 Dan Zhang1,2
Dan Zhang1,2 Kui Li
Kui Li Li-Zhi Gao
Li-Zhi Gao- 1Faculty of Life Science and Technology, Kunming University of Science and Technology, Kunming, China
- 2Faculty of Environmental Science and Engineering, Kunming University of Science and Technology, Kunming, China
- 3Plant Germplasm and Genomics Center, Germplasm Bank of Wild Species in Southwest China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China
Zizania latifolia (Griseb.) Turcz. ex Stapf was once utilized as an important grain in ancient China and has been cultivated as an aquatic vegetable. Here, we report the complete Z. latifolia chloroplast genome sequence obtained through de novo assembly of Illumina paired-end reads generated by directly purified chloroplast (cp) DNA genome sequencing. The Z. latifolia cp genome is 136,501 bp in length, comprising a pair of 20,878-bp inverted repeat regions (IR) separated by small and large single copy regions (SSC and LSC) of 12,590 and 82,155 bp, respectively. The Z. latifolia cp genome encodes 110 unique genes (77 protein-coding genes, 29 tRNA genes, and 4 rRNA genes), of which 15 encompass introns. Sequence analysis identified a total of 39 direct/inverted repeats and 63 simple sequence repeats (SSR) with an average rate of 0.46 SSRs/kb. Our results revealed that the Z. latifolia cp genome is AT-rich (61.02%) and gene codon usage may be largely affected by a low GC content and codon usage bias for A, T-ending codons. We predicted 33 RNA editing sites in the chloroplast of Z. latifolia, all for C-to-U transitions. Comparative analyses with other available Oryzeae plastid genomes showed that the coding and IR sequences were more conserved than the single-copy and non-coding regions, suggesting that the indels should be cautiously employed in phylogenetic studies. Phylogenetic analysis of 52 complete grass chloroplast genomes including the reported Z. latifolia cp genome in this study yielded an identical tree topology as previous plastid-based trees, providing strong support for a sister relationship between Bambusoideae+Pooideae and Ehrhartoideae in the BEP (Bambusoideae, Ehrhartoideae, Pooideae) clade of the grass family.
Introduction
The grass family (Poaceae) is one of the most diverse angiosperm families comprised of approximately 700 genera and more than 10,000 species (Clayton and Renvoize, 1986; GPWG, 2001). It comprises of many economically important cereals in the grass family, such as rice (Oryza sativa), corn (Zea mays), wheat (Triticum aestivum), and sorghum (Sorghum bicolor). An evolutionary framework has been thoroughly established at the family level based on multiple phylogenetic studies (Clark et al., 1995; GPWG, 2001; Duvall et al., 2007; Bouchenak-Khelladi et al., 2008; Vicentini et al., 2008; Kellogg, 2009). Poaceae has been classified into some basal lineages as well as two main lineages including the PACMAD clade (Panicoideae, Arundinoideae, Chloridoideae, Micrairoideae, Aristidoideae, and Danthonioideae) and the BEP clade (Bambusoideae, Ehrhartoideae, and Pooideae) (GPWG, 2001; Duvall et al., 2007; Bouchenak-Khelladi et al., 2008; Kellogg, 2009). Nuclear and chloroplast DNA sequences have been extensively employed to determine phylogenetic relationships at both higher and lower taxonomic levels in the grass family (Doebley et al., 1990; Barker et al., 1995; Clark et al., 1995; Soreng and Davis, 1998; GPWG, 2001; Bouchenak-Khelladi et al., 2008; Kellogg, 2009) Clark et al. (1995) first reconstructed a phylogenetic tree with two major groups (the PACC and BEP clades) using the plastid ndhF sequences and found a strongly supported PACC clade (Panicoids, Arundinoids, Chloridoids, and Centothecoids), a weakly supported BEP clade (Bambusoideae, Ehrhartoideae, and Pooideae), and two isolated clades that were successive sisters to the rest. Within the BEP clade, the evolutionary relationships between Bambusoideae, Ehrhartoideae, and Pooideae have long been controversial (Zhang, 2000; GPWG, 2001; Bouchenak-Khelladi et al., 2008). Some workers reported a (B,E)P relationship with Bambusoideae and Ehrhartoideae being more closely related than Pooideae (GPWG, 2001; Vicentini et al., 2008; Sungkaew et al., 2009). However, other studies proposed either an (E, P)B relationship (Mason-Gamer et al., 1998; Mathews et al., 2000) or a (B,P)E relationship (Zhang, 2000; Bouchenak-Khelladi et al., 2008; Leseberg and Duvall, 2009; Peng et al., 2010; Wu and Ge, 2012).
Chloroplasts initially originated from an endosymbiotic relationship between independent cyanobacteria and a nonphotosynthetic host (Dyall et al., 2004). Every chloroplast has its own genome that is typically nonrecombinant and uniparentally inherited (Birky, 1995). The majority of cp genomes of higher plants possess a conserved quadripartite genomic structure composed of two copies of a large inverted repeat (IR) and two sections of unique DNA, which are referred to as large and small single copy regions (LSC and SSC, respectively) (Jansen et al., 2005). After the first complete chloroplast DNA sequences were reported in Nicotiana tabacum (Shinozaki et al., 1986) and Marchantia polymorpha (Ohyama et al., 1986), complete chloroplast DNA sequences have been determined for numerous plant species. Next-generation sequencing techniques have revolutionized DNA sequencing due to high-throughput capabilities and relatively low costs (Shendure and Ji, 2008), making it more convenient than ever to obtain a large number of cp genome sequences. To date, approximately 824 plant chloroplasts genomes are publicly available in the National Center for Biotechnology Information (NCBI) database.
Zizania latifolia (Griseb.) Turcz. ex Stapf is a perennial herbaceous aquatic plant that is one of four species in the wild rice genus Zizania L. (Poaceae) that grow throughout eastern Asia (Wu et al., 2006). The genus Ziania belongs to the rice tribe Oryzeae and is an aquatic/wetland genus with four species disjunctly distributed between Eastern Asia (Z. latifolia) and North America (Z. aquatica, Z. palustris, and Z. texana) (Terrell et al., 1997). Both Z. palustris and Z. aquatica have served as a traditional food staple for Native Americans for centuries [9] and as a specialty commercial crop more recently (Hayes et al., 1989; Oelke, 1993). Z. latifolia was once an important grain in ancient China and has been cultivated as an aquatic vegetable because the young shoots become swollen, soft, and edible after being infected by the fungus Ustilago esculenta P. Henn (Thrower and Chan, 1980; Zhai et al., 2001; Guo et al., 2007).
Previous phylogenetic studies supported the placement of Zizania in Oryzeae (Zhang and Second, 1989; Duvall et al., 1993; Ge et al., 2002; Guo and Ge, 2005; Tang et al., 2010). Zizania is most closely related to the South American genus Rhynchoryza Baill. At the intrageneric level, the Asian Z. latifolia was described as being well differentiated from the North American species based on chromosome numbers and karyotype (Duvall, 1987; Terrell et al., 1997). However, the evolutionary relationships among the three North American species are not fully resolved. As an economic plants and important wild relative of cultivated rice, it is essential to improve the knowledge of the genetic basis of these species and develop a number of genetic markers to enhance germplasm exploration and accelerate modern breeding programs.
Chloroplast DNA (cpDNA) sequences are increasingly used to resolve the deep phylogeny of flowering plants because of their low nucleotide substitution rates and relatively conserved genomic structural variation (Soltis et al., 2004; Jansen et al., 2007; Moore et al., 2010; Wang et al., 2013). The completion and availability of a large number of cp genome sequences have made it possible to speedily move gene-based phylogenetics into genome-established phylogenomics that has been proven powerful to determine the evolutionary relationships in higher plants (Jansen et al., 2007; Moore et al., 2010).
In the present study, we sequenced, de novo assembled, and characterized the complete Z. latifolia chloroplast genome. Comparative analyses of the five Oryzeae and a total of 52 completely sequenced grass plastomes, including the Z. latifolia plastome sequenced in this study, were further performed to gain in-depth insights into the overall evolutionary dynamics of chloroplast genomes in the tribe Oryzeae and better determine phylogenetic relationships in the grass family.
Materials and Methods
Plant Materials
Fresh leaves were harvested from the plants of wild rice Z. latifolia grown at Kunming Institute of Botany, Chinese Academy of Sciences (CAS), and the studied specimens were deposited in Herbarium of Kunming Institute of Botany (KUN), CAS. All necessary permits were obtained from Wei-bang Sun, Director of Kunming Botanical Garden, Kunming Institute of Botany, Chinese Academy of Sciences.
Chloroplast DNA Isolation, Amplification, and Sequencing
We collected 50–100 g fresh young leaves to isolate cpDNA using an improved high salt method that we previously reported (Shi et al., 2012). The isolated cpDNA was then sequenced with Illumina sequencing platforms. Approximately 40 mg cpDNA was used for the fragmentation by the nebulization with compressed nitrogen gas and constructed a 500 bp insert size library following the manufacturer's protocol. The sequence reads of 2 × 100 bp paired end were generated by using Illumina's Genome Analyzer at the Germplasm Bank of Wild Species in Southwest China, Kunming Institute of Botany, Chinese Academy of Sciences.
Genome Assembly and Annotation
To control the proportion of cpDNA we mapped sequence reads to the cp genome of O. sativa ssp. japonica (NC_001320) as a reference (Hiratsuka et al., 1989) to exclude nonchloroplast genome reads using Bowtie with paired-end alignments and a maximum of 3 mismatches (−v = 3), as the raw sequence reads always include non-cpDNA. Then, raw sequence reads were assembled into contigs with a minimum length of 100 bp using SOAPdenovo (Li et al., 2010) with an overlap length of 27 bp. The contigs were next aligned against the reference genome using BLAST (http://blast.ncbi.nlm.nih.gov/); the aligned contigs (≥90% similarity and query coverage) were afterward ordered based on the reference genome sequence; gaps between the de novo contigs were finally replaced with consensus sequences of raw reads mapped to the reference genome. To close the gaps and verify sequence assembly, genomic regions with ambiguous read mapping (conflicted reads mapped to the same genomic region) and low coverage (≤ 2 reads) were verified by PCR amplifications and Sanger sequencing. The plastid sequence of Z. latifolia was deposited under KM282190 in the NCBI database.
Chloroplast genome annotation was performed using DOGMA (Dual Organellar GenoMe Annotator) (Wyman et al., 2004, http://dogma.ccbb.utexas.edu). This program uses a FASTA-formatted input file of the complete chloroplast genome sequence and identifies putative protein-coding genes by performing BLASTX searches against a custom database containing formerly published chloroplast genomes. The user must select putative start and stop codons for each protein-coding gene and intron and exon boundaries for intron-containing genes. Both tRNAs and rRNAs were identified by BLASTN searches against the same database of chloroplast genomes. The chloroplast genome was drawn using OGDRAW v1.1 (Lohse et al., 2007). Full alignments with annotations were pictured with the VISTA viewer (Figure 1). VISTA-based identity plots show sequence distinctiveness between the five Oryzeae cp genomes using Phyllostachys propinqua as a reference.
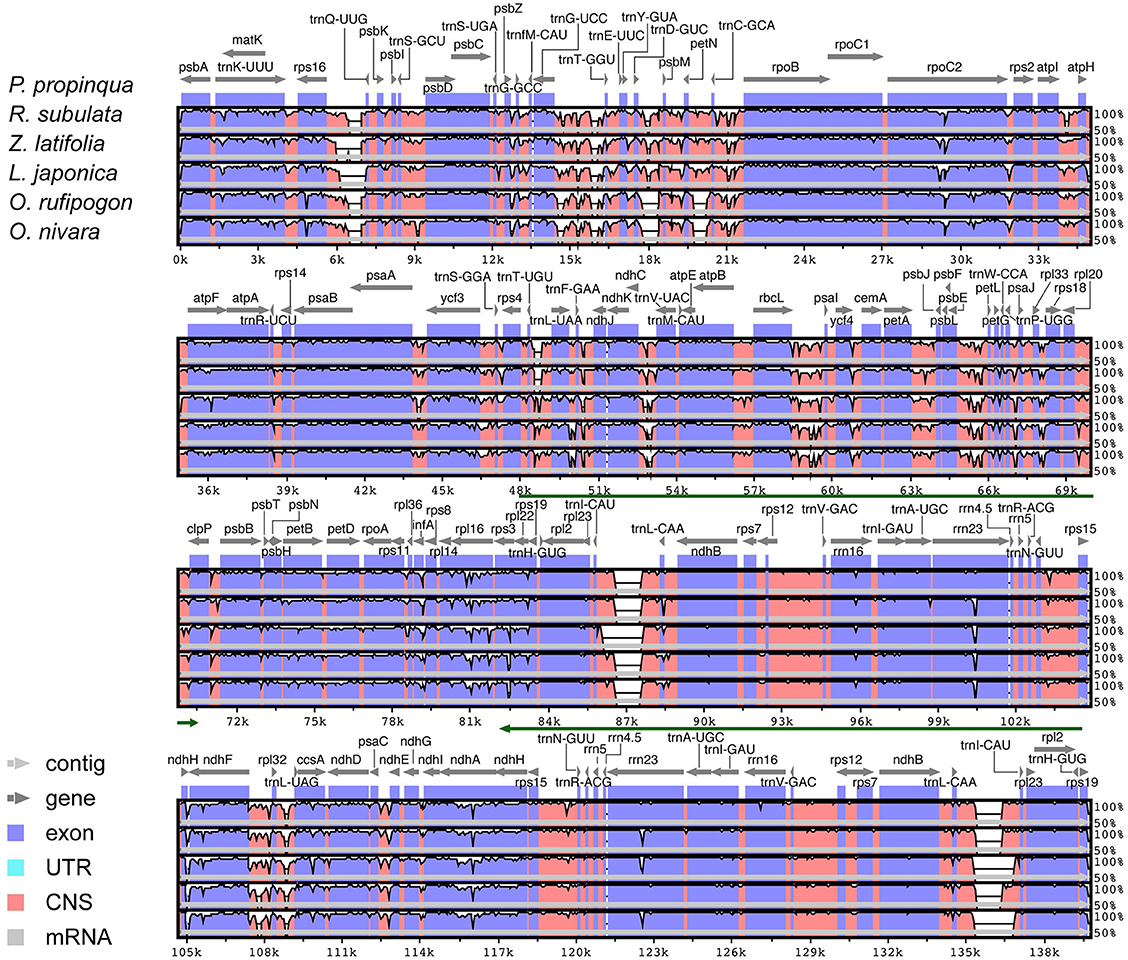
Figure 1. Gene maps of the Z. latifolia chloroplast genome. The inner circle shows the quadripartite structure of the genome including the two copies of the inverted repeat (IRA and IRB) and the large and small single-copy regions (LSC and SSC). Genes inside and outside the circle are transcribed in clockwise and counterclockwise directions, respectively. Genes belonging to different functional groups are color coded. Green lines indicate the inverted repeat (IR) regions. The dashed area in the inner circle indicates the GC content of the chloroplast genome.
Repeat Sequence Analyses
The repeat sequences were classified into the three categories, called as tandem, dispersed, and palindromic repeats. The minimal copy sizes examined were 15 bp for tandem repeat and 20 bp for dispersed and palindromic repeats. We identified these three types of repeats by first applying the program DNAMAN Version 6.0.3.99 (Lynnon Biosoft, Vaudreuil, Quebec, Canada), and then manually filtering the redundant output. The chloroplast simple sequence repeats (SSRs) were searched by using the SSRHunter software (http://www.biosoft.net) (Li and Wan, 2005). The number of dinucleotides ≥8 bp, trinucleotides ≥9 bp, tetranucleotides ≥12 bp, pentanucleotides ≥15 bp, and hexanucleotide ≥18 bp loci were counted individually.
Codon Usage
Synonymous codon usage was analyzed using 53 protein-coding genes (PCGs) with more than 100 codons by measuring codon usage indices. To avoid sampling errors, we only analyzed PCGs that contain more than 100 codons. The GC contents were calculated by using the program CodonW (version 1.4.4, http://mobyle.pasteur.fr/cgi-bin/portal.py - forms::CodonW), including the first (GC1), second (GC2), third codon positions (GC3), and the average; the effective number of codons (Nc); the relative synonymous codon usage (RSCU); and the codon adaptation index (CAI). Nc was often employed to quantify the magnitude of codon bias for an individual gene, yielding values lengthening from 20 (extremely biased, only one synonymous codon in each amino acid family was used) to 61 (totally unbiased, all synonymous codons were equally used) (Wright, 1990). RSCU is the observed frequency of a codon divided by the frequency expected if there is uniform usage within synonymous codon groups (Sharp et al., 1986). If all synonymous codons encoding the same amino acid were used equally, RSCU values were close to 1.0, indicating a lack of bias. CAI was used to estimate the extent of bias toward codons that were recognized to be favored in highly expressed genes. A CAI value is between 0 and 1.0, with a higher value showing stronger codon usage bias and a higher level of gene expression (Sharp and Li, 1987).
Prediction of RNA Editing Sites
The prediction of RNA editing sites was performed using softwares of Prep-Cp (Mower, 2009) and CURE (Du et al., 2009); the prediction parameter threshold (cutoff value) was set to 0.8 to ensure the accuracy.
Phylogenomic Analyses
The phylogeny of the five Oryzeae species was reconstructed for Rhynchoryza subulata (NC_016718), Leersia japonica (KF359922), O. rufipogon (KF359902), O. nivara (KF359901), and Z. latifolia using P. propinqua (NC_016699) as outgroup, and all gaps generated by the alignment were excluded. Phylogenetic analysis of the Poaceae was performed by aligning the whole chloroplast genome sequences from 53 plant taxa that included 52 grasses and Typha latifolia from Typhaceae using as outgroup (Supplemental Table 1).
All of the analyzed cp genome sequences were aligned using the program MAFFT version 5 (Katoh et al., 2005) and adjusted manually where necessary. Maximum likelihood (ML) analyses were implemented in RAxML version 8.0 (Stamatakis, 2014). RAxML searches counted on the general time reversible (GTR) model of nucleotide substitution with the gamma model of rate heterogeneity. Nonparametric bootstrapping as implemented in the “fast bootstrap” algorithm of RAxML used 1000 replicates. Indels were then mapped onto the phylogenetic tree determined by ML analyses of the whole cp genome sequence alignment using the ClustalW1.83 program (Thompson et al., 1994).
Results and Discussion
Sequence, Assembly, and Organization of the Z. latifolia Chloroplast Genome
Whole chloroplast genome sequencing generated 1,865,122 raw reads (~96 bp in read length on average). Illumina paired-end reads were mapped onto to the published cp genome of O. sativa ssp. japonica (NC_001320) as a reference (Shimada and Sugiura, 1991). We collected 804,780 cp-genome-related reads (43.15% of total reads with a mean length of ~98 bp), reaching an average of 575 × coverage over the cp genome. After closing the gaps and validating the sequence assembly with PCR-based experiments, we obtained a complete Z. latifolia cp genome sequence with a total of 136,501-bp. Most cp genomes of higher plants were found to have a conserved quadripartite structure composed of two copies of a large IR and two sections of unique DNA referred to as the LSC and SSC [6]. In this study, the Z. latifolia cp genome is a typical circular double-stranded DNA molecule with a quadripartite structure with LSC regions (82,155 bp) and small single copy regions (12,590 bp) separated by two IR copies (20,878bp) (Figure 2; Table 1).
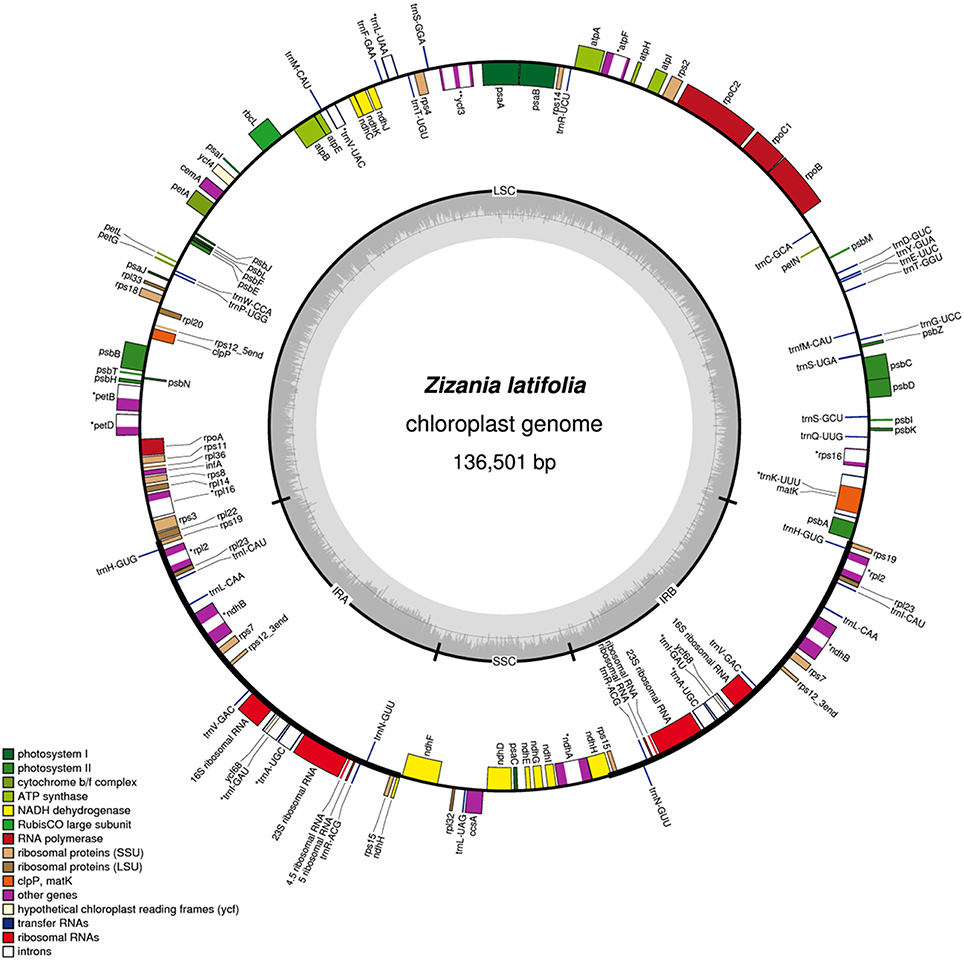
Figure 2. Visualization of alignment of the five chloroplast genome sequences. VISTA-based identity plots show sequence identities between the five Oryzeae cp genomes using P. propinqua as a reference. Genome regions are color-coded as protein- or non-coding sequences; green lines show the inverted repeat (IR) regions.
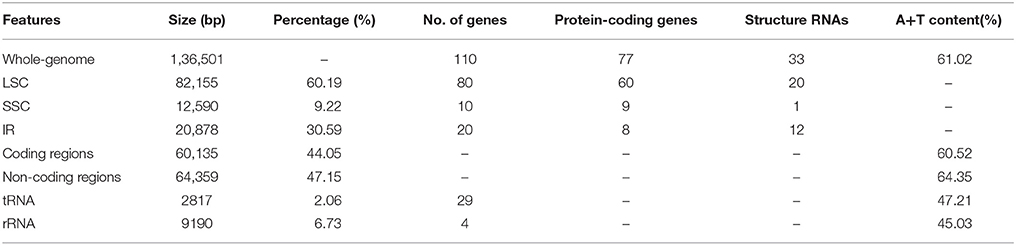
Table 1. Summary of the chloroplast genome features of Z. latifolia.
The gene content and sequence of the Z. latifolia cp genome are relatively conserved, with many characteristics in common with land plant cp genomes (Sugiura, 1992; Raubeson and Jansen, 2005). It encodes 130 predicted protein-coding genes; 110 are unique and 20 are duplicated in the IR regions. Among the 110 annotated unique genes, there are 77 unique protein-coding genes, 29 tRNA genes, and 4 rRNA genes (Figure 2; Supplemental Table 2). Overall, 44.05, 2.06, and 6.73% of the genome sequence encode proteins, tRNAs, and rRNAs, respectively, whereas the remaining 47.15% of the genome is non-coding and filled with introns and intergenic spacers. Similar to other cp genomes (Raubeson et al., 2007; Gao et al., 2009; Yang et al., 2010), the Z. latifolia cp genome is AT-rich (61.02%), and the values vary slightly among defined non-coding, protein-coding, tRNA, and rRNA sequences, with A+T contents of 64.35, 60.52, 47.21, and 45.03%, respectively. The four rRNA genes are all positioned in the IRs. Twenty-one tRNA genes are located in the single-copy region, whereas the others are placed in the IRs. Eighteen genes contain introns, ycf3 comprises two introns, and the rest of genes have an intron; rps12 is trans-spliced, one of its exons is in the LSC region (5′ end) and the other reside in the IR regions (3′ end) separated by an intron. A pair of ndhH genes might be different due to a portion of the 5′ part of ndhH overlapping with the IR/SSC junctions. matK was located within the intron of trnK-UUU, and ycf68 was positioned within the intron of trnI-GAU. In the Z. latifolia cp genome, the gene pairs atpB-atpE, psbC-psbD, and ndhC-ndhK had 4-, 53-, and 10-bp overlapping regions, respectively. The junctions between IR and SSC regions usually vary among chloroplast genomes of higher plants (Zhang et al., 2011; Xu et al., 2012; Wang et al., 2013). In the Z. latifolia cp genome, the distance of rps19 from the LSC/IR junction was 42 bp, while ndhH gene regions extended into the IR region in the junctions between IR and SSC.
Repeat Sequence Analyses
Repeat sequences may play an important role in chloroplast genome rearrangement and the generation of divergent regions via illegitimate recombination and slipped-strand mispairing (Timme et al., 2007; Zhang et al., 2011; Xu et al., 2012; Wang et al., 2013). One of the IR regions was omitted to avoid redundancy in the detection of repeats in the Z. latifolia cp genome. In the three categories, 39 repeats were detected in the Z. latifolia cp genome (Supplemental Table 3). In all of the 39 repeats, the numbers of tandem, dispersed, and palindromic repeats were 11, 17 and 11, respectively. Approximately 84.62% of these repeats ranged between 15 and 29 bp in size (Figure 3A), although the defined smallest size was 20 bp for palindromic and dispersed repeats. The longest repeat was a palindromic repeat of 130 bp. Dispersed repeats, accounting for 43.59% of total repeats, were the most common of the three repeat types. Tandem and palindromic repeats were located in non-coding genomic regions, genes, and tRNAs (Figure 3B) and had the same occupation ratio (28.21% each of total repeats) (Figure 3C). A number of repeats were found in the exons of genes or tRNA such as rps18, rpoC2, rpl23, trnfM-CAU, trnS-GCU, trnS-UGA, trnS-GGA, trnT-GGU, and trnT-UGU. A total of nine repeats were identified in rpoC2, including one tandem repeats and eight dispersed repeats; notably, a 24-bp dispersed repeat (AGAGGAAGACTCAGAGGACGAATA) occurred four times. Three 22-bp dispersed repeats (CCCTTGTCGAAGCCCTTTATGA) were observed in the intergenic region between trnI-CAU and trnL-CAA. A 21-bp repeat (AGAGAGGGATTCGAACCCTCG) was found in both dispersed and palindromic repeats (Supplemental Table 3).
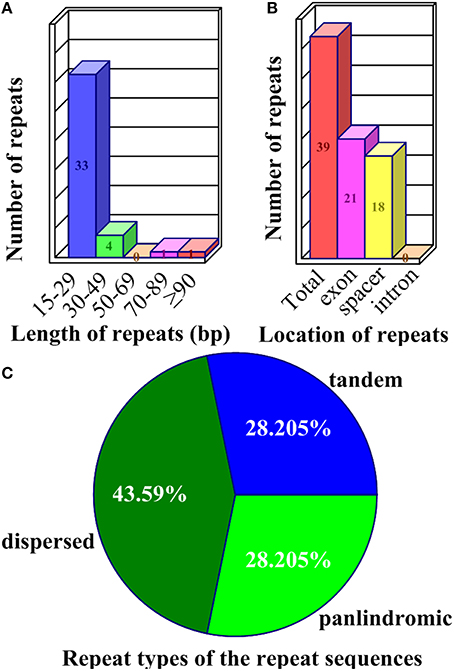
Figure 3. Repeat sequences in the Z. latifolia chloroplast genome. (A) Frequency of repeats by length. (B) Location of all repeats; those that occurred in two regions were counted in both. (C) Repeat sequence types.
SSRs are often used as molecular markers to characterize genetic variability in plant breeding programs and population genetics studies; they usually have high mutation rates compared to other neutral DNA regions due to slipped strand mispairing (slippage) during DNA replication on a single DNA strand. The chloroplast SSRs were initially reported in studies of Pinus radiata and O. sativa (Cato and Richardson, 1996; Powell et al., 1996; Provan et al., 1996), which foreseen the potential use of chloroplast SSR assessments for population genetics. Of the 63 SSR loci characterized in Z. latifolia cp genome, the average rate was 0.46 SSRs/kb (Supplemental Table 4). The numbers of dinucleotides ≥8 bp, trinucleotides ≥9 bp, tetranucleotides ≥12 bp, pentanucleotides ≥15 bp, and hexanucleotides ≥18 bp loci were 18, 33, 10, 1, and 1, respectively. Likewise, the numbers of dinucleotides ≥10 bp, trinucleotides ≥12 bp, tetranucleotides ≥16 bp, and pentanucleotides ≥20 bp loci were 3, 4, 1, 0, and 0, respectively, and the average rate was 0.06 SSRs/kb. The various SSRs motifs exhibited different frequencies, with trinucleotide SSRs being the most abundant (52.38%), and pentanucleotide and hexanucleotide SSRs being the least common (1.59%; Table 2).
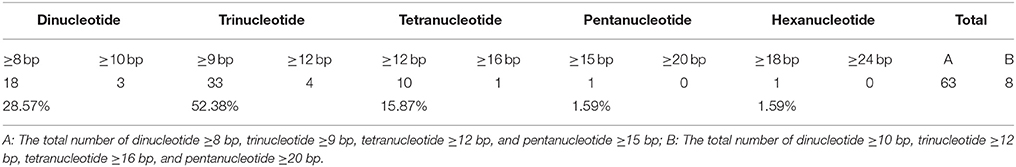
Table 2. Number of SSRs identified in the Z. latifolia chloroplast genome.
Codon Usage
Due to the redundancy of genetic code, most amino acids are coded by two or more synonymous codons. However, these codons often have different frequencies in a genome, a phenomenon termed codon usage bias. In the universal genetic code, multiple codons differ only at the third position or occasionally in the second position in some of amino acids (Ermolaeva, 2001). Plant gene codon frequencies are clearly different, and amino acids also have variable codon ratios. The formation of codon bias is due to complex factors that lead to gene mutation and selection (Wong et al., 2002). Protein-encoded gene structure, function, and expression are closely linked and affected by a variety of evolutionary factors (Chiapello et al., 1998).
In all the 53 PCGs of the Z. latifolia cp genome, the GC contents of the first (GC1), second (GC2), third codon positions (GC3), and the average of the three positions were 47.82, 39.44, 31.03, and 39.43%, respectively. The relatively low GC content of the coding region is consistent with that of the Z. latifolia entire chloroplast genome (38.98%). From the perspective of different codon positions, the base compositions of the three codon positions were not evenly distributed, and the distribution for GC content was GC1>GC2>GC3, with a large difference in GC3, which was only slightly higher than 30%. Overall, the codon usage frequency of Z. latifolia chloroplast genes for A+T-ending codons was higher than for G+C-ending codons. In the 78 protein-coding genes of the Z. latifolia cp genome, the vast majority of translation initiation codons was ATG; only those for matK, rps19, and rpl2 were CTG, GTG, and ACG, respectively. As start codons, CTG, GTG, and ACG are nonsense, but they do code Leu, Val, and Thr; the rest of the codon usage was consistent with that of nuclear genes.
The codon usages for 53 PCGs from Z. latifolia are presented in Supplemental Table 5. To examine codon usage bias at the gene level Nc was calculated that range from 20 (extremely biased) to 61 (no bias). The means of calculated Nc values for each PCG are shown in Supplemental Table 5. The most biased codon usage was detected in rpl16, as indicated by the lowest mean Nc value of 37.27. Nc values of 53 PCGs ranged from 37.27 to 61 (ndhC and infA), with most greater than 44, suggesting that gene codon bias is weak in the Z. latifolia cp genome.
Table 3 shows the cp gene codon usage and RSCU. The RSCU values of 31 codons were >1, indicating that these are biased codons in the Z. latifolia cp genes. In the above codons, only TCC and CCC codons for Ser and Pro are C-ending codons, and the rest are A, T-ending codons. The codon usage for G+C-ending codons showed the opposite pattern; the RSCU values were < 1, showing that they are less common in Z. latifolia cp genes. Stop codon usage was found to be biased toward TAA. By calculating the GC content and RSCU value of Z. latifolia cp protein-coding genes, we found that some gene codon usage bias for A or T base-ending codons is consistent with the analyses of codon usage bias in rice, poplar, and other plant cp genes, suggesting similar codon usage rules (Liu and Xue, 2004; Zhou et al., 2008a,b). Many factors affect codon usage and the reasons for codon usage bias may vary in different species or genes in the same species. In a plant cp gene codon usage factor study, Liu and Xue (2004) identified the level of gene expression and gene base composition in rice. Zhou et al. (2008b) reported that genomic nucleotide mutation bias is a major factor in chloroplast gene codon usage bias of seed plants such as Arabidopsis thaliana and poplar. Morton (2003) considered asymmetric mutation of cpDNA as the main cause of codon usage bias in Euglena gracilis cp gene. Our preliminary result indicates that chloroplast gene codon usage may be largely affected by a low GC content and codon usage bias for A+T-ending codons in Z. latifolia.
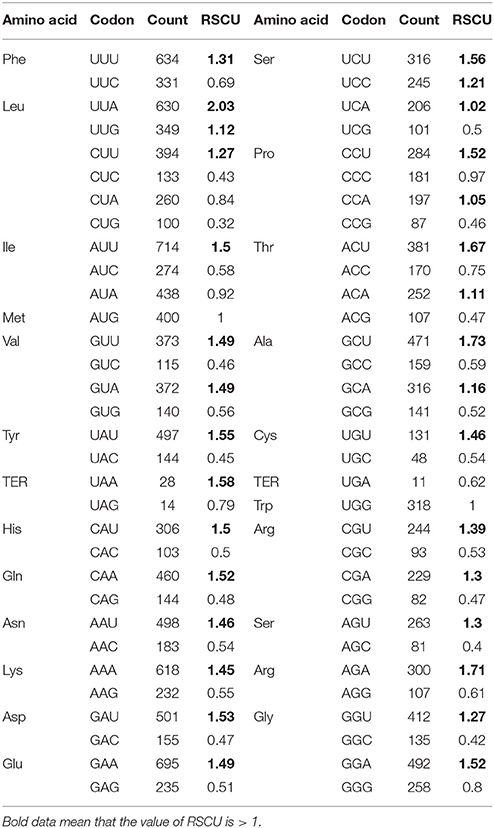
Table 3. The relative synonymous codon usage of the Z. latifolia chloroplast genome.
Predicted RNA Editing Sites in the Z. latifolia Chloroplast Genes
We predicted 33 RNA editing sites in the Z. latifolia cp genome, all of which were C-to-U transitions (Table 4). In chloroplasts and mitochondria of seed plants, a conversion from C to U is the most predominant form (Bock, 2000). These editing sites occurred on 14 genes, with ndhB containing nine. ndhB transcripts were also found to be highly edited in other plants such as maize, sugarcane, rice, barley, tomato, tobacco, and Arabidopsis (Freyer et al., 1995; Kahlau et al., 2006; Chateigner-Boutin and Small, 2007). The genes ndhA and rpoB were predicted to have four editing sites; ropC2 and ndhF, three; ycf3, two; and one each in atpA, ccsA, matK, ndhD, rpl2, rpl20, rps8, and rps14. The locations of editing sites were 3, 30, and 0 in the first, second, and third codons, respectively. Among the 33 sites, 13 were U_A types, which is consistent with previous RNA editing sites found in locations with similar codon bias (Kessel, 1995; Jiang et al., 2011). We also observed that the 33 RNA editing events in Z. latifolia chloroplasts caused amino acid changes for highly hydrophobic residues (e.g., leucine, isoleucine, tyrosin, phenylalanine, and methionine) with conversions from serine to leucine as the most frequent transitions. This observation is consistent with most of the chloroplast RNA editing phenomenon (Jiang et al., 2011), which is a form of post-transcriptional regulation of gene expression in higher plants, occurring mainly in first and second codons (more so in the second), which is indicative of that codon editing follows a certain preference. Notably, our results provide additional evidence supporting previously described chloroplast RNA editing features.
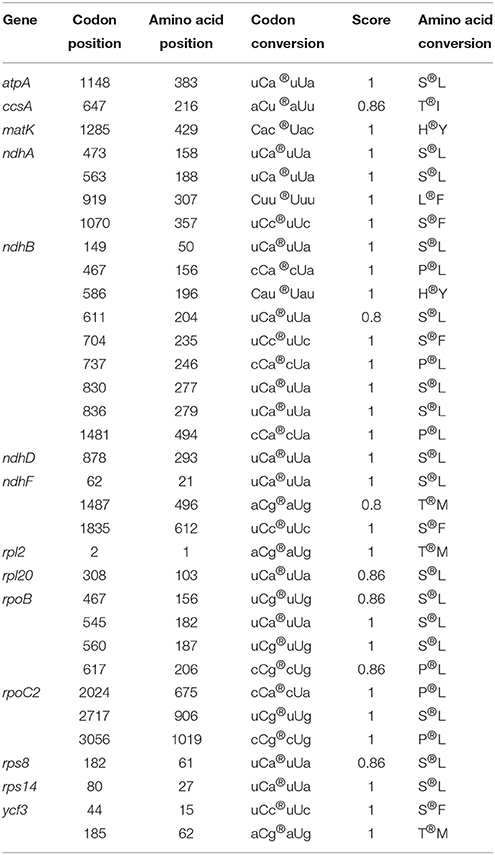
Table 4. The predicted RNA editing site in the Z. latifolia chloroplast genes.
Evolution of the Five Oryzeae Chloroplast Genomes
In plant cp genomes, the coding and IR regions are usually more conserved than single copy and non-coding regions, respectively (Jansen et al., 2005). Global alignment of the five Oryzeae cp genomes detected several genomic variant events (Figure 1). The variation in the alignment confirmed that the IR region was more conserved than single copy regions, and confirmed that genic regions were more conserved than intergenic regions. Although the overall structure, genome size, gene number, and gene order are conserved in higher plant cp genomes (Palmer et al., 1987), the junctions between IR and SSC regions are usually dissimilar. In the tribe Oryzeae, a slight difference in junction positions was observed among these five cp genomes (Figure 4). In the junctions between IR and SSC, for example, ndhH gene regions extended slightly into the IR region, and the distance of rps19 from the junction of LSC/IRb varied from 41 bp in R. subulata to 44 bp in O. rufipogon.
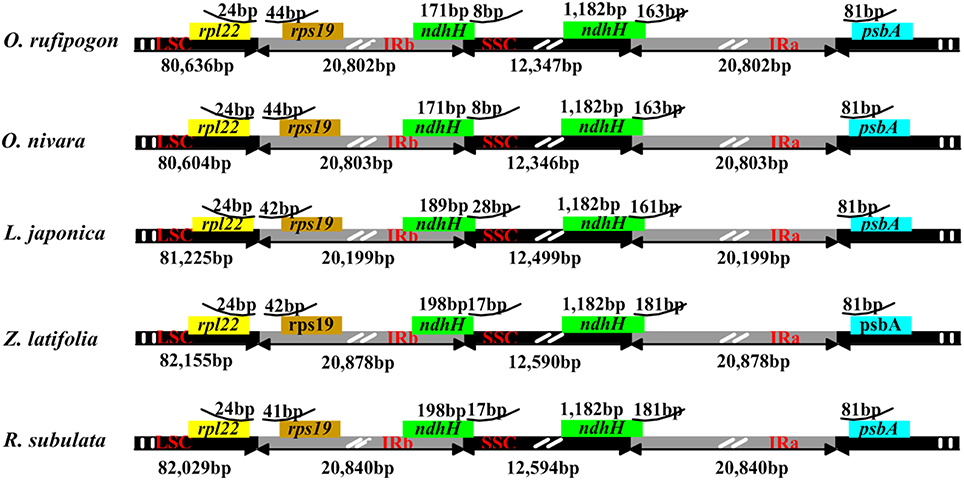
Figure 4. IR junctions of the five Oryzeae chloroplast genomes.
The six published chloroplast genome sequences of the tribe Oryzeae were concatenated into a data set of 143,064 bp in length. Of them, 7601 bp (5.31%), and 2386 bp (1.67%) were variable and phylogenetically informative sites, respectively. Based on the chloroplast genome sequences ML phylogeny was further reconstructed to examine evolutionary relationships of these grass species from the tribe Oryzeae (Figure 5). Our results showed that R. subulata and Z. latifolia formed a monophyletic clade, while the other monophyletic clade included O. nivara, O. rufipogon, and L. japonica with high bootstrap supports, which is in good agreement with a previous study (Tang et al., 2010).
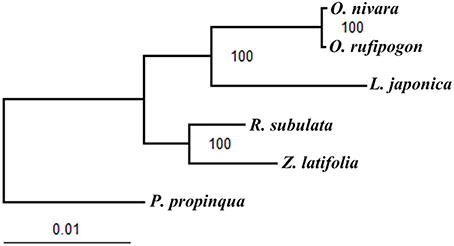
Figure 5. ML phylogeny of the five Oryzeae species inferred from the whole-genome chloroplast sequences. Numbers near branches are the bootstrap values of maximum likelihood (ML).
All the insertions and deletions detected in the exons were mapped onto the phylogenetic tree (Figure 6). The 27 indels were located in 12 genes (ccsA, infA, matK, ndhF, ndhK, psaJ, rpl32, rpoA, rpoC1, rpoC2, rps3, ycf3). Of these, 23 indels mapped to monophyletic groups have been highly supported and thus may be synapomorphies. The remaining four indels may be homoplasies, possibly associated with parallel mutations or back mutations during evolutionary history.
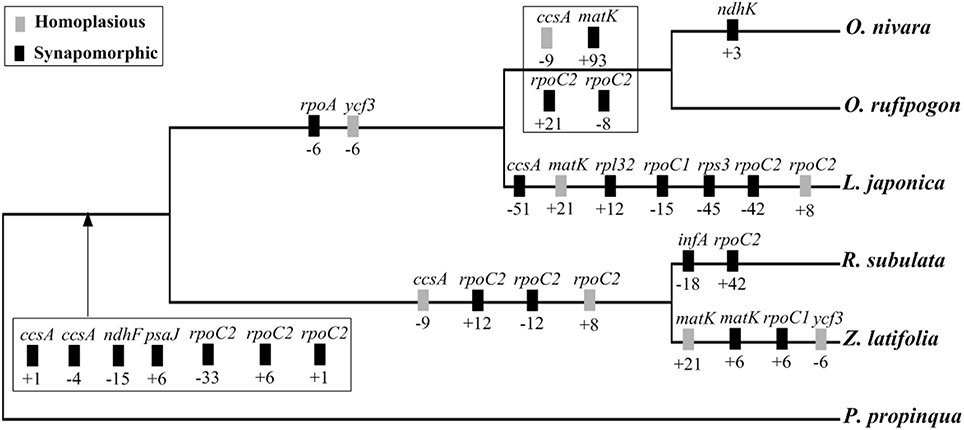
Figure 6. Phylogenetic distribution of exon indels in the five Oryzeae species. Gene names are given above boxes, and sizes of indels (bp) and polarity (+, insertion; –, deletion) are given below boxes. Mutation polarity was determined by comparison to P. propinqua. The topology of the studied species was based on ML analysis of complete cp genomes as shown in Figure 5.
The inclusion of indels as characteristics in phylogenetic studies has gained growing popularity. However, the relative helpfulness of gap characters has been a matter of debate. There is no clear consensus about whether indels should be used for phylogenetic analyses (Bapteste and Philippe, 2002; Egan and Crandall, 2008; Zhang et al., 2011), although most arguments against them are according to studies using one or several DNA fragments. Some researchers have championed indels as phylogenetically reliable (Lloyd and Calder, 1991; Ingvarsson et al., 2003) while others suggest that indels are homoplasious or uninformative (Golenberg et al., 1993; Pearce, 2006). Genes in the cp genome contained both synapomorphic and homoplasious indels. Therefore, genomic structural variations such as indels should be cautiously used in phylogenetic analyses.
Phylogeny Reconstruction of Poaceae Based on Complete Chloroplast Genome Sequences
To understand the evolution of the grass family an improved resolution of phylogenetic relationships have been achieved using these fully sequenced cp genome sequences of 52 Poaceae species using T. latifolia as outgroup (Figure 7). The sequence alignment that was used for phylogenetic analyses comprised 28,594 characters. ML bootstrap values were fairly high, with values ≥95% for 48 of the 50 nodes, and 47 nodes with 100% bootstrap support (Figure 7). Our results showed that Z. latifolia whose cp genome was reported in this study was closely related to R. subulata, which then formed a cluster together with Oryza species and L. tisserantii from Ehrhartoideae with 100% bootstrap supports. Notably, Anomochlooideae was the earliest diverging Poaceae lineage, subsequently followed by Pharoideae and Puelioideae. The Poaceae family diverged into two main clades; one included Arundinoideae and Panicoideae of the PACMAD clade and the other clade being BEP (Bambusoideae, Ehrhartoideae, and Pooideae). The BEP clade has historically been rather weakly supported since it was first identified (Clark et al., 1995). In our obtained phylogenies, the BEP clade was resolved as a monophyletic group with 100% bootstrap support, and Bambusoideae and Pooideae were determined to be more closely related than Ehrhartoideae. Early work supported a sister relationship between Bambusoideae+Ehrhartoideae and Pooideae (GPWG, 2001), while others suggested that Bambusoideae+Pooideae was sister to Ehrhartoideae (Zhang, 2000; Bouchenak-Khelladi et al., 2008; Wu and Ge, 2012). A recent study attempted to resolve BEP clade phylogeny by examining the sequences of 76 chloroplast protein-coding genes from the 22 grass species, and their results supported the (B,P)E hypothesis that Bambusoideae and Pooideae are more closely related than Ehrhartoideae (Wu and Ge, 2012). The PACMAD clade only included whole chloroplast genomes from Arundinoideae and Panicoideae, while other subfamilies such as Chloridoideae are missing in the genome-based grass phylogeny (Figure 7). Based on a recent comprehensive grass phylogeny (GPWG, 2012), however, our phylogenomic analysis of further taxon sampling of 40 grass species belonging to the BEP clade is congruent with the former resolution of BEP clade relationships (Bouchenak-Khelladi et al., 2008; Wu and Ge, 2012). Further, efforts require to include whole chloroplast genome sequences from Chloridoideae and the other missing subfamilies to obtain a fully resolved grass phylogeny based on genome-established phylogenomic analysis.
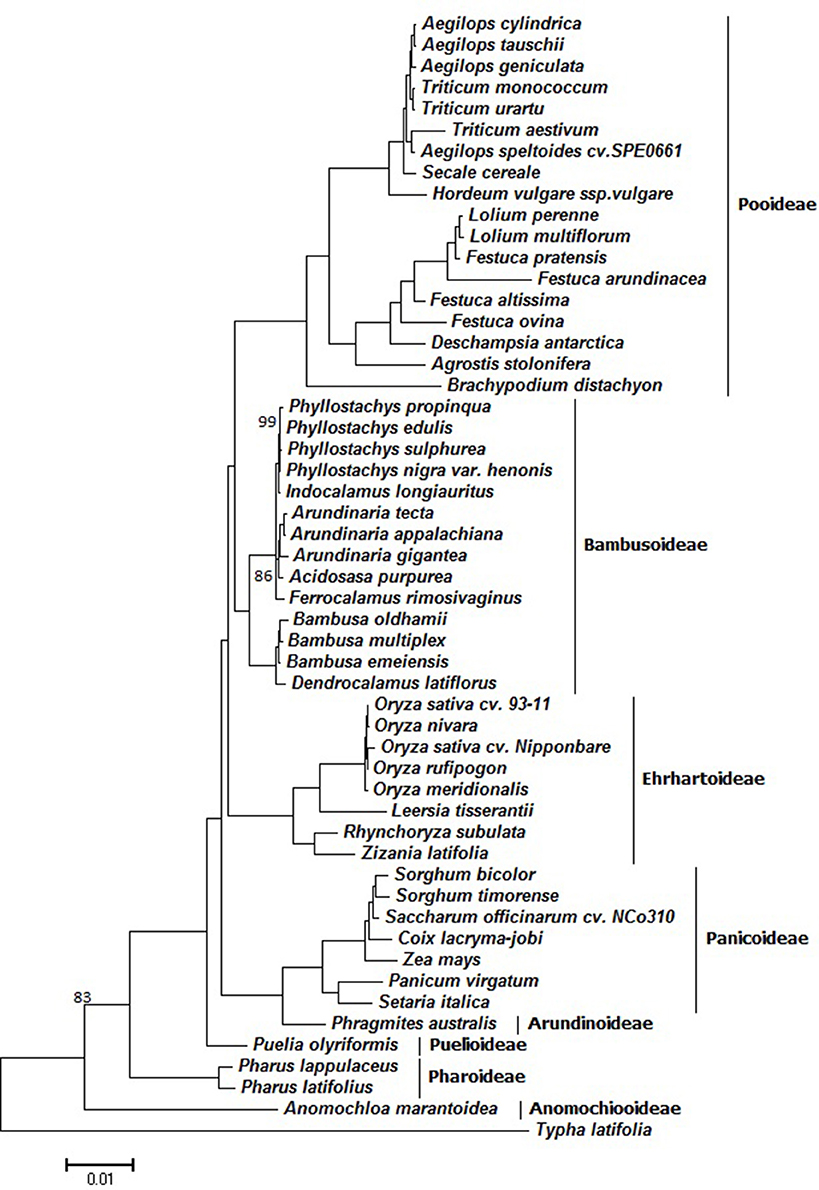
Figure 7. ML phylogeny of the Poaceae species inferred from whole-genome chloroplast sequences. Numbers near branches are bootstrap values of ML. The branches without numbers indicate 100% bootstrap supports.
Conclusions
The wild rice Z. latifolia cp genome sequenced in this study is a typical circular double-stranded DNA molecule with a quadripartite structure common to most land plant genomes, and the genome size, overall structure, gene number, and gene order are well-conserved. Due to the low GC content of this chloroplast genome, the codon usage was biased toward A, T-ending codons. The RNA editing sites in the chloroplast were mainly the C-to-U transitions. Comparative analyses with other five Oryzeae plastid genomes showed that the coding and IR regions were more conserved than the single-copy and non-coding regions. Our comparative chloroplast Oryzeae genomic analysis suggests that genomic structural variations should be used cautiously because synapomorphic and homoplasious indels are both found in cp genes. Phylogenetic analysis based on whole chloroplast genome sequences of 52 grass species yielded a similar topology to previous studies and provided a strong support for the hypotheses that Anomochlooideae was the earliest diverging Poaceae lineage and that Bambusoideae and Pooideae are more closely related than Ehrhartoideae within the BEP clade.
Author Contributions
LG conceived the study, performed data analysis, and drafted and revised the manuscript; DZ drafted the manuscript; DZ, KL, and YL performed data analysis; JG performed the experiments and data analysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the reviewers for their comments on the manuscript. This study was supported by Top Talent Program of Yunnan Province (20080A009), Project of Innovation Team of Yunnan Province, Hundreds Oversea Talent Program of Yunnan Province and Hundreds Talents Program of Chinese Academy of Sciences (CAS) (to LG).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fevo.2016.00088
References
Asano, T., Tsudzuki, T., Takahashi, S., Shimada, H., and Kadowaki, K. (2004). Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 11, 93–99. doi: 10.1093/dnares/11.2.93
Bapteste, E., and Philippe, H. (2002). The potential value of indels as phylogenetic markers: position of trichomonads as a case study. Mol. Biol. Evol. 19, 972–977. doi: 10.1093/oxfordjournals.molbev.a004156
Barker, N. P., Linder, H. P., and Harley, E. H. (1995). Polyphyly of Arundinoideae (Poaceae): evidence from rbcL sequence data. Syst. Biol. 20, 423–435. doi: 10.2307/2419802
Birky, C. W. (1995). Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl. Acad. Sci. U.S.A. 92, 11331–11338. doi: 10.1073/pnas.92.25.11331
Bock, R. (2000). Sense from nonsense: how the genetic information of chloroplasts is altered by RNA editing. Biochimie 82, 549–557. doi: 10.1016/S0300-9084(00)00610-6
Bouchenak-Khelladi, Y., Salamin, N., Savolainen, V., Forest, F., Bank, M., Chase, M. W., et al. (2008). Large multi-gene phylogenetic trees of the grasses (Poaceae), progress towards complete tribal and generic level sampling. Mol. Phylogenet. Evol. 47, 488–505. doi: 10.1016/j.ympev.2008.01.035
Cato, S. A., and Richardson, T. E. (1996). Inter- and intra-specific polymorphism at chloroplast SSR loci and the inheritance of plastids in Pinus radiata D. Don. Theor. Appl. Genet. 93, 587–592. doi: 10.1007/BF00417952
Chateigner-Boutin, A. L., and Small, I. (2007). A rapid high-throughput method for the detection and quantification of RNA editing based on high-resolution melting of amplicons. Nucleic Acids Res. 35, e114. doi: 10.1093/nar/gkm640
Chiapello, H., Lisacek, F., Caboche, M., and Hénaut, A. (1998). Codon usage and gene function are related in sequences of Arabidopsis thaliana. Gene 209, GC1–GC38. doi: 10.1016/s0378-1119(97)00671-9
Clark, L. G., Zhang, W., and Wendel, J. F. (1995). A phylogeny of the grass family (Poaceae) based on ndhF sequence data. Syst. Bot. 20, 436–460. doi: 10.2307/2419803
Clayton, W. D., and Renvoize, S. A. (1986). Genera Graminum, grasses of the world. Kew Bull Additional Series XIII. London: Her Majesty's Stationery Office.
Doebley, J., Durbin, M., Golenberg, E. M., Clegg, M. T., and Ma, D. P. (1990). Evolutionary analysis of the large subunit of carboxylase (rbcL) nucleotide sequence data among the grasses (Poaceae). Evolution 44, 1097–1108. doi: 10.2307/2409569
Du, P. F., Jia, L. Y., and Li, Y. D. (2009). CURE-Chloroplast: a chloroplast C-to-U RNA editing predictor for seed plants. BMC Bioinformatics 10:135. doi: 10.1186/1471-2105-10-135
Duvall, M. R. (1987). A Systematic Evaluation of the Genus Zizania (Poaceae). Ph.D. dissertation, University of Minnesota, St. Paul.
Duvall, M. R., Davis, J. I., Clark, L. G., Noll, J. D., Goldman, D. H., and Sánchez-Ken, J. G. (2007). Phylogeny of the grasses (Poaceae) revisited. Aliso 23, 237–247. doi: 10.5642/aliso.20072301.18
Duvall, M. R., Peterson, P. M., Terrell, E. E., and Christensen, A. H. (1993). Phylogeny of North American Oryzoid grasses as construed from maps of plastid DNA restriction sites. Am. J. Bot. 80, 83–88. doi: 10.2307/2445123
Dyall, S. D., Brown, M. T., and Johnson, P. J. (2004). Ancient invasions: from endosymbionts to organelles. Science 304, 253–257. doi: 10.1126/science.1094884
Egan, A. N., and Crandall, K. A. (2008). Incorporating gaps as phylogenetic characters across eight DNA regions: ramifications for North American Psoraleeae (Leguminosae). Mol. Phylogenet. Evol. 46, 532–546. doi: 10.1016/j.ympev.2007.10.006
Freyer, R., Lopez, C., Maier, R. M., Martin, M., Sabater, B., and Kossel, H. (1995). Editing of the chloroplast ndhB encoded transcript shows divergence between closely related members of the grass family (Poaceae). Plant Mol. Biol. 29, 679–684. doi: 10.1007/BF00041158
Gao, L., Yi, X., Yang, Y. X., Su, Y. J., and Wang, T. (2009). Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 9:130. doi: 10.1186/1471-2148-9-130
Ge, S., Li, A., Lu, B. R., Zhang, S. Z., and Hong, D. Y. (2002). A phylogeny of the rice tribe Oryzeae (Poaceae) based on matK sequence data. Am. J. Bot. 89, 1967–1972. doi: 10.3732/ajb.89.12.1967
Golenberg, E. M., Clegg, M. T., Durbin, M. L., Doebley, J., and Ma, D. P. (1993). Evolution of a non-coding region of the chloroplast genome. Mol. Phylogenet. Evol. 2, 52–64. doi: 10.1006/mpev.1993.1006
GPWG, (Grass Phylogeny Working Group). (2001). (Grass Phylogeny Working Group). Phylogeny and subfamilial classification of the grasses (Poaceae). Ann. Mol. Bot. Gard. 88, 373–457. doi: 10.2307/3298585
GPWG, (Grass Phylogeny Working Group II). (2012). New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 193, 304–312. doi: 10.1111/j.1469-8137.2011.03972.x
Guo, H. B., Li, S. M., Peng, J., and Ke, W. D. (2007). Zizania latifolia Turcz. cultivated in China. Genet. Resour. Crop Evol. 54, 1211–1217. doi: 10.1007/s10722-006-9102-8
Guo, Y. L., and Ge, S. (2005). Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. Am. J. Bot. 92, 1548–1558. doi: 10.3732/ajb.92.9.1548
Hayes, P. M., Stucker, R. E., and Wandrey, G. G. (1989). The domestication of American wild-rice (Zizania palustris, Poaceae). Econ. Bot. 43, 203–214. doi: 10.1007/BF02859862
Hiratsuka, J., Shimada, H., Whittier, R., Ishibashi, T., Sakamoto, M., Mori, M., et al. (1989). The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 217, 185–194. doi: 10.1007/BF02464880
Ingvarsson, P. K., Ribstein, S., and Taylor, D. R. (2003). Molecular evolution of insertions and deletion in the chloroplast genome of Silene. Mol. Biol. Evol. 20, 1737–1740. doi: 10.1093/molbev/msg163
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jansen, R. K., Raubeson, L. A., Boore, J. L., Pamphilis, C. W., Chumley, T. W., Haberle, R. C., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Meth. Enzymol. 395, 348–384. doi: 10.1016/S0076-6879(05)95020-9
Jiang, Y., He, Y., Fan, S. L., Yu, J. N., and Song, M. Z. (2011). The identification and analysis of RNA editing sites of 10 chloroplast protein-coding genes from virescent mutant of Gossypium hirsutum. Cotton Sci. 23, 3–9.
Kahlau, S., Aspinall, S., Gray, J. C., and Bock, R. (2006). Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J. Mol. Evol. 63, 194–207. doi: 10.1007/s00239-005-0254-5
Katoh, K., Kuma, K., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Kellogg, E. (2009). The evolutionary history of ehrhartoideae, Oryzeae, and Oryza. Rice 2, 1–14. doi: 10.1007/s12284-009-9022-2
Kessel, H. (1995). Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 251, 614–628. doi: 10.1006/jmbi.1995.0460
Leseberg, C. H., and Duvall, M. R. (2009). The complete chloroplast genome of Coix lachyrma-jobi and a comparative molecular evolutionary analysis of plastomes in cereals. J. Mol. Evol. 69, 311–318. doi: 10.1007/s00239-009-9275-9
Li, Q., and Wan, J. M. (2005). SSRHunter: development of a local searching software for SSR sites. Yi Chuan 27, 808–810.
Li, R., Zhu, H., and Ruan, J. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Liu, Q. P., and Xue, Q. Z. (2004). Codon usage in the chloroplast genome of rice (Oryza sativa L. ssp. japonica). Acta Agron. Sin. 30, 1220–1224.
Lloyd, D. G., and Calder, V. L. (1991). Multi-residue gaps, a class of molecular characters with exceptional reliability for phylogenetic analyses. J. Evol. Biol. 4, 9–21. doi: 10.1046/j.1420-9101.1991.4010009.x
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Mason-Gamer, R. J., Weil, C. F., and Kellogg, E. A. (1998). Granule-Bound starch synthase: structure, function, and phylogenetic utility. Mol. Biol. Evol. 15, 1658–1673. doi: 10.1093/oxfordjournals.molbev.a025893
Mathews, S., Tsai, R. C., and Kellogg, E. A. (2000). Phylogenetic structure in the grass family (Poaceae), evidence from the nuclear gene Phytochrome, B. Am. J. Bot. 87, 96–107. doi: 10.2307/2656688
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Morton, B. R. (2003). The role of context dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 56, 616–629. doi: 10.1007/s00239-002-2430-1
Mower, J. P. (2009). The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucl. Acids Res. 37, W253–W259. doi: 10.1093/nar/gkp337
Oelke, E. A. (1993). “Wild rice: domestication of a native North American genus,” in New Crops, eds J. Janick and J. E. Simon (New York, NY: Wiley), 235–243.
Ohyama, K., Fukuzawa, H., Kohchi, T., Shirai, H., Sano, T., Sano, S., and at al. (1986). Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322, 572–574. doi: 10.1038/322572a0
Palmer, J. D., Nugent, J. M., and Herbon, L. A. (1987). Unusual structure of geranium chloroplast DNA: a triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc. Natl. Acad. Sci. U.S.A. 84, 769–773. doi: 10.1073/pnas.84.3.769
Pearce, J. M. (2006). Minding the gap: frequency of indels in mtDNA control region sequence data and influence on population genetic analyses. Mol. Ecol. 15, 333–341 doi: 10.1111/j.1365-294x.2005.02781.x
Peng, Z. H., Lu, T. T., Li, L. B., Liu, X. H., Gao, Z. M., Hu, T., et al. (2010). Genome-wide characterization of the biggest grass, bamboo, based on 10,608 putative full-length cDNA sequences. BMC Plant Biol. 10:116. doi: 10.1186/1471-2229-10-116
Powell, W., Morgante, M., Andre, C., Hanafey, M., Vogel, J., Tingey, S., et al. (1996). The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 2, 225–238. doi: 10.1007/BF00564200
Provan, J., Corbett, G., Waugh, R., McNicol, J. W., Morgante, M., and Powell, W. (1996). DNA fingerprints of rice (Oryza sativa) obtained from hypervariable simple sequence repeats. Proc. R. Soc. Lond. Ser. B 263, 1275–1281. doi: 10.1098/rspb.1996.0187
Raubeson, L. A., and Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Diversity and Evolution of Plants; Genotypic and Phenotypic Variation In Higher Plants, ed R. Henry (London: CABI Publishing), 45–68.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Fourcade, H. M., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Sharp, P. M., and Li, W. H. (1987). The codon adaptation index-A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.3.1281
Sharp, P. M., Tuohy, T. M. F., and Mosurski, K. R. (1986). Codon usage in yeast cluster-analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14, 5125–5143. doi: 10.1093/nar/14.13.5125
Shendure, J., and Ji, H. (2008). Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145. doi: 10.1038/nbt1486
Shi, C., Hu, N., Huang, H., Gao, J., Zhao, Y. J., and Gao, L. Z. (2012). An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE 7:e31468. doi: 10.1371/journal.pone.0031468
Shimada, H., and Sugiura, M. (1991). Fine structural features of the chloroplast genome: comparison of the sequenced chloroplast genomes. Nucleic Acids Res. 19, 983–995.
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049. doi: 10.1007/bf02669253
Soltis, D. E., Albert, V. A., Savolainen, V., Hilu, K., Qiu, Y. L., Chase, M. W., et al. (2004). Genome-scale data, angiosperm relationships, and “ending incongruence”, a cautionary tale in phylogenetics. Trends Plant Sci. 9, 477–483. doi: 10.1016/j.tplants.2004.08.0008
Soreng, R. J., and Davis, J. I. (1998). Phylogenetics and character evolution in the grass family (Poaceae): simultaneous analysis of morphological and chloroplast DNA restriction site character. Bot. Rev. 64, 1–85. doi: 10.1007/BF02868851
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sungkaew, S., Stapleton, C. M. A., Salamin, N., and Hodkinson, T. R. (2009). Non-monophyly of the woody bamboos (Bambusinae; Poaceae), a multi-gene region phylogenetic analysis of Bambusoideae s. s. J. Plant Res. 122, 95–108. doi: 10.1007/s10265-008-0192-6
Tang, L., Zou, X. H., Achoundong, G., Potgieter, C., Second, G., Zhang, D. Y., et al. (2010). Phylogeny and biogeography of the rice tribe (Oryzeae): evidence from combined analysis of 20 chloroplast fragments. Mol. Phylogenet. 54, 266–277. doi: 10.1016/j.ympev.2009.08.007
Terrell, E. E., Peterson, P. M., Reveal, J. L., and Duvall, M. R. (1997). Taxonomy of North American species of Zizania (Poaceae). SIDA 17, 533–549.
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). Improved sensitivity of profile searches through the use of sequence weights and gap excision. CABIOS 10, 19–29. doi: 10.1093/bioinformatics/10.1.19
Thrower, L. B., and Chan, Y. S. (1980). Gansun: a cultivated host-parasite combination from China. Econ. Bot. 34, 20–26. doi: 10.1007/BF02859552
Timme, R. E., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2007). A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am. J. Bot. 94, 302–312. doi: 10.3732/ajb.94.3.302
Vicentini, A., Barber, J. C., Aliscioni, A. A., Giussani, L. M., and Kellogg, E. A. (2008). The age of the grasses and clusters of origins of C4 photosynthesis. Global Change Biol. 14, 2693–2977. doi: 10.1111/j.1365-2486.2008.01688.x
Wang, S., Shi, C., and Gao, L. Z. (2013). Plastid genome sequence of a wild woody oil species, Prinsepia utilis, provides insights into evolutionary and mutational patterns of Rosaceae chloroplast genomes. PLoS ONE 8:e73946. doi: 10.1371/journal.pone.0073946
Wong, G. K., Wang, J., Tao, L., Tan, J., Zhang, J., Passey, D. A., et al. (2002). Compositional gradients in Gramineae genes. Genome Res. 12, 851–856. doi: 10.1101/gr.189102
Wright, F. (1990). The effective number of codons used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Wu, Z. Q., and Ge, S. (2012). The phylogeny of the BEP clade in grasses revisited: evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet. Evol. 62, 573–578. doi: 10.1016/j.ympev.2011.10.019
Wu, Z. Y., Raven, P. H., and Hong, D. Y. (2006). Floral of China: Poaceae. Beijing; St. Louis: Science Press; Missouri Botanical Garden Press.
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xu, Q., Xiong, G., Li, P., He, F., Huang, Y., Wang, K., et al. (2012). Analysis of complete nucleotide sequences of 12 Gossypium chloroplast genomes: origin and evolution of allotetraploids. PLoS ONE 7:e37128. doi: 10.1371/journal.pone.0037128
Yang, M., Zhang, X., Liu, G., Yin, Y., Chen, K., Yun, Q., et al. (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 5:e12762. doi: 10.1371/journal.pone.0012762
Zhai, C. K., Lu, C. M., Zhang, X. Q., Sun, G. J., and Lorenz, K. J. (2001). Comparative study on nutritional value of Chinese and North American wild rice. J. Food Comp. Anal. 14, 371–382. doi: 10.1006/jfca.2000.0979
Zhang, S. H., and Second, G. (1989). Phylogenetic analysis of the tribe Oryzeae: total chloroplast DNA restriction fragment analysis (a preliminary report). Rice Genet. Newsl. 6, 76–80.
Zhang, W. (2000). Phylogeny of the grass family (Poaceae) from rpl16 intron sequence data. Mol. Phylogenet. Evol. 15, 135–146. doi: 10.1006/mpev.1999.0729
Zhang, Y. J., Ma, P. F., and Li, D. Z. (2011). High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 6:e20596. doi: 10.1371/journal.pone.0020596
Zhou, M., Long, W., and Li, X. (2008a). Analysis of synonymous codon usage in chloroplast genome in Populus alba. J. Forest. Res. 19, 293–297. doi: 10.1007/s11676-008-0052-1
Keywords: chloroplast genome, comparative chloroplast genomics, grass evolution, Oryzeae, wild rice, Zizania latifolia
Citation: Zhang D, Li K, Gao J, Liu Y and Gao L-Z (2016) The Complete Plastid Genome Sequence of the Wild Rice Zizania latifolia and Comparative Chloroplast Genomics of the Rice Tribe Oryzeae, Poaceae. Front. Ecol. Evol. 4:88. doi: 10.3389/fevo.2016.00088
Received: 05 September 2015; Accepted: 14 July 2016;
Published: 03 August 2016.
Edited by:
Debashish Bhattacharya, Rutgers University, USAReviewed by:
Khidir W. Hilu, Virginia Polytechnic Institute and State University, USAFerhat Celep, Gazi University, Turkey
Copyright © 2016 Zhang, Li, Gao, Liu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Zhi Gao, bGdhb0BtYWlsLmtpYi5hYy5jbg==