 Ines Sanchez-Donoso1,2*
Ines Sanchez-Donoso1,2* Jisca Huisman3
Jisca Huisman3 Jorge Echegaray1
Jorge Echegaray1 Manel Puigcerver4
Manel Puigcerver4 José D. Rodríguez-Teijeiro2
José D. Rodríguez-Teijeiro2 Frank Hailer5,6
Frank Hailer5,6 Carles Vilà1*
Carles Vilà1*- 1Conservation and Evolutionary Genetics Group, Integrative Ecology Department, Estación Biológica de Doñana - Consejo Superior de Investigaciones Científicas (EBD-CSIC), Seville, Spain
- 2Department of Animal Biology, Faculty of Biology, University of Barcelona, Barcelona, Spain
- 3Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, UK
- 4Departament de Didàctica de les Ciències Experimentals i la Matemàtica, Facultat de Formació del Professorat, University of Barcelona, Barcelona, Spain
- 5LOEWE Biodiversity and Climate Research Centre (BiK-F), Ecological Genomics, Senckenberg Gesellschaft für Naturforschung, Frankfurt am Main, Germany
- 6Center for Conservation and Evolutionary Genetics, Smithsonian Conservation Biology Institute, National Zoological Park, Washington, DC, USA
Interbreeding of two species in the wild implies introgression of alleles from one species into the other only when admixed individuals survive and successfully backcross with the parental species. Consequently, estimating the proportion of first generation hybrids in a population may not inform about the evolutionary impact of hybridization. Samples obtained over a long time span may offer a more accurate view of the spreading of introgressed alleles in a species' gene pool. Common quail (Coturnix coturnix) populations in Europe have been restocked extensively with farm quails of hybrid origin (crosses with Japanese quails, C. japonica). We genetically monitored a common quail population over 15 years to investigate whether genetic introgression is occurring and used simulations to investigate our power to detect it. Our results revealed that some introgression has occurred, but we did not observe a significant increase over time in the proportion of admixed individuals. However, simulations showed that the degree of admixture may be larger than anticipated due to the limited power of analyses over a short time span, and that observed data was compatible with a low rate of introgression, probably resulting from reduced fitness of admixed individuals. Simulations predicted this could result in extensive admixture in the near future.
Introduction
Interspecific hybridization and its evolutionary consequences are often studied by analysis of samples obtained over a relatively short time period, without aiming to monitor genetic changes over time. This approach may allow identification of admixed individuals, but it may not be informative about the evolutionary impact of introgressive hybridization. For instance, the presence of first generation hybrids does not necessarily imply the spread of invasive alleles in the native population (introgression). When hybrids have reduced fitness or are infertile, hybridization might not have a large impact on the genetic composition of the parental species. Hybridization in such cases will still imply a loss of intraspecific mating opportunities, reducing the effective breeding population (accelerating genetic drift). Alternatively, if alien alleles do introgress into the gene pool of the native population, hybridization can have long-term fitness consequences (Rhymer and Simberloff, 1996; Haygood et al., 2003).
To estimate the impact and rate of admixture, the proportion of first generation hybrids and subsequent backcrosses can be assessed from analysis with a large panel of molecular markers (Vähä and Primmer, 2006). If the analyzed population sample includes individuals with varying degrees of admixture, this implies that first generation hybrids are interbreeding with the parental species, leading to alleles from one species introgressing into the gene pool of the other (Rhymer and Simberloff, 1996). However, such data can only offer a snapshot of the evolutionary process, without informing about changes over time. Furthermore, inference about long-term effects are susceptible to limitations due to small sample sizes (is the sample representative of the entire population?) and low frequency of interbreeding (is it possible to estimate the frequency of rare interbreeding events?). In addition, an important and often overlooked source of error is associated with varying levels of admixture from year to year (is the level of admixture changing over time?).
Samples obtained over several generations can help to assess temporal changes in the level of admixture and can offer a long-term view on the introgression of invasive alleles, more meaningful from a conservation and evolutionary point of view. The robust characterization of slow introgression requires the use of long time series to allow tracking the arrival of new alleles into the population.
Common quail (Coturnix coturnix) populations offer the opportunity to obtain long temporal series of samples to investigate the consequences of admixture of divergent lineages. Its European breeding population is estimated to consist of several millions of individuals (8,400,000–14,100,000, BirdLife International, 2004) that move nomadically across the breeding range looking for suitable habitat (Puigcerver et al., 1989; Rodríguez-Teijeiro et al., 2009) and mates (Rodríguez-Teijeiro et al., 2006). This game species is of high economic and recreational interest in southern Europe; millions of quails are killed every hunting season (Guyomarc'h, 2003). Due to this interest, every year wild populations are extensively restocked prior to the game season with farm-reared quails. For example, in Catalonia (Northeast Spain), the wild common quail population has been estimated to range between 5000 and 20,000 males (Rodríguez-Teijeiro et al., 2004), and about 57,000 farm-reared individuals have been released yearly from 1990 to 2011 (Hunting order plans of the Regional Government of Catalonia). The number, size, timing, and location of the releases change from year to year and very limited information is available for most of them. While quails for game restocking are sold as be common quail, recent genetic analyses have shown that they are, actually, hybrids between common quails and domestic Japanese quails (C. japonica) that have been interbreeding for a number of generations in game farms (Sanchez-Donoso et al., 2012).
Captive breeding represents a general relaxation of selective forces and promotes traits that are maladaptive in the wild (Lynch and O'Hely, 2001; Frankham, 2008; Baskett and Waples, 2013). Thus, supplementation programs based on the release of captive-bred individuals have the potential to deeply alter the genetic composition of native populations (Ford, 2002). These large-scale releases can lead to loss of genetic diversity, breakdown of adaptations and change in the population genetic structure (Eldridge and Naish, 2007; Laikre et al., 2010; Marie et al., 2010). Specifically, several authors have pointed out that restocking with domestic Japanese quails and hybrids can pose serious threats to the genetic integrity and survival of common quails (Guyomarc'h, 2003; Barilani et al., 2005; Chazara et al., 2006, 2010; Puigcerver et al., 2007). Their interbreeding can lead to introgression of maladaptive domestic Japanese quail alleles into the common quail population, potentially leading to alterations or loss of migratory behavior, and a decline in fitness in native quails (Guyomarc'h, 2003). A decrease in average fitness means a lower population growth rate, which could even become negative. However, even in the case that fitness of admixed individuals was not diminished, the massive releases of farm-reared quails could lead to a counter-intuitive reduction in the effective population size through Ryman-Laikre effects: a large portion of the breeding population could potentially derive from a small number of breeders from farms, resulting in a reduction of the genetic diversity and evolutionary potential (Ryman and Laikre, 1991). As a result of these concerns, the EU Quail Action plan has emphasized the need to preserve common quail genetic integrity (Perennou, 2009).
Several lines of evidence support the existence of introgressive hybridization from farm quails into wild common quails. Japanese and admixed quails have vocalizations that differ from those of common quails, with first generation hybrid male crows being intermediate between domestic Japanese quail and common quail (Derégnaucourt et al., 2001), and this has often been used to characterize the identity of free-ranging quail males (Guyomarc'h and Guyomarc'h, 1996; Collins and Goldsmith, 1998; Barilani et al., 2005; Puigcerver et al., 2007). Using this approach, hybrid quails were first detected in Catalonia by members of our research group in 1990 in an area close to the study site (Rodríguez-Teijeiro et al., 1993). Since then, and following the popularization of farm quails for restocking, hybrid individuals have been detected, using genetic methods, in common quail breeding areas in multiple European countries including Portugal, France, Italy, and Spain (Rodríguez-Teijeiro et al., 1993; Guyomarc'h, 2003; Barilani et al., 2005). Experiments in captivity have shown that common and Japanese quails present only partial mate discrimination (Derégnaucourt and Guyomarc'h, 2003), and that the two species interbreed easily and hybrids do not have reduced survival (Derégnaucourt et al., 2002). These observations suggest that Japanese quail alleles may have been introgressing into the European common quail population since the early 1990s.
However, the occurrence and frequency of interbreeding under natural conditions between native and released quails is not known. Furthermore, while hybrids do not show reduced survival in captivity, selection against hybrids in the wild could potentially lead to strongly reduced rates of introgression. For this reason, it has been suggested that individuals identified in nature as hybrids could correspond to farm released animals, without requiring any hybridization in the wild at all (Puigcerver et al., 2007, 2012). This is in part supported by the radio-monitoring of female farm quails, which showed an increased mortality and predation risk for their clutches (Puigcerver et al., 2014). However, this same study also revealed their success at attracting wild males and reproducing in the wild. Consequently, it remains unclear to what degree these massive releases are affecting European common quail populations.
Here we studied the genetic composition of wild quails sampled in a single locality in Northeast Spain across a 15-year period (about 15 generations), to evaluate to what extent the releases of farm quails are affecting the genetic composition of the local population. We combined genetic analyses with simulations to test the power of our markers to detect introgression of farm alleles. We also carried out simulations assuming different levels of fitness for the admixed individuals to investigate which values are compatible with the observed data. We then used these values in further simulations to forecast changes in the genetic composition of the local population in the near future under different scenarios.
Materials and Methods
Samples and Datasets
The field survey was carried out in an area of about 1 km2 in Northeast Spain (Mas Esplugues, 41° 25′ N, 1° 23′ E, 628 m above sea level), a short, narrow valley with winter cereal crops (mainly barley and wheat) which constitute suitable breeding habitat for quails. The breeding population has been continuously monitored for over 25 years in the framework of a long-term project to study the biology and mating behavior of common quails (José D. Rodríguez-Teijeiro. and Manel Puigcerver). It is an open population and individuals continuously arrive during the breeding season, spend some time, and leave in search of suitable habitat patches and mates (Puigcerver et al., 1989; Rodríguez-Teijeiro et al., 2006, 2009). This is a game area, and the hunting season starts in mid-August, after the end of the breeding season (from about mid-April to early July). The great majority of quails breeding in the area were estimated to be 1 year old, with very few breeders being aged two or more (Puigcerver et al., 1992). For this reason, we considered generation time to be about 1 year.
A feather or a blood sample was obtained from 468 wild quails, captured during several breeding seasons (i.e., prior to the majority of farm releases), following protocols approved by an authorized Ethics Committee (for sampling methods see Puigcerver et al., 2007). Our sample represents a very large proportion of the quails breeding in the study area every season, but is very difficult to have precise count of the number of breeders. These wild quails were genotyped at 9–13 autosomal microsatellite loci. Genotypes were generated as three different datasets (Table 1) produced by different researchers, in different labs and using different instruments. Dataset I included quail samples collected during the breeding seasons of 1996 and 1997, genotyped by Ines Sanchez-Donoso. A subset of these genotypes was included in a previous study (Sanchez-Donoso et al., 2012). Dataset II included quail samples collected during the breeding seasons of 1999, 2002 and 2005, genotyped by Jisca Huisman as part of her MSc thesis at the University of Uppsala, Sweden (Huisman, 2006). Dataset III included quail samples obtained during the breeding seasons from the period 2007–2010, genotyped by Ines Sanchez-Donoso for this study.
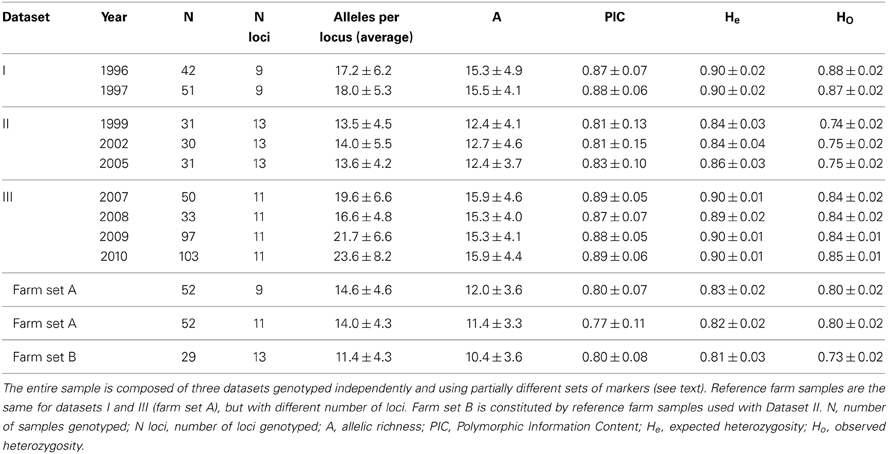
Table 1. Genetic diversity (averaged across loci, ± standard deviation) of wild and farm quails at autosomal microsatellites.
Quail samples from Spanish game farms were analyzed as reference for the three wild quail datasets: 52 individuals from 5 farms (farm set A, sampled in 2009 and 2010, included in Sanchez-Donoso et al., 2012) were used as a reference in analysis with datasets I and III, while 29 individuals from 3 game farms (farm set B, sampled in 2006) were used with Dataset II (see Table 1). All farm individuals were intended for restocking. In a previous study, no genetic differentiation was observed among individuals from the different farms (Sanchez-Donoso et al., 2012, including some of the samples studied here), suggesting that all of them derive from the same stock and/or that the exchange between them is extensive. For this reason, and considering the large size of the overall population in farms and the lack of precise information about the origin and number of breeders, in the simulations we assumed that the allele frequencies in the farm population remained constant throughout the study period (see below).
Joining the three datasets offered the opportunity to compile a time series spanning 15 years. They could not be fully integrated, as the sets of genetic markers used were not identical (see below) and genotype scoring was done by different researchers. Since many DNA samples derived from small feathers, each providing only a small amount of DNA, it was not possible to re-analyze them under a unique set of conditions. Datasets I and III could be merged when restricting analyses to the markers used in both cases after ensuring the consistency of the allele call by genotyping a panel of reference samples.
Laboratory Procedures
For some individuals, DNA was extracted from blood samples (100 μl, stored at −20°C in 95% ethanol until analysis) using DNeasy Blood and Tissue Kit (Qiagen) following manufacturer's protocols, or following standard phenol-chloroform extraction procedures. For the rest of individuals, DNA was extracted from feathers (stored at −20°C in 95% ethanol) using a Chelex resin protocol (Walsh et al., 1991).
To characterize the maternal ancestry of quails and to determine directionality of hybrid matings, a 453 base pair (bp) fragment of the 5′ of the mitochondrial DNA (mtDNA) control region was sequenced for 26 wild quails from Dataset II (7 from 1999, 7 from 2002 and 12 from 2005) and the 29 farm quails from farm set B. The wild quails were selected to include diverse phenotypes and, potentially, admixture. Having admixed individuals of farm and wild origin could allow inferring if both sets of admixed birds had the same maternal origin. DNA samples were amplified via polymerase chain reaction (PCR) using primers PHDL and PH-H521 (Barilani et al., 2005). Sequencing reactions were carried out using the same forward and reverse primers, and were run on a MegaBACE 1000™ instrument (Amersham Biosciences) following manufacturer's protocols. From the two sequences obtained, a consensus was built using SEQUENCHER v. 5.1 (Gene Codes Corporation, Ann Arbor, MI, USA).
Individuals were genotyped for autosomal microsatellite loci originally developed for Japanese quail (Kayang et al., 2000, 2002) (genotype data sets in Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.kv733). Some loci (marked below with an asterisk) were analyzed for datasets I and III using primers modified to facilitate multiplexing (see Supplementary Material Table S1). The number and combination of loci typed varied between datasets. Dataset I was genotyped for 9 unlinked markers (each marker was located on a different chromosome): GUJ01, GUJ17*, GUJ39, GUJ44, GUJ57*, GUJ65*, GUJ85, GUJ93, and GUJ97*. Dataset II was typed for these same markers, excluding GUJ85, plus five additional markers: GUJ14, GUJ28, GUJ33, GUJ62, and GUJ68 (13 loci in total). Although some loci used for Dataset II are on the same chromosome (Kayang et al., 2004), previous analyses on this dataset showed they are not in linkage disequilibrium (Huisman, 2006). Dataset III was genotyped for the same 9 loci as Dataset I, plus GUJ28 and GUJ74 (11 loci in total, located on different chromosomes). Since datasets I and III were scored by the same researcher, and using a panel of reference samples to ensure that all allele calls were consistent, they could be analyzed together for the 9 shared markers.
Some markers were PCR-amplified in a multiplex, while others were amplified separately. Detailed protocols are available upon request. PCR products from dataset I and III, and from their reference farm set, were electrophoresed on an ABI 3730 sequencer (Applied Biosystems) following manufacturer's protocols, and alleles were sized and scored using the software GeneMapper v3.5 (Applied Biosystems). Electrophoresis of Dataset II PCR products, and of their reference farm set, was performed on a MegaBace 1000 capillary instrument (GE Healthcare) following manufacturer's protocols. The associated software, Genetic Profiler 2.2, was used for the allele calling.
Data Analysis
Mitochondrial DNA sequences were aligned with Muscle in MEGA v5.1 (Tamura et al., 2011). The same program was used to compare sequences and for phylogenetic reconstructions using maximum likelihood (ML) with the Tamura-Nei model of sequence evolution and a gamma distribution rate of variation across loci, and neighbor-joining (NJ) based on the maximum composite likelihood model. Support for nodes was assessed with 500 bootstrap replicates.
Average number of alleles per locus and allelic richness (mean number of alleles per locus corrected for the minimum sample size; Leberg, 2002) were calculated using FSTAT version 2.9.3.2 (Goudet, 1995). In order to measure marker informativeness we calculated the Polymorphic Information Content (PIC; Botstein et al., 1980) which takes into account the number of alleles per locus and the frequency of these alleles, using EXCEL MICROSATELLITE TOOLKIT 3.1.1 (Park, 2001). We used the same software to calculate observed (Ho) and expected (gene diversity, He) heterozygosities (Nei, 1987). Overall differentiation between wild and farm samples (for each sample set) was estimated using Weir and Cockerham's (1984) estimate of the fixation index Fst in GENETIX (Belkhir et al., 1996–2004), Hedrick's (2005) standardized measure of genetic differentiation (G'ST) and Jost'D estimator of actual differentiation (Dest, Jost, 2008) with SMOGD (Crawford, 2010).
Genetic composition of wild and farm quails was analyzed using the Bayesian clustering procedure implemented in STRUCTURE 2.3.4 (Pritchard et al., 2000). Other likelihood approaches could have been used to infer population structure, but we chose the approach implemented in STRUCTURE because its use is widespread for hybridization studies, and because it is easy to scale up to analyze thousands of simulated datasets across multiple computer processors (see below). Since our goal was to track the introgression of farm alleles into the wild population we chose to always use the same analytical approach and we avoided using approaches aimed at identifying individual hybrids and backcrosses (for example NEWHYBRIDS, Anderson and Thompson, 2002).
For a given number (K) of genetically distinct clusters, STRUCTURE provides an estimate of the fraction of the genome (qi) that belongs to each one of the clusters. Analyses were performed under the admixture model, as each individual may have ancestry in more than one parental population, and were run with correlated allele frequencies and without population or sampling location information (USEPOPINFO and LOCPRIOR set to 0). Analyses were done for K = 2 to separate farm and wild clusters, since previous analyses showed that this is the highest level of population structure (Sanchez-Donoso et al., 2012). Analyses were run for 100,000 steps following a burn-in period of 100,000 steps, and were replicated five times. Results did not vary significantly with longer runs and likelihood values were observed to converge during the runs. STRUCTURE proportion of membership to each cluster (qi) and 90% posterior probability intervals were estimated for each individual. For consistency across real and simulated datasets, STRUCTURE was always run using these parameters.
All STRUCTURE analyses included wild-caught quails together with quails from farms used as reference to allow the identification of two clusters that would facilitate tracking farm and wild origins. The value of qı = 0.90 was used as an arbitrary threshold above which individuals were assigned to cluster i as pure (see Barilani et al., 2005). Individuals that showed lower values (qi < 0.9 for both clusters) were considered as potentially admixed.
To study if there was an increase through time in the proportion of farm alleles in the wild population, we focused on the estimated yearly proportion of wild-caught individuals classified as admixed (0.1 < qfarm < 0.9) in the population, rather than the degree of admixture for each individual. The classification as being admixed or not for each individual was the response variable (binary) and was analyzed using Generalized Linear Models (GLM). Sampling year was the continuous explanatory variable, since the interest was to test whether there was a trend in the ratio of admixed individuals over time. A binomial error structure and logit link function was used, and the models were fitted in R (version 2.15.2; R Core Team, 2012) using the glm() function. The factor “dataset” was included to account for differences due to investigator and marker set when analyzing the three different data sets. All models were tested for overdispersion to avoid false positives and for collinearity between “year” and “dataset.” With these models we investigated if the probability of being classified as admixed was different between years and, if so, if it was increasing over time.
Simulation of Farm Allele Introgression
Initial analyses of the observed data showed low levels of admixture between wild and farm quails. To test if these analyses had enough statistical power to detect an increase of admixture over time, we performed STRUCTURE analyses on data simulated with a series of scripts (available at http://www.consevol.org). These simulations were intended to represent the population at the breeding season (when field surveys took place). The scenarios that we investigated involved varying the proportion of farm individuals in the breeding population every generation, and different fitness values for admixed individuals born in the wild.
We first simulated genotypes for a wild population of quails where every generation a certain proportion of the potential breeders originated from farm releases. The simulations thus portrayed the composition of our study population during the breeding season. Because wild quails in the area show high mobility (Rodríguez-Teijeiro et al., 1992) and are part of a much bigger population (millions of individuals), we simulated a large population with a constant size of 10,000 breeding individuals to minimize the effects of random genetic drift. Genotypes at generation 0 were generated by randomly drawing two alleles for each locus according to their observed frequency. Starting allele frequencies were based on Dataset III, after excluding individuals whose genetic composition was clearly dominated by farm alleles (qfarm ≥ 0.40 according to STRUCTURE analyses of that dataset alone; this threshold was selected to exclude individuals born in farms—qfarm close to 1—and first generation admixed individuals—qfarm close to 0.5). Similarly, 10,000 genotypes were generated for farm quails by randomly drawing pairs of alleles for each locus based on the observed allele frequencies from farm set A. Since we had selected unlinked markers (see above), alleles at one locus did not influence allele selection at other loci (markers are assumed to recombine freely). Also, since our simulations only cover a limited time span (a few decades at the most), we did not consider the effect of mutations. Although Datasets III and farm set A may have an allelic composition different to the allelic composition of the populations before any admixture had taken place in the wild, we consider that these datasets may be an appropriate overall representation of the degree of differentiation between wild and farm quails and can offer useful insight on how introgression can be expected to proceed.
Successive generations (up to 20 generations to parallel the time period since the first hybrids were detected in the study area) of wild quails involved 10,000 new individuals per generation. To simulate the continuous restocking of quails, each of these new individuals had a certain probability of originating from farm releases (between 1 and 5%, according to the genetic analyses of our field samples; see below) and, in this case, was directly drawn from the file of farm genotypes. The genotypes for the rest of the individuals were generated by drawing alleles from two parental genotypes randomly selected from the previous generation. The simulation was run for 20 generations. We assumed non-overlapping generations and a generation time of 1 year. We did not assign sex to the genotypes and any pair of genotypes could act as parents for a genotype in the following generation. A schematic representation of these simulations is portrayed in Figure 1.
Figure 1. Schematic representation of the population simulations (gray arrows) and analysis of simulated data (black arrows). Boxes represent simulated populations or datasets. Simulations were used to assess the effect over time (across different generations) of the proportion of potential breeders of farm origin and varying fitness of admixed individuals (variables affecting the population simulations, marked with discontinuous circles). The first set of simulations was carried out assuming that all individuals, independently of having farm ancestry or not, had the same probability of contributing to the next generation (fitness = 1). For details, see text.
Initial simulations assumed that all individuals had the same chances of producing offspring in the following generation (fitness = 1) independently of their origin (farm, wild, or admixed). However, it is likely that farm individuals contribute maladaptive traits (Lynch and O'Hely, 2001; Frankham, 2008; Baskett and Waples, 2013), and that they and their offspring have a lower fitness. To introduce this factor into a second set of simulations, we tracked for each individual the proportion of the genome expected to come from the farm population (1 for farm individuals, 0 for pure common quails, 0.5 for first generation admixed individuals). Each time that a new genotype was generated and one or both of the parents had a proportion of farm ancestry of 1 or 0.5, its probability of survival to the breeding season was determined by a predefined fitness value varying between 1 and 0 (1 when the probability of contributing to the next generation for admixed individuals was the same as for wild common quails, 0 when they never contributed; in the simulations we used the following fitness values: 1, 0.8, 0.6, 0.4, or 0.2). To simplify, we assumed that the fitness value was the same independently of the individual's proportion of farm ancestry. If the genotype did not survive, it was not included among the genotypes for a given generation (this would only include those that could participate as potential breeders), and a completely new genotype was generated to replace it and to keep the total population size constant. We assumed that lower fitness affected the chances of survival for the first two generations of admixed individuals, which are the ones that are most likely to suffer loss of extrinsic or intrinsic adaptation (i.e., presence of maladaptive alleles or breakup of co-adapted gene complexes, Laikre et al., 2010). After two generations genetic incompatibilities may be less likely and those individuals were considered as if they were pure wild quails for subsequent generations (proportion of farm ancestry: 0).
Analysis of Simulated Data and Power Tests
For a visual examination of the level of introgression in the simulated population, we randomly sampled individuals from multiple generations (50 individuals per generation for a few predefined generations) and analyzed them with STRUCTURE (K = 2) together with a simulated farm dataset (50 farm individuals). The same analysis was also carried out breaking the temporal series into three sets of samples, covering a shorter time span each, mirroring the three datasets resulting from the field work (Table 1). In addition, in order to compare the results from the simulated data with the results obtained for the field datasets and to assess the power to detect introgression with the markers used, we prepared a program to replicate our field sampling pattern. Since the first hybrid individual in a neighboring area was detected by its vocalizations in 1990 (Rodríguez-Teijeiro et al., 1993), several years before the first of the wild-captured quails analyzed in this study, individuals from 1996 were assumed similar to the 5th simulated generation, 1997 to the 6th, and so on. The program produced 1000 sets of simulated data extracting genotypes from the simulated population of 10,000 individuals per generation and with the same number of individuals per year as in real data (see Table 1). Each one of these simulated datasets was analyzed with STRUCTURE as described above (Figure 1).
For each STRUCTURE run we calculated the proportion of simulated genotypes that were not classified as pure wild (qfarm > 0.10; admixed or pure farm). The 95% confidence interval for this proportion, for a given combination of potential breeders coming from farms and fitness value, was based on the 2.5th and 97.5th percentiles over each set of 1000 STRUCTURE runs. The proportion observed for the real data was then compared with these confidence intervals to assess which simulated scenarios were compatible with the real data (analysis of field samples).
Results
To clarify analyses and results, a flow chart of the analyses and conclusions reached with them is provided as Supplementary Material (Figure S1).
Mitochondrial DNA
Since mtDNA is maternally inherited and common quail and Japanese quail have well differentiated sequences (Barilani et al., 2005), this marker is useful for establishing directionality in hybrid matings. ML and NJ phylogenetic trees showed the existence of two clearly differentiated clades (Figure 2). The control region sequences of 29 farm quails revealed 8 haplotypes and all of them were located within the same clade, with pairwise differences ranging between 1 and 10 substitutions (prefix F in Figure 2). One of the haplotypes (F1W1 in Figure 2) was observed in 22 individuals, while the remaining 7 were observed in one individual each. In contrast, the diversity observed in wild quails was much higher, with 13 haplotypes in 26 quails, mostly located within a second clade (prefix W plus F1W1 in Figure 2). Five of the wild haplotypes were observed in more than one individual. One of these (F1W1), present in five wild individuals, was the only one shared with farm quails and corresponded to the most frequent farm haplotype. A GenBank BLAST search showed that F1W1 was identical to sequences previously identified in Japanese quails, supporting the hypothesis that farm quails derive from captive domestic Japanese quail females used as breeders, as suggested by Sanchez-Donoso et al. (2012). These sequences constitute the Japanese quail clade in Figure 2. Importantly, all analyzed farm individuals carried mtDNA sequences from this clade.
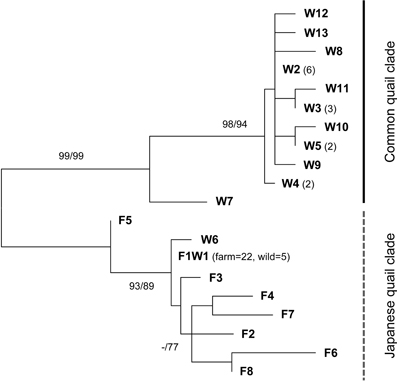
Figure 2. Unrooted maximum likelihood (ML) phylogenetic tree of mitochondrial control region haplotypes identified in wild (indicated with the prefix W) and farm (F) quails. Only one haplotype was shared between wild and farm quails (F1W1) (it was found in 22 farm quails and 5 wild quails). Number of quails sequenced with each haplotype is indicated in parenthesis when larger than 1. Bootstrap support for ML and NJ (in this order) is indicated at the nodes when higher than 70%. Two clades are found, separating common quail (Coturnix coturnix) from Japanese quail (C. japonica) sequences with high support.
Two of the haplotypes found in wild quails were either identical to (F1W1, in five individuals) or clustered with (W6, in one individual) farm haplotypes within the Japanese quail clade. The remaining sequences made up the second clade that did not include any farm sample (common quail clade), with differences between haplotypes also ranging between 1 and 10 substitutions. Despite the short size of the fragment studied here, sequences from the two clades were clearly differentiated by 17 to 24 substitutions. The existence of mitochondrial sequences from the Japanese clade in some wild quails confirms the presence of genetic material from quails of farm origin among the wild-sampled quails (all of these individuals were confirmed as being of farm origin or admixed in the microsatellite analyses, see below).
Identification of Admixed Individuals
All studied microsatellite loci were highly variable in both farm and wild quails. Despite the high polymorphism, previous paternity studies (not shown) revealed that the markers and protocols used did not result in false alleles. All individuals were successfully genotyped for at least 70% of the markers. The number of alleles per locus was high for all datasets and markers, with an average number of alleles per locus and allelic richness higher than 10 for all years and datasets (Table 1). As expected, the average number of alleles per locus was closely associated with sample size, but allelic richness remained practically constant from year to year within datasets. PIC values were high in all cases (with average PIC over 0.81 in wild quails and over 0.77 in farm quails), showing that the allelic variation was potentially very informative of population structure. Expected heterozygosity (He) was above 0.8 in all cases. This high heterozygosity results from the high number of alleles and similar values have been observed in other Galliform species with high allele diversity (He et al., 2008; Hassen et al., 2009; Bei et al., 2012). Observed heterozygosity (Ho) was slightly lower than He in all cases, which could be due to the presence of sets of alleles of different origin (farm and wild) that had not reached Hardy-Weinberg equilibrium. The values for He and Ho were almost constant within datasets, even when comparing datasets I and III (typed by the same person and equipment), which supports our initial assumption of negligible genetic drift.
Game farm quails showed lower average number of alleles per locus and allelic richness than wild quails analyzed with the same markers (Table 1). There was significant genetic differentiation between the wild (including possibly admixed individuals) and farm populations, and Fst values between them were very similar across datasets: Fst for Dataset I = 0.05, for Dataset II = 0.06, for Dataset III = 0.05 (p < 0.05 in all cases). Due to the high heterozygosity of our loci and the upper limit this imposes on Fst [1–(average within population He); Hedrick, 2005], these relatively low values actually represent a pronounced differentiation: Hedrick's standardized G'ST values were between 0.35 and 0.41 (ca. 40% of the maximum differentiation possible), and Dest between 0.25 and 0.31.
We used the software STRUCTURE to divide each dataset (together with the corresponding farm set) in two clusters (K = 2) in order to identify the contribution of the two quail lineages (wild or farm) to the genome of each individual. In each analysis (all five replicates produced practically identical output), a farm cluster and a wild cluster appeared clearly separated, and several individuals were identified as admixed (Figure 3). Using qfarm ≥ 0.90 as a threshold for the proportion of farm ancestry, between 79 and 90% of farm animals in each analysis were classified as pure farm, and none appeared as pure wild (Figure 4A and Supplementary Material Table S2A). Admixed individuals in the farm (between 10 and 21% of the farm animals) may represent descendants of crosses between farm quails and common quails (males) that are regularly introduced into farms as breeders (Sanchez-Donoso et al., 2012). Of the quails sampled in the wild, between 81 and 94% were identified as pure wild (qfarm ≤ 0.10). Some wild individuals (up to 10%) had very high qfarm, compatible with pure farm origin, and likely represent quails from restocking events. Overall, between 3 and 16% of wild-caught birds were classified as admixed (0.10 < qfarm < 0.90).
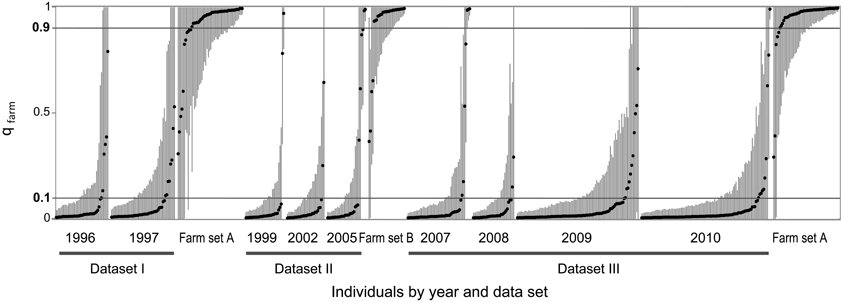
Figure 3. STRUCTURE analyses for K = 2 for wild and farm quails for three datasets from different time periods. Dots represent individual proportion of membership to farm cluster (qfarm) and vertical lines are 90% highest posterior density intervals. Individuals are sorted by dataset, year, and qfarm value. Horizontal lines at qfarm = 0.10 and 0.90 indicate threshold values used to separate pure (qfarm ≤ 0.10 for pure common quails, qfarm ≥ 0.90 for pure farm quails) and admixed individuals (0.10 < qfarm < 0.90; see text).
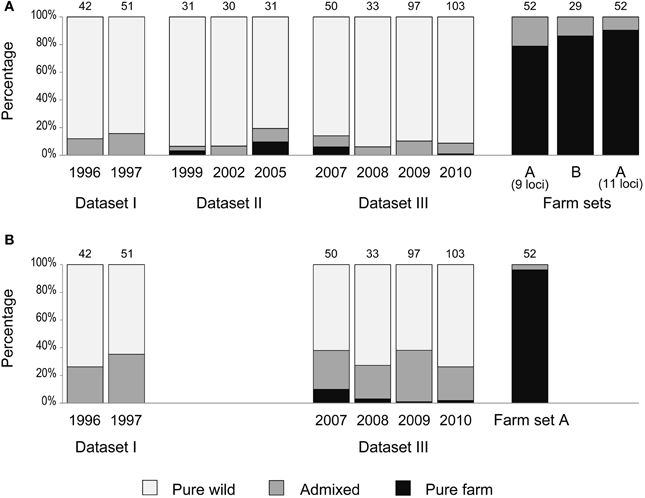
Figure 4. Percentage of individuals classified as pure wild (pale gray), pure farm (black) and admixed (dark gray) according to qfarm values obtained from STRUCTURE (K = 2). Number of samples per year is indicated above each bar. (A) Analysis carried out separately for each dataset (as in Figure 3 and Supplementary Material Table S2A). (B) Datasets I and III analyzed together (as in Figure 6 and Supplementary Material Table S2B).
Since we identified some individuals with admixed nuclear genotypes among farm quails, the presence of admixed individuals in the wild is not, per se, indicative of matings between restocked farm quails and native wild quails; these individuals could represent admixed animals released from farms. However, there are two lines of evidence that point to interbreeding in the wild. First, the ratio between admixed and pure farm, as classified by STRUCTURE, was much higher in the wild sample than in the farm sample (Figure 4A, Supplementary Material Table S2A; Yates Chi-square = 67.21, df = 1, p < 0.0001). We would expect the same ratio in farms and in the wild if no interbreeding occurred in natural conditions; the observed admixed birds in the wild would just have resulted from their release from farms. Therefore, the inflated proportion of admixed quails in the wild compared to the proportion of freely roaming pure farm quails points to possible crosses in the wild between restocked and wild quails. However, the observed pattern would also emerge without crosses in the wild if restocked pure farm quails had lower survival in the wild than admixed quails, thus leading to an over-representation of the admixed individuals in the field sample.
The second line of evidence derives from the mtDNA analysis. For the 26 wild samples from Dataset II used for mtDNA analyses, 6 were identified as admixed by their nuclear DNA and out of these, 4 had common quail mtDNA (3 had haplotype W2, and 1 had W3; Table 2, Figure 2). The other 2 had Japanese quail mtDNA, as had the 4 wild-caught individuals assigned as farm quails by their autosomal genotypes. These last individuals probably represent animals released from farms. Since none of the farm individuals had common quail mtDNA (0 out of 29) and female common quails are known to reproduce very poorly in captivity (Caballero de la Calle and Peña Montañés, 1997), finding four admixed individuals in the wild population with common quail mtDNA by chance is highly unlikely (Fisher exact test: p = 0.008) if they just represented animals released from farms. Our findings therefore strongly indicate successful mating in the wild between restocked quails (males) and native wild quails (females).

Table 2. Classification of 26 wild quails from Dataset II, based on autosomal microsatellites (as suggested by STRUCTURE: pure wild: qfarm ≤ 0.10; admixed: 0.10 < qfarm < 0.90; pure farm: qfarm ≥ 0.90), and mtDNA (common quail or Japanese quail mtDNA, Figure 2).
Changes Through Time in the Level of Introgression
During the 15 years encompassed by our study, there was no clear increase in the proportion of admixed individuals in the wild (Figures 3, 4A). GLM analyses using the year of sampling as independent variable and controlling for dataset failed to show a significant trend in the proportion of admixed individuals through time (β = 0.122, SE = 0.131, p = 0.352). Results did not change when we did not control for dataset (β = −0.028, SE = 0.030, p = 0.347).
To assess our power to detect an increase of the proportion of admixture in the wild, we simulated 15 generations of a population where about 1.5% of the potential breeders every generation were quails restocked from farms. This proportion is below the estimated average from our field data (2.2%; see Figure 4A and Supplementary Material Table S2A) and our simulations could thus exemplify the changes that are expected to occur in the population with a low-moderate contribution of farm alleles every generation. We randomly selected 50 simulated genotypes from different generations and 50 simulated farm genotypes. The proportion of admixed genotypes significantly increased over time (β = 0.133, SE = 0.035, p < 0.001; Figure 5A) when the simulated dataset spanning 15 generations was analyzed as a whole. Ten additional sets of simulated genotypes were generated and all of them showed a significant increase in the proportion of admixed genotypes (p < 0.001 in all cases). However, when the same data was analyzed subdivided into three datasets, mirroring the analysis of real data (Figure 3), the degree of introgression was underestimated. A smaller proportion of the simulated wild population appeared as admixed in later generations, and this proportion did not show changes over time (β = −0.031, SE = 0.039, p = 0.432; Figure 5B). The comparison of the qfarm values for the same simulated genotypes from the last generations (12 to 15) showed that fragmenting the data into shorter time periods led to an underestimation of qfarm in wild quails in later generations, and of the proportion of admixed individuals in the population (Figure 5D). The same comparison for the first simulated generations (1 and 2) did not show the same differences in qfarm values (Figure 5C). Thus, limiting the time span analyzed greatly reduced our ability to detect the progressive introgression of alleles from the farm population.
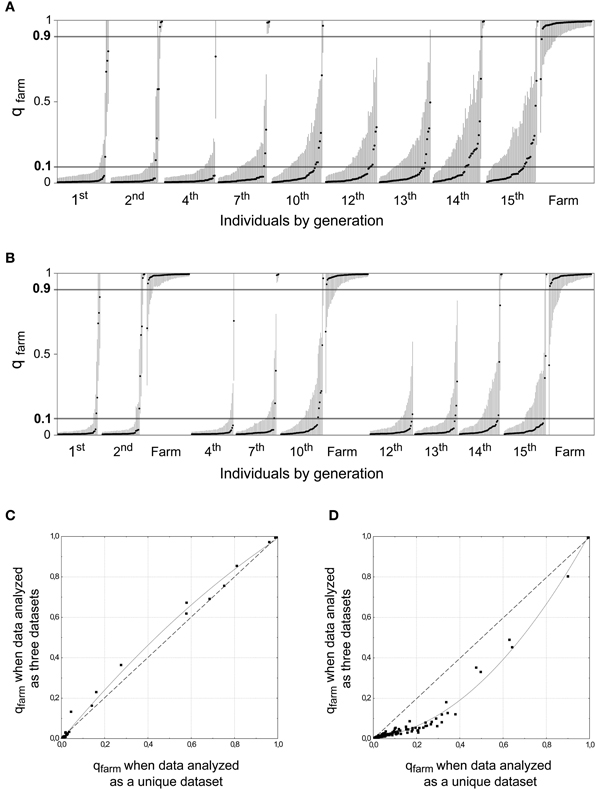
Figure 5. STRUCTURE analyses (K = 2) for simulated data assuming that about 1.5% of the potential breeders are of farm origin. Fifty genotypes were sampled from each generation and 50 simulated farm genotypes were included in the analyses. (A) All data included in the analysis as a unique dataset. (B) Same simulated data as above, but analyzed in three datasets separately, analogous to those in Figure 3 (same 50 simulated farm genotypes included in all analyses). Generations 12 to 15 do not show the degree of introgression that was inferred above. (C) Scatterplot comparing qfarm values for individuals from generations 1 and 2 obtained in (a) (X axis) B and (b) (Y axis). The curve represents the polynomial curve that best fits the data: qfarm values were almost identical for the two axes. (D) Similar scatterplot for individuals from generations 12, 13, 14, and 15. In this case, intermediate values of qfarm are underestimated when analyzed using short time series and more individuals appear as pure wild.
Similarly, the field dataset that spanned the longest time period (Dataset II, 6 years, Table 1) did not show a change in the proportion of admixed individuals over time (β = 0.184, SE = 0.183, p = 0.314); the same result was obtained for a different wild population located over 100 km north of our study site monitored during the same 6 years (data not shown, see Supplementary Material Text S1). We used simulations based on Dataset II to investigate the power to detect an increase of admixed individuals over a period of 6 years using the same number of markers and samples. We found that increased introgression of farm alleles would not be consistently detectable (in at least 90% of the cases) unless the proportion of potential breeders coming from farms was above 3% (see Supplementary Material Text S2), higher than the proportion observed in our study area.
Since the simulations showed that the ability to detect introgression of farm alleles into the wild population (with the sample size available) depends on the length of the time series considered, we joined datasets I and III for a STRUCTURE analysis covering a longer time span (although reducing the loci used to the nine shared by both datasets). This resulted in wider confidence intervals for the estimates of qfarm (Figure 6A). In agreement with the simulations (Figure 5A compared to Figure 5B), joining datasets I and III indicated larger admixture (Figure 6B). The proportion of individuals classified as pure wild decreased compared to previous analyses (Figure 4B and Supplementary Material Table S2B). Again, no noticeable change was observed in the proportion of admixed individuals through time (β = −0.007 SE = 0.021, p = 0.737): about 31% for the first two years of the series (years 1996 and 1997) and 28% for the last four years (years 2007–2010).
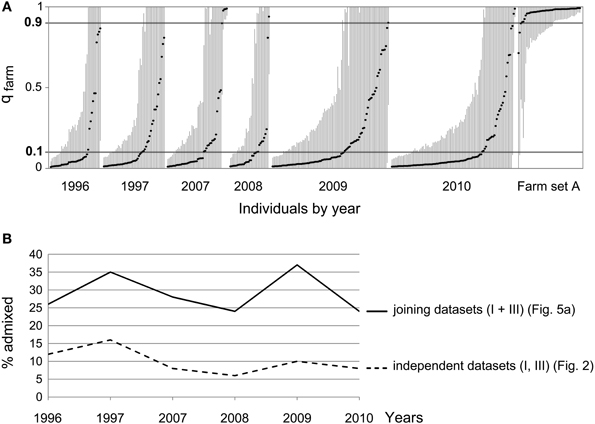
Figure 6. (A) Joint STRUCTURE analysis (K = 2) for wild quails from datasets I and III. (B) Proportion of admixed individuals (0.10 < qfarm < 0.90) per year. Continuous line: datasets I and III analyzed together (as above). Dashed line, datasets I and III analyzed independently (as in Figure 3). No increase in the proportion of admixed individuals is observed.
In conclusion, the genetic analysis of the field data did not show an increase in the proportion of admixed individuals in the population, even after joining datasets I and III to overcome limitations associated with a short time series. However, these analyses suggested that perhaps the level of admixture had been underestimated by considering only short time periods in each analysis.
Introgression as a Function of the Proportion of Farm Breeders and Fitness of Admixed Individuals
One limitation of our simulations was that we considered a fixed proportion of the potential breeders to originate from farms (1.5%) while in real data this proportion was variable between years. It was not clear if a lower proportion would also produce a detectable signal of introgression, nor what level of introgression could be expected if the fitness of admixed individuals was lower than that of the native common quail population. To assess the likely fraction of restocked farm individuals among the potential breeders that could produce the observed patterns of low admixture, we simulated 20 generations with fractions of 1, 1.5, 2, 2.5, 3, 4, or 5% per generation. For each case we generated 1000 datasets of simulated genotypes analogous to the field sampling (same number of samples per year as in datasets I + III) and we analyzed them with STRUCTURE. Since the proportion of individuals with qfarm > 0.10 in Dataset III was 32.4% (when combining datasets I and III; Figure 4B and Supplementary Material Table S2B), we investigated in which cases this value was inside the 95% confidence interval obtained from the simulations. The observed value was compatible with the simulations only when the proportion of potential breeders coming from farms was 1% (Figure 7, not shown for values higher than 3%). However, this value was almost a third of the observed average in the field sample (2.7%; Figure 4B and Supplementary Material Table S2B).
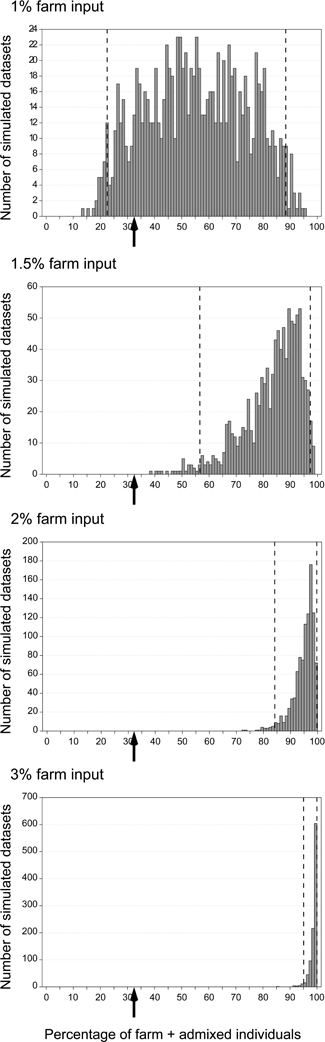
Figure 7. Distribution of the proportion of non pure wild individuals (farm or admixed, qfarm > 0.10) in 1000 simulated datasets representing a sampling pattern analogous to that of Dataset III (2007-2010) assuming various proportions of potential breeders coming from farms: 1, 1.5, 2, and 3%. Dashed lines show 2.5th and 97.5th percentiles. Arrows indicate proportion observed in the real data: 32.4% (Figure 4B and Supplementary Material Table S2B).
We suggest two possible explanations for this mismatch: first, the percentage of farm individuals potentially contributing to the breeding population may have been overestimated. Alternatively, farm and admixed individuals could exhibit lower fitness in the wild than pure common quails, slowing down the spread of farm alleles in the wild population. In simulations incorporating a variety of fitness values (1, 0.8, 0.6, 0.4, and 0.2) and assuming that between 2–3% of the potential breeders come from farms, the observed proportion of admixed (and pure farm) quails in the field was compatible with relative fitness values as low as 0.6 (Table 3). If the proportion of farm individuals among potential breeders was larger than 3%, the data would be compatible with even lower fitness values for admixed birds. These results indicate that admixed individuals may have lower chances of contributing offspring to the next generation than common quails. Interestingly, our data was not compatible with the simulated scenarios in which fitness values of admixed individuals was 0.2. Introgression would only be completely negligible if the results were compatible with fitness = 0. This implies that some introgression must be taking place.
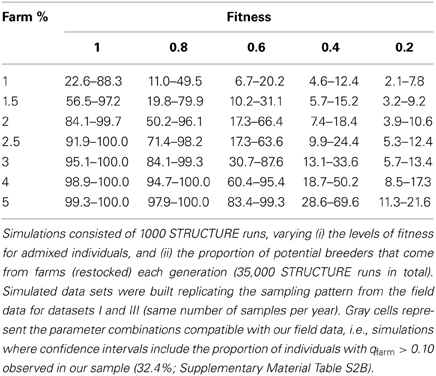
Table 3. Ninety five percentage confidence intervals of the proportion of not pure wild (admixed and pure farm individuals, qfarm > 0.10) for the simulated data corresponding to Dataset III.
Long-Term Effects
Our simulations showed that even though we did not observe a clear increase over time in the proportion of admixed individuals, the genetic analysis of field samples was compatible with slow but accumulating introgression (Table 3). In order to investigate the effects of such introgression on wild quail populations in the near future, we simulated populations evolving under two scenarios compatible with field data: (i) 1% of potential breeders deriving from restocked quails and hybrids without diminished fitness, (ii) 2% of the potential breeders coming from farms and a fitness of 0.6 for hybrids (Figures 8A,B, respectively). In both cases, running the simulations for 20 more generations (40 generations in total) translated into extensive introgression of farm alleles and more than 20% of the wild individuals reached values of qfarm indicative of admixture (0.10 < qfarm < 0.90; Figure 8C).
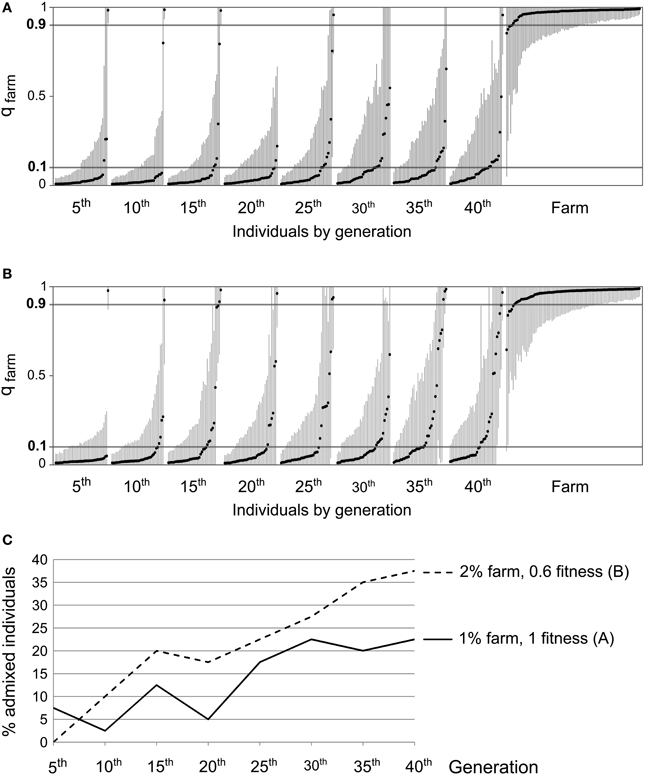
Figure 8. Examples of results from an analysis using STRUCTURE (K = 2) on data simulated over 40 generations, with 50 individuals randomly sampled every 5 generations, along with 100 simulated farm individuals. (A) 1% of potential breeders derived from farms, fitness of 1 for all individuals. (B) 2% derived from farms, fitness of 0.6 for first and second generation admixed individuals. (C) Proportion of admixed individuals (0.10 < qfarm < 0.90) over time under scenarios (A) (continuous line) and (B) (dashed line). In both cases, a clear increase in the introgression of farm alleles into the wild population is observed.
Discussion
Studies on the population-wide impact of introgression have a long history in areas like fisheries research and aquaculture, where massive restocking practices are common (see, e.g., Hedgecock and Coykendall, 2007; Waples et al., 2007; Waples and Drake, 2008). On the other hand, only little is known about the genetic effects of large scale releases for supportive breeding in other wildlife groups (Laikre et al., 2010). Studies on birds and mammals often focus on the individual identification of hybrids and the estimation of their proportion in the population. From this information, however, it is not straightforward to infer the rate of introgression and its potential impact on the population. In this study we integrated three separate datasets to cover a large time period of 15 years (ca. 15 generations), which could allow separating hybridization and introgression, and thus monitor the advance of introduced alleles into the focal population and assess the fitness of hybrids.
In the study region (Catalonia, NE Spain), the number of farm quails restocked every year was 3–11 times larger than the estimated wild population (from Rodríguez-Teijeiro et al., 2004 and Hunting order plans of the Regional Government of Catalonia). However, only a small proportion of the released farm quails become potential breeders: those that escape hunting and predation and those that also survive the winter, with a probably depressed immune system response (due to relaxed selection in captivity) and in breeding grounds with limited food resources, or that successfully migrate to wintering latitudes and return the next breeding season (which seems unlikely due to the reduced migratory impulse they may have inherited from domestic Japanese quails; Derégnaucourt, 2000; Derégnaucourt et al., 2005). Since our results indicate that only 2–3% of the potential breeders in the study area were of farm origin, approximately only one out of every 100–500 farm quails released can potentially contribute to the wild breeding population. However, our results show that those individuals could have an important impact on the wild population.
Based on the proportion of quails with vocalizations indicating hybrid origin, Puigcerver et al. (2007) suggested that the proportion of admixed birds in the population had remained relatively constant, and proposed that those individuals could be animals of farm origin that survived in the field for some time but failed to reproduce. However, the proportion of admixed relative to pure farm in the wild is much larger than in captivity and this, together with the capture of admixed birds with common quail mtDNA, indicating that the mother was a wild quail, confirm that interbreeding has happened in the wild. Field experiments carried out by Puigcerver et al. (2014) have shown that farm and wild individuals can mate in the wild, but our study provides the first evidence that this in occurring outside the experimental conditions.
The detection of offspring from wild × farm quail crosses does not necessarily imply a progressive build-up of Japanese quail alleles in the common quail population. Continued backcrossing of the crossbred individuals with common quails is required to produce an increase in the proportion of admixed individuals and introgression of alleles. An analysis of three separated studies of field samples did not show such increase (Figure 3). However, not seeing such a trend could either be due to its non-existence or to the lack of power to detect it, showing that the observed data is inconclusive. Our simulations pointed to the latter, as they revealed that analyses covering short time frames (as those generated when analyzing the three datasets separately) lack the power to detect an increase in introgression, especially when the proportion of potential breeders coming from farms is relatively small. In these cases, STRUCTURE analyses underestimated the degree of admixture (Figure 5D), possibly due to an inaccurate estimation of the ancestral allele frequencies (before introgression). After a few generations of admixture, some introgressed alleles may have spread in the native population, complicating the identification of their alien origin. This is an especially important outcome of our simulations since studies of hybridization and introgression are generally based on samples collected over a short time period, and this could result in similar biases in the estimation of the rate of admixture. It is possible that increasing sample sizes during the different time periods or genotyping a larger number of genetic markers would also increase the power to detect introgression. However, this is often not possible or too expensive. Overall, our results illustrate how power tests are essential in determining if a study can robustly test the hypotheses presented.
To increase our power to detect changes through time, we jointly analyzed the first and last datasets, spanning 15 years. This implied reducing the power to individually classify samples as pure or admixed and increased confidence intervals for the proportion of farm ancestry qfarm (see Figures 3, 6A). This cannot be just result of reducing the number of genetic markers included in the analysis since it remains the same for Dataset I (9 loci) but may be indicating important admixture.
Simulations replicating the sampling pattern of datasets I and III indicated that the power of the markers and samples analyzed should be sufficient to detect increased introgression in the case that as little as just 1.5% of the potential breeders came from farms. However, the real data did not show an increase in the proportion of admixed individuals even though the average proportion of farm animals detected in the study site every generation was about twice as much. This suggests that either the proportion of farm animals contributing to the reproduction is well below 1.5%, or that farm and admixed individuals have a lower fitness and, thus, have a lower contribution to the following generations than pure common quails. This would be possible if most of the admixed individuals were not involved in the reproduction. However, this does not seem likely because field experiments have shown that female quails of farm origin are more successful than wild common quails in attracting and mating wild common quail males (Puigcerver et al., 1999, 2013, 2014). The limited introgression of farm alleles into the wild population may be due to the lower fitness of the released birds and their descendants, which is also supported by experimental observations in the wild that point to a decreased survival and increased nest predation for females of farm origin (Puigcerver et al., 2014). Our simulations indicate that their overall fitness could easily be 40% lower than the fitness of wild common quails.
Common and Japanese quails have evolved under different selective forces and have experienced different demographic changes. Consequently, it is highly possible that alleles of Japanese quail origin present in the farm population are maladaptive and contribute to a lower fitness in common quail populations. However, probably more important than this is the change in selective forces imposed first by the domestication process in Japanese quails, and later by the captive breeding with common quails for restocking. These further promote the fixation of maladaptive alleles and traits (Lynch and O'Hely, 2001; Frankham, 2008; Baskett and Waples, 2013) that could, for example, contribute to reduce anti-predatory behavior, flight restlessness or ability to explore alternative food sources. As a result, restocking practices often have very low success rates (Gortázar et al., 2000; Guyomarc'h, 2003). This could contribute to the reduced fitness of admixed birds.
Interestingly, real data was not compatible with simulated scenarios where the fitness of hybrids was very low (0.2). When we simulated data with this very low fitness for admixed individuals, fewer individuals from the simulated datasets were identified as admixed than in the real data, suggesting that farm alleles may be slowly, but progressively, integrating into the native common quail gene pool. Our simulations were simple and relied on several assumptions (large effective population size, no population structure, generation time of 1 year, non-overlapping generations, etc); the simulations were also based on the assuming that farm quail releases were initiated at the time our group first detected them (around 1990) and took place at a constant rate (constant proportion of farm individuals added every generation), but this may not be the case. Also, fitness effects could be introduced into the simulations in a number of ways (for example, varying fitness depending on the proportion of the genome originating in the farm, or reduced survival could be modeled at different stages in the life cycle). Consequently, the fitness estimations obtained can only be taken as rough approximations, but we believe that they reflect population trends and indicate that although admixed individuals have reduced fitness some introgression is taking place every generation.
Our results highlight the possible consequences of continuing the current restocking practices. While we do not detect an increase in the proportion of admixed individuals over a 15 year time period, our simulations show that this does not guarantee that introgression is not taking place. Two simulated scenarios, compatible with estimates from real data, predict extensive introgression of farm alleles in the near future (Figure 8). Of course, these simulations are based on estimates obtained from a very small area, and the proportion of potential breeders coming from farms is likely to vary from place to place and from year to year. However, considering the high mobility of common quails and how widespread restocking practices are, we do not have reasons to expect very different patterns in other areas within the western distribution of the common quail.
Conclusions
Extensive restocking with farm-reared quails does not yet appear to have had large impacts on the genetic integrity of common quails in Northeast Spain. However, we here provide evidence that interbreeding has already happened in the wild and that overall admixture levels may be slowly increasing over time. Although some degree of selection against introgressed alleles appears to exist, our simulations predict a marked risk of genetic swamping of the common quail population, if release practices are maintained. Our results therefore support initiatives currently being taken to control quail restocking practices across Europe.
More generally, our results highlight that the impacts of introgression on wild populations can be difficult to detect, especially when hybridization occurs over long time periods and at low frequency. In such cases, data collected over a short time period may be insufficient and lead to an underestimation of the degree of admixture. Our study therefore underlines the need for long time series to reliably assess the consequences of hybridization in natural populations and the need for power tests.
Data Accessibility
Newly obtained mitochondrial DNA sequences have been submitted to GenBank (accession numbers: KF410830-KF410849). Microsatellite data files are deposited in Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.kv733. Scripts were set to run in a computer with a Linux operative system (Ubuntu 12.10) and are available at http://www.consevol.org.
Author Contributions
This study is part of a long-term study on quail populations led by Manel Puigcerver and José D. Rodríguez-Teijeiro. It is part of the doctoral thesis of Ines Sanchez-Donoso (at Barcelona University, Spain), supervised by José D. Rodríguez-Teijeiro, Manel Puigcerver and Carles Vilà, and the master thesis of Jisca Huisman (at Uppsala University, Sweden), supervised by Carles Vilà and Frank Hailer, Carles Vilà and Ines Sanchez-Donoso conceived the genetic study. Ines Sanchez-Donoso, Manel Puigcerver and José D. Rodríguez-Teijeiro did field work. Ines Sanchez-Donoso, Jisca Huisman, Frank Hailer, and Jorge Echegaray performed laboratory work. Ines Sanchez-Donoso and Carles Vilà did genetic and statistical analyses and simulations, with input from Jisca Huisman and Frank Hailer. All authors contributed to the text.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are most grateful to the Department of Evolutionary Biology at Uppsala University for offering logistical support to Jisca Huisman during her MSc thesis. Logistical support was also provided by “Laboratorio de Ecología Molecular” from “Estación Biológica de Doñana” (LEM-EBD), and “Serveis Científico-Tècnics” (SCT) from University of Barcelona to Ines Sanchez-Donoso. We thank R. Casanovas and J.M. López, from the “Àrea d'Activitats Cinegètiques” of the Catalan Government, for providing information about the Catalan hunting order plans. We also thank F. Sardà-Palomera, A. Domínguez, C. Extremera, M. García, I. Jiménez, V. Bonet-Arbolí, E. Pérez-Masdeu, G. Molina-Vacas, A. Barroso, S. Gallego, the Pons family and all field-work volunteers for technical assistance in the field work; J.A. Leonard, V. Muñoz-Fuentes and A. Amador for support during the laboratory work, J.A. Leonard also for reviewing the English text; M. González-Suárez for statistical advice. The Catalan “Direcció General de la Recerca” (2009-SGR-481), the Spanish Science Ministry (CGL2004-05308, CGL2007-63199 and CGL2010-21250 projects), the “Federación de Caza de Euskadi” (2007 to 2009), the “Programa para la Captación del Conocimiento para Andalucía” (C2A) of the Andalusian Government, the Catalan Government for the “Ajuts per a la formació i contractació de personal investigador novell” (FI grant), the Catalan Department of Environment, the Smithsonian Restricted Endowment funds (Frank Hailer), the Hesse's “LOEWE Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz” (Frank Hailer) and the Lawskis Foundation Research Predoctoral Fellowship (Jorge Echegaray) gave financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fevo.2014.00015/abstract
References
Anderson, E. C., and Thompson, E. A. (2002). A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160, 1217–1229.
Barilani, M., Deregnaucourt, S., Gallego, S., Galli, L., Mucci, N., Piombo, R., et al. (2005). Detecting hybridization in wild (Coturnix c. coturnix) and domesticated (Coturnix c. japonica) quail populations. Biol. Conserv. 126, 445–455. doi: 10.1016/j.biocon.2005.06.027
Baskett, M. L., and Waples, R. S. (2013). Evaluating alternative strategies for minimizing unintended fitness consequences of cultured individuals on wild populations. Conserv. Biol. 27, 83–94. doi: 10.1111/j.1523-1739.2012.01949.x
Bei, Y., Li, J., Meng, S., Li, G., and Sun, B. (2012). Isolation and characterization of 12 novel microsatellite loci in Hume's Pheasant, Syrmaticus humiae. Conserv. Genet. Resour. 5, 19–21. doi: 10.1007/s12686-012-9719-1
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N., and Bonhomme, F. (1996–2004). GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions. CNRS UMR 5171. University of Montpellier II, Montpelier, France. Available online at: http://kimura.univ-montp2.fr/genetix/. (Accessed January 15 2014).
BirdLife International. (2004). “Coturnix coturnix,” in IUCN 2013. IUCN Red List of Threatened Species. Version 2013.1. Available online at: http://www.iucnredlist.org/. (Accessed January 15 2014).
Botstein, D., White, R. L., Skolnick, M., and Davis, R. W. (1980). Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331.
Caballero de la Calle, J. R., and Peña Montañés, J. C. (1997). “La explotación cinegética de la codorniz,” in Zootecnia. Bases de producción animal. Tomo XII. Producciones cinegéticas, apícolas y otras, ed C. Buxadé (Madrid: Ediciones Mundi-Prensa), 109–123.
Chazara, O., Lumineau, S., Minvielle, F., Roux, D., Feve, K., Kayang, B. B., et al. (2006). Étude des risques d'introgression génétique de la caille des blés (Coturnix coturnix coturnix) par la caille japonaise (C. c. japonica): comparaison et intégration des données comportementales et moléculaires obtenues dans le sud-est de la France. Les Actes du BRG 6, 317–334.
Chazara, O., Minvielle, F., Roux, D., Bed'hom, B., Feve, K., Coville, J.-L., et al. (2010). Evidence for introgressive hybridization of wild common quail (Coturnix coturnix) by domesticated Japanese quail (Coturnix japonica) in France. Conserv. Genet. 11, 1051–1062.
Collins, S. A., and Goldsmith, A. R. (1998). Individual and species differences in quail calls (Coturnix c. japonica, Coturnix c. coturnix and a hybrid). Ethology 104, 977–990. doi: 10.1111/j.1439-0310.1998.tb00047.x
Corander, J., and Marttinen, P. (2006). Bayesian identification of admixture events using multi-locus molecular markers. Mol. Ecol. 15, 2833–2843. doi: 10.1111/j.1365-294X.2006.02994.x
Crawford, N. G. (2010). SMOGD: software for the measurement of genetic diversity. Mol. Ecol. Resour. 10, 556–557. doi: 10.1111/j.1755-0998.2009.02801.x
Derégnaucourt, S. (2000). Hybridization entre la caille des blés (Coturnix c. coturnix) et la caille japonaise (Coturnix c. japonica): Mise en evidence des risques de pollution génetique des populations naturelles par les cailles domestiques. Ph.D. thesis. University of Rennes I, Rennes.
Derégnaucourt, S., and Guyomarc'h, J.-C. (2003). Mating call discrimination in female European (Coturnix c. coturnix) and Japanese quail (Coturnix c. japonica). Ethology 119, 107–119. doi: 10.1046/j.1439-0310.2003.00854.x
Derégnaucourt, S., Guyomarc'h, J.-C., and Aebischer, N. (2002). Hybridization between European Quail Coturnix coturnix and Japanese Quail Coturnix japonica. Ardea 90, 15–21.
Derégnaucourt, S., Guyomarc'h, J.-C., and Belhamra, M. (2005). Comparison of migratory tendency in European Quail Coturnix c. coturnix, domestic Japanese Quail Coturnix c. japonica and their hybrids. Ibis 147, 25–36. doi: 10.1111/j.1474-919x.2004.00313.x
Derégnaucourt, S., Guyomarc'h, J.-C., and Richard, V. (2001). Classification of hybrid crows in quail using artificial neural networks. Behav. Processes 56, 103–112. doi: 10.1016/S0376-6357(01)00188-7
Eldridge, W. H., and Naish, K. A. (2007). Long-term effects of translocation and release numbers on fine-scale population structure among coho salmon (Oncorhynchus kisutch). Mol. Ecol. 16, 2407–2421. doi: 10.1111/j.1365-294X.2007.03271.x
Ford, M. J. (2002). Selection in captivity during supportive breeding may reduce fitness in the wild. Conserv. Biol. 16, 815–825. doi: 10.1046/j.1523-1739.2002.00257.x
Frankham, R. (2008). Genetic adaptation to captivity in species conservation programs. Mol. Ecol. 17, 325–333. doi: 10.1111/j.1365-294X.2007.03399.x
Gortázar, C., Villafuerte, R., and Martín, M. (2000). Success of traditional restocking of red-legged partridge for hunting purposes in areas of low density of northeast Spain Aragon. Zeitschrift für Jagdwissenschaft 46, 23–30. doi: 10.1007/BF02240661
Goudet, J. (1995). Fstat version 1.2: a computer program to calculate F-statistics. J. Hered. 86, 485–486.
Guyomarc'h, J.-C. (2003). Elements for a Common Quail Coturnix c. coturnix management plan. Game Wildl. Sci. 20, 1–92.
Guyomarc'h, J.-C., and Guyomarc'h, C. (1996). Vocal communication in European quail; Comparison with Japanese quail. Life Sci. 319, 827–834.
Hassen, H., Neser, F. W. C., Kock, A., and Marle-Köster, E. (2009). Study on the genetic diversity of native chickens in northwest Ethiopia using microsatellite markers. Afr. J. Biotechnol. 8, 1347–1353.
Haygood, R., Ives, A. R., and Andow, D. A. (2003). Consequences of recurrent gene flow from crops to wild relatives. Proc. R. Soc. B Biol. Sci. 270, 1879–1886. doi: 10.1098/rspb.2003.2426
He, K., Zhang, P., Fang, S.-G., and Wan, Q.-H. (2008). Development and characterization of 14 novel microsatellite markers from the golden pheasant (Chrysolophus pictus). Conserv. Genet. 10, 511–513. doi: 10.1007/s10592-008-9552-y
Hedgecock, P., and Coykendall, K. (2007). “Genetic risks of marine hatchery enhancement: the good, the bad, and the unknown,” in Ecological and Genetic Implications of Aquaculture Activities, ed T. M. Bert (Netherlands: Springer), 85–101. doi: 10.1007/978-1-4020-6148-6_5
Hedrick, P. W. (2005). A standardized genetic differentiation measure. Evolution 59, 1633–1638. doi: 10.1111/j.0014-3820.2005.tb01814.x
Huisman, J. (2006). Hybridization Between European Quail (Coturnix coturnix) and Released Japanese Quail (C. japonica). M.Sc. thesis. Uppsala University, Uppsala.
Jost, L. (2008). GST and its relatives do not measure differentiation. Mol. Ecol. 17, 4015–4026. doi: 10.1111/j.1365-294X.2008.03887.x
Kayang, B. B., Inoue-Murayama, M., Hoshi, T., Matsuo, K., Takahashi, H., Minezawa, M., et al. (2002). Microsatellite loci in Japanese quail and cross-species amplification in chicken and guinea fowl. Genet. Sel. Evol. 34, 233–253. doi: 10.1186/1297-9686-34-2-233
Kayang, B. B., Inoue-Murayama, M., Nomura, A., Kimura, K., Takahashi, H., Mizutani, M., et al. (2000). Fifty microsatellite markers for Japanese quail. J. Hered. 91, 502–505. doi: 10.1093/jhered/91.6.502
Kayang, B. B., Vignal, A., Inoue-Murayama, M., Miwa, M., Monvoisin, J. L., Ito, S., et al. (2004). A first-generation microsatellite linkage map of the Japanese quail. Anim. Genet. 35, 195–200. doi: 10.1111/j.1365-2052.2004.01135.x
Laikre, L., Schwartz, M. K., and Waples, R. S. (2010). Compromising genetic diversity in the wild: unmonitored large-scale release of plants and animals. Trends Ecol. Evol. 25, 520–529. doi: 10.1016/j.tree.2010.06.013
Leberg, P. L. (2002). Estimating allelic richness: effects of sample size and bottlenecks. Mol. Ecol. 11, 2445–2449. doi: 10.1046/j.1365-294X.2002.01612.x
Lynch, M., and O'Hely, M. (2001). Captive breeding and the genetic fitness of natural populations. Conserv. Genet. 2, 363–378. doi: 10.1023/A:1012550620717
Marie, A. D., Bernatchez, L., and Garant, D. (2010). Loss of genetic integrity correlates with stocking intensity in brook charr (Salvelinus fontinalis). Mol. Ecol. 19, 2025–2037. doi: 10.1111/j.1365-294X.2010.04628.x
Park, S. D. E. (2001). Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. Ph.D. thesis, University of Dublin, Dublin.
Perennou, C. (2009). European Union Management Plan 2009-2011. Common Quail, Coturnix Coturnix. Luxembourg: Office for Official Publications of the European Communities.
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Puigcerver, M., Gallego, S., and Rodríguez-Teijeiro, J. D. (1992). Survival and mean life span of the quail Coturnix c. coturnix. Bird Study 39, 120–123. doi: 10.1080/00063659209477108
Puigcerver, M., Rodríguez-Teijeiro, J. D., D'Amico, S., and Randi, E. (1999). “Hybridization and introgression of Japanese quail mitochondrial DNA in Common quail populations: a preliminary study,” in Perdix VIII - International Symposium on Partritges, Quails and Pheasants in the Western Palearctic and Nearctic. Hungarian Small Game Bulletin (Sopron), 129–136.
Puigcerver, M., Rodríguez-Teijeiro, J. D., and Gallego, S. (1989). ¿Migración y/o nomadismo en la codorniz (Coturnix c. coturnix)? Etología 1, 39–45.
Puigcerver, M., Sanchez-Donoso, I., Vilà, C., Sardà-Palomera, F., García-Galea, E., and Rodríguez-Teijeiro, J. D. (2014). Decreased fitness of restocked hybrid quails prevents fast admixture with wild common quails. Biol. Conserv. 171, 74–81. doi: 10.1016/j.biocon.2014.01.010
Puigcerver, M., Sanchez-Donoso, I., Vilà, C., Sardà-Palomera, F., Morales-Rodríguez, P. A., Caballero de la Calle, J. R., et al. (2013). Hybridization between the common quail (Coturnix coturnix) and farm-reared quails: state of the art of a conservation problem. Ecosistemas 22, 48–53. doi: 10.7818/ECOS.2013.22-2.08
Puigcerver, M., Sardà-Palomera, F., and Rodríguez-Teijeiro, J. D. (2012). Determining population trends and conservation status of the common quail (Coturnix coturnix) in Western Europe. Anim. Biodivers. Conserv. 35, 343–352.
Puigcerver, M., Vinyoles, D., and Rodríguez-Teijeiro, J. D. (2007). Does restocking with Japanese quail or hybrids affect native populations of common quail Coturnix coturnix? Biol. Conserv. 136, 628–635. doi: 10.1016/j.biocon.2007.01.007
R. Core Team. (2012). R: A Language and Environment for Statistical Computing. Vienna, R Foundation for Statistical Computing. Available online at: http://www.R-project.org/ (Accessed January 15, 2014).
Rhymer, J. M., and Simberloff, D. (1996). Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 27, 83–109. doi: 10.1146/annurev.ecolsys.27.1.83
Rodríguez-Teijeiro, J. D., Barroso, A., Gallego, S., Puigcerver, M., and Vinyoles, D. (2006). Orientation-cage experiments with the European Quail during the breeding season and autumn migration. Can. J. Zool. 84, 887–894. doi: 10.1139/z06-070
Rodríguez-Teijeiro, J. D., Puigcerver, M., and Gallego, S. (1992). Mating strategy in the European quail (Coturnix c. coturnix) revealed by male population density and sex ratio in Catalonia (Spain). Gibier Faune Sauvage 9, 377–386.
Rodríguez-Teijeiro, J. D., Puigcerver, M., and Gallego, S. (2004). “Guatlla Coturnix coturnix,” in Atles dels ocells nidificants de Catalunya, 1999–2002. ICO/Lynx edicions, eds S. Herrando, L. l. Brotons, J. Estrada, S. Guallar, and M. Anton (Barcelona: ICO/Lynx edicions), 112–113.
Rodríguez-Teijeiro, J. D., Rodrigo-Rueda, F. J., Puigcerver, M., Gallego, S., and Nadal, J. (1993). Codornices japonesas en nuestros campos. Trofeo 277, 48–52.
Rodríguez-Teijeiro, J. D., Sardà-Palomera, F., Nadal, J., Ferrer, X., Ponz, C., and Puigcerver, M. (2009). The effects of mowing and agricultural landscape management on population movements of the common quail. J. Biogeogr. 36, 1891–1898. doi: 10.1111/j.1365-2699.2009.02109.x
Ryman, N., and Laikre, L. (1991). Effects of supportive breeding on the genetically effective population size. Conserv. Biol. 5, 325–329. doi: 10.1111/j.1523-1739.1991.tb00144.x
Sanchez-Donoso, I., Vilà, C., Puigcerver, M., Butkauskas, D., Caballero de la Calle, J. R., Morales-Rodríguez, P. A., et al. (2012). Are farm-reared quails for game restocking really common quails (Coturnix coturnix)?: a genetic approach. PLoS ONE 7:e39031. doi: 10.1371/journal.pone.0039031
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Vähä, J.-P., and Primmer, C. R. (2006). Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol. Ecol. 15, 63–72. doi: 10.1111/j.1365-294X.2005.02773.x
Walsh, P. S., Metzger, D. A., and Higuchi, R. (1991). Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10, 506.
Waples, R. S., and Drake, J. (2008). “Risk/Benefit considerations for marine stock enhancement: a Pacific salmon perspective,” in Stock Enhancement and Sea Ranching: Developments, Pitfalls and Opportunities, 2nd Edn, eds K. M. Leber, S. Kitada, H. L. Blankenship, and T. Svåsand (Oxford: Blackwell Publishing Ltd), 206–260.
Waples, R. S., Ford, M. J., and Schmitt, D. (2007). “Empirical results of salmon supplementation in the Northeast Pacific: a preliminary assessment,” in Ecological and Genetic Implications of Aquaculture Activities, ed T. M. Bert (Netherlands: Springer), 383–403. doi: 10.1007/978-1-4020-6148-6_21
Keywords: Coturnix, hybridization, introgression, modeling, restocking, temporal sampling
Citation: Sanchez-Donoso I, Huisman J, Echegaray J, Puigcerver M, Rodríguez-Teijeiro JD, Hailer F and Vilà C (2014) Detecting slow introgression of invasive alleles in an extensively restocked game bird. Front. Ecol. Evol. 2:15. doi: 10.3389/fevo.2014.00015
Received: 23 January 2014; Accepted: 12 April 2014;
Published online: 30 April 2014.
Edited by:
Sonia Consuegra, Swansea University, UKReviewed by:
Tina T. Hu, Princeton University, USALan Zhu, Oklahoma State University, USA
Patricia Lee, Swansea University, UK
Giorgio Bertorelle, University of Ferrara, Italy
Mairi Elspeth Knight, University of Plymouth, UK
Copyright © 2014 Sanchez-Donoso, Huisman, Echegaray, Puigcerver, Rodríguez-Teijeiro, Hailer and Vilà. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ines Sanchez-Donoso and Carles Vilà, Integrative Ecology Department, Estación Biológica de Doñana - Consejo Superior de Investigaciones Científicas (EBD-CSIC), Avda. Américo Vespucio, s/n, 41092 Seville, Spain e-mail:aW5lcy5zYW5jaGV6ZG9ub3NvQGdtYWlsLmNvbQ==;Y2FybGVzLnZpbGFAZWJkLmNzaWMuZXM=