid electrolyte membranes for developing solid state magnesium ion cells. Eur. Polym. J. 89, 249–262. doi:10.1016/j.eurpolymj.2017.02.004
Attias, R., Salama, M., Hirsch, B., Goffer, Y., and Aurbach, D. (2019). Anode-electrolyte interfaces in secondary magnesium batteries. Joule. 3, 27–52. doi:10.1016/j.joule.2018.10.028
Aurbach, D., Gofer, Y., Lu, Z., Schechter, A., Chusid, O., Gizbar, H., et al. (2001). A short review on the comparison between Li battery systems and rechargeable magnesium battery technology. J. Power Sources 97-98, 28–32. doi:10.1016/S0378-7753(01)00585-7
Aurbach, D., Levi, E., and Gofer, Y. (2009). On the way to rechargeable Mg batteries: the challenge of new cathode materials. Chem. Mater. 22 (3), 860–868. doi:10.1021/cm9016497
Aurbach, D., Lu, Z., Schechter, A., Gofer, Y., Gizbar, H., Turgeman, R., et al. (2000). Prototype systems for rechargeable magnesium batteries. Nature 407 (23), 724–727. doi:10.1038/35037553
Bieker, G, Winter, M, and Bieker, P. (2015). Electrochemical in situ investigations of SEI and dendrite formation on the lithium metal anode. Phys. Chem. Chem. Phys. 17, 8670–8679. doi:10.1039/c4cp05865h
Canepa, P., Sai Gautam, G., Hannah, D. C., Malik, R., Liu, M., Gallagher, K. G., et al. (2017). Odyssey of multivalent cathode materials: open questions and future challenges. Chem. Rev. 117 (5), 4287–4341. doi:10.1021/acs.chemrev.6b00614
Deivanayagam, R., Ingram, B. J., and Shahbazian-Yassar, R. (2019). Progress in development of electrolytes for magnesium batteries. Energy Storage Materials 21, 136–153. doi:10.1016/j.ensm.2019.05.028
Deng, X., Xu, Y., An, Q., Xiong, F., Tan, S., Wu, L., et al. (2019). Manganese ion pre-intercalated hydrated vanadium oxide as a high-performance cathode for magnesium ion batteries. J. Mater. Chem. 7 (17), 10644–10650. doi:10.1039/B819629J
Deog, H. Y, Shterenberg, I., Gofer, Y., Gershinsky, G., Pour, N., and Aurbach, D. (2013). Mg rechargeable batteries: an on-going challenge. Energy Environ. Sci. 6 (8), 2245–2550. doi:10.1039/C3EE40871J
Gaddum, L. W., and French, H. E. (1927). The electrolysis OF grignard solutions. J. Am. Chem. Soc. 49 (5), 1295–1299.
Gautam, G. S., Canepa, P., Abdellahi, A., Urban, A., Malik, R., and Ceder, G. (2015a). The intercalation phase diagram of Mg in V2O5 from first-principles. Chem. Mater. 27 (10), 3733–3742.
Gautam, GS, Canepa, P, Malik, R, Liu, M, Persson, K, and Ceder, G. (2015b). First-principles evaluation of multi-valent cation insertion into orthorhombic V2O5. Chem Commun (Camb). 51 (71), 13619–13622. doi:10.1039/c5cc04947d
Gautam, GS, Canepa, P, Richards, WD, Malik, R, and Ceder, G. (2016). Role of structural H2O in intercalation electrodes: the case of Mg in nanocrystalline xerogel-V2O5. Nano Lett. 16 (4), 2426–31. doi:10.1021/acs.nanolett.5b05273
Henry, H. K., Johnston, B., Liau, D., Sahadeo, E., and Lee, S. B. (2020). Dual effect of structure and hydration on magnesium-ion insertion into electrodeposited V2O5 thin films. J. Electrochem. Soc. 167 (11), 110523. doi:10.1149/1945-7111/aba36e
Imamura, D., Miyayama, M., Hibino, M., and Kudo, T. (2003). Mg intercalation properties into V[sub 2]O[sub 5] gel/carbon composites under high-rate condition. J. Electrochem. Soc. 150 (6), A753. doi:10.1149/1.1571531
Jaschin, P. W., Gao, Y., Li, Y., and Bo, S.-H. (2020). A materials perspective on magnesium-ion-based solid-state electrolytes. J. Mater. Chem. 8 (6), 2875–2897. doi:10.1039/C9TA11729F
Kozen, A. C., Lin, C. F., Pearse, A. J., Schroeder, M. A., Han, X., Hu, L., et al. (2015). Next-generation lithium metal anode engineering via atomic layer deposition. ACS Nano. 9 (6), 5884–5892. doi:10.1021/acsnano.5b02166
Le, D. B., Passerini, S., Coustier, F., Guo, J., Soderstrom, T., Owens, B. B., and Smyrl, W. H. (1998). Intercalation of polyvalent cations into V2O5 aerogels. Chem. Mater. 10 (3), 682–684. doi:10.1021/cm9705101
Levi, E., Gofer, Y., and Aurbach, D. (2010). On the way to rechargeable Mg batteries: the challenge of new cathode materials. Chem. Mater. (2010). 22 (3), 860–868. doi:10.1021/cm9016497
Levi, E., Gofer, Y., Vestfreed, Y., Lancry, E., and Aurbach, D. (2002). Cu2Mo6S8 Chevrel phase, A promising cathode material for new rechargeable Mg Batteries: a mechanically induced chemical reaction. Chem. Mater. 14 (6), 2767–2773. doi:10.1021/cm021122o
Lim, S. C., Lee, J., Kwak, H. H., Heo, J. W., Chae, M. S., Ahn, D., et al. (2017). Unraveling the magnesium-ion intercalation mechanism in vanadium pentoxide in a wet organic electrolyte by structural determination. Inorg. Chem. 56 (14), 7668–7678. doi:10.1021/acs.inorgchem.7b00204
Liu, J., Wang, Q., Reeja-Jayan, B., and Manthiram, A. (2010). Carbon-coated high capacity layered Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathodes. Electrochem. Commun. 12 (6), 750–753. doi:10.1016/j.elecom.2010.03.024
Liu, X., Zeng, J., Yang, H., Zhou, K., and Pan, D. (2018). V2O5-Based nanomaterials: synthesis and their applications. RSC Adv. 8, 4014–4031. doi:10.1039/C7RA12523B
Lu, Z., Schechter, A., Moshkovich, M., and Aurbach, D. (1999). On the electrochemical behavior of magnesium electrodes in polar aprotic electrolyte solutions. J. Electroanal. Chem. 466 (2), 203–217.
Ma, W, Zhang, C, Liu, C, Nan, X, Fu, H, and Cao, G. (2016). Impacts of surface energy on lithium ion intercalation properties of V2O5. ACS Appl. Mater. Interfaces 8 (30), 19542–19549. doi:10.1021/acsami.6b06359
Morita, M., Yoshimoto, N., Yakushiji, S., and Ishikawa, M. (2001). Rechargeable magnesium batteries using a novel polymeric solid electrolyte. Electrochem. Solid State Lett. 4 (11), A177. doi:10.1149/1.1403195
Muldoon, J, Bucur, CB, and Gregory, T. (2014). Quest for nonaqueous multivalent secondary batteries: magnesium and beyond. Chem Rev. 114 (23), 11683–11720. doi:10.1021/cr500049y
Ni, D., Shi, J., Xiong, W., Zhong, S., Xu, B., and Ouyang, C. (2019). The effect of protons on the Mg2+ migration in an α-V2O5 cathode for magnesium batteries: a first-principles investigation. Phys. Chem. Chem. Phys. 21 (14), 7406–7411. doi:10.1039/C9CP00528E
Novák, P. (1993). Electrochemical insertion of magnesium in metal oxides and sulfides from aprotic electrolytes. J. Electrochem. Soc. 140 (1), 140. doi:10.1149/1.2056075
Novák, P. (1995). Electrochemical insertion of magnesium into hydrated vanadium bronzes. J. Electrochem. Soc. 142 (8), 2544.
Novák, P., Shklover, V., and Nesper, R. (1994). Magnesium insertion in vanadium oxides: a structural study. Z. Phys. Chem. 185 (1), 51–68.
Novak, P., Imhof, R., and Haas, O. (1999). Magnesium insertion electrodes for rechargeable nonaqueous batteries - a competitive alternative to lithium? Electrochim. Acta. 45, 351–367.
Peng, X., Zhang, X., Wang, L., Hu, L., Cheng, S. H.-S., Huang, C., et al. (2016). Hydrogenated V2O5 nanosheets for superior lithium storage properties. Adv. Funct. Mater. 26 (5), 784–791. doi:10.1002/adfm.201503859
Pereira-Ramos, J. P., Messina, R., and Perichon, J. (1987). Electrochemical formation of a magnesium vanadium bronze MgxV2O5 in sulfone-based electrolytes at 150°C. J. Electroanal. Chem. Interfacial Electrochem. 218 (1), 241–249. doi:10.1016/0022-0728(87)87019-5
Perera, S. D., Archer, R. B., Damin, C. A., Mendoza-Cruz, R., and Rhodes, C. P. (2017). Controlling interlayer interactions in vanadium pentoxide-poly(ethylene oxide) nanocomposites for enhanced magnesium-ion charge transport and storage. J. Power Sources 343, 580–591. doi:10.1016/j.jpowsour.2017.01
Roedern, E, Kühnel, RS, Remhof, A, and Battaglia, C. (2017). Magnesium ethylenediamine borohydride as solid-state electrolyte for magnesium batteries. Sci. Rep. 7 (1), 46189. doi:10.1038/srep46189
Sa, N., Kinnibrugh, T. L., Wang, H., Gautam, G. S., Chapman, K. W., Vaughey, J. T., et al. (2016). Structural evolution of reversible Mg insertion into a bilayer structure of V2O5 center dot nH(2)O xerogel material. Chem. Mater. (2016). 28 (9), 2962–2969. doi:10.1021/acs.chemmater.6b00026
Sa, N., Wang, H., Proffit, D. L., Lipson, A. L., Key, B., Liu, M., et al. (2016). Is alpha-V2O5 a cathode material for Mg insertion batteries? J. Power Sources 323, 44–50. doi:10.1016/j.jpowsour.2016.05.028
Saha, P., Datta, M. K., Velikokhatnyi, O. I., Manivannan, A., Alman, D., and Kumta, P. N. (2014). Rechargeable magnesium battery: current status and key challenges for the future. Prog. Mater. Sci. 66, 1–86. doi:10.1016/j.pmatsci.2014.04.001
Sahadeo, E, Song, J, Gaskell, K, Kim, N, Rubloff, G, and Lee, SB. (2018). Investigation of the water-stimulated Mg2+ insertion mechanism in an electrodeposited MnO2 cathode using X-ray photoelectron spectroscopy. Phys. Chem. Chem. Phys. 20 (4), 2517–2526. doi:10.1039/c7cp06312a
Shao, J, Li, X, Wan, Z, Zhang, L, Ding, Y, Zhang, L, Qu, Q, and Zheng, H. (2013). Low-cost synthesis of hierarchical V2O5 microspheres as high-performance cathode for lithium-ion batteries. ACS Appl Mater Interfaces., 5, 7671. doi:10.1021/am401854v
Shao, Y., Rajput, N. N., Hu, J., Hu, M., Liu, T., Wei, Z., et al. (2015). Nanocomposite polymer electrolyte for rechargeable magnesium batteries. Nanomater. Energy. 12, 750–759. doi:10.1016/j.nanoen.2014.12.028
Shklover, V., Haibach, T., Ried, F., Nesper, R., and Novák, P. (1996). Crystal structure of the Product of Mg2+Insertion into V2O5 single crystals. J. Solid State Chem. 123 (2), 317–323. doi:10.1006/jssc.1996.0186
Shterenberg, I., Salama, M., Gofer, Y., and Levi, E. (2014). The challenge of developing rechargeable magnesium batteries | MRS Bulletin | Cambridge Core. MRS Bull. 39 (5), 453–460. doi:10.1557/mrs.2014.61
Son, S. B., Gao, T., Harvey, S. P., Steirer, K. X., Stokes, A., Norman, A., Wang, C., et al. (2018). An artificial interphase enables reversible magnesium chemistry in carbonate electrolytes. Nat. Chem. 10 (5), 532–539. doi:10.1038/s41557-018-0019-6
Song, J., Noked, M., Gillette, E., Duay, J., Rubloff, G., and Lee, S. B. (2015). Activation of a MnO2 cathode by water-stimulated Mg(2+) insertion for a magnesium ion battery. Phys. Chem. Chem. Phys. (2015). 17 (7), 5256–5264. doi:10.1039/c4cp05591h
Su, J., Tsuruoka, T., Tsujita, T., Nishitani, Y., Nakura, K., and Terabe, K. (2019). Atomic layer deposition of a magnesium phosphate solid electrolyte. Chem. Mater. 31 (15), 5566–5575. doi:10.1021/acs.chemmater.9b01299
Tang, K., Du, A. B., Dong, S. M., Cui, Z. L., Liu, X., Lu, C. L., et al. (2020). A stable solid electrolyte interphase for magnesium metal anode evolved from a bulky anion lithium salt. Adv. Mater. 32 (6), e1904987. doi:10.1002/adma.201904987
Tepavcevic, S., Connell, J. G., Lopes, P. P., Bachhav, M., Key, B., Valero-Vidal, C., et al. (2018). Role of structural hydroxyl groups in enhancing performance of electrochemically-synthesized bilayer V2O5. Nanomater. Energy (2018). 53, 449–457. doi:10.1016/j.nanoen.2018.09.005
Tepavcevic, S, Liu, Y, Zhou, D, Lai, B, Maser, J, Zuo, X, et al. (2015). Nanostructured layered cathode for rechargeable Mg-ion batteries. ACS Nano. 9 (8), 8194–8205. doi:10.1021/acsnano.5b02450
Wang, B, Xu, B, Liu, T, Liu, P, Guo, C, Wang, S, et al. (2014). Mesoporous carbon-coated LiFePO4 nanocrystals co-modified with graphene and Mg2+ doping as superior cathode materials for lithium ion batteries. Nanoscale 6 (2), 986–995. doi:10.1039/c3nr04611g
Wang, Y., Lin, C. F., Rao, J. C., Gaskell, K., Rubloff, G., and Lee, S. B. (2018). Electrochemically controlled solid electrolyte interphase layers enable superior Li-S batteries. Acs Applied Materials & Interfaces 10 (29), 24554–24563. doi:10.1021/acsami.8b07248
Whittingham, M. S. (1976). The role of ternary phases in cathode reactions. J. Electrochem. Soc. 123 (3), 315–320. doi:10.1149/1.2132817
Xu, Y. N., Deng, X. W., Li, Q. D., Zhang, G. B., Xiong, F. Y., Tan, S. S., et al. (2019). Vanadium oxide pillared by interlayer Mg2+ ions and water as ultralong-life cathodes for magnesium-ion batteries. Inside Chem. 5 (5), 1194–1209. doi:10.1016/j.chempr.2019.02.014
Yu, L, and Zhang, X. (2004). Electrochemical insertion of magnesium ions into V2O5 from aprotic electrolytes with varied water content. J. Colloid Interface Sci. 278 (1), 160–165. doi:10.1016/j.jcis.2004.05.028
Zhang, R., and Ling, C. (2016). Status and challenge of Mg battery cathode. MRS Energy & Sustainability 3 (1), E1. doi:10.1557/mre.2016.2
Zhang, X.-F., Wang, K.-X., Wei, X., and Chen, J.-S. (2011). Carbon-Coated V2O5 nanocrystals as high performance cathode material for lithium ion batteries. Chem. Mater. 23 (24), 5290–5292. doi:10.1021/cm202812z
Keywords: vanadium oxide, magnesium ion, water, hydrated, solvated, thin film
Citation: Johnston B, Henry H, Kim N and Lee SB (2021) Mechanisms of Water-Stimulated Mg2+ Intercalation in Vanadium Oxide: Toward the Development of Hydrated Vanadium Oxide Cathodes for Mg Batteries. Front. Energy Res. 8:611391. doi: 10.3389/fenrg.2020.611391
Received: 29 September 2020; Accepted: 23 November 2020;
Published: 25 January 2021.
Edited by:
Jin Xie, ShanghaiTech University, ChinaReviewed by:
Seung-Tae Hong, Daegu Gyeongbuk Institute of Science and Technology, South KoreaXiaogang Yang, Xuchang University, China
Copyright © 2021 Johnston, Henry, Kim and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nam Kim, bnNrMDI0OEB1bWQuZWR1; Sang Bok Lee, c2xlZUB1bWQuZWR1