 Zbigniew Łodziana
Zbigniew Łodziana- Institute of Nuclear Physics Polish Academy of Sciences, Krakòw, Poland
Hydrogen in the solid state compounds is still considered as a safe method of energy storage. The ultimate metal hydrides or other materials that can be used for this purpose remain unknown. Such metal hydrides shall have favorable thermodynamics and kinetics of hydrogen ad/desorption, and it shall be resistant to contamination of H2 and should not constitute any environmental hazards. Theoretical investigations, based on quantum mechanics approach, have a well-established position in modern materials research; however, their application for design of new alloys with tailored properties for reversible hydrogen storage is rarely present in the literature. The mainstream research deals with accurate prediction of thermodynamic and structural properties of hydrides as a function of composition or external parameters. On the other hand, the kinetic effects related to hydrogen transport or interaction between solid and pure or contaminated H2 are more demanding. They cannot be easily automated. We present calculations of the equilibrium crystal shapes for LaNi5 and TiFe—two important materials that show reversible hydrogen cycling near ambient conditions. Understanding of the surface properties is crucial for development of materials with better cyclability or resistance to hydrogen impurities. Indeed, the calculated adsorption energy of carbon oxides or water is stronger than hydrogen. These molecules block the active sites for H2 dissociation, leading to formation of surface oxides. Particularly strong adsorption of CO/CO2 on TiFe explains large degradation of hydrogen storage capacity of this compound by carbon oxides. Over-representation of La on exposed facets of LaNi5 is related to formation of La2O3 and La(OH)3. Such examples show that the present development of computational methods allows reliable studies of intermetallic properties related to their surface or novel catalytic applications.
1 Introduction
Alloys and intermetallic compounds have been for a long time and they still are considered as promising hydrogen storage materials (Ivey and Northwood, 1983; Sakintuna et al., 2007; Joubert et al., 2021). These claims and hopes are based on the fact that in contact with the surface of these compounds, the bond between hydrogen atoms in the H2 molecule is broken [H2 dissociation energy 4.477 eV (Herzberg and Monfils, 1961)], and hydrogen atoms diffuse into the bulk forming a stable compound—a metal hydride. This process can be reversed at mild conditions and gaseous hydrogen is extracted from the hydride. Multiple repetitions of such procedure, without significant degradation of kinetic and thermodynamic parameters, are very appealing for practical applications. As a matter of fact, many decades of metal hydride research, numerous real live applications, and enormous literature on this subject justify hopes for applications (Fukai, 2005; Sakintuna et al., 2007; Züttel et al., 2008; Mohtadi and Orimo, 2017; Fuel Cells Bulletin, 2018; Modi and Aguey-Zinsou, 2021). Hydrogen is able to form stable hydrides with variety of metals as well as intermetallic compounds. These compounds are traditionally denoted as AB, AB2, A2B, or AB5. The key aspect of hydride formation is related to the so-called pressure composition isotherms that reveal enthalpy of formation (ΔH0) and entropy of formation (ΔS0) as well as the hydride stoichiometry or an insight into kinetic parameters. There are, however, very stable hydrides such as AlH3 (Saitoh et al., 2008), MgH2 (Baran and Polański, 2020), or LiH (Jain et al., 2016) that cannot be formed by simple exposition of metals to hydrogen because the dissociation of H2 does not easily occur at the surface, and pressures of dozens of kbar with elevated temperature are required in order to form a hydride.
The mainstream literature on metal hydrides (Fukai, 2005; Züttel et al., 2008; Broom, 2011) details all aspects of general reaction
Metal hydrides have great potential to be used as distributed stationary hydrogen (energy) storage; however, some aspects not directly related to their hydrogen capacity, operating conditions, or thermodynamic properties gain importance. As an example, one can mention cyclic stability, cycle life, or sensitivity to hydrogen impurities. It was recognized in the early years of the hydride research that intermatallics and alloys are vulnerable to hydrogen contamination by impure gases such as CO, CO2, O2, H2O, CH4, and others (Sandrock and Goodell, 1980; Block and Bahs, 1983). Their minimal content (on ppm level) in H2 may diminish hydrogenation kinetics or even irreversibly block formation of the hydride (Sandrock and Goodell, 1984; Corré et al., 1997; Schweppe et al., 1997; Hanada et al., 2015; Dematteis et al., 2021). Hydrogen purification is an energetically expensive process, especially on the small scale (Du et al., 2021), and thus, understanding of the problems related to alloy and hydride degradation is crucial in practical applications. The dissociation of H2 requires that active sites for this process are present at the surface, and they are accessible to hydrogen (Züttel et al., 2008). For example, the ideal clean surface of Pd requires at least three empty surface sites to allow hydrogen dissociation (Mitsui et al., 2003; Lopez et al., 2004). As the hydrogen impurity molecules interact with the surface, they might block the active sites for H2 dissociation or a protective layer, that is, oxide, hydroxide, and sulfide might be formed. Such a layer prevents transport or dissociation/association of H2 molecules (Sandrock and Goodell, 1980).
Metal hydrides might act as a catalyst, for example, the for hydrogenation reaction of C2H4 over LaNi5 (Soga et al., 1977). It was reported that the reaction rate was two orders of magnitude higher on hydrogenated alloy (LaNi5H2.4). LaNi4X (X = Ni, Cr, Al, and Cu) were reported as catalysts for CO2 methanation (Ando et al., 1995) with significant activity of LaNi5 and LaNi4Cr. The activity is postulated due to the presence of Ni, known as the methanation catalyst (Aziz et al., 2015). To best of our knowledge, no atomistic understanding of mechanisms beyond these catalytic aspects or poisoning mechanism of LaNi5 exists. Empirical models of LiNi5 surfaces and a chemical analysis indicate that the surface is enriched in La, La2O3, and La(OH)3 (Wallace et al., 1979; Selvam et al., 1991). This oxide layer is permeable for hydrogen; thus, H2 can reach Ni atoms underneath, dissociate there, and form a hydride. Similar situation occurs for TiFe; however, permeability of the oxide layer is low for hydrogen (Edalati et al., 2013). One important difference between two compounds is that unlike LaNi5 TiFe is far more sensitive to CO or CO2, even a small amount of these gasses passivates or poisons this compound. Energetically demanding heat treatment is required to reactivate TiFe (Sandrock and Goodell, 1980; Block and Bahs, 1983). The so-called activation process required for utilization of the alloy’s full potential for reversible hydrogen storage is mostly related to formation of exposed active sites for hydrogen dissociation by perturbing any protective layer that covers them (Schlapbach and Brundle, 1981; Kisi et al., 1992; Edalati et al., 2013). The alloy contamination primarily occurs at the surface or, in general, at the interface between solid and gaseous hydrogen. Therefore, a reliable picture of the alloy’s surface is required for understanding its interaction with gasses.
Gas interactions with surfaces are well studied for heterogeneous catalytic and electrocatalytic systems, where insight from theoretical methods already guides discoveries of new catalysts (Greeley et al., 2006; Nørskov et al., 2009; Seh et al., 2017; Pérez-Ramírez and López, 2019). In particular, it is well known that surface modifications by alloying (restricted to top atomic layers) have a strong influence on surface reactivity (Christensen et al., 1997; Greeley et al., 2006). However, atomistic investigations of the bulk alloy surfaces are at an early stage, in particular, due to the difficulties related to segregation, dealloying, and other processes occurring at the surface (Cao et al., 2019; Mamun et al., 2019).
The quantum mechanics–based research in metal hydrides is rather scarce, taking into account a growing impact of theoretical, atomistic calculations on materials science. Such calculations for compounds relevant for hydrogen storage were pioneered decades ago (Malik et al., 1982; Satterthwaite and Jena, 1983). New developments or predictions of metal hydride properties are still far beyond these for secondary batteries or even the so-called complex hydrides, where hydrogen atoms are incorporated into a molecule or ion like
Hereby, we want to pave the way toward the description of the surface properties of LaNi5 and TiFe by calculating the surface energy for a variety of low index facets and an analysis of the simplest gas adsorption processes for exposed facets. This requires a series of practical simplifications that will be described below. In the era of advanced computational screening and machine learning methods, these examples might seem trivial. In fact, they are not as the formulation of the realistic models of the surface cannot yet be automated, except for elemental metals or non-metallic elements (Tran et al., 2016). After presentation of methods, the surface energies and the equilibrium crystal shapes for LaNi5 and TiFe are presented for the first time. Adsorption energies for simple molecules like H2, CO, CO2, and H2O complement the surface calculations.
2 Methods
LiNi5 has a hexagonal structure (symmetry P6/mmm) (Kisi et al., 1992) with a fraction of Ni located at 2c Wyckoff positions, sharing the (ab)-plane with La, and there are three La nearest neighbors 2.9 Å apart from Ni. The second site occupied by Ni is 3g placed within the (ab)-plane in between La layers, and there are four La nearest neighbors separated by 3.2 Å from Ni (Figure 1). TiFe (cubic, P
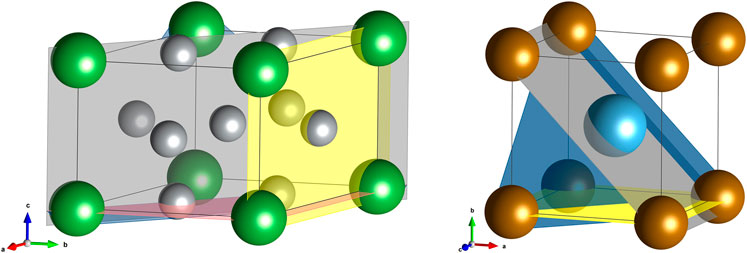
FIGURE 1. (A) The crystal structure of LaNi5. Green spheres are for La and grey for Ni; examples of (0001), (01–10), and (11–20) lattice planes are shown in red, yellow, and grey, respectively. (B) The crystal structure of TiFe. Brown spheres are for Fe and blue one for Ti. Examples of (010), (110), and (111) lattice planes are shown in yellow, grey, and blue, respectively.
All calculations were performed within spin polarized density functional theory (DFT) with a periodic plane wave basis set as implemented in the Vienna ab initio Simulation Package (Kresse and Furthmüller, 1996b,a). The calculation parameters were the cutoff energy for the basis set expansion 500 eV, the k-point sampling with density k⋅a ≥60, the convergence criteria for electronic degrees of freedom 10–6 eV/Å, a conjugated gradient method for atomic relaxation and convergence criteria 10–2 eV/Å, Projected Augmented Wave (PAWs) potentials (Blöchl, 1994; Kresse and Joubert, 1999) for atoms, and the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional (Perdew et al., 1996). The surface calculations were performed in the slab geometry with minimum 12 Å of vacuum separating slab images. The slab geometry for each facet was created using a Pymatgen package (Sun and Ceder, 2013; Tran et al., 2016); the slab thickness was minimum 15 Å. Only stoichiometric slabs were considered; for facets where two or more possible surface terminations exist, all of them were taken into account, and the reported surface energies are for terminations that are the most stable. None of the LaNi5 surface terminations has the inversion symmetry; thus, the surfaces are different on both sides of the slab. We did not consider possible surface reconstructions or off-stoichiometry, and the reported data are the effective surface energies when both surfaces are exposed. Similar procedure was applied for TiFe; however, for this compound, facets with indexes (110), (211), (310), (321), and (332) possess inversion symmetry/mirror plane parallel to the surface. The surface energies were calculated with the approach of (Fiorentini and Methfessel, 1996). Gas molecule adsorption studies were done on the one side of the slab; the adsorption energy is defined as Eads = Etot − (Eslab + EX), where Etot is the energy of the slab with adsorbed X (X = H2, CO, CO2, and H2O molecule), Eslab is the total energy of the slab, and EX is the ground state energy for the X molecule (calculated in cubic box with edge of 12 Å). For adsorption studies, three atomic layers on the bottom side of the slab were frozen and a 2 × 2 surface supercell was used.
3 Results
The calculated ground state lattice parameters for LaNi5 are a = 5.001 Å and c = 3.986 Å. This compares well with experimental data a = 5.0125 Å and c = 3.9873 Å (Kisi et al., 1992) or previous calculations a = 5.008 Å and c = 3.967 Å (Hector et al., 2003). For TiFe, the calculated lattice constant is a = 2.946 Å, and it can be compared to experimental a = 2.9789 Å (Thompson et al., 1989). The optimized structures were used for construction of the surfaces for both compounds and calculations of the equilibrium crystal shapes.
3.1 Surface Energy
Calculated surface energies are presented in Figure 2 and Figure 3. The surface energy for constituent metals is in the range 0.7–0.8 J/m2 for La, 1.9–2.4 J/m2 for Ni, 1.9–2.3 J/m2 for Ti, and 2.4–3.4 J/m2 for Fe (Tran et al., 2016). The weighted average of elemental surface energies for LaNi5 ranges from 1.7 J/m2 to 2.13 J/m2 that covers the range of its surface energies calculated here, with three exceptions (10–10), (02–21), and (11–21). For these facets, lanthanum is over-represented (Figure 1); moreover, such crystal planes are different from the (0001) face often considered in the literature.
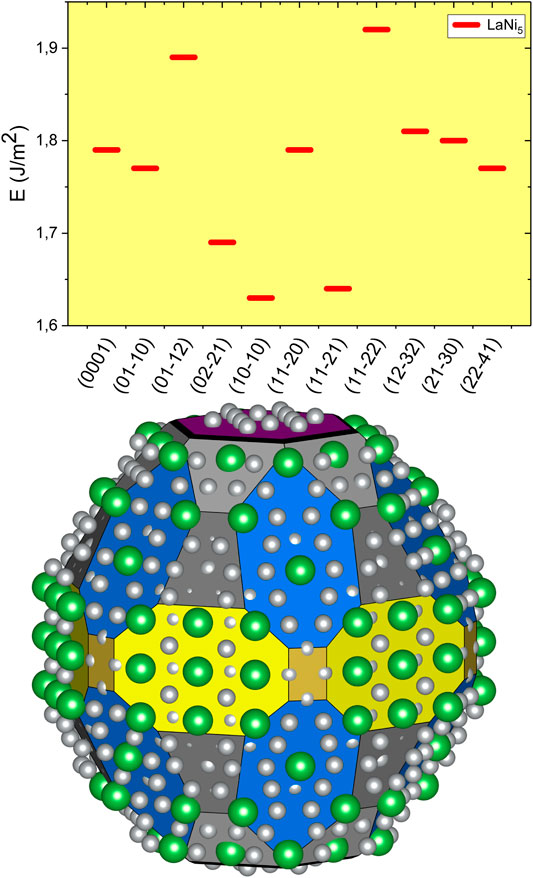
FIGURE 2. Surface energy for selected facets of LaNi5. The equilibrium crystal shape for LaNi5 constructed with the Wulff procedure. Yellow color stands for the (10–10) facet, dark yellow for (11–20), fuchsia for (0001), dark grey for (02–21), grey for (01–11), black for (01–12), and blue for (11–21). The green spheres are for La atoms and small grey ones for Ni.
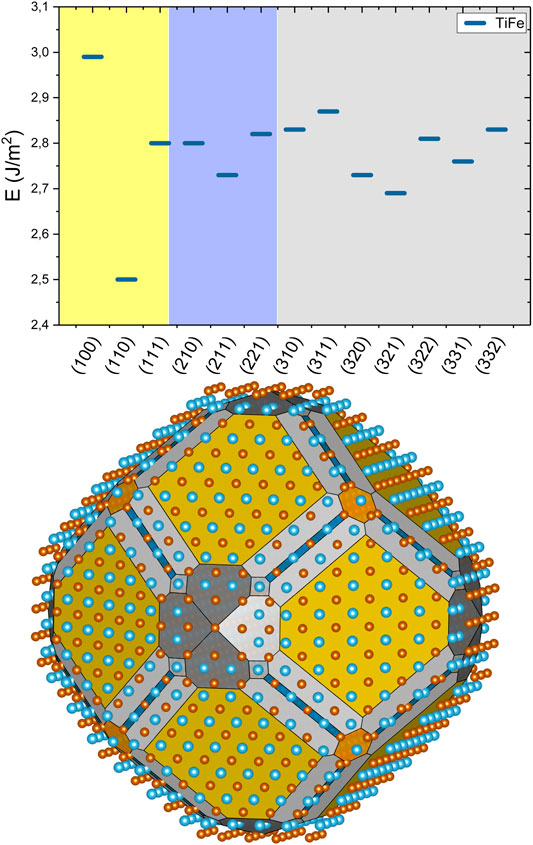
FIGURE 3. Surface energy for TiFe facets with indexes l ≤ 3. Background colors, yellow for (1xx), blue for (2xx), and grey for (3xx) surfaces, correspond to shades used below. The equilibrium shape of the TiFe nanocrystal is constructed with the Wulff procedure. Yellow color stands for (110) facet, orange for (111), dark grey for (310), light grey for (321), light blue for (211), and dark blue for (221). Brown spheres are for Fe and blues ones are for Ti.
Based on the calculated surface energies, we have constructed the equilibrium shape of the LaNi5 nano-crystal with the Wulff method (Wulff, 1901; Barmparis et al., 2015). The crystal symmetry was used for determination of equivalent facets; the nano-crystallite shape is presented in Figure 2. In the equilibrium crystal shape, only 7.7% of the exposed surface belongs to (0001) termination, and the largest fraction of 40.4% is the (11–21) facet. Other exposed surface terminations are (10–10), 20.9%; (01–11), 14.1%; and (02–21), 13.4%. Possible atomic compositions of the surface are shown in Figure 2; for facets like (10–10), La is over-represented at the surface and it might already seed the surface oxidation and formation of La2O3. This can be accompanied by further segregation and La diffusion toward the surface. Observed La2O3 and La (OH)3 can be explained by the composition of most exposed surfaces of LaNi5. The formation of oxide shifts the surface stability toward the stability of the interface, a thermodynamically driven process of self-limiting passivation.
The surface energies of TiFe are generally larger than these for LaNi5, and they fall in the average energies for constituting elements, that is, 2.15 J/m2 to 2.85 J/m2, except the (100) facet.
The (100) surface has usually very large surface energy for bcc metals (Fe, Cr) unlike for alkali metals where the surface energy is very low for all surface terminations (Tran et al., 2016). The lowest surface energy for the (110) crystal plane is not surprising. This is the bcc lattice plane with the densest atom packing. The calculated equilibrium crystal shape is dominated with (110) surface family (50% of all exposed surface); however, surface terminations with higher indexes are largely represented. Such facets expose under-coordinated atoms that are very reactive (Nørskov et al., 2009; Seh et al., 2017) contributing to strong passivation and difficult activation of TiFe alloys. In theoretical calculations, surface terminations with indexes larger than two are rarely used, except for the analysis of highly reactive surfaces (Honkala et al., 2005).
3.2 Gas Adsorption
In order to probe the surface properties, we have calculated the adsorption energies for H2, CO, CO2, and H2O molecules at the (0001) surface of LaNi5 and (110) surface of TiFe, Figure 4. For each molecule, we have considered associative and dissociative adsorption and a variety of adsorption sites. For each compound, it is possible to distinguish on top, bridge, and threefold adsorption sites usually considered in the literature. Due to composition, a top site can be over La, Ni, or Ti, Fe; the bridge site splits into La–Ni and Ni–Ni or Ti–Ti, Fe–Fe, or Ti–Fe. Threefold site consists of 2Ni–La, 2Fe–Ti, or 2Ti–Fe. All adsorption possibilities were considered and the most stable adsorbate configurations are shown in Figure 4. For LaNi5, additional coverage dependence of adsorption energy was considered for H2 and CO.
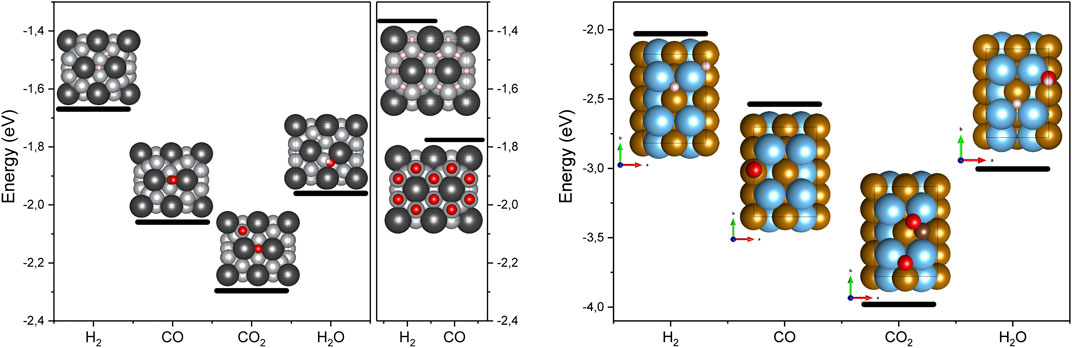
FIGURE 4. The adsorption energies for H2, CO, and CO2 molecules at LaNi5 (A) and TiFe (B). For TiFe, additional H2O was considered. Black spheres are for La, grey for Ni, light blue for Ti, and light brown for Fe. Hydrogen is represented by small light pink spheres, oxygen by red, and C by brown. The adsorption energies are per adsorbed molecule.
For the low coverage limit [
In general, the CO2 dissociation process is very complex. It can lead to formation of nickel carbonyls (NiCO4), formate anions, or radicals (COOH−) or dissociation into elements C and O. Such processes are beyond the scope of this study; however, here we show that dissociative adsorption of CO2 on LaNi5 is thermodynamically stable, and thus, it opens a route for catalytic transformation of this gas and indicates that surface oxidation can be related to the presence of CO/CO2. The adsorption energy for the water molecule is ΔE = −0.37 eV/H2O on top of La, and for dissociation to OH and H, the adsorption energy is ΔE = −1.96 eV/H2O (see Figure 4).
For TiFe, only a low coverage adsorption (
In order to bring calculated adsorption energies into a broader context, one can notice that hydrogen adsorption energies calculated here are generally larger than these calculated for pure elements [−1.29 eV at Ni (100), −1.63 eV at Fe (111), and −2.01 eV at Ti (0001)] (Winther et al., 2019; Billeter et al., 2021). We have found a small activation barrier for H2 dissociation at TiFe (110) (0.07 eV). For CO, reported molecular adsorption energies are of the range −1.53 eV at the Fe (211) surface or −1.96 eV at the Co3Ti (111) facet. For water molecules, adsorption energies strongly depend on particular surface geometry, for example, energies of −0.52 eV and −1.72 eV are reported for Fe (211) and molecular or dissociated (OH + H) state, respectively. For the (111) surface of Fe, these energies are lower: −0.09 eV and −0.60 eV, respectively (Winther et al., 2019). Thus, adsorption energies for carbon oxides and H2O on TiFe (110) calculated here are rather large. However, adsorption of these molecules on binary alloys can result in adsorption energies as large as −5.35 eV for CO2 on FeZr (Mamun et al., 2019). Such large adsorption energies for CO and CO2 are in line with a particularly strong poisoning effect of these gasses on TiFe. In fact, the intrinsic nature of the gas adsorption on binary alloy surfaces still poses many open questions. Two types of atoms constituting an alloy bring the reactivity of dense and flat surfaces toward this of high index facets for late transition metals.
4 Discussion
Calculated surface energies and equilibrium crystal shapes for LaNi5 and TiFe indicate that theoretical models for the surface-related properties in these two intermetallics have to be created with care, as the exposed facets differ from those of elemental metals. Intermetallics and metal hydrides are subject to the mass transport during hydrogen ad/desorption. Within this process, a fresh surface of either phase is formed. The main driving force for the type of surface is related to thermodynamic stability as exemplified with Wulff construction. Thus, the equilibrium nanocrystal shapes are not only a theoretical concept. To the best of our knowledge, there are no reports of direct observation of the nanocrystalline shapes of LaNi5 that allows comparison. However, an indirect indication of preferential exposure of the (10–10) surface of LaNi5 can be deduced from the report of dislocations moving on (10–10) slip planes (Inui et al., 1998). The slip plane must be a low energy surface termination, and indeed, it is such a surface, as presented in Figure 2. For TiFe, one can refer to a detailed transmission electron microscopy study of crystallites as small as 7 nm (Emami et al., 2015). Preferential exposure of the (110) surface family is clearly observed there, and the crystallite shape agrees well with this in Figure 3. Our analysis brings other facets that are exposed at the edges; these facets with high indexes possess under-coordinated atoms that are more reactive (Nørskov et al., 2009) and constitute 50% of the surface. The change of the surface properties between metal and metal hydride is also related to the degradation processes. With repetitive cycling, it can be strongly affected by formation of oxides, hydroxides, or carbides. Further studies are required in this direction.
Calculated adsorption energies for hydrogen, CO, CO2, and H2O molecules indicate that the binding energy for larger molecules is stronger than H2. The physisorption (for weak interactions) or chemisorption (strong interaction) describes interactions of gasses with surfaces. Strong interactions are accompanied by the charge transfer between solid and adsorbed molecules, and once charge donation from the surface to bonding molecular orbitals occurs, the molecule dissociation or transformation might take place. We have performed the charge distribution analysis for adsorbed molecules according to the Bader method (Henkelman et al., 2006) (see Table 1). For dissociative adsorption, the sum of charges for all adsorbed species is reported in Table 1. The charge donation is observed for all adsorbed molecules and for all surfaces considered here. The charge transfer is systematically larger for TiFe as the adsorption energies are larger too.
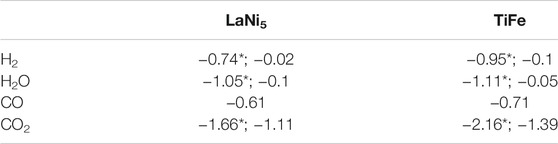
TABLE 1. Bader charges in e calculated for LaNi5 (0001), and TiFe (110) surfaces. The asterisk is for the dissociated state.
Such electron donation leads to bond breaking in H2 (this process is not activated on LaNi5 and calculated activation energy on TiFe is 0.07 eV) or formation of CO2 radical anions. These radical anions are no longer straight, and the O-C-O angle is of the order 130° (Álvarez et al., 2017). In the present calculations, the bend angle of CO2 is 123° for adsorption on LaNi5 and 118° for TiFe. Such large deformation of the CO2 molecule is due to the composition of the surface and geometrical factors. The length of CO2 is 2.32 Å; once adsorbed, the carbon atom locates in the bridge site and oxygen points toward the second element. For LaNi5, the Ni–Ni bridge site for C is preferred and oxygen is oriented toward La, and nearest La atoms are separated by 5.0 Å. On TiFe, the carbon atom is at the Fe–Fe bridge site and oxygen points toward Ti that are separated by 4.3 Å, and the CO2 bending angle is larger for this case. The dissociation of carbon dioxide is an activated process (Liu et al., 2012), and in order to fully understand the interaction of carbon oxides with surfaces considered here, the separate study are required that shall include activation energies and formation of Ni/Fe carbonyls, formate radicals, and other molecules or oxidation.
Adsorption energies calculated here bring important information for the compound property. The firm conclusion from this study is that large adsorption energies for CO, CO2, and H2O shall lead to accumulation of these molecules at the surface, even if they are present in very small concentrations. Such accumulation blocks surface sites that are necessary for H2 dissociation or trigger surface reactions that potentially form even stronger protective layers at the surface. For example, all metals that constitute presented alloys form stable oxides. Their enthalpy of formation varies strongly with the element, and the most stable is La2O3 [ΔH0 = −1791.6 kJ/mol (Konings et al., 2014)]; for TiO2 (rutile), ΔH0 = −944.747 kJ/mol (Chase, 1998), for NiO, ΔH0 = −239.74 kJ/mol (Boyle et al., 1954), and for Fe2O3 ΔH0 = −825.503 kJ/mol (Chase, 1998). Additionally stable carbides like TiC [ΔH0 = −184.096 kJ/mol (Chase, 1998)] can be formed. Dissociative adsorption of water or carbon oxides will, thus, result in very strongly bound oxygen or oxide layers for large coverage. Large adsorption energies for CO and CO2 on TiFe explain sensitivity of this compound to such impurities in H2.
Adsorption of molecules or atoms at transition metal surfaces is successfully described by correlation of the adsorption energy and center of d-band (Newns, 1969; Nørskov et al., 2009). The density of states for the bulk and surface are shown in Figure 5. For LaNi5, it can be seen that the center of the valence band for Ni strongly depends on the exposed facet and the site symmetry of Ni. For the (0001) surface, the Ni valence d-band is shifted to lower energies. This effect is less pronounced for the (10–10) surface. As the La electronic states are located above the Fermi level, the reactivity of the surface is related to exposed nickel and La can be oxidized.
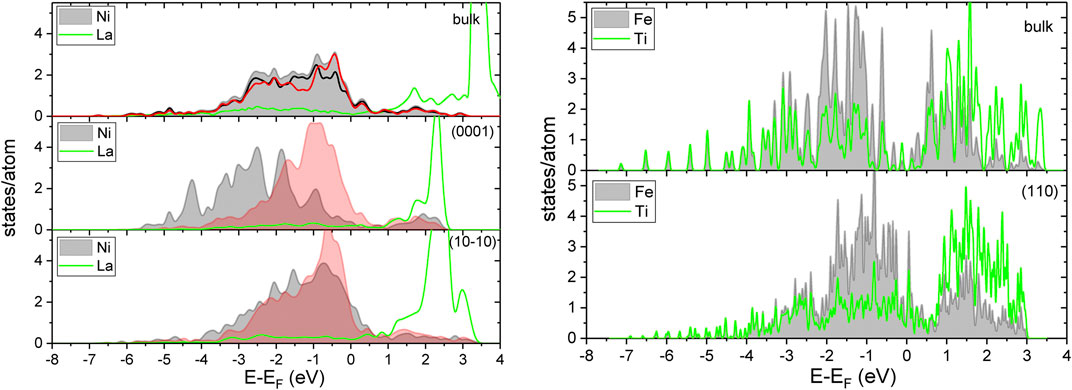
FIGURE 5. The spin averaged density of states for LaNi5 projected on La and Ni for the bulk and atoms exposed at the surfaces. The shaded area is for Ni with red lines for 3g sites and black for 2c (top). The spin averaged density of states projected on Ti and Fe for the bulk and atoms exposed at the (110) surfaces of TiFe.
Besides the stoichiometric composition of the surface where La is over-represented and oxidation of this element occurs, the modification of the electronic structure is an additional factor of the surface reactivity of resistivity for hydrogen contamination. For TiFe, the modification of the valence band at the surface is also present; here, the center of the valence band moves to higher energies at the surface, and the shifts of the band position are similar for Ti and Fe.
5 Summary
In the present study, we have shown an example of the surface energy calculations for alloys relevant for hydrogen storage. The equilibrium crystal shapes were determined for LaNi5 and TiFe. The composition of exposed surfaces of LaNi5 indicates over-representation of La which can be oxidized. The strong binding energies of CO, CO2, and H2O at the surfaces of compounds considered here indicate that they accumulate at the surface, blocking the active sites for hydrogen adsorption/dissociation or became precursors for catalytic activity of intermetallics. Large adsorption energies for carbon oxides on TiFe (110) can explain a very high sensitivity of this material to hydrogen contaminations by CO/CO2. Understanding of the processes at the surface is one of the key points for improvement of the cyclic stability of metal hydrides or finding new applications for catalyzed reactions. The future calculations in this direction shall bring significant advances in material design for efficient hydrogen storage or catalytic transformations with metal hydrides.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This manuscript is the results of the research project funded by the NCBiR project BIOSTRATEG2/297310/13/NCBR/2016.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
CPU allocation at the PL-Grid infrastructure is kindly acknowledged.
References
Álvarez, A., Borges, M., Corral-Pérez, J. J., Olcina, J. G., Hu, L., Cornu, D., et al. (2017). CO2Activation over Catalytic Surfaces. ChemPhysChem 18, 3135–3141. doi:10.1002/cphc.201700782
Ando, H., Fujiwara, M., Matsumura, Y., Miyamura, H., Tanaka, H., and Souma, Y. (1995). Methanation of Carbon Dioxide over LaNi4X-type Intermetallic Compounds as Catalyst Precursor. J. Alloys Compd. 223, 139–141. doi:10.1016/0925-8388(94)01488-4
Aziz, M. A. A., Jalil, A. A., Triwahyono, S., and Ahmad, A. (2015). CO2 Methanation over Heterogeneous Catalysts: Recent Progress and Future Prospects. Green. Chem. 17, 2647–2663. doi:10.1039/C5GC00119F
Baran, A., and Polański, M. (2020). Magnesium-Based Materials for Hydrogen Storage-A Scope Review. Materials 13, 3993. doi:10.3390/ma13183993
Barmparis, G. D., Lodziana, Z., Lopez, N., and Remediakis, I. N. (2015). Nanoparticle Shapes by Using Wulff Constructions and First-Principles Calculations. Beilstein J. Nanotechnol. 6, 361–368. doi:10.3762/bjnano.6.35
Billeter, E., Lodziana, Z., and Andreas, B. (2021). Surface Properties of the Hydrogen - Titanium Nsystem. submitted
Blöchl, P. E. (1994). Projector Augmented-Wave Method. Phys. Rev. B 50, 17953–17979. doi:10.1103/physrevb.50.17953
Block, F. R., and Bahs, H.-J. (1983). Investigation of Selective Absorption of Hydrogen by Lani5 and Feti. J. Less Common Met. 89, 77–84. doi:10.1016/0022-5088(83)90251-5
Boer, F. d., Mattens, W., Boom, R., Miedema, A., and Niessen, A. (1988). Cohesion in Metals. Netherlands: North-Holland.
Boyle, B. J., King, E. G., and Conway, K. C. (1954). Heats of Formation of Nickel and Cobalt Oxides (Nio and Coo) of Combustion Calorimetry. J. Am. Chem. Soc. 76, 3835–3837. doi:10.1021/ja01643a072
Broom, D. P. (2011). Hydrogen Storage Materials. London, United Kingdom: Springer-Verlag London. doi:10.1007/978-0-85729-221-6
Canto, G., and de Coss, R. (2000). Tight-binding Electronic Structure Calculations for the TiFe(001) Surface. Surf. Sci. 465, 59–64. doi:10.1016/s0039-6028(00)00662-2
Cao, L., Niu, L., and Mueller, T. (2019). Computationally Generated Maps of Surface Structures and Catalytic Activities for alloy Phase Diagrams. Proc. Natl. Acad. Sci. USA 116, 22044–22051. doi:10.1073/pnas.1910724116
Chase, M. (1998). NIST-JANAF Thermochemical Tables. 4th Edition. Washington, DC: American Institute of Physics.
Christensen, A., Ruban, A. V., Stoltze, P., Jacobsen, K. W., Skriver, H. L., Nørskov, J. K., et al. (1997). Phase Diagrams for Surface Alloys. Phys. Rev. B 56, 5822–5834. doi:10.1103/PhysRevB.56.5822
Corré, S., Gotoh, Y., Sakaguchi, H., Fruchart, D., and Adachi, G.-Y. (1997). The Hydrogen Confinement in Lani5 and lani5zr0.1 Hydrides Using Poisonous Gases. J. Alloys Compd. 255, 117–121. doi:10.1016/S0925-8388(96)02869-1
Dematteis, E. M., Berti, N., Cuevas, F., Latroche, M., and Baricco, M. (2021). Substitutional Effects in Tife for Hydrogen Storage: a Comprehensive Review. Mater. Adv. 2, 2524–2560. doi:10.1039/D1MA00101A
Du, Z., Liu, C., Zhai, J., Guo, X., Xiong, Y., Su, W., et al. (2021). A Review of Hydrogen Purification Technologies for Fuel Cell Vehicles. Catalysts 11, 393. doi:10.3390/catal11030393
Edalati, K., Matsuda, J., Arita, M., Daio, T., Akiba, E., and Horita, Z. (2013). Mechanism of Activation of TiFe Intermetallics for Hydrogen Storage by Severe Plastic Deformation Using High-Pressure Torsion. Appl. Phys. Lett. 103, 143902. doi:10.1063/1.4823555
Emami, H., Edalati, K., Matsuda, J., Akiba, E., and Horita, Z. (2015). Hydrogen Storage Performance of Tife after Processing by ball Milling. Acta Material. 88, 190–195. doi:10.1016/j.actamat.2014.12.052
Fiorentini, V., and Methfessel, M. (1996). Extracting Convergent Surface Energies from Slab Calculations. J. Phys. Condens. Matter 8, 6525–6529. doi:10.1088/0953-8984/8/36/005
Fuel Cells Bulletin (2018). Doosan Starts Installation of Hydrogen-Fueled 50 Mw Fuel Cell Power Plant in south korea. Fuel Cell Bull. 2018, 1. doi:10.1016/S1464-2859(18)30270-0
Fukai, Y. (2005). The Metal-Hydrogen System. Berlin, Heidelberg: Springer. doi:10.1007/3-540-28883-X
Greeley, J., Jaramillo, T. F., Bonde, J., Chorkendorff, I., and Nørskov, J. K. (2006). Computational High-Throughput Screening of Electrocatalytic Materials for Hydrogen Evolution. Nat. Mater 5, 909–913. doi:10.1038/nmat1752}
Han, S., Zhang, X.-B., Shi, S.-Q., Kohyama, M., Tanaka, H., Kuriyama, N., et al. (2008). CO Adsorption on a LaNi5Hydrogen Storage Alloy Surface: A Theoretical Investigation. ChemPhysChem 9, 1564–1569. doi:10.1002/cphc.200800080
Hanada, N., Nakagawa, T., Asada, H., Ishida, M., Takahashi, K., Isobe, S., et al. (2015). Dependence of Constituent Elements of AB5 Type Metal Hydrides on Hydrogenation Degradation by CO2 Poisoning. J. Alloys Compd. 647, 198–203. doi:10.1016/j.jallcom.2015.05.253
Hector, L. G., Herbst, J. F., and Capehart, T. W. (2003). Electronic Structure Calculations for LaNi5 and LaNi5H7: Energetics and Elastic Properties. J. Alloys Compd. 353, 74–85. doi:10.1016/S0925-8388(02)01324-5
Henkelman, G., Arnaldsson, A., and Jónsson, H. (2006). A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 36, 354–360. doi:10.1016/j.commatsci.2005.04.010
Herzberg, G., and Monfils, A. (1961). The Dissociation Energies of the H2, HD, and D2 Molecules. J. Mol. Spectrosc. 5, 482–498. doi:10.1016/0022-2852(61)90111-4
Honkala, K., Hellman, A., Remediakis, I. N., Logadottir, A., Carlsson, A., Dahl, S., et al. (2005). Ammonia Synthesis from First-Principles Calculations. Science 307, 555–558. doi:10.1126/science.1106435
Hume-Rothery, W., Mabbott, W., GilbertChannel Evans, K. M., and Carpenter, H. C. H. (1934). The Freezing Points, Melting Points, and Solid Solubility Limits of the Alloys of Sliver and Copper with the Elements of the B Sub-groups. Phil. Trans. R. Soc. Lond. A. 233, 1–97. doi:10.1098/rsta.1934.0014
Inui, H., Yamamoto, T., Di, Z., and Yamaguchi, M. (1998). C-type Dislocations Emitted from Cracks Introduced in a Thin Foil of Lani5. J. Alloys Compd. 269, 294–296. doi:10.1016/S0925-8388(98)00164-9
Ivey, D. G., and Northwood, D. O. (1983). Storing Energy in Metal Hydrides: a Review of the Physical Metallurgy. J. Mater. Sci. 18, 321–347. doi:10.1007/BF00560621
Jain, A., Miyaoka, H., and Ichikawa, T. (2016). Destabilization of Lithium Hydride by the Substitution of Group 14 Elements: A Review. Int. J. Hydrogen Energ. 41, 5969–5978. doi:10.1016/j.ijhydene.2016.02.069
Joubert, J.-M., Paul-Boncour, V., Cuevas, F., Zhang, J., and Latroche, M. (2021). LaNi5 Related AB5 Compounds: Structure, Properties and Applications. J. Alloys Compd. 862, 158163. doi:10.1016/j.jallcom.2020.158163
Kisi, E. H., Buckley, C. E., and Gray, E. M. (1992). The Hydrogen Activation of LaNi5. J. Alloys Compd. 185, 369–384. doi:10.1016/0925-8388(92)90484-Q
Konings, R. J. M., Beneš, O., Kovács, A., Manara, D., Sedmidubský, D., Gorokhov, L., et al. (2014). The Thermodynamic Properties of Thef-Elements and Their Compounds. Part 2. The Lanthanide and Actinide Oxides. J. Phys. Chem. Reference Data 43, 013101. doi:10.1063/1.4825256
Kresse, G., and Furthmüller, J. (1996a). Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 6, 15–50. doi:10.1016/0927-0256(96)00008-0
Kresse, G., and Furthmüller, J. (1996b). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186. doi:10.1103/physrevb.54.11169
Kresse, G., and Joubert, D. (1999). From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 59, 1758–1775. doi:10.1103/physrevb.59.1758
Liu, C., Cundari, T. R., and Wilson, A. K. (2012). Co2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis. J. Phys. Chem. C 116, 5681–5688. doi:10.1021/jp210480c
Lopez, N., Łodziana, Z., Illas, F., and Salmeron, M. (2004). When Langmuir Is Too Simple:H2Dissociation on Pd(111) at High Coverage. Phys. Rev. Lett. 93, 146103. doi:10.1103/PhysRevLett.93.146103
Malik, S. K., Arlinghaus, F. J., and Wallace, W. E. (1982). Calculation of the Spin-Polarized Energy-Band Structure of LaNi5and GdNi5. Phys. Rev. B 25, 6488–6491. doi:10.1103/PhysRevB.25.6488
Mamun, O., Winther, K. T., Boes, J. R., and Bligaard, T. (2019). High-throughput Calculations of Catalytic Properties of Bimetallic alloy Surfaces. Sci. Data 6, 76. doi:10.1038/s41597-019-0080-z
Mankovsky, S., Ostroukhov, A., Floka, V., and Cherepin, V. (1997). The Electronic Structure and Magnetic Properties of (100) Surface of TiFe alloy. Vacuum 48, 245–247. doi:10.1016/S0042-207X(96)00265-5
Mitsui, T., Rose, M. K., Fomin, E., Ogletree, D. F., and Salmeron, M. (2003). Dissociative Hydrogen Adsorption on Palladium Requires Aggregates of Three or More Vacancies. Nature 422, 705–707. doi:10.1038/nature01557
Modi, P., and Aguey-Zinsou, K.-F. (2021). Room Temperature Metal Hydrides for Stationary and Heat Storage Applications: A Review. Front. Energ. Res. 9, 128. doi:10.3389/fenrg.2021.616115
Mohtadi, R., and Orimo, S.-i. (2017). The Renaissance of Hydrides as Energy Materials. Nat. Rev. Mater. 2, 16091. doi:10.1038/natrevmats.2016.91
Newns, D. M. (1969). Self-consistent Model of Hydrogen Chemisorption. Phys. Rev. 178, 1123–1135. doi:10.1103/PhysRev.178.1123
Nørskov, J. K., Bligaard, T., Rossmeisl, J., and Christensen, C. H. (2009). Towards the Computational Design of Solid Catalysts. Nat. Chem. 1, 37–46. doi:10.1038/nchem.121
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Pérez-Ramírez, J., and López, N. (2019). Strategies to Break Linear Scaling Relationships. Nat. Catal. 2, 971–976. doi:10.1038/s41929-019-0376-6
Pettifor, D. G. (1986). The Structures of Binary Compounds. I. Phenomenological Structure Maps. J. Phys. C: Solid State. Phys. 19, 285–313. doi:10.1088/0022-3719/19/3/002
Sahara, R., Emura, S., and Tsuchiya, K. (2015). Theoretical Investigation of Effect of Alloying Elements on Phase Stability in Body-Centered Cubic Ti-X Alloys (X=v, Cr, Fe, Co, Nb, and Mo). J. Alloys Compd. 634, 193–199. doi:10.1016/j.jallcom.2015.02.005
Saitoh, H., Machida, A., Katayama, Y., and Aoki, K. (2008). Formation and Decomposition of AlH3 in the Aluminum-Hydrogen System. Appl. Phys. Lett. 93, 151918. doi:10.1063/1.3002374
Sakintuna, B., Lamaridarkrim, F., and Hirscher, M. (2007). Metal Hydride Materials for Solid Hydrogen Storage: A Review. Int. J. Hydrogen Energ. 32, 1121–1140. doi:10.1016/j.ijhydene.2006.11.022
Sandrock, G. D., and Goodell, P. D. (1984). Cyclic Life of Metal Hydrides with Impure Hydrogen: Overview and Engineering Considerations. J. Less Common Met. 104, 159–173. doi:10.1016/0022-5088(84)90452-1
Sandrock, G. D., and Goodell, P. D. (1980). Surface Poisoning of Lani5, Feti and (Fe,mn)ti by O2, Co and H2o. J. Less Common Met. 73, 161–168. doi:10.1016/0022-5088(80)90355-0
Satterthwaite, C., and Jena, P. (1983). Electronic Structure and Properties of Hydrogen in Metals. New York: Springer US.
Schlapbach, L., and Brundle, C. R. (1981). XPS Study of the Chemisorption Induced Surface Segregation in LaNi5 and ThNi5. J. Phys. France 42, 1025–1028. doi:10.1051/jphys:019810042070102500
Schweppe, F., Martin, M., and Fromm, E. (1997). Hydrogen Absorption of Lani5 Powders Precovered with O2, Co, H2s, Co2 or N2. J. Alloys Compd. 253-254, 511–514. doi:10.1016/S0925-8388(96)03002-2
Seh, Z. W., Kibsgaard, J., Dickens, C. F., Chorkendorff, I., Nørskov, J. K., and Jaramillo, T. F. (2017). Combining Theory and experiment in Electrocatalysis: Insights into Materials Design. Science 355, eaad4998. doi:10.1126/science.aad4998
Selvam, P., Viswanathan, B., Swamy, C., and Srinivasan, V. (1991). Surface Properties of LaNi5: A Reinvestigation. Int. J. Hydrogen Energ. 16, 23–33. doi:10.1016/0360-3199(91)90057-P
Soga, K., Imamura, H., and Ikeda, S. (1977). Hydrogenation of Ethylene over Lanthanum-Nickel (LaNi5) alloy. J. Phys. Chem. 81, 1762–1766. doi:10.1021/j100533a010
Sun, W., and Ceder, G. (2013). Efficient Creation and Convergence of Surface Slabs. Surf. Sci. 617, 53–59. doi:10.1016/j.susc.2013.05.016
Tatsumi, K., Tanaka, I., Inui, H., Tanaka, K., Yamaguchi, M., and Adachi, H. (2001). Atomic Structures and Energetics ofLaNi5−Hsolid Solution and Hydrides. Phys. Rev. B 64, 184105. doi:10.1103/PhysRevB.64.184105
Tezuka, A., Wang, H., Ogawa, H., and Ikeshoji, T. (2010). Potential Energy Surface and Hopping Path for Hydrogen inLaNi5. Phys. Rev. B 81, 134304. doi:10.1103/PhysRevB.81.134304
Thompson, P., Reilly, J. J., and Hastings, J. M. (1989). The Application of the Rietveld Method to a Highly Strained Material with Microtwins: TiFeD1.9. J. Appl. Cryst. 22, 256–260. doi:10.1107/S002188988801430X
Tran, R., Xu, Z., Radhakrishnan, B., Winston, D., Sun, W., Persson, K. A., et al. (2016). Surface Energies of Elemental Crystals. Sci. Data 3, 160080. doi:10.1038/sdata.2016.80
Wallace, W. E., Karlicek, R. F., and Imamura, H. (1979). Mechanism of Hydrogen Absorption by Lanthanum-Nickel (LaNi5). J. Phys. Chem. 83, 1708–1712. doi:10.1021/j100476a006
Winther, K. T., Hoffmann, M. J., Boes, J. R., Mamun, O., Bajdich, M., and Bligaard, T. (2019). Catalysis-hub.org, an Open Electronic Structure Database for Surface Reactions. Sci. Data 6, 75. doi:10.1038/s41597-019-0081-y
Wulff, G. (1901). XXV. Zur Frage der Geschwindigkeit des Wachsthums und der Auflösung der Krystallflächen. Z. Kristallogr. Cryst. Mater. 34, 449–530. doi:10.1524/zkri.1901.34.1.449
Züttel, A., Schlapbach, L., and Borgschulte, A. (2008). Hydrogen as a Future Energy Carrier. Weinheim: John Wiley & Sons. doi:10.1002/9783527622894.fmatter
Keywords: metal hydride, surface energy, LaNi5 alloy, TiFe alloy, DFT, hydrogen storage
Citation: Łodziana Z (2021) Surface Properties of LaNi5 and TiFe—Future Opportunities of Theoretical Research in Hydrides. Front. Energy Res. 9:719375. doi: 10.3389/fenrg.2021.719375
Received: 02 June 2021; Accepted: 10 September 2021;
Published: 20 October 2021.
Edited by:
Ah-Hyung Alissa Park, Columbia University, United StatesReviewed by:
Terry David Humphries, Curtin University, AustraliaSukanta Dash, Pandit Deendayal Petroleum University, India
Copyright © 2021 Łodziana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zbigniew Łodziana, WmJpZ25pZXcuTG9kemlhbmFAaWZqLmVkdS5wbA==