 Francisca C. Almeida1†‡
Francisca C. Almeida1†‡ Alejandro Sánchez-Gracia1‡
Alejandro Sánchez-Gracia1‡ Kimberly K. O. Walden2
Kimberly K. O. Walden2 Hugh M. Robertson2
Hugh M. Robertson2 Julio Rozas1*
Julio Rozas1*- 1Departament de Genètica and Institut de Recerca de la Biodiversitat, Universitat de Barcelona, Barcelona, Spain
- 2Department of Entomology, University of Illinois at Urbana-Champaign, Urbana, IL, USA
The recent publication of a centipede (Strigamia maritima) genome has revealed that most members of the chemosensory gene families of ionotropic (IR) and gustatory (GR) receptors do not have identifiable orthologs in insect species. In other words, the diversity of these chemoreceptors in centipedes appears to have evolved after its split from other arthropod lineages. Here we investigate the role of adaptive evolution in S. maritima chemoreceptor diversification using an approach that allows us to discuss functional aspects of such diversification. We applied codon substitution models in a phylogenetic framework to obtain the distribution of selective constraints across the different domains in the IR and GR proteins, and to assess the impact of positive selection in the evolution of these chemoreceptors. We found low selective constraints in most IR and GR duplicates and significant evidence for the presence of positively selected amino acids in two of the four IR, and in six of the GR recent specific expansions. Mapping the sites with high posterior probability of positive selection in protein structure revealed a remarkable uneven distribution of fast-evolving sites across protein domains. Most of these sites are located in extracellular fragments of these receptors, which likely participate in ligand recognition. We hypothesize that adaptive evolution in ligand-binding domains was a major force driving the functional diversification of centipede chemoreceptors.
Introduction
The chemosensory system of arthropods is an interesting subject to study evolution due to its adaptive value. Chemosensation is necessary for finding food, avoiding predators, and locating and choosing mates. The system is composed of proteins encoded by small to medium-sized gene families of two main types: chemosensory (membrane) receptors and ligand-binding proteins (Pelosi et al., 2006; Sánchez-Gracia et al., 2009, 2011; Touhara and Vosshall, 2009). First studied in Drosophila melanogaster, three chemoreceptor gene families were identified: Olfactory receptors (OR), Gustatory receptors (GR), and Ionotropic receptors (IR) (Robertson et al., 2003; Benton et al., 2009). With the sequencing of other arthropod genomes, it became evident that the ORs are unique to insects (Peñalva-Arana et al., 2009; Chipman et al., 2014). Members of the GR and IR gene families were, in contrast, found in all arthropod genomes sequenced so far. In fact, the IRs and GRs seem to have an even older origin; they are present in non-arthropod animals, but GRs probably did not always have a chemosensory function (Croset et al., 2010; Rytz et al., 2013; Saina et al., 2015; Hugh Robertson unpublished). In D. melanogaster, olfaction, or the perception of airborne or volatile chemicals, is mediated by the ORs. GRs are implicated in the perception of soluble chemicals and CO2, while IRs have been implicated in the detection of both soluble and airborne cues (Vosshall and Stocker, 2007; Rytz et al., 2013). Arthropods have a marine ancestry, but there were several independent invasions of land, such as in the Chelicerates, Myriapods, and Insects (Rota-Stabelli et al., 2013). Since the ORs are not present in the Chelicerates and Myriapods (Chipman et al., 2014; Frías-López et al., 2015; Hugh Robertson unpublished results, Julio Rozas unpublished results), an evident question is which genes encode receptors for air-borne cues in these land arthropods.
Here, we begin to investigate this question by analyzing the evolution of the GR and IR families in a myriapod that has recently had its genome sequence published: Strigamia maritima (Chipman et al., 2014). This species is found along the coastline of northwestern Europe and has been used as a model system in developmental studies (Kettle et al., 2003; Arthur and Chipman, 2005; Chipman and Akam, 2008; Green and Akam, 2013). Interestingly, it belongs to a centipede Order (Geophilimorphs) that includes animals that have completely lost their eyes, which suggests they rely heavily on other sensory systems. A search for GR and IR orthologs in the S. maritima genome identified 76 GRs (13 of which are pseudogenes) and 54 IRs (3 of which are pseudogenes) (Chipman et al., 2014). These numbers are well within the range of gene family members in other arthropod species. In both families, however, only a few 1:1 orthologous relationships were found with other arthropod chemoreceptors (Chipman et al., 2014). Instead, phylogenetic analyses for each chemoreceptor family revealed that all S. maritima GR and most IR genes clustered together in a single clade without close relationships to orthologs in other arthropod species. This result suggests that the observed diversity of myriapod chemosensory receptors evolved after this lineage split from its last common ancestor with other arthropods. The only genes with an identifiable candidate ortholog in other arthropods were three IRs (SmarIR25a, SmarIR8a, and SmarIR49) with antennal expression in D. melanogaster, which points to a role in olfaction in flies (Chipman et al., 2014). Namely, IR25a is a highly conserved gene in Protostomia and appears to have kept its chemosensory function throughout the evolution of this group (Croset et al., 2010). In fact, this gene is broadly expressed in Drosophila olfactory tissues and might represent a common subunit in different IR complexes with a function analogous to that of Orco (Sato et al., 2008; Croset et al., 2010).
To gain insights into the evolution of functional properties of the S. maritima GR and IR repertoires, we investigated the selective pressures acting during the diversification of duplications that led to the observed diversity of lineage specific genes. It is generally believed that gene duplications allow relaxed evolution of one or both gene copies for at least some time following the duplication event (Ohno, 1970; Innan and Kondrashov, 2010). Over time, while some copies may accumulate deleterious mutations and eventually cease to be functional, others may instead evolve under positive selection for an acquired beneficial mutation. In the latter case, the copy may be maintained due to its functional differentiation in a process called neofunctionalization (Ohno, 1970). Another, non-exclusive explanation for the lineage specific expansions observed in the chemosensory gene families is the stochastic nature of the gene birth and death process. According to this hypothesis, the maintenance, and loss of duplicated gene copies can be mostly explained by an entirely random process dubbed random genomic drift, which is characterized by only two parameters: gene birth and death rates (Nei, 2007; Nei et al., 2008). Here we make use of state-of-the-art phylogenetic methods that allow for the exploration of genomic data to understand very specific details of gene family functional evolution. We used these methods to test whether positive selection was involved in GR and IR paralog divergence and in this way evaluate the role of selection in the evolution of these gene families in S. maritima. This approach permits the identification of amino acid residues likely under positive selection and consequently which gene regions and amino acid sites are under selective pressure for diversification. This study is a fundamental genomics contribution to the functional aspects of the chemosensory receptors of S. maritima and lays the ground for further examination of S. maritima's chemosensory system through experimental approaches.
Materials and Methods
Dataset
All gene sequences used in this study were manually curated by us and published in Chipman et al. (2014). In brief, we ran a number of similarity searches on the S. maritima genome to identify candidate S. maritima GR and IR genes. We then superimposed the results of these searches, available EST information, and the automated gene predictions on the genome to manually annotate each chemosensory gene and obtain the dataset we used in the present study (see details in Chipman et al., 2014; S. maritima IR and GR CDS sequences are included in the Supplementary Material).
The methodological approach we used herein to detect signs of positive selection depends on sequence variation and its accuracy is reduced if it is applied to very divergent paralogs. To circumvent this problem, we first identified recent clades based on synonymous divergence in both the GR and the IR gene families within which separate analyses would be carried out (Figure 1). For that, we first obtained a multiple alignment of amino acid sequences with MAFFT v. 7 (Katoh and Standley, 2013) using default parameters and used it in a maximum likelihood search to obtain a gene tree using the program RAxML v.8 (Stamatakis, 2006) with the PROTGAMMAWAG model. We were aware that the S. maritima GRs and IRs most likely do not represent species-specific chemosensory expansions since we have low phylogenetic coverage in this part of the arthropod tree (the most recent common ancestor of centipede and insects from which we have IR sequences was ~700 mya); chemoreceptors from other myriapod species are expected to spread out across the S. maritima clades. Nevertheless, some of the considered sub-clades (Figure 1) are relatively recent and they could in fact represent recent paralogous expansions. Both gene families showed expanded clades with more than five recently diverged paralogs (Figure 1). In the GR family, six main clades were identified, while in the IR family there were four clades. One IR clade (Clade 1, Figure 1A) was further subdivided into two subclades to maximize the number of analyzed positions and the power of the ML analyses. Notice that these clades only include the paralogous genes belonging to the centipede specific expansion. We then obtained separate multiple alignments of the nucleotide sequences of the genes included in each clade with the program MAFFT (Katoh and Standley, 2013) for the statistical analyses. Pseudogenes, i.e., fragments with early stop codons and/or frameshift mutations, were excluded from the analysis since they have inflated mutation rates that are not adaptive.
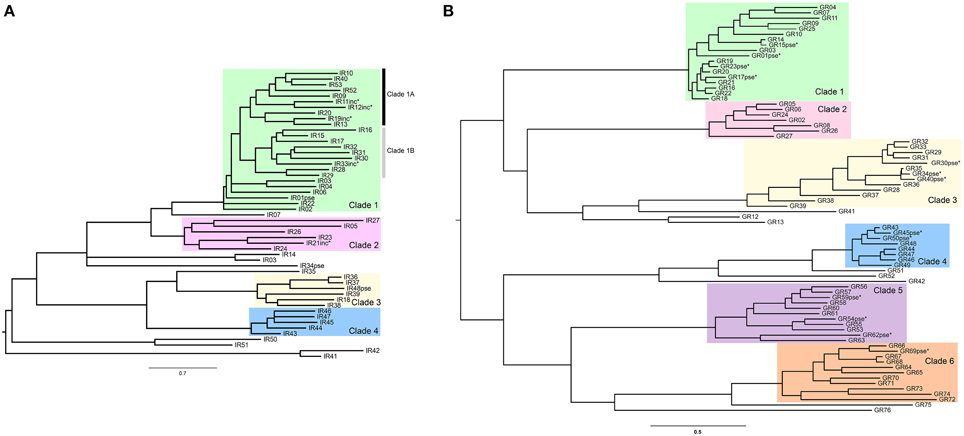
Figure 1. Maximum likelihood phylogenetic trees highlighting (in colored boxes) the Clades with IR and GR genes used in codon substitution model-based analyses. Putative pseudogenes and incomplete copies (indicated with asterisks) were not used for the analysis. (A) IR tree. (B) GR tree.
Statistical Tests for Positive Selection
We used the codon-based substitution models (Nielsen and Yang, 1998; Yang et al., 2000) implemented in codeml program (PAML 4.4; Yang, 2007) to estimate non-synonymous to synonymous substitution rate (dN/dS) ratios (ω) across paralog sequences. To determine whether there is significant evidence of evolution by positive selection in recently diverged paralogs, we used codeml to estimate the goodness of fit of different models to the observed data using a maximum likelihood approach. First, we fit to the data a model with one single average ω (Model M0) in order to obtain the branch lengths to be used as initial values for more complex models. Then we estimated model parameters and the log likelihood (L) of the data under two alternative models: M7, a model with ten classes of sites with beta distributed ω-values in the interval (0–1) (no positively selected sites allowed) and M8, where an extra class of sites (with proportion ps) constrained to have ωs≥1 is added to the beta model. We compared the L of these two models by using the likelihood ratio test (LRT; α = 0.05 after controlling for the false discovery rate—FDR; Benjamini and Hochberg, 1995). As pointed out by Swanson et al. (2003) the M7 vs. M8 comparison may result (in some particular cases) in a high proportion of false positives (significant tests in the absence of positive selection). In fact, the test only indicates if there is a class of sites with a ω> 1 but not if this ratio is significantly greater than 1. Hence, we also used the M8a model, which was proposed as an alternative null hypothesis in which the extra class in M8 is fixed to ω = 1, making a more refined (and conservative) test for the existence of positively selected sites (when compared to model M8). We assumed that the asymptotic null distribution of the LRT statistic is a c2 (df = 2) for the M8 vs. M7 comparison and a 50:50 mixture of point mass 0 and c2 (df = 1) for M8 vs. M8a comparison (Self and Liang, 1987; Swanson et al., 2003). All analyses were repeated using different ω starting values to avoid local optima in the maximum likelihood calculation. The Bayes Empirical Bayes (BEB; Yang et al., 2005) analysis under model M8 was used to identify codons under positive selection. Briefly, this method calculates, for each site in the alignment, the posterior probability (PP) of belonging to each of the different omega site classes defined in the model (11 classes in M8). Sites with PP > 0.5 of belonging to the class of sites with ω> 1 are candidates to have been under positive selection.
Delimiting Functional Domains
To evaluate whether sites with a probability of being under positive selection have a random or patterned distribution across the different protein domains, we mapped the amino acids corresponding to these codons in the predicted functional domains of the IR and GR receptors. We used different pieces of information to delimit these domains. For IRs, we first predicted the transmembrane segments in the multiple sequence alignment of each clade using TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/) with default settings. Then we delimited the pore loop, S1 and S2 ligand binding domains of S. maritima IRs using the alignments of the amino acid sequences of Drosophila and human iGluRs and Drosophila IRs in Benton et al. (2009) as a guide (based on the sequence similarity with some highly conserved residues).
For GRs, the prediction of the transmembrane domains is less clear-cut because not all of the domains are as prominently hydrophobic as most transmembrane proteins such as GPCRs, channels, and transporters, and various transmembrane prediction programs regularly under-predict them and sometimes over-predict them; they consequently also often indicate the incorrect orientation in the membrane (e.g., Benton et al., 2006; Smart et al., 2008; Zhang et al., 2011; Hull et al., 2012). The orientation in the membrane is known for the conserved Odorant Receptor Co-Receptor or Orco protein and other ORs (Benton et al., 2006; Lundin et al., 2007; Smart et al., 2008; Tsitoura et al., 2010; Hull et al., 2012), which are clearly related to the GR family in the insect chemoreceptor superfamily (Robertson et al., 2003), as well as for one GR (Zhang et al., 2011). It is the opposite of the GPCR orientation, with the N-terminus internal to the cell. The transmembrane domains were therefore defined on the basis of a combination of hydrophobic regions highlighted in CLUSTALX (Larkin et al., 2007) alignments and Kyte-Doolittle hydropathy plots (Kyte and Doolittle, 1982).
We tested whether amino acid sites with PP > 0.5 of being one of the positive selected sites in the BEB analysis were homogeneously distributed across functional domains. For that, we built a contingency table (2 × n table, where n is the number of domains defined in each receptor) to compare the relative number of sites with PP > 0.5 with the number of sites with PP < 0.5 across domains. In this way, the total number of sites of each domain is implicitly taken into account, removing the effect of domain length. We used the Fisher's exact test and FDR (False Discovery Rate—when doing multiple comparisons across clades) to obtain P-values.
Results and Discussion
Functional Constraints
The number of genes per clade varied from 5 to 20 in the IRs and the total tree length in number of substitutions per codon varied from 2.59 to 13.24 (Table 1). These medium-to-high levels of nucleotide divergence have been found to maximize power and accuracy of the LRT (Anisimova et al., 2001). The ω-values across alignment (codeml model M0) never reached 1, being largest (ω = 0.579) for the IR Clade 1A (Table 1). The lowest value (ω = 0.221) was observed in Clade 2, which was also the oldest clade as inferred from the longer branch lengths separating its members (Figure 1A). Among the GRs, the number of genes per analyzed clade varied from 7 to 14 and the maximum tree length was 7.99 in GR Clade 6 (Table 1). The ω-values were in a similar range (0.337–0.646) to that observed in the IR family, although a little higher, on average (Table 1). Interestingly, this is in accordance with the GR family having on average the lowest selective constraint among chemosensory families observed in Drosophila (Sánchez-Gracia et al., 2009).
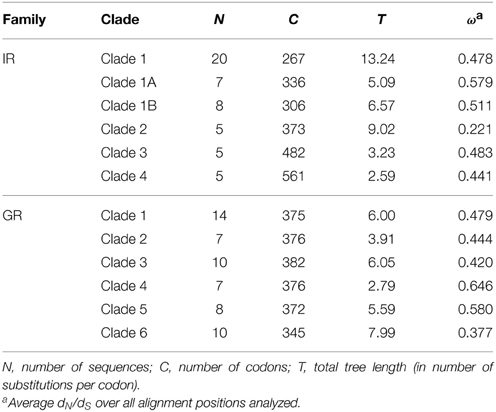
Table 1. Characteristics of the codon sequence alignments per clade.
All the estimated ω-values are relatively high as compared to values found in ortholog comparisons across several gene families in Drosophila (Clark et al., 2007). Chemosensory genes, however, may have extraordinarily low functional constraints, with high ω-values even in among-ortholog comparisons as compared to the ω-values obtained for other Drosophila gene families (Sánchez-Gracia et al., 2009, 2011). In some Drosophila chemosensory ortholog comparisons, ω-values were as high as the ones observed here among paralogs; these were outliers, however, as gene family averages of ω estimates from ortholog comparisons were never higher than 0.25 (Sánchez-Gracia et al., 2011). The relatively high ω-values we observed among S. maritima IR and GR paralogs is in accordance with a relaxation of purifying selection as expected in duplicated genes (Innan and Kondrashov, 2010) and observed in chemosensory gene families in other species (Smadja et al., 2009; Almeida et al., 2014). On the other hand, ω-values below one suggest that although selective constraints are relaxed and probably some codons might be under positive selection, most codons are evolving under purifying selection in these genes.
Tests for Positive Selection
The presence of positively selected sites accounting for the increased ω-values observed in the S. maritima paralogs comparisons was statistically supported in IR Clade 1 and IR Clade 4, and all six GR clades (Table 2). Among the IR clades, the strongest signal of positive selection was in the larger and more recently expanded clade of centipede IRs (Clade 1, LR P-values in Table 2), both considering the clade as whole and separately for each of the two main sub-clades (Clade 1A and 1B, Figure 1A). The range of ω-values of the positively selected sites in IRs (1.2–2.7), estimated under the M8 model, is slightly smaller but similar to those estimated for GRs (1.4–5.5). The proportion of sites estimated to have an ω> 1 ranged from p1 = 4.6–26.7% in IRs and p1 = 2.1–20.8% in GRs, which indicates high levels of positive selection acting during the diversification of these receptors. Evidence of positive selection in recently duplicated chemosensory genes has also been found in Drosophila using similar site-specific methods (Croset et al., 2010) or using a branch-model approach (Almeida et al., 2014). The application of the latter methods demonstrated that the evidence of positive selection is stronger in the divergence among recent paralogs than in orthologous diversification. In the present work we applied for the first time a model accounting for heterogeneous selective pressure across sites to study the diversification of chemoreceptor paralogs in a non-insect species.

Table 2. Results of likelihood ratio tests and parameters estimates under the best-fitting model for each clade.
Distribution of Positively Selected Sites
Given the highly significant LRT attesting for the presence of positively selected sites in the analyzed paralogs, we used the BEB analysis to predict the putative location of such sites (Supplementary Tables S1–S10). The BEB analysis, however, had a low performance in pinpointing the specific codon sites affected (i.e., positions with strong posterior probability (PP) to belong to the ω > 1 site class). The number of sites with PP > 0.95 in IRs was very low (e.g., 3 out of 21 sites with probability to be under positive selection in IR proteins of Clade 1), with no site with PP > 0.99 (the sites with PP > 0.95 and PP > 0.7 belonging to the positively selected class were 9 and 22, respectively). In the GR family, the number of sites with high probability is somewhat higher; the GR Clade 4 had the highest number of sites with PP > 0.95 (8), followed by GR Clade 6 (7).
To investigate how selective pressure is distributed across functional elements, we mapped the location of all amino acid sites under positive selection (PP > 0.5) in the predicted functional domains of the IR and GR proteins. The structure of the IRs is characterized by an N-terminal domain (N-term), which is highly variable in size, a bipartite ligand-binding domain (LDB), three transmembrane domains (M1–M3), an ion channel domain composed of a pore loop (P-loop) located between M1 and M2 and a short C-terminal domain (C-term) (Croset et al., 2010; Traynelis et al., 2010). The LDB recognizes specific ligands that trigger the opening of the ion channel pore (Armstrong et al., 1998). The structure of the GRs consists of seven transmembrane domains (M1-7), three extracellular short loops (ECL1-3), three intracellular long loops (ICL1-3), an extracellular C-terminus (C-term), and an intracellular N-terminus (N-term) (Clyne et al., 2000). The GRs are evolutionarily related to the ORs, sharing the same basic structure. Although little is known about the specific function of the GRs' domains, some information has been gathered on the ORs' domains that could be extrapolated. For instance, several studies have shown that the outer edge of different transmembrane domains affect ligand specificity (Nichols and Luetje, 2010; Pellegrino et al., 2011; Leary et al., 2012; Hughes et al., 2014).
In a lineage specific diversification of chemosensory genes, such as the observed expansions of IRs and GRs in centipedes, it would be expected that the main domains under positive selection would be the ones affecting ligand recognition/specificity. Positive selection for diversification in these domains would lead to an increase in the number of chemicals able to be sensed by the system. In agreement with this expectation, using a branch-site model, Croset et al. (2010) identified a positively selected site (PP > 0.95) in the LBD of a recently duplicated IR of Drosophila mojavensis. Here, we found that the vast majority (87%) of the IR residues predicted to be under positive selection (including all sites predicted with PP > 0.95) are in the extracellular loops (Figures 2A–D, Supplementary Tables S1–S4). In the proteins of IR Clade 1, most of these residues were located in the S1 and S2 domains, while those of the IR Clade 4 preferentially accumulated in the large N-terminal domain. We also inferred a number of positively selected sites in the P-loop domain, suggesting that such structure might also have an important role in the functional diversification of IR paralogs. This pattern is specific to this kind of ion channel receptor. Importantly, the accumulation of positively selected sites in specific domains (in this case the LBD and P-loop domains) is significant and not explained by the relative length (in number of amino acids) of these domains within the protein (P = 1.27×10−15 or P = 6.28×10−12 taking together the sites analyzed for proteins of IR Clade 1 and IR Clade 4, or IR Clade 1A, IR Clade 1B and IR Clade 4, respectively), according to the results of the Fisher's exact test. Considering each clade separately, we found a significant departure from a homogeneous distribution of candidate sites across domains in all cases (P<0.006, after controlling for FDR), except for IR Clade1A (P = 0.424). The LBD and the P-loop, therefore, have made a major contribution to the functional diversification of IR paralogs in centipedes.
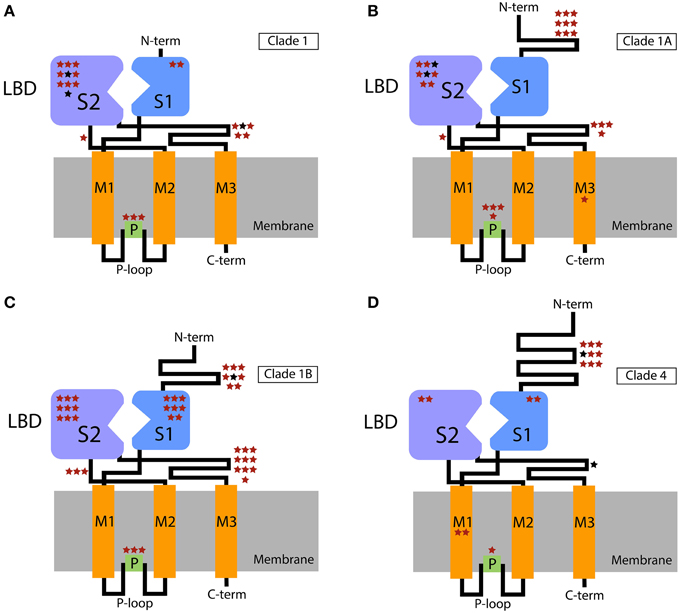
Figure 2. Schematic representation of the IR domains with the extra- and intracellular regions shown above and below the cellular membrane, respectively. Stars show the putative location of positively selected sites across functional domains. The location of these sites within the domains is arbitrary. (A) Clade 1; (B) Clade 1A; (C) Clade 1B; (D) Clade 4. Sites with PP > 0.5 and PP > 0.95 in the BEB analysis are indicated in red and black, respectively. P-loop, ion channel pore loop; M, transmembrane domains; LBD, ligand-binding domain (S1 + S2 lobes); C-term, C-terminal; N-term, N-terminal.
Similarly, in the GR family the majority of the candidate sites are located in outer parts of the proteins (Figures 3, 4). Overall, the ECL1-3 and the outer sections of the transmembrane domains accumulate more candidate sites than expected given their relative length within the protein (P = 0.021). This is very evident for GR Clades 1, 3, 5, and 6 (P<0.02 after controlling for FDR; Figures 3B,D–F, Supplementary Tables S5–S10). In these proteins the sites with PP > 0.95 also grouped in the ECL1-3 and transmembrane domains (P = 1.61×10−12; Figure 4), indicating an important functional role of these amino acids. Although there is no functional study focused on GRs, several authors have identified specific amino acids that are involved in ligand specificity of particular ORs. For instance, Nichols and Luetje (2010) found that the outer edge of TM3 of DmOr85b affects ligand specificity; Pellegrino et al. (2011) found that a Val91Ala polymorphism at the outer edge of TM2 of DmOr59b affects ligand-specificity; Leary et al. (2012) found that an Ala148Thr substitution at the outer edge of TM3 in a moth pheromone receptor mediated the ability to detect a new pheromone component; and Hughes et al. (2014) found that mutation of Ala195 on the outer edge of TM4 in Anopheles gambiae Or15 greatly affects ligand specificity. Hence, we expect that the positively selected sites we found in GR proteins at positions equivalent to those in the ORs have similar ligand-binding functions.
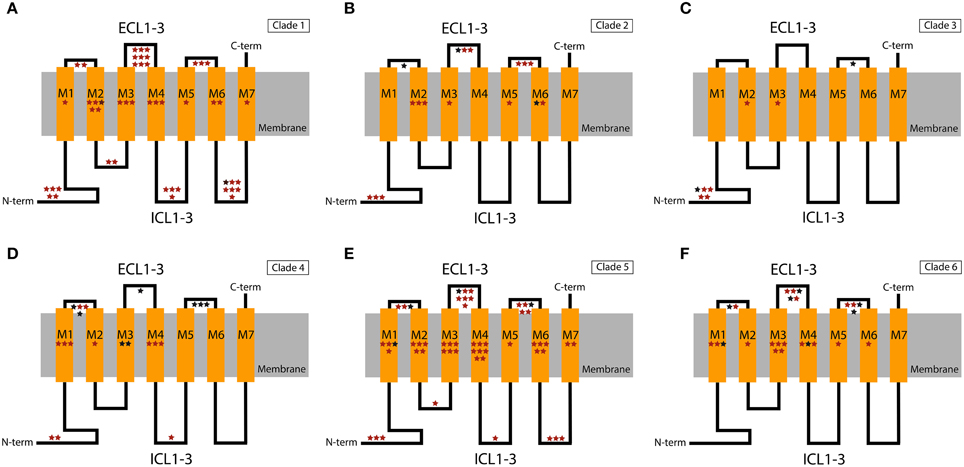
Figure 3. Schematic representation of the GR domains with the extra- and intracellular regions shown above and below the cellular membrane, respectively. Stars show the putative location of positively selected sites in the different functional domains. The location of these sites within the domains is arbitrary. (A) Clade 1; (B) Clade 2; (C) Clade 3; (D) Clade 4; (E) Clade 5; (F) Clade 6. Sites with PP > 0.5 and PP > 0.95 in the BEB analysis are indicated in red and black, respectively. ECL1-3, extracellular loops 1, 2, and 3; ICL1-3, intracellular loops 1, 2, and 3; C-term, C-terminal; N-term, N-terminal.
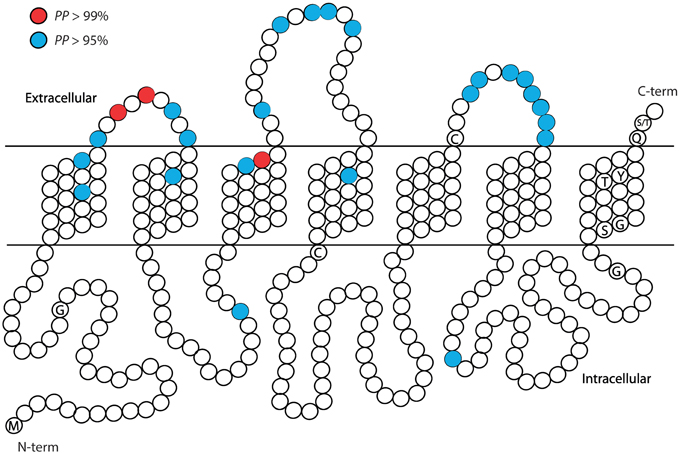
Figure 4. Summary of the approximate positions of the amino acids with high probability to be under positive selection (PP > 0.95 in blue; PP > 0.99 in red) across the six recent paralogous expansions of S. maritima GRs. Each amino acid position is represented with a circle. Characteristic conserved amino acid residues are indicated with the one letter code inside the circle.
The present results are remarkable in view of the notable differences in the molecular structure and transmembrane configuration of these two receptor families (GRs have seven transmembrane receptors with an extracellular C-terminal domain, while IRs have an inverted topology with three transmembrane helices and an intracellular C-terminus). Similar results were obtained with the aphid GRs, in which more than 70% of positively selected sites in paralog comparison were identified in the extracellular region that includes the putative ligand-binding domains (Smadja et al., 2009). Such uneven distribution of positively selected sites across these proteins is reinforcing evidence of selective processes acting on key functional domains. Hence, we can hypothesize that functional diversification of chemosensory receptors would have been largely driven by adaptive changes affecting the ligand-binding specificity and/or sensitivity. Importantly, our results point to specific codons that can, in the future, be targeted in functional experiments to determine ligand specificity and channel characteristics in the GR and IR genes of S. maritima.
Very little is known about the molecular apparatus of the chemosensory system of non-insect arthropods. This is a first contribution that explores the potential of molecular evolutionary analyses to contribute to the understanding of functional diversification in chemoreceptors. We found that positive selection has had a role in the evolution of the GR and IR repertoires in S. maritima and that adaptive diversification has happened mostly in the ligand-binding interacting domains of these receptors. The next steps in the study of these gene families are the characterization of their expression patterns and of their ligand specificity. The identification of chemosensory genes in other non-insect arthropods would also greatly contribute to our understanding of chemosensation in these less studied organisms.
Author Contributions
FA identified and manually annotated the IR genes. AS did the codon substitution models-based analyses for the IR family and prepared most figures. KW and HR identified and annotated the GR genes and did all the analyses for this gene family. FA was responsible for the primary writing of the manuscript with input from all other authors. All authors participated in the design of the study and interpretation of the data. JR, AS, and FA prepared the final version of the manuscript, which was read and approved by all authors. JR supervised the project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Ministerio de Economia y Competitividad of Spain (BFU2010-15484 and CGL2013-45211), and from the Comissió Interdepartamental de Recerca I Innovació Tecnològica of Catalonia, Spain (2009SGR-1287 and 2014SGR-1055). JR was partially supported by ICREA Academia (Generalitat de Catalunya), AS by a grant under the program Beatriu de Pinós (Generalitat de Catalunya, 2010-BP-B 00175) and FA by the Juan de la Cierva fellowship (Ministerio de Economia y Competitividad of Spain; JCI-2008-3456).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fevo.2015.00079
References
Almeida, F. C., Sánchez-Gracia, A., Campos, J. L., and Rozas, J. (2014). Family size evolution in Drosophila chemosensory gene families: a comparative analysis with a critical appraisal of methods. Genome Biol. Evol. 6, 1669–1682. doi: 10.1093/gbe/evu130
Anisimova, M., Bielawski, J. P., and Yang, Z. (2001). Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 18, 1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945
Armstrong, N., Sun, Y., Chen, G. Q., and Gouaux, E. (1998). Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 395, 913–917. doi: 10.1038/27692
Arthur, W., and Chipman, A. D. (2005). The centipede Strigamia maritima: what it can tell us about the development and evolution of segmentation. Bioessays 27, 653–660. doi: 10.1002/bies.20234
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300.
Benton, R., Sachse, S., Michnick, S. W., and Vosshall, L. B. (2006). Atypical membrane topology and heteromeric function of Drosophila odorant receptors in vivo. PLoS Biol. Feb 4:e20. doi: 10.1371/journal.pbio.0040020
Benton, R., Vannice, K. S., Gomez-Diaz, C., and Vosshall, L. B. (2009). Variant ionotropic glutamate receptors as chemosensory receptors in Drosophila. Cell 136, 149–162. doi: 10.1016/j.cell.2008.12.001
Chipman, A. D., and Akam, M. (2008). The segmentation cascade in the centipede Strigamia maritima: involvement of the Notch pathway and pair-rule gene homologues. Dev. Biol. 319, 160–169. doi: 10.1016/j.ydbio.2008.02.038
Chipman, A. D., Ferrier, D. E., Brena, C., Qu, J., Hughes, D. S., Schröder, R., et al. (2014). The first myriapod genome sequence reveals conservative arthropod gene content and genome organisation in the centipede Strigamia maritima. PLoS Biol. 12:e1002005. doi: 10.1371/journal.pbio.1002005
Clark, A. G., Eisen, M. B., Smith, D. R., Bergman, C. M., Oliver, B., Markow, T. A., et al. (2007). Evolution of genes and genomes on the Drosophila phylogeny. Nature 450, 203–218. doi: 10.1038/nature06341
Clyne, P. J., Warr, C. G., and Carlson, J. R. (2000). Candidate taste receptors in Drosophila. Science 287, 1830–1834. doi: 10.1126/science.287.5459.1830
Croset, V., Rytz, R., Cummins, S. F., Budd, A., Brawand, D., Kaessmann, H., et al. (2010). Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS Genet. 6:e1001064. doi: 10.1371/journal.pgen.1001064
Frías-López, C., Almeida, F. C., Guirao-Rico, S., Vizueta, J., Sánchez-Gracia, A., Arnedo, M. A., et al. (2015). Comparative analysis of tissue specific transcriptomes in the funnel-web spider Macrothele calpeiana (Aranae, Hexathelidae). PeerJ 3:e1064. doi: 10.7717/peerj.1064
Green, J., and Akam, M. (2013). Evolution of the pair rule gene network: insights from a centipede. Dev. Biol. 382, 235–245. doi: 10.1016/j.ydbio.2013.06.017
Hughes, D. T., Wang, G., Zwiebel, L. J., and Luetje, C. W. (2014). A determinant of odorant specificity is located at the extracellular loop 2-transmembrane domain 4 interface of an Anopheles gambiae odorant receptor subunit. Chem. Senses 39, 761–769. doi: 10.1093/chemse/bju048
Hull, J. J., Hoffmann, E. J., Perera, O. P., and Snodgrass, G. L. (2012). Identification of the western tarnished plant bug (Lygus hesperus) olfactory co-receptor Orco: expression profile and confirmation of atypical membrane topology. Arch. Insect. Biochem. Physiol. 81, 179–198. doi: 10.1002/arch.21042
Innan, H., and Kondrashov, F. (2010). The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108. doi: 10.1038/nrg2689
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kettle, C., Johnstone, J., Jowett, T., Arthur, H., and Arthur, W. (2003). The pattern of segment formation, as revealed by engrailed expression, in a centipede with a variable number of segments. Evol. Dev. 5, 198–207. doi: 10.1046/j.1525-142X.2003.03027.x
Kyte, J., and Doolittle, R. (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Leary, G. P., Allen, J. E., Bunger, P. L., Luginbill, J. B., Linn, C. E., Macallister, I. E., et al. (2012). Single mutation to a sex pheromone receptor provides adaptive specificity between closely related moth species. Proc. Natl. Acad. Sci. U.S.A. 109, 14081–14086. doi: 10.1073/pnas.1204661109
Lundin, C., Käll, L., Kreher, S. A., Kapp, K., Sonnhammer, E. L., Carlson, J. R., et al. (2007). Membrane topology of the Drosophila OR83b odorant receptor. FEBS Lett. 581, 5601–5604. doi: 10.1016/j.febslet.2007.11.007
Nei, M. (2007). The new mutation theory of phenotypic evolution. Proc. Natl. Acad. Sci. U.S.A. 104, 12235–12242. doi: 10.1073/pnas.0703349104
Nei, M., Niimura, Y., and Nozawa, M. (2008). The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat. Rev. Genet. 9, 951–963. doi: 10.1038/nrg2480
Nichols, A. S., and Luetje, C. W. (2010). Transmembrane segment 3 of Drosophila melanogaster odorant receptor subunit 85b contributes to ligand-receptor interactions. J. Biol. Chem. 285, 11854–11862. doi: 10.1074/jbc.M109.058321
Nielsen, R., and Yang, Z. (1998). Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148, 929–936.
Pellegrino, M., Steinbach, N., Stensmyr, M. C., Hansson, B. S., and Vosshall, L. B. (2011). A natural polymorphism alters odour and DEET sensitivity in an insect odorant receptor. Nature 478, 511–514. doi: 10.1038/nature10438
Pelosi, P., Zhou, J.-J., Ban, L. P., and Calvello, M. (2006). Soluble proteins in insect chemical communication. Cell Mol. Life Sci. 63, 1658–1676. doi: 10.1007/s00018-005-5607-0
Peñalva-Arana, D. C., Lynch, M., and Robertson, H. M. (2009). The chemoreceptor genes of the waterflea Daphnia pulex: many GRs but no ORs. BMC Evol. Biol. 9:79. doi: 10.1186/1471-2148-9-79
Robertson, H. M., Warr, C. G., and Carlson, J. R. (2003). Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 100(Suppl. 2), 14537–14542. doi: 10.1073/pnas.2335847100
Rota-Stabelli, O., Daley, A. C., and Pisani, D. (2013). Molecular timetrees reveal a Cambrian colonization of land and a new scenario for Ecdysozoan evolution. Curr. Biol. 23, 392–398. doi: 10.1016/j.cub.2013.01.026
Rytz, R., Croset, V., and Benton, R. (2013). Ionotropic Receptors (IRs): chemosensory ionotropic glutamate receptors in Drosophila and beyond. Insect Biochem. Mol. Biol. 43, 888–897. doi: 10.1016/j.ibmb.2013.02.007
Saina, M., Busengdal, V., Sinigaglia, C., Petrone, L., Oliveri, P., Rentzsch, F., et al. (2015). A cnidarian homologue of an insect gustatory receptor functions in developmental body patterning. Nat. Commun. 6, 1–12. doi: 10.1038/ncomms7243
Sánchez-Gracia, A., Vieira, F. G., Almeida, F. C., and Rozas, J. (2011). Comparative Genomics of the Major Chemosensory Gene Families in Arthropods. Chichester: eLS, John Wiley & Sons, Ltd.
Sánchez-Gracia, A., Vieira, F. G., and Rozas, J. (2009). Molecular evolution of the major chemosensory gene families in insects. Heredity 103, 208–216. doi: 10.1038/hdy.2009.55
Sato, K., Pellegrino, M., Nakagawa, T., Nakagawa, T., Vosshall, L. B., and Touhara, K. (2008). Insect olfactory receptors are heteromeric ligand-gated ion channels. Nature 452, 1002–1006. doi: 10.1038/nature06850
Self, S. G., and Liang, K.-Y. (1987). Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J. Am. Stat. Assoc. 82, 605–610. doi: 10.1080/01621459.1987.10478472
Smadja, C., Shi, P., Butlin, R. K., and Robertson, H. M. (2009). Large gene family expansions and adaptive evolution for odorant and gustatory receptors in the pea aphid, Acyrthosiphon pisum. Mol. Biol. Evol. 26, 2073–2086. doi: 10.1093/molbev/msp116
Smart, R., Kiely, A., Beale, M., Vargas, E., Carraher, C., Kralicek, A. V., et al. (2008). Drosophila odorant receptors are novel seven transmembrane domain proteins that can signal independently of heterotrimeric G proteins. Insect. Biochem. Mol. Biol. 38, 770–780. doi: 10.1016/j.ibmb.2008.05.002
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Swanson, W. J., Nielsen, R., and Yang, Q. (2003). Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 20, 18–20. doi: 10.1093/oxfordjournals.molbev.a004233
Touhara, K., and Vosshall, L. B. (2009). Sensing odorants and pheromones with chemosensory receptors. Ann. Rev. Physiol. 71, 307–332. doi: 10.1146/annurev.physiol.010908.163209
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Tsitoura, P., Andronopoulou, E., Tsikou, D., Agalou, A., Papakonstantinou, M. P., Kotzia, G. A., et al. (2010). Expression and membrane topology of Anopheles gambiae odorant receptors in lepidopteran insect cells. PLoS ONE 5:e15428. doi: 10.1371/journal.pone.0015428
Vosshall, L. B., and Stocker, R. F. (2007). Molecular architecture of smell and taste in Drosophila. Annu. Rev. Neurosci. 30, 505–533. doi: 10.1146/annurev.neuro.30.051606.094306
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Nielsen, R., Goldman, N., and Pedersen, A. M. K. (2000). Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155, 431–449.
Yang, Z., Wong, W. S. W., and Nielsen, R. (2005). Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Keywords: positive selection, functional domains, chemosensory function, gustatory receptor, ionotropic receptor
Citation: Almeida FC, Sánchez-Gracia A, Walden KKO, Robertson HM and Rozas J (2015) Positive selection in extra cellular domains in the diversification of Strigamia maritima chemoreceptors. Front. Ecol. Evol. 3:79. doi: 10.3389/fevo.2015.00079
Received: 16 April 2015; Accepted: 07 July 2015;
Published: 22 July 2015.
Edited by:
William Benjamin Walker, Swedish University of Agricultural Sciences, SwedenReviewed by:
Jeremy J. Heath, North Carolina State University, USAJing-Jiang Zhou, Rothamsted Research, UK
Copyright © 2015 Almeida, Sánchez-Gracia, Walden, Robertson and Rozas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julio Rozas, Departament de Genètica, Universitat de Barcelona, Av. Diagonal 643, Barcelona 08028, Spain,anJvemFzQHViLmVkdQ==
†Present Address: Francisca C. Almeida, Departamento de Ecología, Genética y Evolución, Consejo Nacional de Investigaciones Científicas y Tecnológicas, Universidad de Buenos Aires, Buenos Aires, Argentina
‡These authors have contributed equally to this work.