 Piotr M. Kowalski1,2*
Piotr M. Kowalski1,2* Thomas Bornhake1,2,3
Thomas Bornhake1,2,3 Oskar Cheong1,2,3
Oskar Cheong1,2,3 Noah Dohrmann4
Noah Dohrmann4 Andre Luiz Koch Liston5Shannon Kimberly Potts6Alison Shad7
Andre Luiz Koch Liston5Shannon Kimberly Potts6Alison Shad7 Rebekka Tesch1,2,3
Rebekka Tesch1,2,3 Yin-Ying Ting1,2,3
Yin-Ying Ting1,2,3- 1Forschungszentrum Jülich GmbH, Institute of Energy and Climate Research—IEK-13: Theory and Computation of Energy Materials, Jülich, Germany
- 2Jülich Aachen Research Alliance JARA Energy & Center for Simulation and Data Science (CSD), Jülich, Germany
- 3Chair of Theory and Computation of Energy Materials, Faculty of Georesources and Materials Engineering, RWTH Aachen University, Aachen, Germany
- 4Department of Chemistry, University of Chicago, Chicago, IL, United States
- 5Department of Chemistry, Princeton University, Princeton, NJ, United States
- 6Forschungszentrum Jülich GmbH, Institute of Energy and Climate Research—IEK-6: Nuclear Waste Management, Jülich, Germany
- 7Walter Scott Jr. College of Engineering, Colorado State University, Fort Collins, CO, United States
Efficient electrochemical energy storage and conversion require high performance electrodes, electrolyte or catalyst materials. In this contribution we discuss the simulation-based effort made by Institute of Energy and Climate Research at Forschungszentrum Jülich (IEK-13) and partner institutions aimed at improvement of computational methodologies and providing molecular level understanding of energy materials. We focus on discussing correct computation of electronic structure, oxidation states and related redox reactions, phase transformation in doped oxides and challenges in computation of surface chemical reactions on oxides and metal surfaces in presence of electrolyte. Particularly, in the scope of this contribution we present new simulated data on Ni/Co and Am/U-bearing oxides, and Pb, Au and Ag metal surface materials. The computed results are combined with the available experimental data for thoughtful analysis of the computational methods performance.
1 Introduction
Energy transition requires cost efficient, compact and durable materials for energy production, conversion and storage (Grey and Tarascon, 2017; Stamenkovic et al., 2017). There is a race in finding materials with increased energy and/or power density for energy storage devices (Grey and Tarascon, 2017). Energy fuels of the future such as hydrogen require efficient electrocatalysts that are economically viable and available for mass production of the fuels. Fuel cells used in conversion of hydrogen to electricity, besides electrocatalyst material, require durable and efficient electrolytes that can offer enhanced ionic conduction and withstand different operational conditions (Wang et al., 2020). Last, but not least, nuclear energy is still foreseen as clean energy resource. Deep understanding of behavior of nuclear fuel during and post reactor operation requires enhanced knowledge of the phase formations, redox reactions and oxidation states that prevail in uranium oxide materials. Understanding the phase transitions in uranium oxides is essential for assuring safety and security of nuclear technology, including efficient, post operational management of nuclear waste (Neumeier et al., 2017a; Bosbach et al., 2020).
In the last two decades, atomistic modeling became a popular research tool in various research fields, including energy materials (Chroneos et al., 2013; Jahn and Kowalski, 2014; Wu et al., 2019). Steady advancements in high performance computing and computational software enable investigation of complex systems containing hundreds of atoms from first principles (Jahn and Kowalski, 2014). Over the past decade, ab initio methods have been intensively applied, including our own studies, for computational investigation of various classes of energy materials, including these of importance in electrochemistry, energy storage and nuclear energy production, delivering information on: the structural (Rustad, 2012; Feng et al., 2013; Blanca-Romero et al., 2014; Beridze et al., 2016; Connor et al., 2021), the electronic structure (Blanca-Romero et al., 2014; Kowalski et al., 2017a; Lee et al., 2017; Kowalski et al., 2021; Murphy et al., 2021; Cui et al., 2022; Tesch and Kowalski, 2022), the elastic (Wang et al., 2005; Feng et al., 2013; Ali et al., 2016; Kowalski and Li, 2016; Ji et al., 2017a; Kowalski et al., 2017b), the thermodynamic (Mogilevsky, 2007; Feng et al., 2013; Li et al., 2014; Kowalski et al., 2015; Kowalski et al., 2016; Ji et al., 2017b; Neumeier et al., 2017b; Eremin et al., 2019; Kowalski et al., 2021), the thermochemical (Rustad, 2012; Beridze et al., 2016; Kowalski, 2020) parameters, properties of electrochemical interfaces (Krishnamurthy et al., 2004; Lee et al., 2017; Tesch et al., 2021) and the radiation damage resistance (Kowalski et al., 2016; Li et al., 2016; Ji et al., 2017c; Jolley et al., 2017; Cui et al., 2022), to name but a few. Energy materials often contain d and f elements (e.g., transition metals (TM), lanthanides (Ln), actinides (An)), which play an active part in determining the materials properties. These contain strongly correlated electrons, which represent a challenge to the computational quantum chemistry (Vogiatzis et al., 2019). In a series of papers we have demonstrated that only with proper accounting for the electronic correlation effects, beyond standard methods such as the DFT + U approach, one can deliver correct information on the molecular scale properties of energy materials (Beridze and Kowalski, 2014; Blanca-Romero et al., 2014; Kowalski et al., 2015; Li and Kowalski, 2018; Murphy et al., 2021; Tesch and Kowalski, 2022). Among other aspects, we found the importance of derivation of the Hubbard U parameter for the cations in different oxidation states and structural arrangements, and careful choice of projectors for the estimation of occupancy of d and f orbitals within the DFT + U scheme (Maxisch and Ceder, 2006; Kvashnina et al., 2018; Kick et al., 2019; Kowalski et al., 2021; Murphy et al., 2021). In particular, we apply the linear response method for computation of the Hubbard U parameter (Cococcioni and de Gironcoli, 2005) and Wannier-type functions as representation of d or f orbitals (Kvashnina et al., 2018; Kowalski et al., 2021). Here we will demonstrate the impact of these procedures on the correct computation of the TM oxides materials that are considered as electrodes in energy storage devices. In addition, we will discuss certain aspects of computation of electrochemical interfaces, including simulations under realistic conditions such as presence of aqueous electrolyte and the applied potential (Tesch et al., 2021).
In this contribution we provide an overview of our recent atomistic modeling activities on various aspects of energy materials, focusing on the studies that allowed for better characterization of these materials, including electronic structure, interface charging relations and thermodynamics aspects of surface chemistry in the presence of electrolyte. Besides such an overview, we present new results on computation of common TM and actinide oxides, and different aspects of Au, Ag, Pb and Pt metal surfaces under realistic electrochemical conditions.
2 Computational approach
Most of the ab initio calculations discussed and performed here were performed with the density functional theory (DFT)-based plane wave Quantum-ESPRESSO simulation package (Giannozzi et al., 2009)1. To represent the core electrons of atoms we applied the ultrasoft pseudopotentials (Vanderbilt, 1990). The plane-wave energy cutoff of 50 Ryd was sufficient to obtain converged results. Because we are specifically interested in correct computation of structural data, we applied the PBEsol exchange-correlation functional (Perdew et al., 2008). The DFT + U calculations were performed with the Hubbard U parameter values computed from first principles using the linear response method of Cococcioni and de Gironcoli (2005), as in our previous studies (Blanca-Romero et al., 2014; Li et al., 2015; Beridze et al., 2016; Murphy et al., 2021). We applied the poormanwannier. x tool implemented in Quantum-ESPRESSO to construct realistic projectors for occupations of d orbitals. This computational setup was successfully applied in our previous studies [e.g., Blanca-Romero et al. (2014); Beridze et al. (2016); Finkeldei et al. (2017); Murphy et al. (2021); Kowalski et al. (2021)].
For the structural models used in calculations of LixNiO2 in Section 3.2.1 we selected the most favorable arrangement of Li atoms and vacancies, which give the most negative (most stable) Coulomb energies among all possible configurations. The Coulomb energies were calculated using the package designed by Okhotnikov et al. (2016).
The DFT-based calculations of the 3 × 3 Pb(100) surface discussed in Section 3.3.2 were performed with the PBE exchange correlational functional using the VASP package (Perdew et al., 1996; Kresse and Hafner, 1993; Kresse and Furthmüller, 1996b; a). We used the projector augmented wave (PAW) method to describe the core electrons and the plane-wave cutoff energy was set to 500 eV. The 3 × 3 Pb(100) surface was represented by a five-layers slab and the model was created using the atomic simulation environment (ASE) (Hjorth Larsen et al., 2017). In order to mimic the bulk-like environment at the bottom of the slab, the two bottom Pb layers were kept fixed during the geometry optimization, while the other three layers were allowed to relax. In order to avoid any undesired interaction between the periodically repeated surface, in the direction perpendicular to the surface, a
where EHCOO/COOH+surface, Esurface, ECO2,
The effective screening medium reference interaction site method (ESM-RISM) implementation applied in Section 3.3.3 is that implemented in Quantum-ESPRESSO code. Within the ESM framework (Otani and Sugino, 2006), a potentiostat is implemented (Bonnet et al., 2012) that allows to grand-canonically vary the charge of the electrode, and thus to simulate it at an applied electrode potential. The RISM setup was identical to the one applied by Tesch et al. (2021). The DFT calculations of Au(111) surface were performed with the PBE exchange-correlation functional (Perdew et al., 1996), with the optimized lattice constant of
For the calculation of molar entropies of molecules presented in Section 3.3.4 we applied the two-phase thermodynamic (2PT) method (Lin et al., 2010; 2003; Pascal et al., 2011). The molecular dynamics (MD) trajectories necessary for this approach were simulated with the LAMMPS software package (Plimpton, 1995). The OPLS AA/L (Jorgensen and Tirado-Rives, 1988) and the Interface Force Field (IFF) (Heinz et al., 2013) were applied to describe interaction between the solutes and metal surfaces atoms, respectively. The SPC water model (Berendsen et al., 1987) was used to represent water molecules. The random initial configuration of solvent molecules was created using the PACKMOL package (Martínez et al., 2009). MD simulations of the solute molecule (HCOOH) in a solvent were 200 ps long. For the 2PT analysis, we performed a NPT ensemble equilibration run followed by a 20 ps NVT ensemble production run. This setup follows from previous studies of Lin et al. (2003), who have shown that 20 ps long trajectory is sufficient for obtaining accurate thermodynamic properties. The longer applied equilibration runs are essential to assure equilibrated water structure at metal surfaces (Cheong et al., 2022).
3 Results and discussion
3.1 Computation of electronic structure
Correct computation of the electronic structure of TM elements bearing materials is a challenge for computational electrochemistry. This is because the underlying chemistry is driven by the strongly correlated d electrons (Vogiatzis et al., 2019). DFT often fails to predict the electronic structure of these materials, also on the qualitative level. Wide-band gap materials considered as electrodes in energy storage, conversion or catalysts devices are often predicted by DFT or DFT + U methods to be metals. These include, for instance, NiOOH electrocatalyst (Zaffran and Caspary Toroker, 2016). Below we discuss specific aspects of computation of the electronic structure of TM elements-bearing compounds.
3.1.1 NiO and CoO
As a test case we computed here the simple oxides: NiO and CoO, both containing TM-cation in + 2 oxidation state. The Hubbard U parameters derived for TMs and the resulting band gaps in these two compounds are provided in Table 1. CoO and NiO are wide band gap charge transfer Mott insulators (Zhang et al., 2021). For such materials, the band gap should be equivalent to half of the Hubbard U parameter (expected shift of the unoccupied d states). The Hubbard U parameters computed here for these compounds are large and indicate wide band gaps (Table 1). The band gaps computed with the DFT + U method are consistent with the measurements. This is, however, somewhat misleading, as it results from incorrect occupations of d states. Because the atomic orbitals are applied as projectors, these result in significant, fractional occupations of empty orbitals (Table 2) and overestimated total occupancy of the d states. This shortcoming is corrected with the Wannier-functions as projectors. The resulting occupation matrix and number of d electrons are also reported in Table 2. These reflect the expected values, which has also significant impact on the computed band gaps. In set of previous studies we obtained similar improvement for occupations of d and f orbitals of various other elements [e.g., Murphy et al. (2021); Kowalski et al. (2021)]. The band gaps computed with the DFT + U(WF) method are larger than the ones predicted with the standard DFT + U approach. We note that for this case, the hybrid functionals such as HSE06 also predict correct band gaps of the considered oxide materials (Seo et al., 2015). This is opposite to the case of lithiated transition metal oxides, which show significant level of d elements delocalization and overestimation of the band gaps of these materials by the hybrid functionals approaches (Seo et al., 2015).
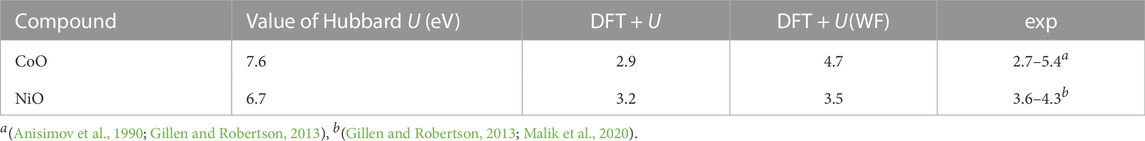
TABLE 1. The Hubbard U parameter and the band gap values (direct band gap) of CoO and NiO compounds computed with different methods, and measured. We note that for the two compounds we obtained the same Hubbard U parameter values with both considered d orbitals projector types.
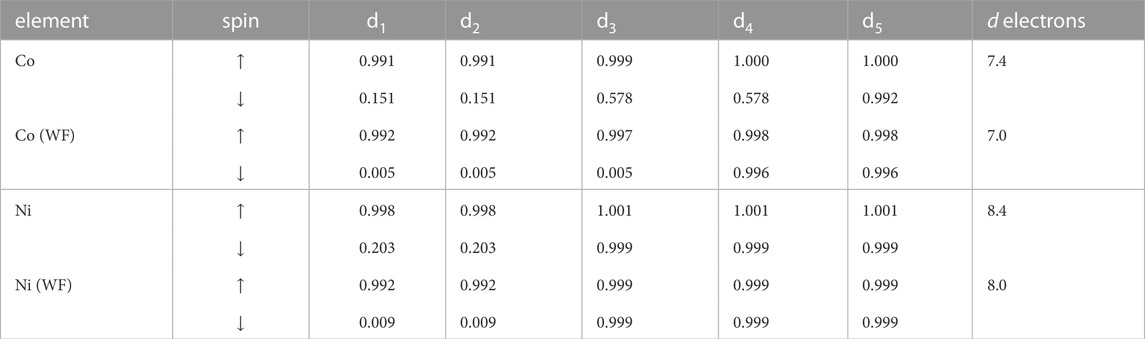
TABLE 2. The occupancies of 3d orbitals obtained with the atomic orbital- and Wannier functions (WF)-based projectors. The resulting number of d electrons is reported in the last column.
3.1.2 Metals with DFT + U
Metal phases consisting of transition metal elements play a key role in the electrochemical devices. The strong correlation character of d electrons these systems contain has been illustrated by the computation of large Hubbard U parameters in a series of computational studies (Schnell et al., 2002; Nakamura et al., 2006; Şaşıoğlu et al., 2011; Tesch and Kowalski, 2022). This would imply the importance of Hubbard model corrections also for these systems. On the other hand, it has been demonstrated that the standard DFT method results in very good prediction of metal properties, including good description of the x-ray photoelectron spectroscopy (XPS) spectra by the resulting density of states (DOSes) (Hofmann et al., 2012). This apparent contradiction could be explained by the delocalized character of d electrons in metals and inapplicability of the fully localized limit (FLL) of standard DFT to metals. An “around mean field” (AMF) version of DFT has been proposed instead for computation of metals (Czyżyk and Sawatzky, 1994; Petukhov et al., 2003; Himmetoglu et al., 2014; Ryee and Han, 2018). In this approach, the occupations are forced to reproduce the mean values, instead of the preference of fully occupied or unoccupied orbitals, as in the standard DFT + U approach. This causes no overall change in the DOSes when the AMF version of the DFT + U method is applied. Nevertheless, it has been also proposed that the correct computation of metals should be done with a combination of the FLL and AMF methods (Petukhov et al., 2003). Having wide popularity of the FLL version of the DFT + U approach, it is thus of importance to understand the performance of this method when applied to metallic systems.
Tesch and Kowalski (2022) derived the Hubbard U parameters for all the 3d, 4d and 5d transition metals applying the linear response method (Cococcioni and de Gironcoli, 2005). The values (up to 11 eV) and trends of increasing the U parameter values with increasing number of valence electrons, observed in previous studies (Şaşıoğlu et al., 2011), have been reproduced. These show that the 3d metals exhibit the largest Hubbard U parameter values, followed by 4d and 5d metals. Having these results, Tesch and Kowalski (2022) performed a thoughtful analysis of the performance of the DFT + U method for metals. When computing transition metals with the DFT + U approach, they observed a shift of the d-band to lower energies, comparing to the experimental XPS spectra. Interestingly, they found a very similar behavior when hybrid functionals (like PBE0) were applied. As mentioned, this behavior could be prevented when the AMF approach is applied. However, Tesch and Kowalski (2022) also demonstrated the importance of the projectors of d orbitals for the overall results. When the Wannier functions-based projectors of the occupancy of d states were applied, the spurious shift of the d-band was prevented. A correct description of metal d-bands is extremely important, since they are often used as descriptors for catalytic activity, like in the famous d-band model (Hammer and Norskov, 1995; Ruban et al., 1997).
3.2 Voltage of electrode materials
3.2.1 LiNiO2
Layered oxides are the most widely used electrode materials in rechargeable lithium-ion batteries. Among them, LiCoO2 (LCO) is one of the successfully commercialized cathodes (Blomgren, 2016). However, the increased demand of cobalt resulted in a high cost of such materials, triggering a widespread research effort to find alternative cobalt-free cathode materials, applicable especially for automotive applications (Olivetti et al., 2017). In the past decades, LiNiO2 (LNO) has been considered as an alternative cathode material for rechargeable lithium batteries. This is because of its high availability and associated low cost, as well as safety and energy-density when compared to the commercial cobalt counterpart (Kalyani and Kalaiselvi, 2005; Mukai et al., 2010). If accurately described by first principles methods, a valuable, molecular-level insight into the properties that determine the cycle stability and charge capacity, when these materials are used in electrochemical compounds, could be obtained (Chakraborty et al., 2018). There exist several first-principle studies on LNO [e.g., Yoshida et al. (2019); Zhu et al. (2011); Chen et al. (2011); Mock et al. (2021))]. However, DFT fails to correctly predict the electronic structure of these systems: the band gap is severely underestimated and oxidation states of cations are incorrectly described (Pavarini et al.,2012).
The DFT + U method improves the materials description in the outlined aspects (Seo et al., 2015; Kowalski et al., 2021). However, even this correction can fail for the TM elements with more than five d-electrons, such as iron or nickel (Kowalski et al., 2021). The description of d-electrons in nickel can be further improved by representing the orbitals with strongly correlated electrons with Wannier-type functions (Marzari et al., 2012; Gu et al., 2020; Kowalski et al., 2021; Murphy et al., 2021).
Here, we computed the Hubbard U parameter for Ni in the layered rhombohedral
FIGURE 1. The partial and total DOSes computed for LNO with the derived (right) and rescaled (left) Hubbard U parameters.
In order to track the effect of delithiation, series of LixNiO2 compounds was computed with different Li concentration, namely with x = 1.00, 0.75, 0.50, 0.25 and 0.00. The geometries were relaxed and the Hubbard U parameter was computed and rescaled for all the considered structures. Beyond the expected oxidation of Ni3+ into Ni4+ (three atoms at a time with increasing Li content), no disproportionation of Ni3+ into Ni2+ and Ni4+ species was observed during delithiation process, after wannierization scheme was applied. This suggests that the instability of Ni4+ reported in literature is due to other chemical forces during the synthesis of the material rather than to an inherent instability of the final structure itself (Li et al., 2022). The DFT + U(WF) method resulted in overall more stable structures, lower in energy by at least 10 kJ/mol, and the correct occupations of d orbitals, as in the case of NiO and CoO.
The intercalation voltage was estimated using standard approach (Aydinol et al., 1997; Dixit et al., 2016):
with dx = 0.25 as a step change of Li content, E (LixNiO 2) the total energy per formula unit at Li content x, and E (Libcc) the total energy per formula unit of Li in the bulk material. As shown in Figure 2, the average calculated value of 4.17 eV is in good agreement with the measured value (Ohzuku et al., 1993).
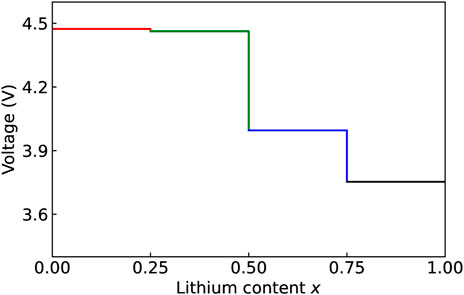
FIGURE 2. Computed specific voltage of LixNiO2 as a function of Li content x.
In order to test the performance of the applied method for predicting the formation enthalpy of LNO, we also computed the Li2O and NiO compounds, and the molecular O2. Assuming the following LNO formation reaction:
we estimate the formation energy of LNO from oxide to be 65.12, kJ/mol. When the entropy of gas phase (oxygen) is considered (S = 205 J/mol/K at 293 K), the resulting free energy of formation is 50.31, kJ/mol, is in good agreement with the measured value of 53.35, kJ/mol (Wang and Navrotsky, 2004).
During the delithiation process, as a result of oxidation of Ni atoms, removal of Li causes a linear increase of the Hubbard parameter U (Figure 3). Such a dependence of the Hubbard U parameter on the oxidation state is consistent with our previous studies [e.g., Beridze et al. (2016); Beridze and Kowalski (2014); Kowalski et al. (2021)]. The change in the oxidation state of Ni from + 3 to + 4 results in change in the DOS and the band gap (Figure 4), and in shortening of the Ni-Ni, Ni-O and O-O bond lengths, and increase of Ni-Li distances, as shown in Table 3. The cell parameters of the optimized supercell show excellent agreement with the crystallographic data.
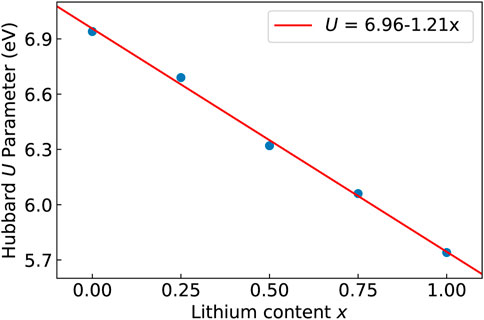
FIGURE 3. The Hubbard U parameter obtained with the linear response approach as a function of Li content. The linear fit to the data is presented in upper right corner.
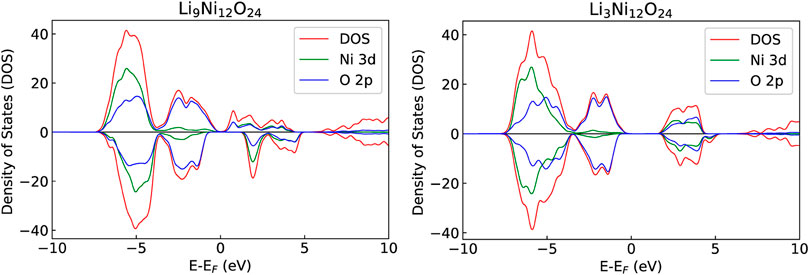
FIGURE 4. The partial and total DOSes of LixNiO2 with x = 0.75 (left) and 0.25 (right). The overlap between Ni d-bands and O p-bands is more pronounced upon delithiation, as expected.
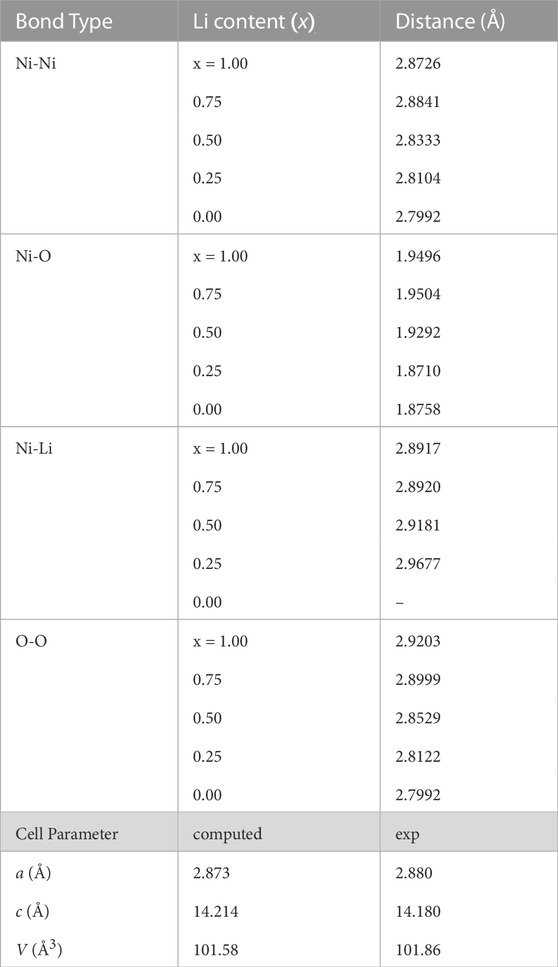
TABLE 3. Different bond lengths computed for LNO compound (LixNiO2) at different delithiation level. The experimental data are those of Molenda et al. (2002).
It is worth noting that, while the applied Wannierization scheme contributes to the correct description of the oxidation states of Ni cations, it also replicates the small experimental band gap of 0.4 eV in LiNiO2 (Laubach et al., 2009; Shishkin and Sato, 2016), as indicated in Figure 4. On the other hand, in most of the computational studies, perfect-layered LNO was computed as half-metal even with the computationally intensive hybrid functionals (Laubach et al., 2009; Shishkin and Sato, 2016; Chakraborty et al., 2018). In our studies, the accurate computation of occupation of d orbitals was prioritized, as the changes in the oxidation state of Ni are crucial to correctly describe the delithiation process.
3.2.2 LiCoO2
Similar computations were performed for LiCoO2 in the layered rhombohedral
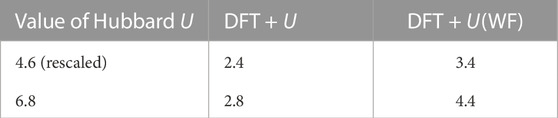
TABLE 4. The band Gap of LCO computed with different methods and U parameters (computed and rescaled). The values are provided in eV.
Applying the same computational approach as for LNO, we also computed the formation energy of LCO from oxides, following the reaction:
We obtained the value of 128.50 kJ/mol. Though less accurate than the one obtained for LNO, the computed LCO formation enthalpy from oxides shows reasonable agreement with the experimental value of 140.18 kJ/mol (Wang and Navrotsky, 2004; Takahashi et al., 2007).
3.3 Thermodynamic aspects of energy materials
3.3.1 Am-doped U3O8
Spent nuclear fuel contains significant amount of fission products or minor actinides (Np, Am, Cm), elements which can alter the material structure and performance (Bosbach et al., 2020). Doping of a phase with an element of other oxidation state can trigger a phase transition and stabilization of the new phase. This is for instance the case of yttria-stabilized zirconia, where doping of zirconium dioxide phase with ∼8% of tri-valent element like yttrium, stabilizes the cubic phase of that compound, which is one of the fastest known ionic conductors (Kowalski et al., 2021). Here we investigate the phase transition upon doping U3O8 phase (realized as a phase with the space group C2mm) with small amount of Am. This system has been investigated experimentally by Caisso et al. (2016). They found stabilization of a phase with the space group P62m phase upon doping with
First, we computed the Hubbard U parameters for U and Am in both structures. The resulting values are reported in Table 5. These are consistent with our previous studies (Beridze and Kowalski, 2014; Beridze et al., 2016). We notice, however, that the values derived for U species in U3O8 are larger than the ones obtained in Kvashnina et al. (2018). This is because in those studies another structure of U3O8 was investigated.
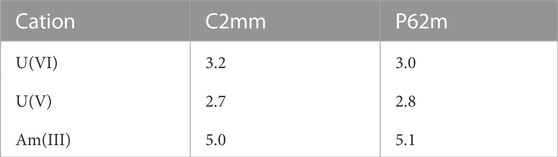
TABLE 5. The Hubbard U parameters derived for Am doped U3O8 compounds. The values are given in eV.
The computed formation enthalpy difference between the phases with space groups P62m and C2mm as a function of Am content are shown in Figure 5. Following studies of Caisso et al. (2016) we assumed that Am is incorporated as +3 species on U +5 site and the charge is compensated by the conversion of the two U +5 species to the oxidation state of +6. We predict the phase transition at very low content of Am of
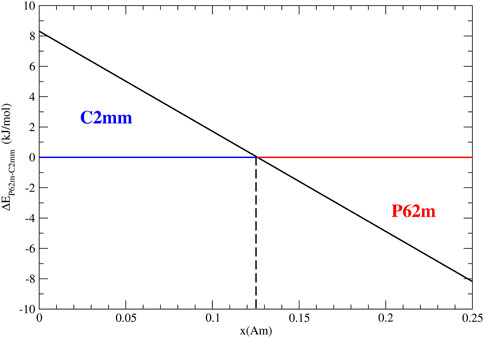
FIGURE 5. The computed difference in enthalpy between the C2mm and P62m phases of U3O8. The predicted phase transition is at the content of Am of 1.2%.

TABLE 6. The computed and measured volumes of pure and Am doped U3O8 (at Am/U ratio of 1/9). The measured data are those of Caisso et al. (2016). The values are in Å3 per two formula units.
3.3.2 Solvent effects on surface chemistry
To show the importance of solvation for computation of surface electrochemical pathways, as a test case we investigated the effect of explicit and implicit solvation on the CO2 reduction reaction towards CO and HCOOH on the Pb(100) surface. We considered the following reaction pathways for the formation of CO and HCOOH species:
We assumed that the CO2 forms either a HCOO* intermediate, which reacts further to formic acid (HCOOH), or COOH* which further reacts to CO and water.
Figure 6 shows the energy diagram for both pathways computed in three ways: (M1) assuming no solvation, (M2) in the presence of explicit solvation and (M3) by applying the continuum solvation model. In the approach M2, the aqueous phase was modeled by explicitly adding 12 water molecules. The case M3 was computed using the implicit solvation model implemented in VASPsol method (Mathew et al., 2019). The adsorption energies for HCOO* and COOH* intermediates computed with M1 are -0.45 eV and 0.73 eV, respectively. In the presence of implicit solvation, the HCOO* and COOH* adsorption energy increases to −0.61 eV and 0.52 eV. These indicated bonded HCOO* and unbonded COOH*. Applying method M2, the adsorption energy of the two species increases by 0.27 eV and 1.00 eV, respectively. These results show the importance of including solvent effects, when computing chemical pathways on metal surfaces.
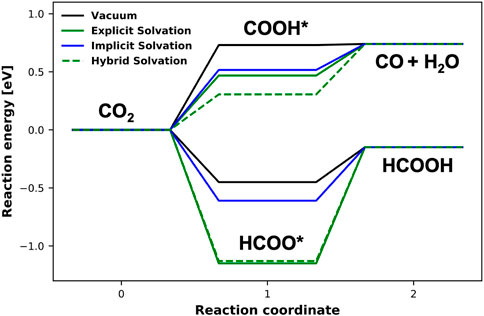
FIGURE 6. Reaction energy diagram for CO2 reduction towards HCOOH and CO on the Pb(100) surface, computed in vacuum as well as with the implicit/explicit/hybrid solvation models.
The method M3 is a fast way to include solvent effects in the DFT calculations. In the considered cases, it results in increase in the adsorption energies, but significantly smaller than the explicit solvation schemes. The adsorption energies are changing by 0.1 eV only.
In Figure 6, we also show the results of hybrid solvation approach, where on top of the 12 explicit water molecules we have applied the implicit solvation, to preserve continuity of the solvent medium. While for the HCOO* intermediate, the effect of implicit solvation leads to an insignificant change, COOH* is stabilized by more than 0.2 eV. This shows that for the adsorbed species, the effect of first hydration shell dominates the solvent effect on the bonding energy between the adsorbate and the metal surface. In case of the weak bonding, the long-range interaction between the species and the solvent plays an important role.
3.3.3 Metal interfaces with the ESM-RISM approach
Simulation of electrochemical solid/liquid interfaces at an applied electrode potential is a challenge in computational electrochemistry (Schwarz and Sundararaman, 2020). It requires an approach that can consistently describe charging of the electrode as well as effects of an electrolyte solution. Both conditions are realized by the DFT-based effective screening medium reference interaction site method (ESM-RISM) (Nishihara and Otani, 2017). The RISM (Chandler and Andersen, 1972; Hansen and McDonald, 2013) is an implicit solvent model that relies on the classical theory of liquids. It computes in a statistical way correlations between electrolyte species and thus takes into account the electrolyte structure. Electrolyte–electrolyte as well as electrode–electrolyte interactions are described by the parameterized interparticle interaction potentials. The method requires a choice of classical water model for description of the aqueous electrolyte solution, a set of Lennard–Jones parameters for the electrode–electrolyte interaction and the partitioning of the system into quantum-mechanically and classically treated parts (i.e., treatment of near-surface water layers at the level of DFT) (Nishihara and Otani, 2017; Tesch et al.,2021).
We applied this method in our previous study for computation of the partially oxidized Pt(111)/electrolyte interface (Tesch et al., 2021). Taking into account the potential-dependent oxygen coverage of Pt(111) surface, we were able to accurately describe the chemisorption and charging state of the interface, correctly reproducing the chemisorption-induced non-monotonic charging relation of this specific interface [see Figure 8 of Tesch et al. (2021)]. Here, we applied the ESM-RISM approach to model the interface between the Au(111) electrode and an 0.1 M aqueous HCl solution. Figure 7 shows the obtained interface structure of the electrolyte solution as represented by the density distribution functions of water and electrolyte ions species. The near-surface structure of water solvent shows alternating shells of water, with the main peaks positions that agree well with the AIMD simulations (Goldsmith et al., 2021). The effects of the applied potential and the resulting surface charge on excess or depletion of ions near the interface, as expected from the electrostatic arguments, are clearly seen. This indicates that the electrode charge is correctly balanced by the arrangement of electrolyte ions. The relation between the surface charge and the applied potential is linear (see Figure 8). This is expected because the Au(111) surface under these conditions (the considered potential range) is characterized by the formation of double layer and is not covered by adsorbed species, as indicated by the cyclic voltammetry (Hamelin, 1996). Other computational studies have derived very similar charging relation (Letchworth-Weaver and Arias (2012); Goldsmith et al. (2021), see in Figure 8). The trends are consistent with the experimental findings of linearly decreasing (i.e. opposite) charge at the outer Helmholtz plane (Saha and Zenyuk, 2021). The double layer capacitance, as derived from the slope of linear fit, is 23.4 μF/cm2 and compares well to the experimental (Vasiljevic et al., 2004; Garlyyev et al., 2018) and other computational (Kastlunger et al., 2018; Hörmann et al., 2019; Goldsmith et al., 2021) results.
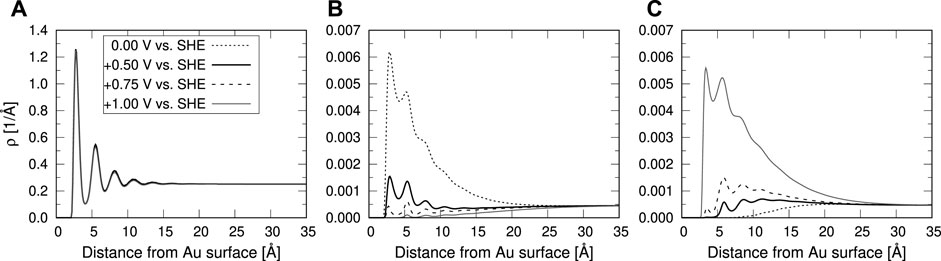
FIGURE 7. Density profiles as a function of distance from the Au(111) surface. Different panels show results for: (A) water molecules, (B) H3O+ electrolyte ions and (C) Cl− electrolyte ions. Different lines represent the results computed with the ESM–RISM approach at different electrode potentials. The potential of zero charge (pzc) is located at 0.5 V vs. SHE.
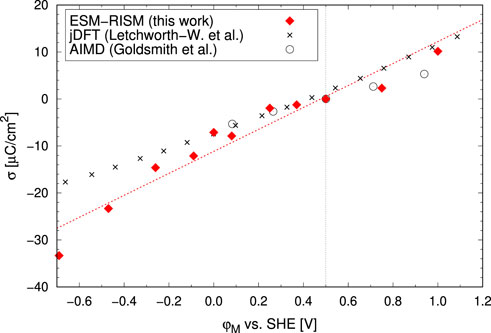
FIGURE 8. The computed surface charge vs. the applied electrode potential for the Au(111)/electrolyte interface in 0.1 M aq. HCl solution (our data), in pure water by Goldsmith et al. (2021) and in an electrolyte of 1 M ionic strength by Letchworth-Weaver and Arias (2012). The dashed red line represents a linear fit to our data. The vertical dashed line shows the pzc assumed here [0.5 V vs. SHE (Le et al., 2017)].
3.3.4 Entropy of solvation
The reliable computation of surface chemical reactions at electrochemical conditions requires correct estimates of thermodynamic parameters. In particular, entropy effects are usually omitted in atomistic modeling studies [e.g., Klinkova et al. (2016))]. However, most of the electrocatalytic reactions on a solid catalyst surface happen in the presence of a dense electrolyte. The entropic contributions from the solvation effects can have a non-negligible effect on the reactions free energy. There are ways to compute the entropy of species in a solvent phase. These are based either on the theoretical consideration [e.g., Garza (2019)] or simulation based approaches, such as thermodynamic integration (Baranyai, 2018) or analysis of the velocity autocorrelation function (Pascal et al., 2011). Garza (2019) proposed a simplified approach to the computation of entropy of liquid phases and obtained agreements with the measured values within 12 J/mol/K. Regarding entropy at the interfaces, Jung et al. (2021) applied the two-phase thermodynamics (2PT) approach (Lin et al., 2010; 2003; Pascal et al., 2011) and computed entropy of aqueous phase as a function of distance from the Pt(111) interface. Here we applied the 2PT approach to evaluate different entropy contributions for HCOOH molecules submerged in aqueous phase, in the bulk and at the Pb(100) and Ag (100) surfaces. The so estimated total entropy is given in Table 7.
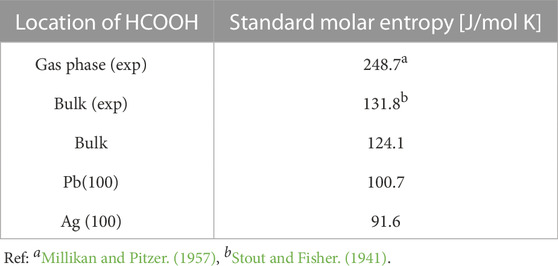
TABLE 7. Standard molar entropies of HCOOH in bulk solution and at the Pb(100) and Ag (100) surfaces. All entropy values except the experimental value are calculated using the 2PT method.
The computed molar entropy of HCOOH in bulk water is in good agreement with the experimental values. For HCOOH on Pb(100) surface and Ag (100) surface, we observe a reduction in entropy by
4 Conclusion
In this contribution we presented an overview of the atomistic modeling research on energy materials we have performed in the last decade. We focused on discussing various challenges associated with the computation of molecular-level materials properties. In particular, we discussed the computation of electronic structure, phase transitions, surface chemistry and electrochemical interfaces, focusing on accounting for the presence of electrolyte phase. For the class of electrode materials we show the performance of the parameter-free DFT + U approach as well as the importance of application of realistic projectors for counting the occupancy of orbitals containing strongly correlated d and f electrons for obtaining correct description of electronic structure, and the related parameters, such as formation energies, specific voltage or phase stability. Materials important for electrochemistry and energy sector, such as LCO, LNO, U3O8 or metal surfaces have been considered, with some parameters essential for understanding of their performance computed. Last but not least, we discussed the importance of entropic effects on the computational surface (electro) chemistry.
Besides discussing various successful applications of atomistic modeling to computation of atomic-scale properties of energy materials, we outlined challenges encountered by computational electrochemistry. We highlight the importance of thoughtful analysis of the computed results vs. the available experimental data. Overcoming the challenges faced by the atomistic simulations of energy materials will lead to accurate, simulation-based materials design, that will facilitate development of more efficient materials for fulfilling future energy demands.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
Y-YT, AKL, ND, and PK performed calculations of the transition metal and actinide oxides. RT performed analysis of the electronic structure and calculations of metal surfaces with the ESM-RISM method. TB, OC, and AS performed calculation of the chemistry at metal surfaces. OC performed analysis of the entropies at metal surfaces. SP contributed expertise on doped uranium oxide materials. PK led the project and performed collective analysis of the data and editing of the manuscript. All parties contributed to writing the manuscript. The authors list after PK is arranged according to alphabetical order and name positioning does not reflect individual contributions.
Acknowledgments
The authors gratefully acknowledge the computing time granted by the JARA Vergabegremium and provided on the JARA Partition part of the supercomputers JURECA at Forschungszentrum Jülich and CLAIX at RWTH Aachen University (Project cjiek61).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1In this contribution we call DFT methods an ab initio approach as the exchange-correlation functionals utilized in our studies were designed based on pure-theoretical considerations.
References
Aydinol, M. K., Kohan, A. F., Ceder, G., Cho, K., and Joannopoulos, J. (1997). Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys. Rev. B 56, 1354–1365. doi:10.1103/PhysRevB.56.1354
Ali, K., Arya, A., Ghosh, P. S., and Dey, G. K. (2016). A first principle study of the pressure dependent elastic properties of monazite LaPO4. AIP Conf. Proc. 1728, 020090. doi:10.1063/1.4946141
Anisimov, V. I., Korotin, M. A., and Kurmaev, E. Z. (1990). Band-structure description of Mott insulators (NiO, MnO, FeO, CoO). J. Phys. Condens. Matter 2, 3973–3987. doi:10.1088/0953-8984/2/17/008
Baranyai, A. (2018). Thermodynamic integration for the determination of nonequilibrium entropy. J. Mol. Liq. 266, 472–477. doi:10.1016/j.molliq.2018.05.113
Berendsen, H. J. C., Grigera, J. R., and Straatsma, T. P. (1987). The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271. doi:10.1021/j100308a038
Beridze, G., Birnie, A., Koniski, S., Ji, Y., and Kowalski, P. M. (2016). DFT+U as a reliable method for efficient ab initio calculations of nuclear materials. Prog. Nucl. Energy 92, 142–146. doi:10.1016/j.pnucene.2016.07.012
Beridze, G., and Kowalski, P. M. (2014). Benchmarking the DFT+U method for thermochemical calculations of uranium molecular compounds and solids. J. Phys. Chem. A 118, 11797–11810. doi:10.1021/jp5101126
Blanca-Romero, A., Kowalski, P. M., Beridze, G., Schlenz, H., and Bosbach, D. (2014). Performance of DFT+U method for prediction of structural and thermodynamic parameters of monazite-type ceramics. J. Comput. Chem. 35, 1339–1346. doi:10.1002/jcc.23618
Blomgren, G. E. (2016). The development and future of lithium ion batteries. J. Electrochem. Soc. 164, A5019–A5025. doi:10.1149/2.0251701jes
Bonnet, N., Morishita, T., Sugino, O., and Otani, M. (2012). First-principles molecular dynamics at a constant electrode potential. Phys. Rev. Lett. 109, 266101. doi:10.1103/PhysRevLett.109.266101
Bosbach, D., Brandt, F., Bukaemskiy, A., Deissmann, G., Kegler, P., Klinkenberg, M., et al. (2020). Research for the safe management of nuclear waste at Forschungszentrum Jülich: Materials chemistry and solid solution aspects. Adv. Energy Mat. 22, 1901417. doi:10.1002/adem.201901417
Caisso, M., Roussel, P., Den Auwer, C., Picart, S., Hennig, C., Scheinost, A. C., et al. (2016). Evidence of trivalent Am substitution into U3O8. Inorg. Chem. 55, 10438–10444. doi:10.1021/acs.inorgchem.6b01672
Chakraborty, A., Dixit, M., and Major, D. T. (2018). Accurate cathode properties of LiNiO2, LiCoO2, and LiMnO2 using the SCAN meta-GGA density functional. Npj Comput. Mat. 4, 60. doi:10.1038/s41524-018-0117-4
Chandler, D., and Andersen, H. C. (1972). Optimized cluster expansions for classical fluids. II. Theory of molecular liquids. J. Chem. Phys. 57, 1930–1937. doi:10.1063/1.1678513
Chen, Z., Zou, H., Zhu, X., Zou, J., and Cao, J. (2011). First-principle investigation of Jahn–Teller distortion and topological analysis of chemical bonds in LiNiO2. J. Solid State Chem. 184, 1784–1790. doi:10.1016/j.jssc.2011.05.024
Cheong, O., Eikerling, M. H., and Kowalski, P. M. (2022). Water structures on Pb(100) and (111) surface studied with the Interface force field. Appl. Surf. Sci. 589, 152838. doi:10.1016/j.apsusc.2022.152838
Chroneos, A., Rushton, M., Jiang, C., and Tsoukalas, L. (2013). Nuclear wasteform materials: Atomistic simulation case studies. J. Nucl. Mat. 441, 29–39. doi:10.1016/j.jnucmat.2013.05.012
Cococcioni, M., and de Gironcoli, S. (2005). Linear response approach to the calculation of the effective interaction parameters in the LDA + U method. Phys. Rev. B 71, 035105. doi:10.1103/PhysRevB.71.035105
Connor, T., Cheong, O., Bornhake, T., Shad, A. C., Tesch, R., Sun, M., et al. (2021). Pyrochlore compounds from atomistic simulations. Front. Chem. 9, 733321. doi:10.3389/fchem.2021.733321
Cui, Y., Sukkurji, P. A., Wang, K., Azmi, R., Nunn, A. M., Hahn, H., et al. (2022). High entropy fluorides as conversion cathodes with tailorable electrochemical performance. J. Energy Chem. 72, 342–351. doi:10.1016/j.jechem.2022.05.032
Czyżyk, M. T., and Sawatzky, G. A. (1994). Local-density functional and on-site correlations: The electronic structure of La2CuO4 and LaCuO3. Phys. Rev. B 49, 14211–14228. doi:10.1103/PhysRevB.49.14211
Dixit, M., Kosa, M., Lavi, O. S., Markovsky, B., Aurbach, D., and Major, D. T. (2016). Thermodynamic and kinetic studies of LiNi0.5Co0.2Mn0.3O2 as a positive electrode material for Li-ion batteries using first principles. Phys. Chem. 18, 6799–6812. doi:10.1039/C5CP07128C
Eremin, N. N., Marchenko, E. I., Petrov, V. G., Mitrofanov, A. A., and Ulanova, A. S. (2019). Solid solutions of monazites and xenotimes of lanthanides and plutonium: Atomistic model of crystal structures, point defects and mixing properties. Comput. Mat. Sci. 157, 43–50. doi:10.1016/j.commatsci.2018.10.025
Fan, M., Eslamibidgoli, M. J., Garbarino, S., Tavares, A. C., Eikerling, M. H., and Guay, D. (2020). A computational-experimental investigation of the mechanisms responsible for the enhanced CO2 electrochemical reduction of dendritic Sn1Pb3 alloy. J. Electrochem. Soc. MA2020-01, 2630–17979. doi:10.1149/ma2020-01462630mtgabs
Feng, J., Xiao, B., Zhou, R., and Pan, W. (2013). Anisotropy in elasticity and thermal conductivity of monazite-type REPO4 (RE=La, Ce, Nd, Sm, Eu and Gd) from first-principles calculations. Acta Mater 61, 7364–7383. doi:10.1016/j.actamat.2013.08.043
Finkeldei, S., Kegler, P., Kowalski, P., Schreinemachers, C., Brandt, F., Bukaemskiy, A., et al. (2017). Composition dependent order-disorder transition in NdxZr1−xO2−0.5x pyrochlores: A combined structural, calorimetric and ab initio modeling study. Acta Mater 125, 166–176. doi:10.1016/j.actamat.2016.11.059
Galakhov, V., Kurmaev, E., Uhlenbrock, S., Neumann, M., Kellerman, D., and Gorshkov, V. (1996). Degree of covalency of LiCoO2: X-Ray emission and photoelectron study. Solid State Commun. 99, 221–224. doi:10.1016/0038-1098(96)00251-7
Garlyyev, B., Xue, S., Watzele, S., Scieszka, D., and Bandarenka, A. S. (2018). Influence of the nature of the alkali metal cations on the electrical double-layer capacitance of model Pt(111) and Au(111) electrodes. J. Phys. Chem. Lett. 9, 1927–1930. doi:10.1021/acs.jpclett.8b00610
Garza, A. J. (2019). Solvation entropy made simple. J. Chem. Theory Comput. 15, 3204–3214. doi:10.1021/acs.jctc.9b00214
Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., et al. (2009). Quantum espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502. doi:10.1088/0953-8984/21/39/395502
Gillen, R., and Robertson, J. (2013). Accurate screened exchange band structures for the transition metal monoxides MnO, FeO, CoO and NiO. J. Phys. Condens. Matter 25, 165502. doi:10.1088/0953-8984/25/16/165502
Goldsmith, Z. K., Calegari Andrade, M. F., and Selloni, A. (2021). Effects of applied voltage on water at a gold electrode interface from ab initio molecular dynamics. Chem. Sci. 12, 5865–5873. doi:10.1039/D1SC00354B
Grey, C. P., and Tarascon, J. M. (2017). Sustainability and in situ monitoring in battery development. Nat. Mat. 16, 45–56. doi:10.1038/NMAT4777
Gu, Y., Zhu, S., Wang, X., Hu, J., and Chen, H. (2020). A substantial hybridization between correlated Ni-d orbital and itinerant electrons in infinite-layer nickelates. Commun. Phys. 3, 84–89. doi:10.1038/s42005-020-0347-x
Hamelin, A. (1996). Cyclic voltammetry at gold single-crystal surfaces. Part 1. Behaviour at low-index faces. J. Electroanal. Chem. 407, 1–11. doi:10.1016/0022-0728(95)04499-X
Hammer, B., and Norskov, J. K. (1995). Why gold is the noblest of all the metals. Nature 376, 238–240. doi:10.1038/376238a0
Hansen, J., and McDonald, I. (2013). Theory of simple liquids: With applications to soft matter. Elsevier Science. doi:10.1016/C2010-0-66723-X
Heinz, H., Lin, T.-J., Kishore Mishra, R., and Emami, F. S. (2013). Thermodynamically consistent force fields for the assembly of inorganic, organic, and biological nanostructures: The INTERFACE force field. Langmuir 29, 1754–1765. doi:10.1021/la3038846
Heinz, H., Vaia, R. A., Farmer, B. L., and Naik, R. R. (2008). Accurate simulation of surfaces and interfaces of face-centered cubic metals using 126 and 96 Lennard-Jones potentials. J. Phys. Chem. C 112, 17281–17290. doi:10.1021/jp801931d
Himmetoglu, B., Floris, A., de Gironcoli, S., and Cococcioni, M. (2014). Hubbard-corrected DFT energy functionals: The LDA+U description of correlated systems. Int. J. Quantum Chem. 114, 14–49. doi:10.1002/qua.24521
Hjorth Larsen, A., Jørgen Mortensen, J., Blomqvist, J., Castelli, I. E., Christensen, R., Dułak, M., et al. (2017). The atomic simulation environment—A python library for working with atoms. J. Phys. Condens. Matter 29, 273002. doi:10.1088/1361-648X/aa680e
Hofmann, T., Yu, T. H., Folse, M., Weinhardt, L., Bär, M., Zhang, Y., et al. (2012). Using photoelectron spectroscopy and quantum mechanics to determine d-band energies of metals for catalytic applications. J. Phys. Chem. C 116, 24016–24026. doi:10.1021/jp303276z
Hörmann, N. G., Andreussi, O., and Marzari, N. (2019). Grand canonical simulations of electrochemical interfaces in implicit solvation models. J. Chem. Phys. 150, 041730. doi:10.1063/1.5054580
Jahn, S., and Kowalski, P. M. (2014). Theoretical approaches to structure and spectroscopy of Earth materials. Rev. Mineral. Geochem. 78, 691–743. doi:10.2138/rmg.2014.78.17
Ji, Y., Beridze, G., Bosbach, D., and Kowalski, P. M. (2017a). Heat capacities of xenotime-type ceramics: An accurate ab initio prediction. J. Nucl. Mat. 494, 172–181. doi:10.1016/j.jnucmat.2017.07.026
Ji, Y., Beridze, G., Li, Y., and Kowalski, P. M. (2017b). Large scale simulation of nuclear waste materials. Energy Procedia 127, 416–424. doi:10.1016/j.egypro.2017.08.108
Ji, Y., Kowalski, P. M., Neumeier, S., Deissmann, G., Kulriya, P. K., and Gale, J. D. (2017c). Atomistic modeling and experimental studies of radiation damage in monazite-type LaPO4 ceramics. Nucl. Instrum. Methods Phys. Res. Sect. B 393, 54–58. doi:10.1016/j.nimb.2016.09.031
Jolley, K., Asuvathraman, R., and Smith, R. (2017). Inter-atomic potentials for radiation damage studies in CePO4 monazite. Nucl. Instrum. Methods Phys. Res. B 393, 93–96. doi:10.1016/j.nimb.2016.10.016
Jorgensen, W. L., and Tirado-Rives, J. (1988). The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666. doi:10.1021/ja00214a001
Jung, C. K., Braunwarth, L., Sinyavskiy, A., and Jacob, T. (2021). Thermodynamic description of interfaces applying the 2PT method on ReaxFF molecular dynamics simulations. J. Phys. Chem. C 125, 24663–24670. doi:10.1021/acs.jpcc.1c07327
Kalyani, P., and Kalaiselvi, N. (2005). Various aspects of LiNiO2 chemistry: A review. Sci. Technol. Adv. Mater. 6, 689–703. doi:10.1016/j.stam.2005.06.001
Kastlunger, G., Lindgren, P., and Peterson, A. A. (2018). Controlled-potential simulation of elementary electrochemical reactions: Proton discharge on metal surfaces. J. Phys. Chem. C 122, 12771–12781. doi:10.1021/acs.jpcc.8b02465
Kick, M., Reuter, K., and Oberhofer, H. (2019). Intricacies of DFT+U, not only in a numeric atom centered orbital framework. J. Chem. Theory Comput. 15, 1705–1718. doi:10.1021/acs.jctc.8b01211
Klinkova, A., De Luna, P., Dinh, C.-T., Voznyy, O., Larin, E. M., Kumacheva, E., et al. (2016). Rational design of efficient palladium catalysts for electroreduction of carbon dioxide to formate. ACS Catal. 6, 8115–8120. doi:10.1021/acscatal.6b01719
Kowalski, P. M. (2020). Formation enthalpy of Ln2B2O7-type (B=Ti, Sn, Hf, Zr) compounds. Scr. Mater 189, 7–10. doi:10.1016/j.scriptamat.2020.07.048
Kowalski, P. M., Beridze, G., Ji, Y., and Li, Y. (2017a). Towards reliable modeling of challenging f electrons bearing materials: Experience from modeling of nuclear materials. MRS Adv. 2017, 491–497. doi:10.1557/adv.2017.46
Kowalski, P. M., Beridze, G., Li, Y., Ji, Y., Friedrich, C., Sasioglu, E., et al. (2016). Feasible and reliable ab initio approach to computation of materials relevant for nuclear waste management. Ceram. Trans. 258, 205–217. doi:10.1002/9781119236016.ch21
Kowalski, P. M., Beridze, G., Vinograd, V. L., and Bosbach, D. (2015). Heat capacities of lanthanide and actinide monazite-type ceramics. J. Nucl. Mat. 464, 147–154. doi:10.1016/j.jnucmat.2015.04.032
Kowalski, P. M., He, Z., and Cheong, O. (2021). Electrode and electrolyte materials from atomistic simulations: Properties of LixFePO4 electrode and zircon-based ionic conductors. Front. Energy Res. 9. doi:10.3389/fenrg.2021.653542
Kowalski, P. M., Ji, Y., Li, Y., Arinicheva, Y., Beridze, G., Neumeier, S., et al. (2017b). Simulation of ceramic materials relevant for nuclear waste management: Case of La1−xEuxPO4 solid solution. Nucl. Instrum. Methods Phys. Res. Sect. B 393, 68–72. doi:10.1016/j.nimb.2016.09.029
Kowalski, P. M., and Li, Y. (2016). Relationship between the thermodynamic excess properties of mixing and the elastic moduli in the monazite-type ceramics. J. Eur. Ceram. Soc. 36, 2093–2096. doi:10.1016/j.jeurceramsoc.2016.01.051
Kresse, G., and Furthmüller, J. (1996a). Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15–50. doi:10.1016/0927-0256(96)00008-0
Kresse, G., and Furthmüller, J. (1996b). Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186. doi:10.1103/PhysRevB.54.11169
Kresse, G., and Hafner, J. (1993). Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561. doi:10.1103/PhysRevB.47.558
Krishnamurthy, R., Yoon, Y.-G., Srolovitz, D. J., and Car, R. (2004). Oxygen diffusion in yttria-stabilized zirconia: A new simulation model. J. Am. Ceram. 87, 1821–1830. doi:10.1111/j.1151-2916.2004.tb06325.x
Kvashnina, K. O., Kowalski, P. M., Butorin, S. M., Leinders, G., Pakarinen, J., Bès, R., et al. (2018). Trends in the valence band electronic structures of mixed uranium oxides. Chem. Commun. 54, 9757–9760. doi:10.1039/C8CC05464A
Laubach, S., Laubach, S., Schmidt, C. P., Ensling, D., Schmid, S., Jaegermann, W., et al. (2009). Changes in the crystal and electronic structure of LiCoO2 and LiNiO2 upon Li intercalation and de-intercalation. Phys. Chem. Chem. Phys. 11, 3278–3289. doi:10.1039/B901200A
Le, J., Iannuzzi, M., Cuesta, A., and Cheng, J. (2017). Determining potentials of zero charge of metal electrodes versus the standard hydrogen electrode from density-functional-theory-based molecular dynamics. Phys. Rev. Lett. 119, 016801. doi:10.1103/PhysRevLett.119.016801
Lee, K., Youn, Y., and Han, S. (2017). Identification of ground-state spin ordering in antiferromagnetic transition metal oxides using the Ising model and a genetic algorithm. Sci. Technol. Adv. Mater 18, 246–252. doi:10.1080/14686996.2017.1300046
Letchworth-Weaver, K., and Arias, T. A. (2012). Joint density functional theory of the electrode-electrolyte interface: Application to fixed electrode potentials, interfacial capacitances, and potentials of zero charge. Phys. Rev. B 86, 075140. doi:10.1103/PhysRevB.86.075140
Li, X., Wang, Q., Guo, H., Artrith, N., and Urban, A. (2022). Understanding the onset of surface degradation in linio2 cathodes. ACS Appl. Energy Mat. 5, 5730–5741. doi:10.1021/acsaem.2c00012
Li, Y., Kowalski, P. M., Beridze, G., Birnie, A. R., Finkeldei, S., and Bosbach, D. (2015). Defect formation energies in A2B2O7 pyrochlores. Scr. Mat. 107, 18–21. doi:10.1016/j.scriptamat.2015.05.010
Li, Y., Kowalski, P. M., Beridze, G., Blanca-Romero, A., Ji, Y., Vinograd, V. L., et al. (2016). Atomistic simulations of ceramic materials relevant for nuclear waste management: Cases of monazlte and pyrochlore. Ceram. Trans. 255, 165–175. doi:10.1002/9781119234531.ch15
Li, Y., Kowalski, P. M., Blanca-Romero, A., Vinograd, V., and Bosbach, D. (2014). Ab initio calculation of excess properties of solid solutions. J. Solid State Chem. 220, 137–141. doi:10.1016/j.jssc.2014.08.005
Li, Y., and Kowalski, P. M. (2018). Energetics of defects formation and oxygen migration in pyrochlore compounds from first principles calculations. J. Nucl. Mat. 505, 255–261. doi:10.1016/j.jnucmat.2017.11.005
Lin, S.-T., Blanco, M., and Goddard, W. A. (2003). The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: Validation for the phase diagram of Lennard-Jones fluids. J. Chem. Phys. 119, 11792–11805. doi:10.1063/1.1624057
Lin, S.-T., Maiti, P. K., and Goddard, W. A. (2010). Two-phase thermodynamic model for efficient and accurate absolute entropy of water from molecular dynamics simulations. J. Phys. Chem. B 114, 8191–8198. doi:10.1021/jp103120q
Malik, R., Tomer, V. K., Mishra, Y. K., and Lin, L. (2020). Functional gas sensing nanomaterials: A panoramic view. Appl. Phys. Rev. 7, 021301. doi:10.1063/1.5123479
Martínez, L., Andrade, R., Birgin, E. G., and Martínez, J. M. (2009). Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164. doi:10.1002/jcc.21224
Marzari, N., Mostofi, A. A., Yates, J. R., Souza, I., and Vanderbilt, D. (2012). Maximally localized Wannier functions: Theory and applications. RMP 84, 1419–1475. doi:10.1103/RevModPhys.84.1419
Mathew, K., Kolluru, V. S. C., Mula, S., Steinmann, S. N., and Hennig, R. G. (2019). Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 151, 234101. doi:10.1063/1.5132354
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A., and Hennig, R. G. (2014). Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106. doi:10.1063/1.4865107
Maxisch, T., and Ceder, G. (2006). Elastic properties of olivine LixFePO4 from first principles. Phys. Rev. B 73, 174112. doi:10.1103/PhysRevB.73.174112
Millikan, R. C., and Pitzer, K. S. (1957). Infrared spectra and vibrational assignment of monomeric formic acid. J. Chem. Phys. 27, 1305–1308. doi:10.1063/1.1743996
Mock, M., Bianchini, M., Fauth, F., Albe, K., and Sicolo, S. (2021). Atomistic understanding of the LiNiO2–NiO2 phase diagram from experimentally guided lattice models. J. Mat. Chem. A 9, 14928–14940. doi:10.1039/D1TA00563D
Mogilevsky, P. (2007). On the miscibility gap in monazite–xenotime systems. Phys. Chem. Min. 34, 201–214. doi:10.1007/s00269-006-0139-1
Molenda, J., Wilk, P., and Marzec, J. (2002). Structural, electrical and electrochemical properties of LiNiO2. Solid State Ion. 146, 73–79. doi:10.1016/S0167-2738(01)00992-4
Monkhorst, H. J., and Pack, J. D. (1976). Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188–5192. doi:10.1103/PhysRevB.13.5188
Mukai, K., Sugiyama, J., and Aoki, Y. (2010). Structural, magnetic, and electrochemical studies on lithium insertion materials LiNi1−xCoxO2 with 0 < x < 0.25. J. Solid State Chem. 183, 1726–1732. doi:10.1016/j.jssc.2010.05.019
Murphy, G. L., Zhang, Z., Tesch, R., Kowalski, P. M., Avdeev, M., Kuo, E. Y., et al. (2021). Tilting and distortion in rutile-related mixed metal ternary uranium oxides: A structural, spectroscopic, and theoretical investigation. Inorg. Chem. 60, 2246–2260. doi:10.1021/acs.inorgchem.0c03077
Nakamura, K., Arita, R., Yoshimoto, Y., and Tsuneyuki, S. (2006). First-principles calculation of effective onsite coulomb interactions of 3d transition metals: Constrained local density functional approach with maximally localized wannier functions. Phys. Rev. B 74, 235113. doi:10.1103/PhysRevB.74.235113
Neumeier, S., Arinicheva, Y., Ji, Y., Heuser, J. M., Kowalski, P. M., Kegler, P., et al. (2017a). New insights into phosphate based materials for the immobilisation of actinides. Radiochim. Acta 105, 961–984. doi:10.1515/ract-2017-2819
Neumeier, S., Kegler, P., Arinicheva, Y., Shelyug, A., Kowalski, P. M., Schreinemachers, C., et al. (2017b). Thermochemistry of La1−xLnxPO4 monazites (Ln=Gd, Eu). J. Chem. Thermodyn. 105, 396–403. doi:10.1016/j.jct.2016.11.003
Nishihara, S., and Otani, M. (2017). Hybrid solvation models for bulk, interface, and membrane: Reference interaction site methods coupled with density functional theory. Phys. Rev. B 96, 115429. doi:10.1103/physrevb.96.115429
Ohzuku, T., Ueda, A., and Nagayama, M. (1993). Electrochemistry and structural chemistry of LiNiO2 (R3m) for 4 volt secondary lithium cells. J. Electrochem. Soc. 140, 1862–1870. doi:10.1149/1.2220730
Okhotnikov, K., Charpentier, T., and Cadars, S. (2016). Supercell program: A combinatorial structure-generation approach for the local-level modeling of atomic substitutions and partial occupancies in crystals. J. Cheminformatics. 8, 17. doi:10.1186/s13321-016-0129-3
Olivetti, E. A., Ceder, G., Gaustad, G. G., and Fu, X. (2017). Lithium-ion battery supply chain considerations: Analysis of potential bottlenecks in critical metals. Joule 1, 229–243. doi:10.1016/j.joule.2017.08.019
Otani, M., and Sugino, O. (2006). First-principles calculations of charged surfaces and interfaces: A plane-wave nonrepeated slab approach. Phys. Rev. B 73, 115407. doi:10.1103/PhysRevB.73.115407
Pascal, T. A., Lin, S.-T., and Goddard, W. A. (2011). Thermodynamics of liquids: Standard molar entropies and heat capacities of common solvents from 2PT molecular dynamics. Phys. Chem. Chem. Phys. 13, 169–181. doi:10.1039/C0CP01549K
Pavarini. E, Koch. E, Anders. F, and Jarrell. M (Editors) (2012). Correlated electrons: from models to materials. lecture notes of the Autumn School Correlated Electrons 2012: at Forschungszentrum Jülich, 3-7 September 2012. No. Band 2 in Schriften des Forschungszentrums Jülich. Reihe Modeling and Simulation (Jülich: Forschungszentrum Jülich, Zentralbibliothek, Verl)for Advanced Simulation, I., and for Simulation Sciences, G. R. S.
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Perdew, J. P., Ruzsinszky, A., Csonka, G. I., Vydrov, O. A., Scuseria, G. E., Constantin, L. A., et al. (2008). Erratum: Restoring the density-gradient expansion for exchange in solids and surfaces [phys. Rev. Lett.100, 136406 (2008)]. Phys. Rev. Lett. 100, 039902. doi:10.1103/physrevlett.102.039902
Petukhov, A. G., Mazin, I. I., Chioncel, L., and Lichtenstein, A. I. (2003). Correlated metals and the LDA + U method. Phys. Rev. B 67, 153106. doi:10.1103/PhysRevB.67.153106
Plimpton, S. (1995). Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19. doi:10.1006/jcph.1995.1039
Ruban, A., Hammer, B., Stoltze, P., Skriver, H., and Nørskov, J. (1997). Surface electronic structure and reactivity of transition and noble metals1Communication presented at the First Francqui Colloquium, Brussels, 19–20 February 1996.1. J. Mol. Catal. A Chem. 115, 421–429. doi:10.1016/S1381-1169(96)00348-2
Rustad, J. R. (2012). Density functional calculations of the enthalpies of formation of rare-Earth orthophosphates. Am. Mineral. 97, 791–799. doi:10.2138/am.2012.3948
Ryee, S., and Han, M. J. (2018). The effect of double counting, spin density, and Hund interaction in the different DFT+U functionals. Sci. Rep. 8, 9559. doi:10.1038/s41598-018-27731-4
Saha, P., and Zenyuk, I. V. (2021). Electrokinetic streaming current method to probe polycrystalline gold electrode-electrolyte interface under applied potentials. J. Electrochem. Soc. 168, 046511. doi:10.1149/1945-7111/abf4aa
Şaşıoğlu, E., Friedrich, C., and Blügel, S. (2011). Effective coulomb interaction in transition metals from constrained random-phase approximation. Phys. Rev. B 83, 121101. doi:10.1103/PhysRevB.83.121101
Schnell, I., Czycholl, G., and Albers, R. C. (2002). Hubbard-U calculations for Cu from first-principle Wannier functions. Phys. Rev. B 65, 075103. doi:10.1103/PhysRevB.65.075103
Schwarz, K., and Sundararaman, R. (2020). The electrochemical interface in first-principles calculations. Surf. Sci. Rep. 75, 100492. doi:10.1016/j.surfrep.2020.100492
Seo, D.-H., Urban, A., and Ceder, G. (2015). Calibrating transition metal energy levels and oxygen bands in first principles calculations: Accurate prediction of redox potentials and charge transfer in lithium transition metal oxides. Phys. Rev. B 92, 115118. doi:10.1103/PhysRevB.92.115118
Shishkin, M., and Sato, H. (2016). Self-consistent parametrization of DFT + U framework using linear response approach: Application to evaluation of redox potentials of battery cathodes. Phys. Rev. B 93, 085135. doi:10.1103/PhysRevB.93.085135
Stamenkovic, V. R., Strmcnik, D., Lopes, P. P., and Markovic, N. M. (2017). Energy and fuels from electrochemical interfaces. Nat. Mat. 16, 57–69. doi:10.1038/NMAT4738
Stout, J. W., and Fisher, L. H. (1941). The entropy of formic acid. The heat capacity from 15 to 300°K. Heats of fusion and vaporization. J. Chem. Phys. 9, 163–168. doi:10.1063/1.1750869
Takahashi, Y., Kijima, N., Dokko, K., Nishizawa, M., Uchida, I., and Akimoto, J. (2007). Structure and electron density analysis of electrochemically and chemically delithiated LiCoO2 single crystals. J. Solid State Chem. 180, 313–321. doi:10.1016/j.jssc.2006.10.018
Tesch, R., Kowalski, P. M., and Eikerling, M. H. (2021). Properties of the Pt(111)/electrolyte electrochemical interface studied with a hybrid DFT-solvation approach. J. Phys. Condens. Matter 33, 444004. doi:10.1088/1361-648X/ac1aa2
Tesch, R., and Kowalski, P. M. (2022). Hubbard U parameters for transition metals from first principles. Phys. Rev. B 105, 195153. doi:10.1103/PhysRevB.105.195153
van Elp, J., Wieland, J. L., Eskes, H., Kuiper, P., Sawatzky, G. A., de Groot, F. M. F., et al. (1991). Electronic structure of CoO, Li-doped CoO, and LiCoO2. Phys. Rev. B 44, 6090–6103. doi:10.1103/PhysRevB.44.6090
Vanderbilt, D. (1990). Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895. doi:10.1103/PhysRevB.41.7892
Vasiljevic, N., Trimble, T., Dimitrov, N., and Sieradzki, K. (2004). Electrocapillarity behavior of Au(111) in SO2−4 and F−. Langmuir 20, 6639–6643. doi:10.1021/la049632f
Vogiatzis, K. D., Polynski, M. V., Kirkland, J. K., Townsend, J., Hashemi, A., Liu, C., et al. (2019). Computational approach to molecular catalysis by 3d transition metals: Challenges and opportunities. Chem. Rev. 119, 2453–2523. doi:10.1021/acs.chemrev.8b00361
Wang, J., Zhou, Y., and Lin, Z. (2005). First-principles elastic stiffness of LaPO4 monazite. Appl. Phys. Lett. 87, 051902. doi:10.1063/1.2005392
Wang, L., Li, J., Lu, G., Li, W., Tao, Q., Shi, C., et al. (2020). Fundamentals of electrolytes for solid-state batteries: Challenges and perspectives. Front. Mat. Sci. 7. doi:10.3389/fmats.2020.00111
Wang, M., and Navrotsky, A. (2004). Enthalpy of formation of LiNiO2, LiCoO2 and their solid solution. LiNi1xCoxO2. Solid State Ionics 166, 167–173. doi:10.1016/j.ssi.2003.11.004
Wu, X., Kang, F., Duan, W., and Li, J. (2019). Density functional theory calculations: A powerful tool to simulate and design high-performance energy storage and conversion materials. Prog. Nat. Sci. 29, 247–255. doi:10.1016/j.pnsc.2019.04.003
Yoshida, T., Hongo, K., and Maezono, R. (2019). First-principles study of structural transitions in LiNiO2 and high-throughput screening for long life battery. J. Phys. Chem. C 123, 14126–14131. doi:10.1021/acs.jpcc.8b12556
Zaffran, J., and Caspary Toroker, M. (2016). Benchmarking density functional theory based methods to model NiOOH material properties: Hubbard and van der waals corrections vs hybrid functionals. J. Chem. Theory Comput. 12, 3807–3812. doi:10.1021/acs.jctc.6b00657
Zhang, Z., Mondal, S., Mandal, S., Allred, J. M., Aghamiri, N. A., Fali, A., et al. (2021). Neuromorphic learning with Mott insulator NiO. Proc. Natl. Acad. Sci. 118, e2017239118. doi:10.1073/pnas.2017239118
Keywords: atomistic simulations, energy materials, electronic structure, electrodes for batteries, thermodynamics, electrolyte, electrochemical conditions
Citation: Kowalski PM, Bornhake T, Cheong O, Dohrmann N, Koch Liston AL, Potts SK, Shad A, Tesch R and Ting Y-Y (2023) Fundamentals of energy storage from first principles simulations: Challenges and opportunities. Front. Energy Res. 10:1096190. doi: 10.3389/fenrg.2022.1096190
Received: 11 November 2022; Accepted: 19 December 2022;
Published: 12 January 2023.
Edited by:
Sheng S. Zhang, United States Army Research Laboratory, United StatesReviewed by:
Sicong Zhu, Wuhan University of Science and Technology, ChinaYujin Tong, University of Duisburg-Essen, Germany
Copyright © 2023 Kowalski, Bornhake, Cheong, Dohrmann, Koch Liston, Potts, Shad, Tesch and Ting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piotr M. Kowalski, cC5rb3dhbHNraUBmei1qdWVsaWNoLmRl